Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées
Institut de chimie des milieux et matériaux de Poitiers - IC2MP (Diplôme National - Arrêté du 7 août 2006)
École doctorale : Sciences pour l'environnement - Gay Lussac (La Rochelle) Secteur de recherche : Chimie organique, minérale et industrielle
Présentée par :
Vanessa Melang Me Nze
Préparation de nouveaux matériaux pour l'élimination
catalytique des composés organiques volatils
Directeur(s) de Thèse : Jacques Barbier, Céline Fontaine Soutenue le 22 juin 2016 devant le jury
Jury :
Président Rachid Brahmi Professeur, Université Chouaïb Doukkali, El Jadida, Maroc Rapporteur Rachid Brahmi Professeur, Université Chouaïb Doukkali, El Jadida, Maroc Rapporteur Pascal Granger Professeur des Universités, Université de Lille 1
Membre Jacques Barbier Professeur des Universités, Université de Poitiers Membre Céline Fontaine Maître de conférences, Université de Poitiers
Pour citer cette thèse :
Vanessa Melang Me Nze. Préparation de nouveaux matériaux pour l'élimination catalytique des composés
organiques volatils [En ligne]. Thèse Chimie organique, minérale et industrielle. Poitiers : Université de Poitiers,
THESE
Pour l’obtention du Grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS
(Faculté des Sciences Fondamentales et Appliquées)
(Diplôme National - Arrêté du 7 août 2006)
Ecole Doctorale : Sciences pour l'environnement-Gay Lussac (Poitiers)
Secteur de Recherche : Chimie organique, minérale et industrielle
Présentée par :
Vanessa MELANG ME NZE
************************
PREPARATION DE NOUVEAUX MATERIAUX POUR L’ELIMINATION
CATALYTIQUE DES COMPOSES ORGANIQUES VOLATILS
************************
Directeur de thèse : Jacques Barbier
Co-directrice de thèse : Céline Fontaine
************************
Soutenue le 22 juin 2016
devant la Commission d’Examen
************************
JURY
Rapporteurs : Rachid Brahmi Professeur des Universités, LCCM, Université Chouaïb
Doukkali, El Jadida, Maroc
Pascal Granger Professeur des Universités, UCCS, Université de Lille 1
Examinateurs : Jacques Barbier Jr. Professeur des Universités, IC2MP, Université de Poitiers
Ce travail a été réalisé au sein de l’équipe du Site Actif au Matériau Catalytique (SAMCat) de l’Institut de Chimie des Milieux et Matériaux de Poitiers (IC2MP).
Je tiens à exprimer ma profonde gratitude à Monsieur Boniface KOKOH et Madame Florence EPRON, pour m'avoir accueilli dans leur équipe au début de cette thèse. J'associe ces remerciements à Mme Catherine ESPECEL, co-responsable actuelle de l'équipe, qui a contribuée de la même manière à mon
épanouissement dans l’équipe.
Je tiens à remercier mon directeur de thèse Monsieur Jacques BARBIER Jr. pour m'avoir fait partager son expérience scientifique tout au long de ce travail. Ces conseils et son soutien moral m’ont permis de surmonter certaines difficultés. Je tiens à exprimer ma profonde reconnaissance à ma co-directrice de thèse Madame Céline FONTAINE car sa disponibilité, son soutien moral, ses conseils scientifiques ainsi que sa sympathie et sa bonne humeur ont contribué au bon déroulement de cette thèse.
Je tiens à remercier l'Agence Nationale des Bourses du Gabon pour leur soutien financier tout au long de cette thèse.
Je veux exprimer ma reconnaissance à Messieurs Rachid BRAHMI et Pascal GRANGER pour avoir accepté de juger ce travail et d'en être rapporteurs.
J’aimerais aussi remercier toutes les personnes qui ont pris le temps de me faire bénéficier de leurs
compétences scientifiques et techniques au cours de ma thèse notamment Messieurs Stéphane PRONIER, Jean-Dominique COMPAROT et mesdames Sandrine ARII-CLACENS et Laurence
VIVIER. J’exprime également toute ma sympathie à tous les membres de l'équipe SAMCat.
Je garde une pensée profonde pour mes nouveaux et anciens collègues doctorants ainsi que mes ami(e)s : Henry-Joël, Davina, Mickaël, Nathaelle, Mélissandre, Alejandra, Nelly, Marie-Laure, Mathias, Fabien, Céline, Samuel, Modibo, Hilma, Maria, Yasuyuki, Halima, Asma et Fatima.
Je tiens à exprimer toute mon affection pour mon compagnon Jean-Sébastien avec qui j'ai partagé mes réussites et mes angoisses tout au long de ce travail.
Pour conclure, je dédie mon manuscrit à ma famille et la remercie infiniment pour son soutien durant
Introduction générale ... 1
Chapitre I : Partie bibliographique ... 7
I. Etat de l’art sur les composés organiques volatils (COV) ... 9
I.1.Définitions et classifications des COV ... 9
I.1.1.Composé organique ... 9
I.1.2.Composé organique volatils (COV) ... 9
I.1.3.Classifications ... 11
I.2.Emissions de COV ... 13
I.β.1.Sources d’émissions ... 13
I.2.2.Quantification des émissions ... 15
I.3.Impacts environnementaux ... 16
I.3.1.Impacts directs ... 16
I.3.2.Impacts indirects ... 17
I.4.Méthodes de traitement des COV ... 18
I.4.1.Techniques de récupération ... 18
I.4.2.Techniques destructives ... 19
I.4.3.Techniques émergeantes ... 20
II. Matériaux catalytiques utilisés pour l’oxydation des COV ... 21
II.1.Morphologie ... 21
II.2.Oxydes métalliques basiques ... 22
II.2.1.Mg1-xAlx(O) ... 24
II.2.2.CeO2 ... 35
III. Elimination catalytique de l'acide acétique considéré comme COV ... 39
III.1.L'acide acétique ... 39
III.2.Décomposition ... 40
III.3.Oxydation voie humide catalysée (OVHC) ... 41
III.4.Oxydation dans l’air humide ... 42
III.4.1.Facteurs influençant l'activité catalytique ... 42
III.4.2.Etudes préliminaires Pt/oxydes ... 45
III.4.3.Choix du catalyseur ... 46
Chapitre II : Partie expérimentale ... 53
I. Préparations des catalyseurs ... 55
I.1.Synthèse par co-précipitation ... 55
I.2.Synthèse par voie sol-gel ... 55
I.3.Imprégnation voie humide ... 56
I.4.Autres synthèses... 56
II.1.Quantification du cérium par analyse ICP-OES ... 57
II.2.Mesure de la surface spécifique par la méthode BET (Brunauer-Emmet-Teller) ... 58
II.3.Diffraction des rayons X (DRX) sur poudre ... 59
II.4.Mesure des Capacités de Stockage en Oxygène (CSO et CMSO) ... 61
II.5.Caractérisations acido-basiques des catalyseurs ... 66
II.5.1.Mesure de la basicité par chimisorption du CO2 ... 66
II.5.2.Thermo-désorption du CO2 suivie par infrarouge ... 67
II.5.3.Thermo-désorption de la pyridine suivie par infrarouge ... 69
II.6.Microscopie Electronique à Transmission (MET) ... 70
II.7.Réduction en température programmée (RTP) ... 71
III. Mesure des performances catalytiques des catalyseurs ... 72
III.1.Configuration du réacteur ... 72
III.2.Descriptif du montage réactionnel ... 73
III.3.Calculs des débits ... 76
III.4.Tests catalytiques ... 78
III.4.1.Protocole expérimental ... 78
III.4.2.Etude préliminaire des limitations diffusionnelles internes et externes ... 80
III.4.2.1.Etude préliminaire des limitations diffusionnelles internes ... 80
III.4.2.2.Etude préliminaire des limitations diffusionnelles externes ... 81
Chapitre III : Préparations et caractérisations des catalyseurs ... 85
I. Préparations des catalyseurs ... 87
I.1.Synthèse des catalyseurs ... 87
I.β.Formation de l’hydrotalcite ... 88
II. Caractérisations des catalyseurs ... 89
II.1.Mesure de la surface spécifique ... 89
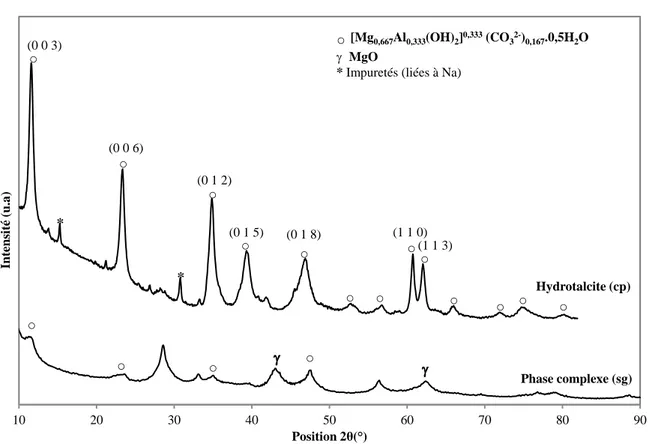
II.2.Identification des phases cristallines par DRX ... 91
II.3.Capacité de stockage de l'oxygène des catalyseurs ... 96
II.4.Influence de la méthode de préparation sur les catalyseurs MgAl ... 98
II.5.Influence de la méthode de préparation sur la dispersion de la cérine ... 100
II.6.Morphologies des catalyseurs : mise en évidence du recouvrement des surfaces ... 102
II.7.Etude de la réductibilité des catalyseurs ... 104
II.8.Propriétés acido-basique des catalyseurs ... 107
II.8.1.Mesure de la basicité par chimisorption du CO2 ... 107
II.8.2.Caractérisation des sites basiques par adsorption du CO2 suivie par infrarouge ... 108
II.8.3.Identification de l'acidité des solides par adsorption de la pyridine suivie par IR.... 112
III. Conclusion ... 114
I. Etudes préliminaires ... 119
I.1. Etude des limitations diffusionnelles externes et internes ... 119
I.2. Décomposition de l’acide acétique en réacteur vide ... 122
I.γ. Oxydation de l’acide acétique en réacteur vide ... 125
I.4. Oxydation de l’acide acétique en présence de cordiérite ... 11288
I.5. Conditions optimales ... 130
II. Etude de la stabilité du catalyseur MgAl ... 130
II.1. Reproductibilité des expériences et stabilité du matériau MgAl_cp ... 130
II.2. Influence des phénomènes d'adsorption ... 133
II.3. Etude de la stabilité en isothermes... 135
III. Influence de la basicité des catalyseurs ... 136
III.1. Influence de la basicité de l’alumine (Al2O3) ... 136
III.2. Utilisation de la cérine (CeO2) ... 139
III.γ. Influence de l’acido-basicité de l’oxyde mixte MgAl ... 141
III.4. Comparaison des activités des catalyseurs ... 143
IV. Influence de l’ajout de la cérine sur les matériaux catalytiques MgAlCey ... 144
IV.1 Echantillons préparés par co-précipitation ... 144
IV.2. Echantillons préparés par voie sol-gel ... 147
V. Evolutions des échantillons après test catalytique ... 149
VI. Conclusion ... 151
Conclusion générale ... 155
1
3
L’air et le climat ont été, du γ0 Novembre au 1β Décembre β01η à Paris, au centre des
discussions de 195 pays du monde pendant la conférence des Nations Unies sur les changements climatiques dite conférence des parties (COP-21). Au cours de ce rendez-vous majeur, un nouvel accord international a été conclu afin de maintenir le réchauffement climatique en dessous de 2°C. Cependant,
il est difficile de ne pas lier la pollution de l’air au changement climatique puisque ces problématiques environnementales sont influencées, toutes les deux, par l’activité humaine. En effet, le changement
climatique est généré par l’augmentation des gaz à effet de serre (ozone, CO2…) tandis que la pollution de l’air, notamment la pollution photochimique, concerne, entre autres, l’augmentation de l’ozone troposphérique. Ce dernier apparait comme un polluant de l’air et du climat dont la formation est due à l’accroissement des composés organiques volatils (formaldéhyde, acide acétique, éthanol…) dans l’atmosphère.
Les Composés Organiques Volatils (COV) sont des composés toxiques et/ou précurseurs
d’ozone, qui sont libérés dans l’atmosphère à température ambiante et contribuent de cette manière à la pollution de l’air [1],[2]. Réduire leurs émissions dans l’atmosphère constitue une préoccupation
environnementale et un réel enjeu de santé publique étant donné leurs effets néfastes (difficultés
respiratoires…) sur l’Homme. D’ailleurs, la signature des protocoles de Montréal (1987), Kyoto (1997)
ou de Göteborg (1999), témoigne de la prise de conscience des différents pays signataires tels que la France dans le but de limiter les émissions polluantes dans l’air [3],[4]. La principale source d’émission
de ces composés est l’activité humaine à travers l’utilisation de produits ménagers dont les constituants sont volatils (éthanol, acide acétique, acétone…). Le secteur des transports y contribue également de
manière équivalente via le développement du parc automobile au cours des deux dernières décennies.
Plusieurs procédés ont été mis en place pour limiter les émissions de COV dans l’atmosphère (biofiltres, oxydation thermique ou catalytique…) [2]. Parmi ces méthodes, l’oxydation catalytique reste une technique de choix pour l’élimination, à faibles concentrations, des COV. En effet, la dépense énergétique est diminuée par l’abaissement des températures de réaction tandis que la formation des NOx, laquelle est générée par la combustion des polluants, n’intervient pas aux basses températures. Toutefois, l’efficacité de cette méthode dépend essentiellement de catalyseurs efficaces dont les
propriétés physico-chimiques concourent à l’amélioration de l’activité catalytique. Parmi celles-ci on peut noter une surface spécifique élevée, une bonne stabilité thermique ainsi que de bonnes propriétés oxydo-réductrices et/ou acido-basiques. Plusieurs études ont été réalisées sur l’oxydation catalytique des composés organiques [5]–[8]. L'élimination de polluants tels que le toluène, le propène ou encore le butanol a déjà été discutée dans la littérature [5],[9],[10]. Cependant, les recherches concernant l’acide
acétique en font rarement référence comme COV, bien qu’il soit nocif pour la santé humaine [11]. En effet, c’est une substance dangereuse, très corrosive, irritante et peu dégradable. Elle est éventuellement
présente dans l'atmosphère via les végétaux dont elle constitue la sève [12], à travers les émissions naturelles issues de l'Homme telles que les urines, la sueur et le lait ou encore à partir des gaz
4
d'échappement des véhicules et/ou de la combustion des plastiques ou d'autres déchets [12]. Toutefois,
l’acide acétique est capable de se décomposer en sous-produits nocifs pour l’Homme et l’environnement
[13]. Les études préliminaires effectuées en oxydation voie humide catalysée (OVHC) mettent en
évidence son caractère réfractaire à l’oxydation d’un grand nombre de composés [14]–[17]. Au sein du
laboratoire IC2MP, une étude effectuée par Lafaye et al. [18] a montré d’ailleurs que l’OVHC de l’acide acétique est limitée en présence de catalyseurs basiques forts et réducteurs. En effet, la basicité contribue à la formation de carbonates tandis que la réductibilité, due à une capacité de stockage de l’oxygène élevée, entraîne la désactivation des catalyseurs. Toutefois, l’acidité (Lewis) des matériaux semblerait avoir un effet positif sur la réaction puisqu’elle augmente la sélectivité en CO2. L’oxydation voie humide catalysée (OVHC) est une technique d’élimination de polluants industriels en phase aqueuse. Elle consiste à oxyder les effluents gazeux (phénol, acide acétique…) à pression et température basses. L’OVHC de l’acide acétique fait l’objet de plusieurs études dans la communauté scientifique. En effet, sous sa forme gazeuse, l’acide acétique est connu comme étant un intermédiaire réactionnel des réactions d’oxydation des composés organiques telles que l’oxydation du phénol [14]. Lors de l’OVHC de l’acide acétique, la molécule présente en phase aqueuse peut facilement, puisque volatile, se retrouver dans la phase gazeuse. De ce fait, l’étude réalisée par H-J Sedjame au sein du laboratoire [6], concernant l’oxydation catalytique de l’acide acétique, a permis d’étudier la réactivité de cette molécule en phase gaz. Contrairement à l’OVHC, il ressort que l’oxydation de l’acide acétique nécessite des catalyseurs basiques et réductibles bien qu’une étude approfondie sur l’influence de ces propriétés sur l’activité catalytique n’ait pas été réalisée.
Ainsi, l’objectif de ce travail est d’approfondir cette étude en utilisant des matériaux catalytiques
novateurs et peu onéreux afin que leurs propriétés acido-basiques et/ou redox aident à l’oxydation totale
de l’acide acétique en phase gaz. Les matériaux catalytiques choisis sont des oxydes mixtes synthétiques
à base de magnésium (Mg) et d'aluminium (Al), obtenus à partir de la synthèse de matériaux hydrotalcites, sur lesquels un dépôt de cérium a été réalisé afin d'améliorer leur réductibilité. En effet,
la littérature montre que selon le mode de préparation et/ou l’ajout d’un métal réducteur fort tel que le cérium, il est possible d’améliorer les performances des catalyseurs [6],[19]. De la même manière, les oxydes mixtes MgAl(O) obtenus après calcination d’hydrotalcites précurseurs possèdent une grande
surface spécifique capable d'améliorer l'activité des catalyseurs, une forte basicité susceptible de
favoriser l’adsorption de l’acide acétique et une résistance thermique élevée. Dans ce travail, une série
de catalyseurs MgAlCey (y : pourcentage molaire de cérium), préparés par co-précipitation et par voie
sol-gel puis caractérisés par différentes techniques ont été testés en oxydation de l’acide acétique à basse
température (T ≤ η00°C) afin de déterminer leurs performances catalytiques. Le premier chapitre de
cette étude sera une étude bibliographique présentant l’état de l’art des composés organiques volatils en
termes de contexte environnemental et de solutions d’abattements connues. Les propriétés des
5
préliminaire effectuée au laboratoire sur l’élimination catalytique de l’acide acétique. Au cours du deuxième chapitre, une présentation de l’ensemble des techniques utilisées pour caractériser les
matériaux synthétisés sera effectuée en même temps que celle du montage d’oxydation catalytique réservé exclusivement pour cette étude. Dans le troisième chapitre seront mises en évidence les
influences des méthodes de préparation et de l’ajout de cérium sur les propriétés acido-basiques et redox
des catalyseurs. Le quatrième chapitre sera consacré à l’étude des performances catalytiques des
catalyseurs au cours de la réaction d’oxydation de l’acide acétique.
Références
[1] A. Casset et F. de Blay, Rev. Mal. Respir., vol. 25, no 4, p. 475‑485, (2008).
[2] P. Le Cloirec, COV (composés organiques volatils). Techniques Ingénieur, (2004).
[3] Protocole de Kyoto, Ministère de l’Environnement, de l’Energie et de la Mer. Disponible sur: http://www.developpement-durable.gouv.fr/Le-Protocole-de-Kyoto,13782.html.
[4] Protocole de Göteborg, Office fédéral de l’environnement OFEV. Disponible sur: http://www.bafu.admin.ch/luft/13769/13785/13786/index.html?lang=fr.
[5] H.-J. Sedjame, G. Lafaye et J. Barbier Jr., Appl. Catal. B Environ., vol. 132‑133, p. 132‑141, (2013). [6] H.-J. Sedjame, C. Fontaine, G. Lafaye et J. Barbier Jr, Appl. Catal. B Environ., vol. 144, p. 233‑242,
(2014).
[7] C. Gennequin, S. Kouassi, L. Tidahy, R. Cousin, J.-F. Lamonier, G. Garcon, P. Shirali, F. Cazier, A. Aboukaïs et S. Siffert, Comptes Rendus Chim., vol. 13, no 5, p. 494‑501, (2010).
[8] R. Mrad, R. Cousin, N. A. Saliba, L. Tidahy et S. Siffert, Comptes Rendus Chim., vol. 18, no 3, p.
351‑357, (2015).
[9] C. Gennequin, R. Cousin, J.-F. Lamonier, S. Siffert et A. Aboukaïs, Catal. Commun., vol. 9, no 7,
p. 1639‑1643, (2008).
[10] S. Gil, J. Garcia-Vargas, L. Liotta, G. Pantaleo, M. Ousmane, L. Retailleau et A. Giroir-Fendler, Catalysts, vol. 5, no 2, p. 671‑689, (2015).
[11] Institut national de recherche et de sécurité (INRS), Fiche toxicologique Acide acétique. Disponible sur : http://www.inrs.fr/publications/bdd/fichetox/fiche.html?refINRS=FICHETOX_24.
[12] L.-J. Thénard, Traité de chimie élémentaire, théorique et pratique, Crochard, (1835).
[13] G. Bozzano, M. Dente et E. Ranzi, XXXVI Meeting of the Italian Section of the Combustion Institute, (2013).
[14] A. E. de los Monteros, G. Lafaye, A. Cervantes, G. Del Angel, J. Barbier Jr. et G. Torres, Catal. Today, vol. 258, Part 2, p. 564‑569, (2015).
[15] D. Duprez, F. Delanoë, J. Barbier Jr, P. Isnard et G. Blanchard, Catal. Today, vol. 29, no 1‑4, p.
317‑322, (1996).
[16] J. Barbier Jr., F. Delanoë, F. Jabouille, D. Duprez, G. Blanchard et P. Isnard, J. Catal., vol. 177, no
2, p. 378‑385, (1998).
[17] P. Gallezot, S. Chaumet, A. Perrard et P. Isnard, J. Catal., vol. 168, no 1, p. 104‑109, (1997).
[18] G. Lafaye, J. Barbier Jr. et D. Duprez, Catal. Today, vol. 253, p. 89‑98, (2015). [19] K. J. František Kovanda, Catal. Today, vol. 176, no 1, p. 110–115, (2011).
7
9
Les composés organiques volatils (COV) sont des polluants néfastes pour la santé et l'environnement. Réduire leurs émissions dans l’atmosphère constitue une préoccupation environnementale et un réel enjeu de santé publique. Dans cette partie, l'état de l'art est présenté sur les COV et leurs différentes voies de traitements ou d'élimination.
I.
Etat de l’art sur les Composés Organiques Volatils (COV)
I.1. Définitions et classifications des COV I.1.1.Composé organique
Pour définir ce qu'est un composé organique volatil, il est préférable de comprendre au préalable la définition d'un composé organique. En effet, un composé organique est un composé chimique dont sa molécule possède un atome de carbone lié directement à un atome d'hydrogène. Cet hydrogène peut être substitué par d'autres atomes tels que l'oxygène, l'azote, le soufre, le phosphore, le silicium ou encore des halogènes (chlore, fluor, brome...) à l'origine des différentes familles de composés organiques volatils.
I.1.2.Composé organique volatil (COV)
Un composé organique volatil peut être défini selon ses différentes caractéristiques physico-chimiques. Ces critères peuvent varier en fonction du pays et/ou encore de la législation appliquée. En général, un COV est une substance chimique très volatile c'est-à-dire capable de s'évaporer à température ambiante et à la pression atmosphérique. Cependant, l'Europe restreint cette définition par une réglementation basée sur des propriétés physico-chimiques précises. En effet, la Directive Européenne IED (Industrial Emission Directive) a adopté, en 2010, la définition de COV suivante : un composé organique volatil est "un composé organique ayant une pression de vapeur de 0,01 kPa ou plus, à une température de 293,15 K (20°C) ou ayant une volatilité correspondante dans les conditions d'utilisation particulières" [1].
La volatilité des COV peut être définie en fonction de la température d'ébullition ou encore de la pression de vapeur saturante selon les conditions d'utilisation. La température d'ébullition, par exemple, correspond à la température du changement d'état du composé de la phase liquide à la phase gaz. Au-delà de cette température, la phase liquide est complètement évaporée et le composé n'existe que sous sa forme gazeuse. Bien que la volatilité des liquides ne soit pas strictement proportionnelle à la température d'ébullition, il apparait clairement qu'un liquide à point d'ébullition bas s'évapore plus rapidement que celui dont le point d'ébullition est plus élevé. Il est donc possible de déterminer la
volatilité d’un composé à partir de de sa température d'ébullition. Dès lors, un composé organique est
10
d’ébullition renvoie à la tension de vapeur saturante du composé. En effet, à une température donnée, la
pression de la phase gazeuse du composé est en équilibre au-dessus de la phase liquide. Cette pression correspond à la pression de vapeur saturante du composé et caractérise sa volatilité. Il semblerait donc que le composé est d'autant plus volatil que sa pression de vapeur saturante est élevée. Une définition hétérogène à la précédente, inscrite dans la directive européenne 2004/42/EC [2] et la norme ISO 16000-6 [3], définit un COV en fonction de sa température d’ébullition. Il apparait dans la directive européenne
β004/4β/EC qu’un COV est "toute substance organique dont le point d'ébullition est inférieur à 250°C
à une pression atmosphérique standard de 101,3 kPa". Celle utilisée par L'OMS (Organisation Mondiale de la Santé), dans la norme ISO 16000-6, décrit qu'un composé organique est dit volatil lorsque sa température d'ébullition se situe entre 50°C-100°C et (240 °C-260°C). Cela correspond à une pression de vapeur saturante supérieure à 100 kPa (25°C).
Aux Etats-Unis et en Allemagne, la règlementation est plus restrictive et la définition est décrite en termes de substances volatiles [4]. En effet, la règlementation allemande définit un COV comme substance organique volatile "dont le point d'ébullition est inférieur à 200°C à une pression atmosphérique standard de 101,3 kPa". La règlementation californienne du SCAQMD (South Coast Air Quality Management District), quant à elle, considère comme COV toute substance volatile dont la température d'ébullition est inférieure à 280°C. Cette définition exclut certains composés tels que
l'acétone, l’éthane, l’acétate de méthyle..., dont la réactivité photochimique est faible.
Il existe également d’autres définitions de COV arbitraires lesquelles excluent des composés organiques volatils à cause de l’origine de leur source d’émission et leur influence sur l’environnement ou encore
de leur degré de volatilité. Parmi ces définitions, la littérature propose les composés organiques non méthaniques (COVNM), les composés organiques très volatils (COTV) ou encore les composés organiques semi volatils (COSV) [5],[6]. Pour les COVNM, le méthane est exclu de la famille des COV
car son influence sur l’environnement (effet de serre) est différente de celle des COV conventionnels dont l’effet néfaste est essentiellement axé sur la pollution photochimique. En général, les COV ont des températures d’ébullition comprises dans les intervalles η0°C < T < 100°C et β40°C < T < βθ0°C. Les COTV sont les composés organiques très volatils c’est-à-dire que leur température d’ébullition est
comprise dans les intervalles 0°C et 50°C. Les COSV sont des composés qui possèdent une faible volatilité. Ils sont dits composés organiques semi-volatils car leur température d’ébullition se situe dans les intervalles 240°C < T < 260°C et 380°C < T < 400°C.
11
I.1.3.ClassificationsUne liste exhaustive des différents COV est difficilement réalisable étant donnée la multiplicité des composés chimiques. Il est donc préférable de classifier les COV en familles chimiques. Ces diverses familles concernent [7] :
– Les hydrocarbures (alcanes, alcènes, alcynes, hydrocarbures aromatiques…) ;
– Les alcools (méthanol, éthanol, propanol…) ;
– Les aldéhydes (formaldéhyde, acétaldéhyde, benzaldéhyde…) ; – Les cétones (acétone, butanone, cyclohexanone…) ;
– Les acides carboxyliques (acide formique, acide acétique, acide butanoïque…) ; – Les esters (formiate de méthyle, acétate de méthyle, propanoate de méthyle) ;
– Les éthers (éther éthylique, éthylène glycol, propylène glycol ;
– Les dérivés chlorés (dichlorométhane…), nitrés (β-nitropropane…) et aminés (éthylamine…) ; – ...
Les COV sont des précurseurs photochimiques c’est-à-dire qu’ils réagissent sous l’effet d’un
rayonnement solaire. Cette réaction chimique contribue à la formation de l’ozone troposphérique (à
basse altitude) [5]. La réactivité des COV est différente d’un composé à un autre. Par exemple, les hydrocarbures sont des composés moins réactifs que certains composés aromatiques tels que le benzène.
La contribution à la formation d’ozone troposphérique été définie sous forme d’échelle relative correspondant au potentiel de création d’ozone photochimique (PCOP). En effet, cette grandeur permet
d'évaluer la participation d'un COV dans les réactions photochimiques responsables de l'augmentation de l'ozone dans l'atmosphère. Il s’exprime en kg d’équivalent éthylène dont le potentiel de réactivité PCOP est égal à 1. Cette référence est due au fait que les alcènes contribuent le plus à la formation de l'ozone. Sur le tableau I.1, sont répertoriés les PCOP de quelques COV [8]. Sur ce tableau, les aldéhydes et les aromatiques tels que l'acétaldéhyde et le toluène présentent des PCOP très élevés. Les autres familles chimiques composées d'acétylène, acétone, acide acétique et formique ont des PCOP faibles. L'acide acétique dont le PCOP est de 0,097 contribue faiblement à la production d'ozone à basse altitude contrairement au formaldéhyde lequel possède un PCOP de 0,519.
A partir des valeurs de PCOP, une deuxième classification a été réalisée sur les COV en fonction de leur
rôle dans la production d’ozone. Les COV ont été classés, dans le tableau I.β, par famille chimique en
fonction du rôle peu important, assez important et très important dans la production d’ozone [7]. Les COV peuvent également être classés en fonction de leur odeur. Il existe, en effet, des COV inodores
et d’autres qui possèdent une odeur plus ou moins caractéristique. Parmi les plus odorants, il y a les
amines, les composés soufrés, les dérivés oxygénés (aldéhyde et cétone) et les composés aromatiques. Le tableau I.3 répertorie quelques COV classés en fonction de leur famille chimique ainsi que de leur caractère odorant [5].
12
Tableau.I.1 :Potentiel de Création d’Ozone Photochimique (PCOP) de quelques COV [7] (Source : INERIS – DRC- 07 – 85842 – 12011A).
Tableau.I.2 : Classification des COV selon leur rôle dans la production d’ozone troposphérique [7]
Famille chimique COV PCOP
Acides organiques Acide acétique Acide formique 0,097 0,032 Alcynes Acétylène 0,085 Aromatiques Benzène Toluène 0,218 0,637 Aldéhydes Formaldéhyde Acétaldéhyde 0,519 0,641 Cétones Acétone 0,094 – – –
Rôle Famille COV
Très important Alcanes (méthane) Alcynes (acétylène) Aromatiques (benzène) Aldéhydes (benzaldéhyde) Cétones (acétone) Alcools (méthanol) Esters (acétate de méthyle)
Hydrocarbures chlorés (méthylchloroforme…) Assez important Alcène Aromatique Alcanes > C6 COV naturels (Isoprène)
Peu important
Alcanes Cétones Alcools (éthanol)
13
Tableau.I.3 : Classement de quelques COV en fonction de leur caractère odorant et leur pression de vapeur saturante [5],[9].
I.2. Emissions de COV I.2.1. Sources d’émissions
Les émissions de COV sont classées en deux catégories : les émissions primaires et les émissions
secondaires. Les émissions primaires correspondent aux COV libérés directement dans l’atmosphère par
évaporation de matériaux (solvants, plastifiants, biocides…). Les émissions secondaires des COV sont généralement issues des différents processus de transformation chimiques des matériaux (combustion,
décomposition…). Ces émissions proviennent de différents secteurs d’activité tels que le transport, l’industrie ou encore de sources biotiques comme la forêt, l’agriculture... La commission européenne a déterminé la formation de l'ozone troposphérique par secteur d’activité au cours des années 2000 et 2012
[10]. Les résultats obtenus sont représentés, sur la figure I.1, en pourcentage des émissions totales de tonnes d'équivalents COVNM ce qui correspond au pourcentage d’ozone produit par les émissions
totales d’une tonne de COVNM. En β000, les secteurs contribuant le plus à la formation de l'ozone à
basse altitude sont les ménages, la fabrication et dans une moindre mesure, le transport. Il a été observé en 2012, une augmentation de 9% des émissions d'ozone issues du secteur des transports au détriment des ménages. Tous les autres secteurs ont enregistré des variations relativement faibles. Il semblerait que cette évolution soit en rapport avec la croissance économique des pays et le développement du parc automobile en Europe [11].
Famille chimique Noms usuels
Pression de vapeur saturante
(Pa à 20 C) Apparence
Acides organiques Acide acétique Acide formique
1,57.103 4,4.103
Liquide incolore, odeur piquante Liquide incolore, odeur
piquante
Alcynes Acétylène 4,5.106 Gaz incolore, inodore
Aromatiques Benzène
Toluène
9,97.103 3,8.103
Liquide incolore, odeur aromatique Liquide incolore, odeur
caractéristique
Aldéhydes Formaldéhyde
Acétaldéhyde
4,4.105
1,01.105
Gaz incolore, odeur piquante Liquide incolore, odeur
fruitée
Cétones Acétone 4,0.105 Liquide incolore, odeur
14
Figure I.1 : Formation d'ozone troposphérique en Europe par secteur d’activité en β000 et β01β [10].
Les émissions des composés organiques volatils peuvent être d’origine anthropique ou naturelle. Les
émissions naturelles sont celles qui proviennent des forêts, des cultures et des prairies. Les COV émis
dans l’atmosphère sont essentiellement issus de la famille des hydrocarbures. Les plus abondants sont l’isoprène et les terpènes. Les émissions anthropiques de type industrielles concernent les secteurs de l’imprimerie, traitement de surface, pharmacie, ... Les COV émis sont issus des familles des esters
(acétate d’éthyle…), alcools (méthanol…) ou encore des aromatiques et des dérivés chlorés
(dichlorométhane). Il apparait dans la littérature que les secteurs de l’industrie manufacturière et du résidentiel/tertiaire contribuent fortement à l’émission des COV dans l’atmosphère [10],[12]. Les données statistiques évaluant la contribution de plusieurs secteurs d’activités ont été réalisées par l’Eurostat laquelle représente la direction générale de la Commission européenne chargée de
l'information statistique à l'échelle communautaire. En France plus particulièrement, les données statistiques ont été réalisées par le CITEPA (Centre Interprofessionnel Technique d'Etudes de la Pollution Atmosphérique). Elles recensent, comme le présente la figure I.2, la contribution de différents
secteurs d’activités dans les émissions de COV entre 1990 et β01β en France [12]. Sur cette figure, les
secteurs d'activités sont répartis selon plusieurs couleurs. En bleu ciel, est représenté le secteur transformation énergie. L'industrie manufacturière correspond à la couleur bleu marine. Les secteurs du résidentiel/tertiaire, de l'agriculture/sylviculture et du transport routier sont respectivement représentés en jaune, vert et rouge. Enfin, les autres transports correspondent à la couleur orange. De 1990 à 2012, les secteurs du résidentiel/tertiaire et de l'industrie manufacturière montrent une évolution croissante contrairement à celui du transport routier lequel diminue au fil des années.
Agriculture, forêt et pêche 10% Mines et carrières 1% Fabrication 19% Électricité, gaz, vapeur et air conditionné 10% Transport 24% Autres services, approvisionnement en eau et la construction 11% Ménages 25% 2012 Agriculture, forêt et pêche 12% Mines et carrières 2% Fabrication 20% Électricité, gaz, vapeur et air conditionné 8% Transport 15% Autres services, approvisionnement en eau et la construction 13% Ménages 30% 2000
15
Figure I.2 : contribution par secteur d’activité des émissions de COV en France entre 1990 et β01β [12]. (Source : CITEPA)
Les autres secteurs d'activité tels que l'agriculture/sylviculture et les autres transports restent stables au fil des années. L'évolution du secteur résidentiel/tertiaire est due à l'utilisation croissante des solvants à usage domestique (peintures, colles...). L'utilisation des peintures a, quant à elle, favorisé les émissions issues de l'industrie manufacturière en France.
I.2.2.Quantification des émissions Par secteur d’activité
Dans le cadre du protocole de Göteborg [13], 26 pays dont la France se sont engagés le 1er
décembre 1999 devant la Commission Economique pour l'Europe des Nations Unies (CEE-NU) dans une politique de réductions des émissions de COV. Afin de respecter les objectifs fixés, les quantifications des émissions de COV sont actualisées chaque année. Les règles de comptabilisation des émissions de COV ont été réalisées conformément aux recommandations de la CEE-NU et de la CCNUCC (Convention Cadre des Nations Unies pour les Changements Climatiques). Ces règles ont été généralisées en France et adoptées par le CITEPA lequel est en charge, au niveau national, de la
réalisation des inventaires d’émissions à la demande du Ministère en charge de l’écologie. En France, conformément à l’arrêté du β4 août β011 et à l’aide du SNIEBA (Système National d’Inventaires d’Emissions et de Bilans dans l’Atmosphère), le CITEPA élabore les inventaires au format SECTEN
(Secteur Economique et Energie) c’est-à-dire sous forme d’image reformulée des données de base.
D’autres critères de mesure sont pris en compte notamment en termes d’émissions totales ou hors-total. En effet, les émissions de COV comptabilisées à l’échelle nationale sont dites " totales ". Celles appelées
16
" hors-total " exclues du total national sont les émissions provenant des secteurs d’activités tels que le secteur maritime (international), les trafics aériens (domestiques et internationaux) et les sources biotiques (agriculture et forêts). En 2011, le CITEPA a comptabilisé les émissions totales de COV de 7
secteurs d’activités (résidentiel/tertiaire, industrie manufacturière…) dont 4γ sous-secteurs [14]. Il en
ressort que pour tous secteurs confondus, 734 kt (kilotonnes) de COV ont été émis sur le plan national.
Par risque sanitaire
La quantification des émissions de COV permet de mesurer leur seuil de toxicité sur l’organisme.
Deux critères d’évaluations des limites de toxicité des COV ont été définis par des valeurs correspondant aux concentrations de COV dans l’atmosphère sans risques d’altération de la santé. Le premier critère est la Valeur Limite d’Exposition (VLE) laquelle correspond au seuil de concentration limite d’exposition d’un individu, durant 1η minutes, sans altération physiologique immédiate. Le deuxième
critère appelé La Valeur limite Moyenne d’Exposition (VME), correspond à la concentration limite
d’exposition d’un individu durant une période de 8h par jour et de 40h par semaine sans altérations à long terme sur l’organisme. Ces deux valeurs sont spécifiques à chaque COV et s’expriment en partie
par million (ppm) ou en milligramme par mètre cube (mg/m3). Par exemple, la VLE de l’acide acétique
est de 10 ppm soit 25 mg/m3 et sa VME est nulle dans la réglementation française. Aux Etats-Unis, la VME et la VLE de l’acide acétique sont respectivement de 10 ppm soit 25 mg/m3 et de 15 ppm soit 37
mg/m3.
I.3. Impacts environnementaux
Les émissions de COV dans l’atmosphère engendrent des impacts directs et indirects sur la santé humaine et l’environnement. Les impacts directs affectent immédiatement la santé tandis que les impacts indirects sont dus à la production de l’ozone dans des conditions particulières de température et de
rayonnement solaire.
I.3.1.Impacts directs
Les effets des COV sur l’Homme varient en fonction de la nature chimique de chaque polluant. Généralement, une gêne des voies respiratoires (nez, gorge) ainsi qu’une irritation des yeux et de la peau
sont constatés après une exposition longue. La gêne olfactive créée par certains composés organiques
volatils (benzène, formaldéhyde…) a des effets mutagènes et cancérigènes [15]. Il est également
constaté une dépression du système nerveux central traduit par des maux de têtes, nausées, vomissements...
Certains COV pourraient également nuire à la reproduction. De fortes concentrations de COV dans
l’organisme limiteraient le développement prénatal et postnatal. Les composés les plus préoccupants à
17
I.3.2.Impacts indirectsLes COV contribuent indirectement à la pollution atmosphérique. Lorsqu’ils sont libérés dans l’atmosphère, ils participent aux réactions photochimiques à l’origine de la formation de l’ozone (O3). L’ozone troposphérique généralement mis en cause a une altitude comprise entre 0 et 10 km. Il est vrai que l’ozone existe naturellement dans l’air à des concentrations comprises entre 0,005 et 0,05 ppm [16]. Cependant, à concentration élevée, la molécule provoque des lésions pulmonaires sévères (œdèmes pulmonaires…) à cause de son action irritante mais également des complications rénales,
neurologiques...
Les COV sont impliqués dans les réactions photochimiques en tant que précurseurs de composés
oxydants. Lorsqu’ils sont libérés dans l’atmosphère, ils se dégradent de manières à perturber les
équilibres chimiques dans l’air. En effet, l’ozone est naturellement formé dans l’air par recombinaison
d’un atome d’oxygène, issu de la photodissociation du dioxyde d’azote (NO2), avec l’oxygène
moléculaire O2. Lorsque ce cycle est perturbé par la présence de COV dans l’atmosphère, les mécanismes d’oxydation de ces composés, via le radical OH• et en présence des molécules d’azote (NOx), augmentent la production d’ozone. Le cycle de Chapman schématisé sur la figure I.γ résume les processus de formation de l’ozone [5].
Parallèlement à ce phénomène, la présence de composés organiques volatils dans l’atmosphère engendre d’une autre manière, un effet de serre au niveau de la troposphère en réfléchissant les rayons infrarouges
émis par le rayonnement solaire à la surface de la Terre.
Figure I.3 : Cycles de Chapman normal et perturbé schématisant la formation de l’ozone troposphérique [5].
18
I.4. Méthodes de traitement des COVLes COV sont traités par différents procédés dont le processus est soit destructif, soit de récupération. Certains traitements utilisés sont, pour la plupart, spécifique à l’élimination d’un COV ou
de familles de COV. D’autres, de manière plus globale, sont utilisés pour le traitement des odeurs. Parmi
les procédés de récupération, on retrouve : la condensation, l’absorption et l’adsorption [5]. Les procédés
destructifs sont essentiellement les procédés d’oxydation thermique, la voie biologique ou l’oxydation
catalytique [17].
I.4.1.Techniques de récupération La condensation
Le traitement par condensation est réservé aux fortes concentrations de COV. Le procédé privilégie les faibles débits (inférieurs à 1000 m3/h) afin de pouvoir récupérer les molécules sans
modification de la composition. La technique de traitement consiste à récupérer le COV sous forme
liquide en abaissant, à l’aide de la température, sa pression de vapeur saturante. On distingue deux types
de techniques :
– La condensation mécanique : limitée aux températures -30°C et -40°C et élaborée à l’aide de
compresseurs ou d’échangeurs
– La condensation cryogénique : effectuée à l’aide d’azote liquide car les températures peuvent atteindre -180°C.
L’avantage d’utiliser cette méthode réside dans la récupération quasi-totale du polluant. Par contre, un
inconvénient apparait au niveau du traitement des faibles débits de COV lesquels limitent les
performances d’analyse. L’absorption
La technique d’absorption des COV consiste en un transfert des molécules gazeuses vers un
liquide de lavage potentiellement réactif. La régénération du liquide se fait généralement par distillation
sous vide et permet la récupération du polluant. La solution utilisée est soit de l’eau, soit une solution
aqueuse acide, basique ou oxydante.
Il est à noter que l’utilisation de cette méthode est relativement simple et rapide. Cependant, les limites de ce procédé sont liées à l’usage de solutions adaptées au COV et au risque de transfert de pollution
par régénération du liquide.
L’adsorption
La technique d’adsorption met en jeu deux étapes dans laquelle le COV est d’abord retenu sur un support sélectif (charbon actif…), puis désorbé sous l’effet d’une diminution de la pression (sous vide) ou d’une augmentation de la température (gaz neutre chaud…) pour être récupéré. Cette technique peut
19
être utilisée pour les COV difficilement traitables tels que les COV chlorés. Cependant, la régénération est indispensable afin de récupérer le COV, ce qui augmenterait le cout d’exploitation.
I.4.2.Techniques destructives
Oxydation thermique
L’oxydation thermique est une technique énergivore car elle consiste à bruler les molécules
polluantes aux températures supérieures à 750°C, afin de les transformer en dioxyde de carbone (CO2)
et en eau (H2O). Elle représente θ0% des traitements mis en place dans l’industrie [5]. Cependant, ce
procédé est limité par son incapacité à traiter les composés organiques volatils possédant un hétéroatome
(soufre, azote…) dans la mesure où l’oxydation favoriserait la formation de gaz toxiques tels le SO2, les
NOx, ou encore Cl2 gazeux...
Oxydation par voie biologique
L’oxydation des COV par traitement biologique représente actuellement η% des méthodes
utilisées pour leur élimination [5]. La technique consiste à utiliser des biofiltres de telle manière qu’au
contact des COV, les microorganismes fixés sur le support biologique (tourbes, copeaux de bois…) dégradent le polluant en présence d’oxygène. Les propriétés physico-chimiques du polluant à dégrader sont directement liées à l’efficacité d’un tel système. En effet, la solubilité du COV et sa biodégradabilité sont des caractéristiques non négligeables. Les COV capables d’être dégradés par cette méthode sont
des familles de composés oxygénés tels que des alcools, cétones, esters.... Les composés organiques
ayant un hétéroatome (chlore, soufre…) sont difficilement biodégradables. Oxydation catalytique
Le principe de la méthode est le même qu’en oxydation thermique. Les composés organiques
volatils sont transformés en molécules inorganiques CO2 et H2O. Cependant, l’oxydation catalytique est une technique de choix car elle nécessite moins d’énergie que l’oxydation thermique [18]. En effet, les conditions de mise en œuvre (débit, concentration, température...) sont adaptées aux traitements d'un
COV où d'un mélange de COV. Les réactions sont réalisées à basse température (T < 500°C) en présence
d’un catalyseur ce qui diminue l’apport énergétique d’une part, et limite, d’autre part, la formation d’oxydes d’azote NOx. Le catalyseur peut être utilisé sous forme de bille poreuse, de nids d’abeilles
monolithiques, de poudre... Il peut être à base de métaux précieux (platine, rhodium, palladium...) ou à base d'oxydes métalliques (cuivre, cobalt, nickel...). Les concentrations des polluants peuvent être très faibles ainsi que les débits de gaz. L'oxydation catalytique des COV est une technique destructive dont la réaction d'oxydation par l'oxygène correspond à l'équation I.1 [17].
CaHb + a+b4 Oβ → aCOβ +bβHβO Equation I.1 Où a et b sont respectivement le nombre de carbone et d'hydrogène de la molécule organique.
20
La vitesse de réaction dépend de la concentration du polluant et de l'oxygène ainsi que de la température. Le mélange polluant/oxygène (O2 en excès) doit être homogène et la température doit être suffisamment
élevée pour initier la réaction. La présence d'un catalyseur permet de diminuer l'énergie d'activation de
la réaction ce qui favorise l'activité catalytique à basse température. Malgré tout, l’inconvénient de ce système réside dans l’éventualité d’un empoisonnement du catalyseur (masquage de sites actifs, effets thermiques, perte de matière…) à l’origine de sa désactivation.
I.4.3.Techniques émergeantes
L’élimination des composés organiques volatils dans l’atmosphère est une problématique majeure
pour les laboratoires de recherche et développement (R&D) spécialisés dans l’environnement. A cet
effet, l’élaboration de nouvelles techniques de traitement à petite échelle présente un intérêt
technologique en termes de coût et de faisabilité en industrie. Parmi ces techniques, on retrouve : la
photocatalyse, les procédés à membranes tels que la perméation gazeuse, ou encore l’oxydation par
plasma froid.
Photocatalyse
La photocatalyse est une technique innovante qui consiste à oxyder le polluant sur un catalyseur semi-conducteur à l’aide d’un rayonnement photonique apporté par la lumière. Son principe s’apparente à de la catalyse hétérogène où le réactif est décomposé à la surface du catalyseur. Les catalyseurs utilisés
sont généralement des oxydes métalliques (oxyde de titane…) supportés sur des supports inertes (alumine, silice…). L’inconvénient de cette technique repose sur l’incapacité à maitriser les sous-produits d’oxydation ainsi que l’empoisonnement du catalyseur.
Plasma froid
L’oxydation des COV par plasma est une technique prometteuse en terme de gain énergétique. En effet, le plasma est formé à partir d’un gaz ionisé à faible énergie lequel est "froid" car sa température
reste proche de la température ambiante (25°C) [19]. La décomposition du COV est réalisée à pression atmosphérique. Les effets induits par le plasma fragilisent les liaisons C-H des COV jusqu’à la formation de composés inorganiques CO2 et H2O. La difficulté de cette technique est relative à sa faisabilité à
grande échelle et à la maitrise des sous-produits d’oxydation.
Perméation gazeuse
La perméation gazeuse est une technique de séparation et de purification des gaz [20]. C’est une
technique de récupération des constituants gazeux, développée et appliquée à l’échelle industrielle. Son
principe est essentiellement basé sur les vitesses de perméation à travers une membrane des composants
21
permettre le transfert à travers la membrane. Elle nécessite une faible consommation d’énergie mais les
puretés des produits ne sont pas toujours très élevées.
En définitive, les applications industrielles des différents traitements des composés organiques volatils dépendent fortement du débit de flux et de la concentration en COV à traiter. Ces caractéristiques
mettent en jeu la faisabilité économique et l’efficacité du procédé. Dans ce travail, c’est la technique d’oxydation catalytique qui a été choisie afin de traiter les composés organiques volatils.
II.
Matériaux catalytiques utilisés pour l’oxydation des COVII.1.Morphologie
L'utilisation d'un catalyseur doit être adaptée au procédé pour lequel il est utilisé. Contrairement à la catalyse homogène, dans laquelle les réactifs et le catalyseur sont sous forme identique (gazeuse ou liquide), en catalyse hétérogène, le catalyseur et les réactifs sont sous différentes phases. En effet, le catalyseur est, en général, sous une forme solide et les réactifs sont soit gazeux, soit liquide. Dans ce cas, la réaction catalytique suppose que les espèces réactives se transforment au contact de la surface du catalyseur afin de former des molécules complètement différentes de celles d'origine. Il semble évident que la surface du catalyseur, également appelée surface active, joue un rôle primordial dans l'efficacité de la réaction. Généralement, un catalyseur est d'autant plus efficace que sa surface active est grande. Cependant, la réactivité des molécules à la surface et les échanges d'espèces actives aux interfaces impliquent d'importantes propriétés physicochimiques des catalyseurs. Celles-ci sont obtenues lorsque le matériau catalytique a été finement préparé de manière à optimiser sa porosité et sa surface d'échange appelée surface spécifique. Toutefois, structurer un catalyseur suppose d'optimiser la densité des grains du matériau catalytique. Autrement dit, il s'agit d'améliorer la taille des grains, le volume poreux et la forme du catalyseur.
Dans un réacteur à lit fixe, le catalyseur peut éventuellement être sous formes de billes, anneaux, pastilles, extrudés ou concassés [21]. Leur forme et la taille des grains de catalyseur influent d'une certaine manière sur les pertes de charges. Il est important qu'elles soient suffisamment élevées pour améliorer la répartition du fluide réactionnel à la surface des catalyseurs. Parmi ces différentes formes, la concassée est celle qui assure au catalyseur une plus grande perte de charge. En effet, l'écoulement des gaz diffuse les réactifs sur la surface externe et interne du catalyseur grâce aux frottements du fluide gazeux au contact des catalyseurs. Les réactifs atteignent donc les sites actifs des catalyseurs sur lesquels ils sont transformés. Cependant, pour une réactivité homogène de la molécule en surface, il est important de structurer de la même manière tous les grains de catalyseur. C'est pourquoi, l'optimisation de la structure des catalyseurs est généralement réalisée à l'aide des études de limitations diffusionnelles intergranulaires (ou internes) et extragranulaires (ou externes) du catalyseur. Ces études permettent de déterminer si le flux de réactifs reste constant tout au long de la réaction car une simple modification de
22
la concentration des réactifs impliquerait un changement de la vitesse de réaction du catalyseur et par conséquent celle de son activité chimique potentielle. Le catalyseur est réalisé de manière à assurer un excellent contact entre mélange réactionnel et la surface mais également à éviter une distribution non homogène du fluide de réactifs dans lit catalytique. Deux paramètres sont donc pris en compte à savoir : la taille des grains de catalyseur (granulométrie) et le volume poreux. La granulométrie du catalyseur informe sur l'existence de limitations diffusionnelles internes tandis que la porosité du solide renseigne sur l'existence de limitations diffusionnelles externes. Lorsqu'un de ces paramètres n'est pas optimisé, l'efficacité de la réaction est diminuée.
II.2.Oxydes métalliques basiques
L’abattement des COV par oxydation catalytique base son efficacité sur les performances des catalyseurs (activité, sélectivité…). Le choix du catalyseur doit être adapté au COV étudié car le
rendement de la réaction dépend des propriétés physicochimiques des solides. Dans la littérature [22]– [25], les matériaux catalytiques utilisés pour l’oxydation des COV sont à base de :
– Métaux nobles (Pt, Pd…),
– Oxydes métalliques : simples (CeO2, Al2O3…) ou mixtes (Co6-xMgxAl2(O), CeO2
-ZrO2…),
– Zéolithes (CsX, NaX…)
– …
Les oxydes métalliques ou les zéolithes peuvent être utilisés seuls ou combinés aux métaux nobles. Ces derniers sont dénommés catalyseurs supportés puisque la phase active en métal est déposée sur les différents supports.
Les catalyseurs supportés à base de métaux nobles sont les plus onéreux. Leurs performances catalytiques sont les plus prometteuses car le site actif propre au métal possède une activité et une sélectivité élevée quelle que soit la réaction d'oxydation catalytique. Cependant, les coûts d'achats des métaux nobles sont élevés [22] ce qui ne facilite pas la faisabilité économique des procédés d'oxydation
catalytique des COV. Toutefois, l’utilisation de matériaux catalytiques peu chers reste une alternative
économique puisque de nombreuses études ont mis en évidence leur capacité à transformer certains COV avec des rendements tout aussi appréciables que ceux des catalyseurs supportés à base de métaux nobles [23],[24]. Les oxydes métalliques à base de métaux de transitions sont des matériaux peu chers aux propriétés physico-chimiques intéressantes.
L’oxydation catalytique des COV nécessite des catalyseurs susceptibles de constituer un dispositif
stable, actif et sélectif au cours de la réaction d’oxydation étant donnée la complexité de la dégradation des molécules [25],[26]. Certains matériaux tels que l’alumine (Al2O3), la cérine (CeO2) ou encore des
23
oxydes mixtes comme CeO2-ZrO2 ou Co6-xMgxAl2(O), ont déjà été étudiés au cours de l'oxydation de
certains composés organiques volatils (butanol, toluène...) [24],[27]–[29], pendant lequel leur activité
catalytique a été suffisamment élevée pour permettre l’abattement du COV étudié. Plusieurs recherches
[23]–[25], [30], ont montré également que les performances catalytiques de certains oxydes métalliques peuvent être améliorées par modification des propriétés physico-chimiques des catalyseurs. Au cours de
l’oxydation catalytique des acides organiques n-butyrique et i-valérique, Ali et al. [25] ont montré que l’activité de l'alumine (Al2O3) est améliorée en présence de cuivre. De la même manière, Sedjame et al.
[23] ont montré que le dopage du catalyseur supporté Pt/Al2O3 par la cérine (CeO2) a un effet bénéfique sur l’activité catalytique au cours de l’oxydation de l'acide acétique. Ce changement d'activité est dû à
la modification du caractère réductible du catalyseur par ajout d'un matériau très réducteur (CuO et CeO2). D’un autre point de vue, les études menées par Parida et Das [31] ainsi que Perera et al. [32] ont
montré que les propriétés acido-basiques des oxydes métalliques augmentent l'interaction des COV à la surface ce qui facilite leur dégradation. Cependant, cette aptitude catalytique peut être influencée par la méthode de préparation du catalyseur laquelle définit ses propriétés physico-chimiques. Il est donc
important d’adapter le choix du catalyseur ainsi que la méthode de préparation à la molécule étudiée. Dans le cas de la réaction d'oxydation de l’acide acétique, nous nous sommes intéressés aux propriétés
physicochimiques des oxydes métalliques CeO2 et Mg1-xAlx(O). Le choix de ces catalyseurs s'est fait
dans le but de constituer des matériaux réducteurs, facilitant l'interaction acido-basique à la surface et présentant les propriétés catalytiques suivantes :
– Une surface spécifique élevée ;
– Une basicité élevée ;
– Une stabilité élevée ;
24
II.2.1. Mg1-xAlx(O) Propriétés structurales
Un hydrotalcite est un hydroxycarbonate de magnésium et d’aluminium de la famille des hydroxydes doubles lamellaires. De manière plus courante, c’est une argile anionique naturelle de
formule générale Mg6Al2(OH)16(CO3).4H2O [33]. Sa structure est de type brucite (Mg(OH)2) laquelle
est composée d'un enchainement d'octaèdres dont les centres sont occupés par les ions Mg2+ et les sommets par des groupements hydroxy (‒OH). Ces groupements situés sur les arêtes des octaèdres de
la structure permettent, via les liaisons hydrogène, de former une chaine infinie de feuillets empilés [34]. La structure de ce matériau est représentée sur la figure 4.
Figure I.4 : Structure en feuillets d’un matériau de type hydrotalcite tel que Mg6Al2(OH)16(CO3).4H2O [34]
L’hydrotalcite synthétique possède la même structure en forme de feuillets laquelle donne accès
à une multitude de compositions. Les feuillets sont constitués de cations métalliques divalents et/ou
trivalents et l’espace interfeuillet est susceptible d’intercaler des anions (OH-, CO
32-…) de dimensions
variables afin de compenser la charge cationique (figure I.4). En effet, les cations Mg2+ de la structure
brucite sont plus ou moins substitués au cours de la synthèse par les cations Al3+ pour constituer des
feuillets chargés positivement. Cavani et al. [33] ont ainsi pu former différentes structures hydrotalcites à partir des cations présentés sur le tableau I.4.
Feuillets métalliques (M2+/M3+)
Anion intercalé (An-) Molécule d’eau H2O
-25
Tableau I.4 : Rayons ioniques de quelques cations ayant formé une structure hydrotalcite
Les anions intercalés dans la structure, tels que les carbonates CO32-, permettent de compenser cette charge positive en assurant ainsi un ensemble électriquement neutre. La formule simplifiée d’un
hydrotalcite synthétique est sous la forme suivante : [MII
1-xMIIIx(OH)2]x+(An-)x/n.mH2O Equation I.2
Où,
– MII correspond au métal divalent (Mg2+, Zn2+…), – MIII représente le métal trivalent (Al3+, Fe3+…),
– x détermine le ratio stœchiométrique MII / MIIId’après la relation x = MIII/(MIII + MII) – An-est l’anion intercalé et n la charge de l’anion
– m la proportion d’eau dans la structure relative à l’anion intercalé.
Elle est obtenue par la relation m = 1 - γ × (x ÷ β) lorsque l’anion intercalé est CO32-. Méthodes de préparations
Pour obtenir un oxyde mixte à partir d'un hydrotalcite, la composition stœchiométrique des anions et des cations du catalyseur doit généralement répondre aux conditions suivantes :
0,2 ≤ MIII/ [MIII +MII] ≤ 0,4
1/n ≤ An-/MIII≤ 1
L’oxyde mixte de type Mg/Al est un solide homogène constitué de deux phases simples MgO et Al2O3.
D'après la littérature, ce catalyseur peut être synthétisé via différentes méthodes telles que :
– La co-précipitation [35]
– L'échange d'anions [36]
– La voie sol-gel [35]
– la croissance hydrothermale [37]
– ... .
Parmi ces méthodes, la co-précipitation et la voie sol-gel sont les plus simples à mettre en œuvre et permettent de réaliser des quantités importantes de catalyseurs. Cependant, la méthode par
co-précipitation implique la formation d’un hydrotalcite [33]. La synthèse par voie sol-gel impose, préalablement à l’obtention du solide homogène, la formation d’un gel constitué des phases MgO et Cation métallique Al3+ Fe3+ Cr3+ Ni3+ Mn3+ Co2+ Mg2+ Cu2+ Zn2+ Ca2+
26
Al2O3. Ces deux méthodes de préparations ont été retenues pour notre étude afin de juger de leur
influence sur les propriétés de surface des catalyseurs.
En ce qui concerne la co-précipitation, Les solutions de réactifs préparées contiennent d'une part les cations métalliques (Mg2+, Al3+...) qui constituent la charge cationique de la structure disposée en
feuillets et d'autre part les anions de compensation (OH-, CO
32-...) intercalés dans la structure. La
technique utilisée impose la précipitation simultanée des cations. En effet, la précipitation est réalisée à l'aide d'une solution basique contenant les agents de précipitation (NaOH, Na2CO3...) dans laquelle est
ajouté le mélange réactif contenant les sels métalliques. Le pH du mélange est maintenu constant à 10 mais peut éventuellement atteindre des valeurs supérieures à 10 et la température peut varier entre 25°C
et 70°C. Le débit d'écoulement du mélange de réactifs est contrôlé de la même manière que l’agitation
du mélange.
Cette étape est susceptible d’influencer la cristallisation des matériaux. Cependant, plusieurs paramètres sont à contrôler afin d’obtenir un matériau bien défini (tableau I.η).
Tableau I.5 : Paramètres influençant la configuration structurale d'un hydrotalcite [33].
L’hydrotalcite contenant les atomes de magnésium et d’aluminium est la plus synthétisée dans la
littérature [31],[33],[35], car sa composition élémentaire est similaire à un hydrotalcite naturel. Les cations Mg2+ et Al3+ composent les feuillets de la structure tandis que l'anion de compensation intercalé
est le carbonate de formule CO32- (figure I.4) [34]. En effet, le cation Al3+ possède un rayon ionique
voisin de celui de Mg2+, ce qui lui donne la capacité de substituer des atomes de magnésium. Ce
phénomène permet à l'aluminium d'occuper les centres des octaèdres de la structure hydrotalcite. L'anion intercalé augmente la distance de l'espace interfeuillet lorsque celui-ci est suffisamment volumineux.
L'acido-basicité du composé hydrotalcite Mg6Al2(OH)16(CO3).4H2O dépend du ratio stœchiométrique Mg/Al. Ce rapport est définit par la valeur x qui indique le taux d'occupation des
cations Mg2+ en surface susceptibles de former l'oxyde de magnésium (MgO) par rapport aux cation
Al3+ capables de former l'alumine Al
2O3. La valeur x est comprise entre 0,2 x 0,33 pour un matériau
Paramètres controlés Impacts
Rapport MII/MIII Propriétés redox de surface
pH et vieillisement Cristallinité
Concentrations des réactifs anioniques Composition de l’espace interfeuillets Concentrations des réactifs cationiques Composition des feuillets
Température et agitation Mélange de cations Lavage, séchage Eliminations d’impuretés
![Figure I.1 : Formation d'ozone troposphérique en Europe par secteur d’activité en β000 et β01β [10]](https://thumb-eu.123doks.com/thumbv2/123doknet/7995239.267867/25.892.132.768.114.382/figure-formation-ozone-troposphérique-europe-secteur-activité-β.webp)
![Figure I.4 : Structure en feuillets d’un matériau de type hydrotalcite tel que Mg 6 Al 2 (OH) 16 (CO 3 ).4H 2 O [34]](https://thumb-eu.123doks.com/thumbv2/123doknet/7995239.267867/35.892.156.714.431.699/figure-structure-feuillets-matériau-type-hydrotalcite-tel-que.webp)
![Figure I.12 : différentes espèces carbonates formées à la surface de la cérine observées par IRTF [70]](https://thumb-eu.123doks.com/thumbv2/123doknet/7995239.267867/48.892.202.702.178.674/figure-espèces-carbonates-formées-surface-cérine-observées-irtf.webp)
![Figure I.13 : Oxydation catalytique de l'acide acétique: Rendements en CO 2 des matériaux [101]](https://thumb-eu.123doks.com/thumbv2/123doknet/7995239.267867/56.892.182.711.420.730/figure-oxydation-catalytique-acide-acétique-rendements-co-matériaux.webp)




![Figure II.12 : montage réactionnel utilisé pour la réaction d’oxydation de l’acide acétique [3]](https://thumb-eu.123doks.com/thumbv2/123doknet/7995239.267867/84.892.185.710.308.761/figure-montage-réactionnel-utilisé-réaction-oxydation-acide-acétique.webp)