HAL Id: tel-02476412
https://tel.archives-ouvertes.fr/tel-02476412
Submitted on 12 Feb 2020
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
An alternative way to look at diffuse large B-cell
lymphoma : the impact of frequent engagement of ReIB
NF-κB subunit on cell survival and patient outcome
Davi Moreira Collares
To cite this version:
Davi Moreira Collares. An alternative way to look at diffuse large B-cell lymphoma : the impact of frequent engagement of ReIB NF-κB subunit on cell survival and patient outcome. Cellular Biology. Université Sorbonne Paris Cité, 2019. English. �NNT : 2019USPCB019�. �tel-02476412�
Université de Paris (Paris Descartes)
École doctorale hématologie, oncogenèse et biothérapies (ED 561)« NF-κB, differentiation and cancer » team, EA 7324
An alternative way to look at diffuse large B-cell lymphoma:
the impact of frequent engagement of RelB NF-κB subunit
on cell survival and patient outcome.
Davi Moreira Collares
PhD thesis on specialty “Hematology”
Directed by Thierry Molina and Véronique Baud
Defended on the 25th of June 2019
Jury composed by:
Christiane Copie-Bergman, PU-PH, Université Paris-Est, Rapporteur Emmanuel Dejardin, Université de Liège, PhD, Rapporteur
Karen Leroy, PU-PH, Université Paris Descartes, Examinateur Maria Chiara Maiuri, PhD, CR1, Université Paris-Sud, Examinateur Thierry Molina, PU-PH, Université Paris Descartes, Director
Véronique Baud, DR2, Université Paris Descartes, Co-director
2 To all trans people who never had a chance.
3 “Behind every exquisite thing that existed, there was something tragic.”
4
ACKNOWLEDGEMENTS
It has been almost 5 years since I left my country to hop on this adventure of learning and personal growth that doing a PhD and living abroad has been. It has been a very long and hard journey and I would have not been able to get here without the help of so many around me. And since in these situations there is no other way to show gratitude than the absolute cliché, I’ll try to keep it simple.
I would like to thank Thierry Molina, my director, for giving me the opportunity to be here today, for seeing potential in me and helping me through all the process, since before my moving to France. Véronique Baud, my co-director, thank you for the warm reception at your lab, for the always opened-door of your office, for the daily guidance and all the support. I truly appreciate what you both have done for me.
Everyday work would have never been the same without the “NF-κB differentiation and cancer” team, the help, coffee/tea time, happy hours and even “pétanque” sometimes. A very special thanks to Stéphanie Nuan-Aliman, who has been my right arm in this project since the very early days, without whom all of it would have been twice as hard, and whose professional growth I had the pleasure to watch. A special thanks also to Baptiste Eluard. Thank you for your friendship, your support, especially in the very beginning, when I was still adapting to my new life, for the cat videos, dog memes and all the shenanigans. Didier Bordereaux for sharing his years of lab experience, for the French lessons and trivia moments. Lastly, as he was the last to join the team, thank you, Leonardo Lordello, for the crucial help with the statistics, the (sometimes
5 nonsense) long discussions, your companionship and the unconditional friendship all these years.
For granting me the scholarships to accomplish this PhD, thanks to Sorbonne Paris Cité and Societé Française d’Hématologie.
I also would like to thank the teams who collaborated with this work. José Angel Martinez-Climent, who has kindly provided the cell lines used in my research; Guido Kroemer’s team, especially Chiara Maiuri and Isabelle Martins, who’s always been extremely available and efficient; Jacqueline Lehmann-Che, for the evaluations regarding p53.
I’d like to acknowledge the work and effort of Alexadra Elbakyan, that is so important for so many young scientists. Thank you for sharing with me and many others the belief that there should be no barriers to knowledge and science.
Mouni Dadoun, Paula Beltrami, Rayane Amaral, Thomas Boudon, Marcos Fernandes, Martha Santos Menezes, Hanna Motta Costa and those who had to leave early, Mariana de Padua and Lucas Menezes: all the “Tetê” group. You were (and are) my family in Paris. Thank you for everything! Thank you, Mariane Miranda, Ambroise Bera and Heglyn Pimenta for all the assistance and special moments. Thanks to Denise and Bertrand Clavel for being always so kind and so helpful with the papers. My second “French” family, Mércia Sousa, Christophe Bahr, Juliana Lobo and little Oliver and Sophie, thank you for the support and for always welcoming me so well at your home.
Elena Perepelitsa, I would have never made it through this final stretch without you. You are much more than I could ever ask for.
6 I’m immensely grateful for those who have supported me from far away, my beloved family and friends who I had to leave behind when I moved to France. Thank you for accepting my absence, for cheering for my success and for trying to be a part of it, even if it is not the same via the internet. My mother and father, each one in their own way, who made the possible and the impossible to assure I would be where I am. Mariana, for being the little sister I never had, to who I have taught everything she knows. Natalia de Castro, for being an inspiration in her battle. And I’m also thankful for Ana Morita’s support from the other side of the Atlantic.
At last, it is never too much to say again that a work this toilful can only be accomplished in collaboration. To all of you my most sincere MUITO OBRIGADO.
7
ABSTRACT
NF-κB transcription factors play critical role in cell proliferation, cell survival and the physiopathology of numerous cancers. Deregulation of the classical NF-κB pathway is known to be involved in at least the ABC subset of diffuse large B-cell lymphoma (DLBCL). However, the activation status of RelB NF-κB alternative pathway subunit and its role remain unclear. We have demonstrated a frequent engagement of RelB in human DLBCL-derived cell lines. RelB activation protected cells from DNA damage and apoptosis induced by doxorubicin, the main drug in DLBCL treatment. RelB also controlled the expression of the anti-apoptotic protein cIAP2. In a cohort of 66 de novo DLBCL patients, we have directly assessed the DNA binding activity of all NF-κB subunits by EMSA coupled with supershift. RelB activation was present in 66.6% of cases regardless of ABC or GCB classification and was an independent predictor of worse outcome. RelB activation status by EMSA allowed the definition of a RelB gene expression signature that was able to confirm RelB’s negative impact on patient outcome when extended to a larger cohort. Altogether, our study indicates that RelB is a potential new biomarker for DLBCL and sheds light on its important role in DLBCL cell biology.
8 TABLE OF CONTENTS ACKNOWLEDGEMENTS ABSTRACT TABLE OF CONTENTS FIGURES AND TABLES LIST OF ABREVIATIONS INTRODUCTION
I.1 - NF-κB family of transcription factors I.1.2 - NF-κB pathways
I.1.2.1 - Classical (or canonical) I.1.2.2 - Alternative (or non-canonical)
I1.2.2.1. RelB regulation and functions I.1.3 - NFκB and B-cell physiology
I.1.4 - NF-κB and apoptosis I.2 - Diffuse large B-cell lymphoma
I.2.1 - Classification
I.2.2 – Frequent genetic alterations I.2.3 - Prognosis and treatment
I.2.3.1 – Doxorubicin I.2.3.2 – Rituximab I.3 - DLBCL and NF-κB
I.3.1 - Alternative pathway and RelB I.3.2 - NF-κB as therapeutic target OBJCTIVE
MATERIALS AND METHODS M.1 - Patient selection and biopsies M.2 - Human DLBCL cell lines M.3 - Protein extraction
M.4 - Electrophoretic mobility shift assays for NF-κB M.5 - Immunoblotting
M.6 - Lentiviral production and transduction M.7 - Anexin V binding assay
M.8 - γH2AX foci
M.9 – RNA extraction and RT-qPCR M.10 - p53 functional status M.11 - Immunohistochemistry M.12 - Statistical analysis M.12.1 – Cell death M.12.2 – Patients Clinical analysis Transcriptomic analysis RESULTS
SECTION I: CELL LINES
4 7 8 10 13 17 18 20 23 27 31 35 35 39 42 42 45 47 49 50 53 54 56 56 56 57 58 59 60 60 61 61 62 62 64
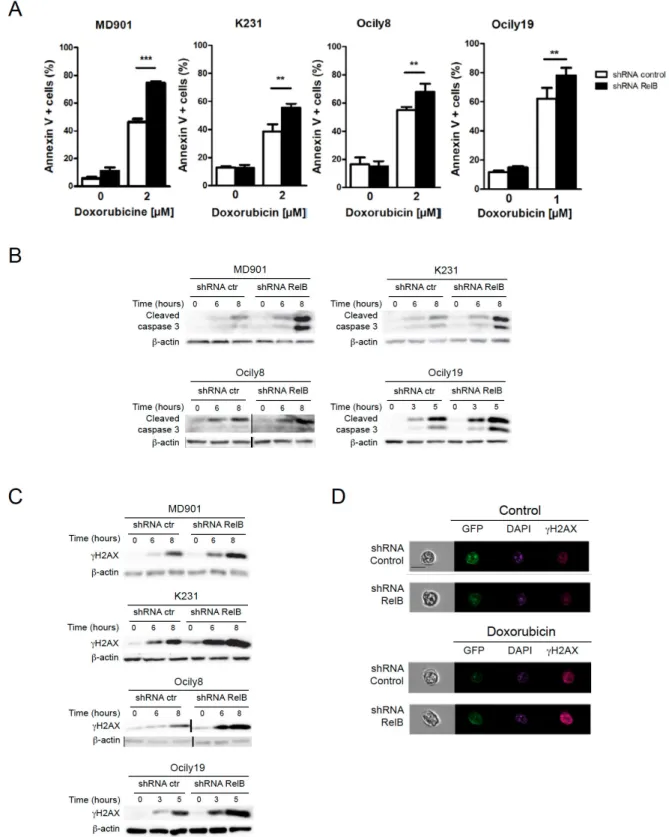
9 1. RelB is frequently activated in DLBCL derived cell lines
2. RelB protects ABC and GCB DLBCL derived cell lines against doxorubicin-induced DNA damage and apoptosis
3. RelB constitutive activation is associated with up-regulation of cIAP2 in DLBCL cells
SECTION II: PATIENTS
1.RelB is frequently activated in both GCB and ABC DLBCL subtypes.
2.RelB constitutive activation is not associated with NF-κB-related mutations or current gene expression signature in DLBCL
3.Establishment of RelB-associated gene expression signature
4.RelB activation has a negative impact on patient overall survival DISCUSSION
CONCLUSIONS BIBLIOGRAPHY ANNEXES
List of publications
Article: Post-Translational Modifications of RelB NF-κB Subunit and Associated Functions
64 66 72 76 80 82 84 93 101 102 126 127
10
FIGURES AND TABLES
Introduction
Figure I1: The NF-κB transcription factor family
Figure I2: The NF-κB classical pathway Figure I3: The NF-κB alternative pathway Figure I4: Overview of some of RelB functions Table I1: Post-translational modifications of RelB Figure I5: B-cell development and NF-κB
Figure I6: Control of CD40 and BAFF-R signaling Figure I7: B-cell survival signaling pathways Figure I8 : Extrinsic and intrinsic apoptosis
Figure I9: NF-κB target genes involved in programmed cell death
Figure I10: Frequent mutations in DLBCL patients
Figure I11: Schematic representation of Doxorubicin mechanisms of action
Figure I12: Mechanism of action of rituximab
Materials and Methods
Table M1: Patients from GHEDI cohort included in the study
Table M2: Clinical trials used to compose the GHEDI cohort
Table M3: Antibodies used for immunoblotting
Table M4: Primer sequences used for RT-qPCR
Results
Table R1: Characterization of human derived DLBCL cell lines Figure R1: NF-κB activation status in chosen DLBCL cell lines
18 21 22 26 26 28 29 30 34 34 41 44 46 55 55 58 61 65 68
11 Figure R2: Verification of efficient knockdown of RelB in DLBCL cell
lines
Figure R3: Effect of doxorubicin on NF-κB DNA binding in DLBCL cell lines
Figure R4: RelB protects DLBCL cell lines against caspase 3-dependent apoptosis and DNA damage accumulation induced by doxorubicin
Table R2: Characterization of TP53 in DLBCL cell lines Figure R5: Effect of RelB on antiapoptotic agents
Table R3: Characterization of the patients from the GHEDI cohort
Figure R6: Analysis of the DNA-binding activity of NF-κB in a cohort of
newly diagnosed DLBCL patients
Table R4: DLBCL frequent mutations according to RelB status
Table R5: NF-κB DLBCL gene expression signature (Davis et al.)
comparison by groups
Figure R7: RelB-associated gene expression signature
Table R5: Definition of RelB activation status by EMSA vs gene expression profile
Figure R8: Prognostic impact of RelB activation defined by EMSA on the
GHEDI cohort of DLBCL patients
Figure R9: Prognostic impact of RelB activation defined by EMSA on R-CHOP treated DLBCL patients from the GHEDI cohort
Figure R10: Prognostic impact of RelB activation defined by RelB gene
expression signature on the GHEDI cohort of DLBCL patients
Table R6: Cox proportional hazard regression for multivariate analyses
in cohorts with RelB status evaluated by EMSA and GEP signature
69 70 71 72 74 78 79 81 82 83 83 86 88 90 91
12 Table R7: Bcl-2 and c-Myc protein expression according to RelB
13 LIST OF ABBREVIATIONS
(aa)IPI (age adjusted) International prognostic index
(γ)H2AX (phosphorylated on serine 139) Histone 2AX
(R)-ACVBP
(Rituximab) doxorubicin, cyclophosphamide, vindesine, bleomycin, prednisone
(R)-CHOP
(Rituximab) cyclophosphamide, doxorubicin, vincristine, and prednisone
ABC Activated B-cell
AICD Activation-induced death
ASCT Autologous stem cell transplantation
ATP Adenosine tri-phosphate
BAFF B-cell activation factor
Bcl-2/6/10 B-cell lymphoma 2/6/10
Bcl-2A1 BCL2 related protein A1
BCR B-cell receptor
BLIMP1 B-cell-induced maturation protein 1
BM Bone marrow
c-FLIP Casp8 and FADD like apoptosis regulator
CARD11 Caspase recruitment domain family member 11
CAT Catalase
CCR7 C-C motive chemokine receptor 7
CD Cluster of differentiation
CI Confidence interval
cIAP1/2 Baculoviral IAP repeat containing protein 2/3
COO Cell of origin
CREBBP CREB binding protein
DAPI 4',6-diaminidino-2-phenylindole
DCF 2'-7' dichlorfluorescein
DD Death domain
14
DNA Deoxyribonucleic acid
DR4/5/6 Death receptor 4/5/6
DTT 1,4-Dithiothreitol
ECOG Eastern Cooperative Oncology Group
ELISA Enzyme-linked immunosorbent assay
EMSA Electrophoretic mobility shift assay
EZH2 Enhancer of zeste homolog 2
FASAY The p53 Yeast Assay
FasL Fas ligand
FFPE Formalin fixed paraffin embedded
GCB Germinal center B-cell
GEP Gene expression profile
GFP Green fluorescent protein
GPX1 Glutathione peroxidase
GRR Glycine-rich region
HIV Human immunodeficiency virus
IGH Immunoglobulin heavy locus
IHC Immunohistochemistry
IKKα/β/γ IκB kinase α/β/γ
IL-1 Interleukin 1
INF-α/β Interferon α/β
IRF4 Interferon Regulatory Factor 4
LDH Lactate dehydrogenase
LMP1 Latent membrane protein 1
LPS Lipopolysaccharide
LTβ Lymphotoxin β
LTR Long terminal repeats
LYSA Lymphoma study association
15
Mdm2 Mouse double minute 2 homolog
MFI Mean fluorescence intensity
MHC Major human histocompatibility complex
MnSOD Mitochondrial antioxidant manganese superoxide dismutase
MYC Avian Myelocytomatosis Viral Oncogene Homolog
MYD88 Myeloid Differentiation Primary Response 88
NF-kB Nuclear factor kappa B
NGS Next generation sequencing
NIK NF-κB inducing kinase
NOS Not otherwise specified
NOS3 Endothelial nitric oxide synthase 3
NPV Negative predictive value
NQO1 NAD(P)H dehydrogenase
PCR Polymerase chain reaction
PMSF phenylmethylsulfonyl fluoride
PNPP para-Nitrophenylphosphate
PPV Positive predictive value
PRDM1 PR domain zinc finger protein 1
RHD Rel homology domain
RNA Ribonucleic acid
ROS Reactive oxygen species
RT Real time
RT-MLPA
Reverse transcriptase multiplex ligation-dependent probe amplification
SDS Sodium dodecyl sulfate
SE Standard error
shRNA Short hairpin RNA
SOD1 Superoxide dismutase 1
16
TBE Tris-borate-EDTA
TBS Tris Buffered Saline
TG-SDS Tris-Glycine SDS
TLR Toll-like receptor
TNF-α Tumor necrosis factor α
TNFAIP3 (A20) TNF Alpha Induced Protein 3
TNFRSF14 TNF receptor superfamily member 14
TOP2A Topoisomerase II
TRAF2/3 TNF receptor-associated factor 2/3
TRAIL TNF-related apoptosis-inducing ligand
ULN Upper limit of normal
UV Ultra-violet
WES Whole-exome sequencing
WHO World Health Organization
XDH Endothelial nitric oxide synthase
17
INTRODUCTION
I.1 - NF-κB family of transcription factors
NF-κB was first described in 1986 as a nuclear factor binding the kappa light chain enhancer in B-cells (1). NF-κB transcription factors family plays a crucial role in the inflammatory and immune responses, cell proliferation and survival (2,3). When deregulated, these transcriptional factors can participate in tumor development, maintenance and even initiation, especially in hematopoietic malignancies (4–7). NF-κB has also been implicated in resistance to cancer treatment (8).
In mammals, this family is composed by five members, RelA (p65), RelB, cRel (Rel), NF-κB1 (p50 and its precursor p105) and NF-κB2 (p52 and its precursor p100) (9) (Figure I1) . All NF-κB family members present a highly conserved region, the Rel Homology Domain (RHD), in the N-terminus domain. It is vital to dimerization, nuclear translocation and DNA binding. Furthermore, RelA, cRel and RelB (but not p105/p50 and p100/p52) possess a transactivation domain (TAD), without which transcriptional activity is not possible. Interestingly, RelB is the only member which has a Leucine Zipper (LZ), a domain in its N-terminal region (10). Cleavage of RelB on the LZ has been described as activation mechanism for RelA-RelB inhibitory complexes (11), however little is known about the functions of RelB LZ.
The precursors p105 and p100 are cleaved in their C-terminal ankyrin repeats to be converted into their active forms p50 and p52, respectively. In their inactive unprocessed forms, they have a crucial inhibition role and behave as IκBs (9,10).
18 These proteins form various homo- and heterodimeric complexes, the only form they can be transcriptionally active. NF-κB activity is regulated by two main pathways: the first one known as the classical or canonical pathway, mainly applies to RelA and/or cRel containing complexes; the second one, so-called alternative or non-canonical pathway leads to the activation of RelB containing complexes (12).
Figure I1: The κB transcription factor family. The five members of the
NF-kB family are presented with their main domains. Proteins p100/p52 and p105/p50 are shown in their full length. DD, death domain; GRR, glycine-rich region; LZ, leucine-zipper; RHD, Rel homology domain; TAD, transactivation domain. From Oeckinghaus and Ghosh, Cold Spring Harb Perspect Biol, 2009.
I.1.2 - NF-κB pathways
I.1.2.1 - Classical (or canonical)
Inflammatory cytokine tumor necrosis factor α (TNF-α), toll-like receptors (TLR), interleukine-1 (IL-1) and lipopolysaccharide (LPS), are some of the stimuli
19 capable of activating this pathway. This pathway regulates mainly RelA/p50 heterodimers, but can also regulate c-Rel/p50 and p50/p50 complexes (9,10).
Among activators of NF-κB classical pathway, TNFR1 signaling pathway is perhaps one of the best characterized. After ligand recognition, TNFR1 cytoplasmic death domain (DD) binds to TRADD (TNFR-associated protein with a DD) and RIP1, which also contains a death domain. TRAF2 is then recruited by TRADD via its TRAF-binding domain, which in turn will recruit cIAP1 and cIAP2. cIAP1/2 act as E3 ubiquitin ligases, recruiting LUBAC (linear ubiquitin assembly complex), which will catalyse linear polyubiquitination of NEMO and subsequent activation of IKK (13). Recruitment of TAK1 results in phosphorylation and activation of IKKβ and IKKα. IKKβ phosphorylates IκBα, leading to its polyubiquitination and degradation by the proteasome. In absence of stimulation, the NFκB dimer is sequestered in the cytoplasm through interaction with inhibitory proteins of the IκB family. Now released, the active dimer translocates to the nucleus and executes its transcription functions (2,9). Some of targets genes of NF-κB are its own negative regulators, such as IκBα and A20. Newly synthesized IκBα binds to NF-κB active dimers shutting them back to the cytoplasm. A20 and CYLD deubiquitinate RIP1, NEMO and TRAFs, ending the signal, thus acting as negative regulator of the duration of NF-κB activity. (Figure I2) (14).
This pathway is typically responsible for a strong and rapid NF-κB activating signal in response to stress situations and plays a crucial role in the regulation of inflammation and innate immunity (2).
20
I.1.2.2 - Alternative (or non-canonical)
This pathway is known to be involved in diverse processes such as proliferation and lymphoid organogenesis (15). It is activated by a subset of TNF family members (e.g. lymphotoxin β (LTβ), B-cell activating factor (BAFF), CD40 ligand, TWEAK) (9,15,16). It is important to state that, except for BAFFR, all activators of the alternative pathway also induce the classical pathway.
In unstimulated cells, TRAF3 and TRAF2 form a complex with cIAP1/2, which ubiquitin tags NF-κB inducing kinase (NIK) for its degradation. Stimulation of activator receptors results in the ubiquitination of TRAF3 by cIAP1/2 and its following degradation. Degradation of TRAF3 by the proteasome allows stabilization of NIK, which will activate IKKα that will phosphorylate p100. Phosphorylation will ultimately conduct to either total p100 degradation and liberation of RelB/p50, or partial degradation, producing free RelB/p52 dimers (9,15,17) (Figure I3).
Activated IKKα has been described to destabilize NIK as negative feedback mechanism. TANK-binding kinase 1 (TBK1) also binds and phosphorylates NIK for degradation (16). The alternative pathway induces the protein OTUD7B, a deubiquitinase that deubiquitinates TRAF3, stabilizing it, reducing the signaling and creating a negative feedback loop (16,18). Recently described as a regulator, OTUB1 is a deubiquitinase that controls p100 stability and prevents abnormal alternative pathway activity (19). Additionally, crosstalk between the classical and alternative pathway has been described, being NEMO responsible for NIK negative regulation (16).
21 The alternative pathway plays a crucial role in innate and adaptive immunological responses, immune system development and maturation and even bone remodeling. It is not surprising that is deregulation is implicated in several inflammatory diseases and cancers (9,16,17,20).
Figure I2: The NF-κB classical pathway. Schematic representation of NF-kB
activation cascade by TNFR1. Engagement of TNFR1 results in activation of TRADD and recruitment of TRAF proteins, which will ultimately result in ubiquitination of NEMO and activation of the IKK complex. This complex will
22 phosphorylate IκBα allowing its degradation and liberation of NF-κB dimer to translocate into the nucleus. Lys-48 linked ubiquitin chains are shown in red, and Lys-63-linked ubiquitin chains are shown in green. Adpted from Wertz et al, Cold
Spring Harb Perspect Biol 2010.
Figure I3: The NF-κB alternative pathway. Activation of NF-κB alternative
pathway through CD40. 48 linked ubiquitin chains are shown in red, and Lys-63-linked ubiquitin chains are shown in green. See text for additional details. Adpted from Wertz et al, Cold Spring Harb Perspect Biol 2010.
23
I1.2.2.1. RelB regulation and functions
RelB gene is located on chromosome 19q13.32 and encodes an mRNA from 11 exons, resulting in a 579-amino acid protein (21). RelB can potentially be regulated by many different transcription factors at its promoter, introns, and exons; among these putative regulatory sites are domains that bind glucocorticoid receptors, c-Jun, c-Fos, sp-1, and B-zip chromatin repressor and insulator CCCTC-binding factor (CTFC), which defines the boundary between euchromatin/heterochromatin and IFN response factor 1 (IRF1).
The best-studied transactivator of RelB is RelA, which can bind proximal promoter NF-κB sites (22). Interestingly, the same study shows that RelB can bind its own promoter and regulate its own mRNA expression. In presence of cytomegalovirus protein IE1, AP-1 regulates RelB expression. It can be inhibited by 1α,25-dihydroxyvitamin D3 blocking dendritic cell differentiation. SIRT1, a cellular bioenergy sensor, can also induce RelB mRNA expression (21).
In addition to the alternative NF-κB signaling cascade (described before), RelB DNA-binding activity is negatively regulated at the nuclear level by several mechanisms, such as trapping in RelA/RelB or p100/RelB complexes, as well as post-translational modifications (discussed ahead). RelB-containing dimers also display DNA-binding specificity. RelB recruitment to some genes correlates with transcriptional downregulation (IL12-p40), whereas inother cases (EBV-induced molecule 1 ligand chemokine (ELC) and macrophage-derived chemokine (MDC)), it increases transcriptional activity (12).
RelB-p50 heterodimers crystal structure researches have suggested that although DNA-interacting residues are highly conserved among NF-κB members, RelB can bind a more diverse set of NF-κB consensus sequences than the other
24 family members (21).
Studies in knockout mice showed that RelB has important roles in development and function of immunological system that cannot be compensated by the present of the other NF-κB subunits (23).
RelB directly controls the transcription of MMP3 on fibroblasts, impacting on cell migration (24); cIAP2 in multiple myeloma cells, influencing cell survival (25); manganese-SOD in prostate cancers (26), as well as EZH2 (27) have also been identified as direct RelB targets. RelB participates in metabolisms and mitochondria biogenesis. RelB-mediated changes in the NADH/NAD+ balance, activate the NAD+-sensing sirtuin family member, SIRT1. In turn, SIRT deactivates nuclear RelA and induces RelB transcription, resulting in an anti-inflammatory phenotype and increased fatty acid flux and transport into mitochondria. RelB can also regulate mitochondrial biomass by regulating PGC-1β. Additionally, RelB supports expression of SIRT3, which localizes to mitochondria and enhances many mitochondrial functions, having implications for oxidative metabolism (21). RelB was shown to participate in the regulation of the circadian rhythm in murine fibroblasts, by direct binding to BMAL1, one of the main regulator of the cycle (28). The regulation of the pathway is also influenced by SIRT1, which gives RelB a second indirect role (21). It also takes part in the xenobiotic-detoxifying pathway in lung fibroblasts by binding to AhR, a transcription factor involved in the xenobiotic response responsible for detoxifying polychlorinated biphenyls and polycyclic aromatic hydrocarbons via increased expression of the cytochrome P450 family. This interaction induces RelB, which provokes a decrease in COX-2 and PG-mediated inflammation. RelB also plays an important role in RANKL-induced osteoclastogenesis that cannot be
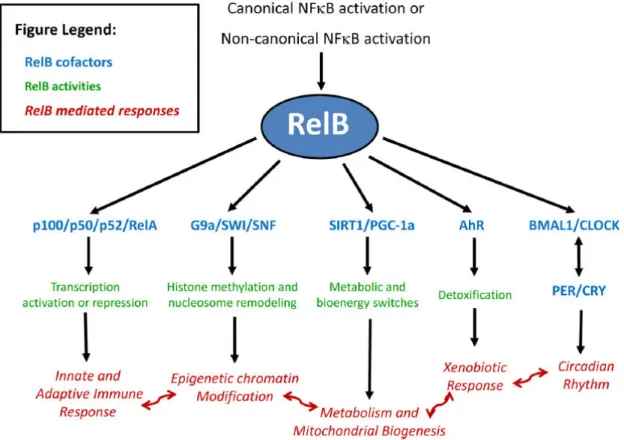
25 compensated for by RelA (12,21). Some of the main functions of RelB are summarized in figure I4.
RelB can also function as a negative regulator of the classical pathway of NF-κB. RelB can bind to RelA, forming an inhibitory complex (29), and cleavage of RelB on it’s LZ can activate the classical pathway of NF-κB (30).
Post-translational modifications are changes or alterations in a protein occurring after the completion of the translational process that act as a mechanism for the specification of proteins and increase their variety. Reported modifications involving NF-κB include phosphorylation, methylation, ubiquitinylation, acetylation, SUMOylation, and isomerization of specific amino acid residues, and target the IKKs, the IκBs, the NF-κB subunits, or critical adaptor proteins that feed into NF-κB. However, until now few of such described modifications have been reported for RelB (see table I1). Remarkably, the best characterized is its phosphorylation on serine 472 by IKKα/IKKβ, which has been reported to induce the transcription of MMP3 and influence on cell migration. (24). Enzymes regulating other RelB modifications are not yet identified and no biological functional has been associated to them for the moment. In some cases, it remains to be uncovered if the described phosphorylation happen in
26
Figure I4: Overview of some of RelB functions. RelB interacts with different
proteins and factors (blue), involved in actions described in green, culminating in the execution of diverse functions (red). From Millet et al, J Leukoc Biol 2013.
Table I1: Post-translational modifications of RelB. From Baud and
27
I.1.3 - NFκB and B-cell physiology
Notably, during B-cell generation and maintenance, NF-κB subunits have many physiological functions and play unique roles in the development and function of mature B lymphocytes (Figure I5) (31).
The primary role of NF-κB in B-cell development is to guarantee cell survival. In early stages, NF-κB protects B-cells from TNFα induced apoptosis by induction of Bcl-xL. It also participates in the negative selection in bone marrow (BM) via both alternative and classical pathways (2,32,33).
After leaving the BM, they complete maturation in the spleen. At this point, both survival, mainly by NF-κB-induced Bcl-2 and A1 expression, and differentiation depend on both NF-κB pathways. Differentiation of marginal zone B-cells (MZB) depends as well on NF-κB. Perturbations in NF-κB, particularly RelB, at this point can result in autoimmunity (2,10).
Activation of the alternative pathway in wild-type immature B cells is likely to occur via BAFF receptor signaling and maturation is at least in part dependent on that pathway. The alternative pathway promotes survival via Bcl-2 expression, and, indirectly, the retention in the cytoplasm of otherwise apoptotic nuclear PKCδ (34).
Classical and alternative pathway activation in stromal cells is crucial for establishing primary and secondary lymphoid organs. This anatomical niches are required for good functioning of the adaptive immune response and RelB seems to have a particularly important role in their establishment (35,36).
28 Mature B-cells express a variety of NF-κB dimers. Constitutive, low nuclear activation of both classical and alternative pathways of NF-κB is thought to mainly serve a survival role in non-activated B-cells (2,33). NF-κB activation via the B-cell receptor (BCR) protects CD40-stimulated B-cells from activation-induced death and is essential for survival and differentiation (37). Different B-cell receptors activate different NF-κB pathways by different signaling. The crosstalk among these receptors is crucial for determination of B-cell response and how NF-κB will influence functionally the B-cell (Figures I6 and I7). NF-κB signaling is also essential for immunoglobulin isotype switching (32).
29
Figure I5: B-cell development and NF-κB. Simplified representation of bone marrow and splenic B-cell development. NF-κB contributions in different stages of differentiation, maturation, formation of marginal zone (MZ B) and follicular mature (FM) B cells (B2 B cells) and entry the peripheral circulation are indicated. The roles of NF-κB in the development of B1 B-cell, a peripherally self-renewing population, are also indicated. Attention must be paid to the participation of NF-κB in formation of proper architecture in spleen, Peyer’s patches and lymph nodes. From Gerondakis et al, Cold Spring Harb Perspect Biol 2010.
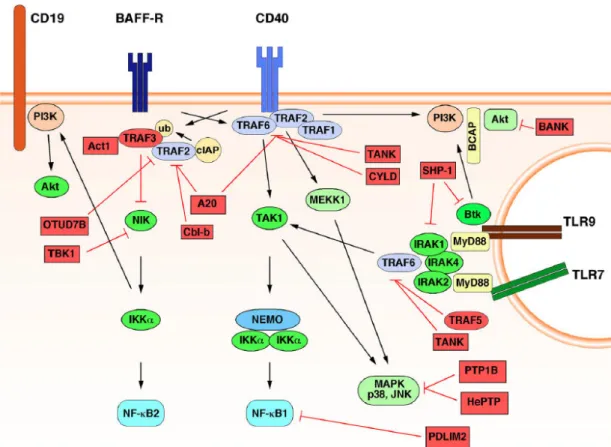
Figure I6: Control of CD40 and BAFF-R signaling. TRAF2 is inhibited by A20
and Cbl-b, preventing the degradation of TRAF3 at either BAFF-R or CD40. This results in the degradation of NIK, which prevents alternative NF-κB activation. OTUD7B and TBK1 are indirectly regulators, through their activities of TRAF2
30 deubiquitination and NIK phosphorylation, respectively. CYLD and TANK inhibit the canonical NF-κB pathway downstream of CD40 through TRAF6. Tyrosine phosphatases PTP1B and HePTP regulate CD40 and BAFF-R signaling by directly dephosphorylating the p38 MAPK. PDLIM2 is a nuclear ubiquitin ligase that induces the degradation of the p65 NF-κBprotein. From Hobeika et al J Mol Med, 2015 (38)
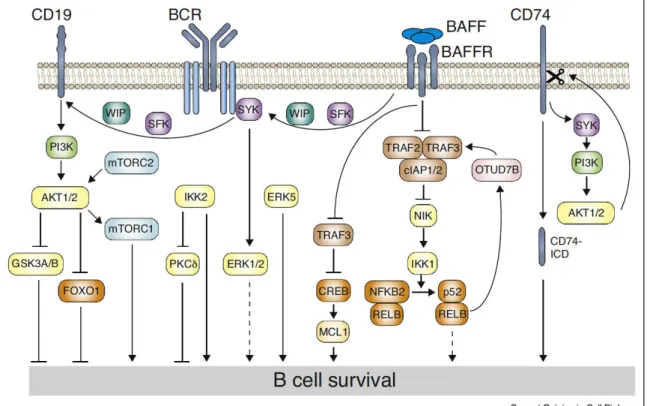
Figure I7: B-cell survival signaling pathways. Signaling from BAFFR to BCR
and SYK depends on SRC-family kinases (SFK). Further signaling to CD19 requires either SFK or SYK. Regulation of the actin cytoskeleton by WIP is required for signal transduction from BAFFR to CD19. Signals from the BCR via SYK lead to activation of ERK1 and ERK2, contributing to cell survival in unclear ways. BAFFR transduces signals via the TRAF2-TRAF3-cIAP1/2 E3 ligases to
31 NIK, leading to processing of p100. TRAF3 also directly enters the nucleus and associates with CREB, leading to its degradation. CREB induces expression of MCL1. The alternative NF-κB pathway induces OTUD7B deubiquitinase, which stabilizes TRAF3 in negative feedback. From Schweighofer et al, Curr Opi Cell
Biol 2018. (18)
I.1.4 - NF-κB and apoptosis
Apoptosis is a physiological process of organized programmed cell death, fundamental for organ development, homeostasis, and elimination of defective or potentially dangerous cells. It can be triggered by a great variety of stimuli and can be devided in two major subtypes: extrinscic and intrinscic (39,40).
Extrinsic apoptosis is mediated by death receptors (e.g., fas cell surface death receptor [FAS] and TNFR1), initiated by caspase 8 and caspase 10; or the withdrawal of dependence receptor ligands, initiated by caspase 9 (Figure I8) (40).
In response to cellular stress, BH3-only proteins promote mitochondria outer membrane permeabilization (MOMP), releasing mitochondrial proteins, such as cytochrome C into the cytosol. MOMP is tightly controlled by the BCL2 family, including pro-apoptotic (e.g., BCL2 associated X, apoptosis regulator [BAX], BCL2 antagonist/killer 1 [BAK1, also known as BAK]), and anti-apoptotic (e.g., BCL2 and BCL2 like 1 [BCL2L1, also known as BCL-XL]) members. Cytochrome C will interact with apoptosis protease activating factor-1 (Apaf-1), recruiting caspase 9, for the assembling of the apoptosome, culminating in cleavage of caspase 3, which will cleave substrates in order to assure cell death.
32 DNA damage, hypoxia, metabolic stress, are some of the inducers of intrinsic apoptosis (Figure I8) (39–41).
In this context, activation of NF-κB (through TNFR for instance) can activate the transcription of regulators of intrinsic and/or extrinsic death signaling cascade (Figure I9) (42). The main targets are inhibitor of apoptosis proteins XIAP, c-IAP1 and c-IAP2, that prevent activation of pro-caspase-9 and blocking caspase-3 and -7 activity; and the antiapoptotic 2 family proteins xL, Bcl-2A1, NR13 and Bcl-2, all of which will ultimately prevent mitochondrial cytochrome c unloading (39,42–44). Other upregulated genes are A20, that prevents recruitment of TRADD and RIP1, and c-FLIP, which regulates caspase 8 (4).
NF-κB can also induce the expression of proapoptotic genes. These include mainly death receptors Fas, TRAIL receptors DR4, DR5 and DR6, and their death-inducing ligands FasL, TNFα and TRAIL; tumor suppressor p53, the proapoptotic Bax and the proapoptotic alternatively spliced form of Bcl-xL, Bcl-xS (42,44). In ultra violet (UV) exposure induced- and chronic exposure to hydrogen peroxide induced-cell death, NF-κB represses prosurvival genes (4).
NF-κB’s role in apoptosis is critical for development and homeostasis in diverse cell systems (42). As an example, it is required for developing B-cells survival, but also for activation-induced cell death (AICD), important mechanism through which immune system homeostasis is maintained (2).
Context can interfere in the role of NF-κB in apoptosis: c-Rel upregulates the expression of MnSOD to protect cells against cell death by converting toxic superoxide anions to H2O2. However, the accumulation of hydrogen peroxide
33 can turn RelA into a transcriptional repressor of antiapoptotic target genes (42,46). Fas-L-induced NF-κB activation can induce c-FLIP, which will in turn downregulate NF-κB and promote FasL-induced cell death (4).
Importantly, TP53 gene, a major regulator of apoptosis and cell cycle, has a κB site in its promoter, and can have its expression induced through this site through TNFα stimulation (47). NF-κB and p53 are also both induced by certain forms of stress that cause DNA damage, such as UV radiation (43). p53 and NF-κB can function together to promote transcription of certain genes, but can also compete for co-activators. p53 activation can lead to NF-κB DNA binding, however suppressing its transcriptional activation (4). NF-κB’s transcriptional activity can result in enhanced degradation of p53 caused by NF-κB-dependent upregulation of Mdm2, culminating in escape from cell death (42). TNF-induced NF-κB protects against UV-induced cell death in p53-expressing cells. In another example, activation of NF-κB by p53, not mediated by TNFα, has been reported to suppress p53-induced cell death. In this context, inhibition of NF-κB in presence of wild-type p53 might enhance cell survival (48).
NF-kB also participates in several non-apoptotic programmed cell death processes. Since RIPK1 phosphorylation prevents apoptosis or necrosome formation with following necroptosis, activation of NF-κB by TNFR1 participates in the regulation of this pathway. In another exemple, induction of inflamasome components through NF-kB, triggered by INF or TLR signaling participate in pyroptosis (40).
In conclusion, regulation of apoptosis through multiple different mechanisms is with no doubt one of the main roles of NF-κB activation.
34 Dependence on NF-κB to escape programmed cell death and resist conventional therapy is a trait shared by numerous human cancers. Therefore, it remains a central topic for understanding cancer genesis and development of cancer therapy.
Figure I8 : Extrinsic and intrinsic apoptosis. Representation of the main steps
in extrinsic and intrinsic apoptosis pathways, and interaction between them via BID. From Tang et al, Cell Research 2019.
35
Figure I9: NF-κB target genes involved in programmed cell death. Activated
NF-κB induces transcription of multiple genes (e.g. IAP proteins, antioxidant proteins such as MnSOD and ferritin heavy chain (FHC), c-FLIP, A20, FBX10, and Bcl-xL) to suppress apoptosis, both in the intrinsic and extrinsic pathways. Adapted from Baldwin, Immun Rev 2012.
I.2 - Diffuse large B-cell lymphoma
Diffuse large B-cell lymphoma (DLBCL) is the most common non-Hodgkin lymphoma. The median age of incidence is the 7th decade of life, nevertheless, it can occur in young adults and more rarely in children. There is a slight male preference. Its most usual clinical presentation is one or multiple fast growing nodal and/or extra-nodal masses (up to 40% of cases), being the gastrointestinal tract the most frequent site (49,50). The most common type of DLBCL, representing 80-85% of all cases, is designated as “not otherwise specified” (NOS) (50). It is the aim of this work and will be referred henceforth simply as “DLBCL”.
I.2.1 - Classification
Gene expression profiles (GEP) allowed cases to be separated in two main groups, based on their cell of origin (COO): germinal center B-cell-like (GCB) and activated B-cell-like (ABC) (51). Shortly after the publication of this study a third group was added. Since its gene expression profile according to Wright’s algorithm does not fit neither within germinal center nor activated B-cell-like profiles, it has been called “type 3” (52), and later “unclassified”. This
36 classification revealed that the ABC group presented a significant worse prognosis among patients treated with R-CHOP (51–54).
Even though the classification brought precious information, GEP is not applicable in routine, thus many different groups tried to develop immunohistochemistry (IHC) surrogates. The most widely used was the Hans algorithm, which is based on the combination of CD10, BCL6 and MUM1 immunohistochemistry markers to differentiating DLBCL into two groups: GCB (germinal center B-cell like) and non-GCB (55). Recently, new technology of GEP using a smaller panel of genes, applicable in formalin fixed paraffin embedded (FFPE) tissue has been developed with higher concordance with the transcriptome (i.e. RT-MLPA (56) and nanostring (57)) and might probably be increasingly used. Nonetheless, there is considerable heterogeneity within each group (58), pointing out for the necessity for further stratification.
For these reasons, many efforts were made to better stratify DLBCL patients by gene expression in order to better treat, predict response to treatment and have a better understanding of the pathogenesis of the disease.
Further characterization, with gene clustering based on the clinical outcome of patients defined four groups: “germinal center B-cell”, associated with better prognosis; “proliferation”, associated with poor outcome, MYC and its target genes; “major histocompatibility complex (MHC) class II”, associated with silencing of MHC II expression and inferior survival; and “lymph node”, which presented a better prognosis and was suggested to be a microenvironment-related signature (52). The “proliferation” profile cormicroenvironment-related fairly with the ABC subtype, while not surprisingly, “germinal center B-cell” correlated with GCB
37 subtype. Interestingly, “MHC II” and “lymph node” profiles did not correlate with any of the COO groups.
In 2005, Monti et al. have established new genetic signatures that separated cases in 3 clusters (59). The first one showed enrichment in mitochondrial function and oxidative phosphorylation-related genes. It was called “OxPhos”. The second cluster had increased expression of cell cycle regulator, DNA repair genes and elements of the BCR signaling cascade. It was named “BCR/proliferation”. Finally, the third one was called “Host response cluster”, for it presented enhancement in T-cell mediated response and complement marker, connective tissue elements, among other host response related genes.
All 3 clusters had similar 5-year survivals and did not overlap with the previous COO classification, suggesting their value is more related to pathogenesis mechanisms and possible development of specific targeted therapy.
In a different approach, Lenz et al. have described two different gene expression signatures of the microenvironment of the tumor (60). The so called “stromal 1” is characterized by monocyte host-reaction and extracellular matrix deposition related genes and is associated with a better prognosis. “Stromal 2” is associated with tumor blood vessel density and angiogenesis and is indicative of a worse outcome.
More recently, Schmitz et al. have analyzed 574 patient samples by means of exome and transcriptome sequencing, deep amplicon resequencing of 372 genes, and DNA copy-number analysis (61). Based on the co-occurrence of genetic abnormalities, they have divided cases in 4 main groups: MCD, BN2, N1
38 and EZB. MCD was defined by CD79B-MYD88 double mutation; BN2, by
NOTCH2 mutation or BCL6 fusion; N1 by NOTCH1 mutation; and EZB was
based on EZH2 mutation or BCL2 translocation. MCD and N1 represented predominantly ABC cases, EZB had a majority of GCB, and BN2 was represented by the three subtypes. They were able to classify only 44% of cases, suggesting the others may share features. Regarding prognosis, the 4 molecular types had significantly different outcomes, being EZB and BN2 of more favorable prognosis than MCD and N1. Additionally, they have adapted this classification for feasibility with next generation sequencing (NGS) in clinic.
Later on, Chapuy et al. (58) have performed whole-exome sequencing (WES) capturing identified low-frequency alterations, recurrent mutations, somatic copy number alterations and structural variants in 304 de novo DLBCL patients, to assess their prognostic value. By consensus clustering of the 158 genetic driver alterations found they were able to define 5 subsets of tumors and a sixth group with no defining genetic alterations. Four of the groups represented two different subsets of ABC and GCB tumors, respectively, with different outcomes and potential targets for therapy, probably reflecting different pathogenesis. The finding reinforces the heterogeneity within COO subgroups. The 2 remaining groups could not be classified in one single COO subgroup.
In the light of recent knowledge, DLBCL complexity in terms of physiopathology and genetic alteration spectrum is evident. In spite of all the outstanding advances made in the past decades, we still lack optimal classification, risk stratification for patients and therapy selection.
39
I.2.2 – Frequent genetics alterations
As stated before, DLBCL is a disease characterized by a complex genetic profile of numerous mutations, translocations, amplifications, deletions and others (58,61,62). In a recent series, a tumor harbored a median of 17 (range: 0– 48) different genetic driver alterations (58). With increasing development of targeted therapy, a best characterization of frequent and less frequent genetic alterations is in our best interests to better understand and cure this disease.
Because of immunoglobulin rearrangements intrinsic to B-cell development, these cells are particularly prone to translocations. Alterations involving BCL6 and its control of IRF4 Blimp1 in the progression to plasma cell differentiation have been implicated in the genesis of DLBCL. As additional oncogenic processes, other alterations involving MYC and BCL2 are reported in DLBCL (62,63).
Frequency of common and less frequent mutations may vary depending on the study. With the improvement of sequencing technologies new alterations are being uncovered. Frequent mutations and their frequencies may be seen in figure I8A.
Some frequent mutations such as MYD88, CARD11, CD79B, PRDM1 and
IRF4 are preferentially encountered in ABC tumors. Mutations in EZH2, CREBBP, TNFRSF14, BCL2 and MYC are most commonly found in GCB cases
(64).
TP53 mutations are present in about 20% of cases of DLBCL and have
been shown to be an independent predictor of poorer prognosis (50,63). They occur in both the GCB and ABC/non-GCB subtypes of DLBCL in approximately
40 equal frequency (50,58,61,63). TRAF3 is also equally mutated in both GCB and ABC DLBCL in up to 15% of cases (65).
In terms of rearrangement, IGH, BCL2, BCL6, and MYC are the most frequently found (58). DLBCLs harboring translocations involving MYC, BCL2 and/or BCL6 are associated with worse prognosis and were recognized in the latest WHO classification as “high grade B-cell lymphoma with rearrangements of
MYC and BCL2 and/or BCL6” (49,66). MYC and BCL2 protein expression
assessed by IHC has also been shown to identify a subset of patients with poor outcome (66).
Concerning NF-κB, known alterations involve mainly the BCR signaling, an important activator of the classical NF-κB pathway in the B-cell (2,67). Loss of function of inhibitors of the cascade, like A20, or gain of function of activators, such as CD79B, MYD88 or CARD11, results in signaling autonomous from the actual BCR activation and constitutive NF-κB activation (62,68–70). Recently, a multiprotein supercomplex formed by MYD88, TLR9 and the BCR has been described to coordinate BCR oncogenic signaling that results in NF-κB pro survival activation (71). In addition to being implicated in the pathogenesis of DLBCL (62,72,73), NF-κB constitutive activation results in survival and proliferation signaling, and also in resistance to treatment, as discussed before. These genetic alterations are found mainly in ABC DLBCL (64,69) (FIigure I8B). Nevertheless, recent studies found several different alterations resulting in abnormal BCR signaling-dependent NF-κB activation in nearly 45% of cases. Events that may affected other NF-κB regulators were present in 66,2% of all cases. Most importantly, these features were not exclusive of ABC DLBCL (61).
41
Figure I10: Frequent mutations in DLBCL patients. (A) The number at the top
of each bar represents the mutation frequency of the indicated gene in a cohort of patients. Bars are subdivided by DLBCL subtype, with segments proportional to mutation frequency. Potentially actionable targets for therapy are highlighted by each mutation. From Dubois et al. Clin Cancer Res 2016. (B) Frequency and overlap of mutations in the “BCR-NF-κB” cascade in a series of ABC DLBCL patients. From Ngo et al. Nature 2011.
42
I.2.3 - Prognosis and treatment
Classical therapy for DLBCL patients is based on CHOP regimen (CHOP: cyclophosphamide, doxorubicin, vincristine, and prednisone). The introduction of rituximab, a monoclonal antibody against CD20, in the late 90’s improved significantly the outcome of these patients (74,75). Nevertheless, refractory/relapse cases can still reach up to 40% (75).
Alternative treatments, have been proposed, all of them with their advantages and flaws. More intensive ones, as ACVBP-based chemotherapy (ACVBP: doxorubicin, cyclophosphamide, vindesine, bleomycin, prednisone) in association with rituximab, are more effective, particularly in non-GCB patients, however have more frequent adverse effects and are therefore frequently restricted to younger patients (76,77).
Because DLBCL remains a heterogeneous disease in clinical presentation, genetic findings and response to therapy, it is difficult to stratify patients and define the best treatment. Rituximab in association with CHOP remains the classic first line therapy for DLBCL. Even though it might not be the best option for all patients, we lack markers for more precise and personalized choices of treatments in most patients.
I.2.3.1 – Doxorubicin
Doxorubicin is the main drug in DLBCL first line chemotherapy (75). It is an anthracycline first extracted from the bacterium species Streptomyces
peucetius in the 1970’s (78). It has two main mechanisms of action: the first one
43 strain breaks; Doxorubicin has also been described to produce DNA adducts provoking transcriptomic and epigenetic changes, and even crosslink lesions (79–81).
The second is the generation of reactive oxygen species (ROS) through multiple mechanisms (78): doxorubicin is metabolized into a semiquinone, an unstable metabolite, being reduced back to doxorubicin in a cycle that generates ROS (78). It also increases iron accumulation in the mitochondria, increasing ROS production (82). ROS can lead to lipid peroxidation and membrane damage, DNA damage, oxidative stress, and triggers apoptotic pathways of cell death (78,83).
Doxorubicin can also interfere in the iron metabolism and ultimately provoke ferropoptosis (84,85). Ferropoptosis is a singular non-apoptotic process of iron-dependent ROS accumulation that results in lipid peroxidation and ultimately cell death (86). Although relatively well characterized as an actor in cardiotoxicity secondary to doxorubicin (84,85), its importance for doxorubicin anti-cancer action remains to be further explored (85,86).
It is important to notice that different doses and times of exposure will result in different consequences regarding compartment distribution (nucleus, mitochondria, cytosol), ROS production, growth arrest and cell death (87). Also, that doxorubicin acts through several other minor mechanisms, once again highlighting its complex mechanism of action, which is briefly presented in figure I9.
44
Figure I11: Schematic representation of Doxorubicin mechanisms of action.
Doxorubicin was shown to cause DNA breaks to interfere with DNA synthesis and poison topoisomerase II. Its translocation into the nucleus is thought to occur via binding to proteasomes. Doxorubicin undergoes a one-electron reduction by several oxidoreductases to form a semiquinone radical. Re-oxidation of semiquinone back to doxorubicin leads to the formation of ROS and hydrogen peroxide. ROS, causing oxidative stress, can be deactivated by glutathione peroxidase, catalase and superoxide dismutase. Topoisomerase II, TOP2A; NAD(P)H dehydrogenase, NQO1 ; xanthine oxidase, XDH ; endothelial nitric oxide synthase, NOS3 ; glutathione peroxidase, GPX1 ; catalase, CAT ; superoxide dismutase, SOD1. From Thorn, CF et al. Pharmacogenet Genomics. 2011
45
I.2.3.2 – Rituximab
Rituximab is a chimeric human/murin anti-CD20 monoclonal antibody, approved for clinical use around 20 years ago that has revolutionized the treatment of B-cell malignancies. Through its affinity with the CD20 transmembrane protein, present in most malignant B-cells, it engages cell death via at least 4 different mechanisms: antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis, complement-dependent cytotoxicity (CDC), and direct antitumor effects via either apoptosis or other cell death pathways (Figure I10) (88).
Despite of its remarkable success and undeniable importance in the treatment of B-cell malignancies, a proportion of patients eventually face resistance and treatment failure (88,89). For this reason new anti-CD20 monoclonal antibodies, such as Obinutuzumab, so called “type II”, are being developed. They present different epitope affinity and different special conformation, serving as promising alternatives for refractory/relapsed patients (89).
46
Figure I12: Mechanism of action of Rituximab. Rrituximab binds to CD20 on
the B-cell surface activating the complement cascade, which generates the membrane attack complex (MAC) and induces B-cell lysis by complement- dependent cytotoxicity (CDC). Rituximab also favors interaction with natural killer (NK) cells through Fc receptors (FcRs), which leads to antibody-dependent cellular cytotoxicity (ADCC). The Fc portion of rituximab and binded complement fragments are recognised by macrophages, which leads to phagocytosis and ADCC. Lastly, interaction of several molecules of rituximab and CD20 in the lipid raft with elements of a signaling pathway involving Src kinases mediates direct apoptosis. FCR Fc receptor, FCcR Fcc receptor. From Salles et al, Adv Ther 2017.
47
I.3 - DLBCL and NF-κB
NF-κB has been reported to participate in the pathogenesis of DLBCL through multiple mechanisms (65,72,73). In addition to NF-κB downstream targets being involved in prevention of apoptosis and proliferation, Calado et al. have described that canonical NF-κB pathway cooperates with disruption of
BLIMP1 in the development of ABC-DLBCL-like lymphoma in mouse models
(73).
The ABC DLBCL subtype was characterized by a GEP associated to BCR activation in peripheral blood B-cells (51,54) and BCR is one of the main upstream activators of NF-κB classical pathway (2,67). In this context, a small set of previously reported NF-κB target genes were found to be preferentially overexpressed in ABC subtype (54), defining a NF-κB-related gene expression signature in DLBCL. However, it is important to stress that BCR activation engages mainly the canonical pathway (90).
Inhibition of the classical NF-κB pathway by means of either an overexpression of a non-degradable form of the inhibitory protein IκBα (super repressor SS32/36AA ΙκBα), or an IKKβ kinase-dead mutant, or the use of a chemical inhibitor of the IKK complex was found to induce cell death in ABC cell lines (54,91). Later works have linked frequent mutations in regulators of the classical NF-κB pathway (i.e. MYD88, TNFAIP3, CD79A, CARD11) with the ABC DLBCL (53,92).
Taken together, these findings led to the concept of NF-κB addiction in the ABC DLBCL, mainly through the activation of the classical NF-κB pathway.
48 However, it is not known if the presence of genetic mutations in regulators of NF-κB or the expression of the described NF-NF-κB target genes correlate with the DNA binding activity per se of the classical pathway of NF-κB (i.e. RelA and cRel). Further, the activation status of the alternative NF-κB pathway (i.e. RelB) subunits remains unclear.
Several attempts have been made in the past 10 years to determine the actual NF-κB activation status, and their impact in DLBCL (65,70,93–101). However, these attempts to evaluate NF-κB in DLBCL patients differed largely in methodology, criteria, technology available at the time and aim of the study. Therefore, there are numerous discrepancies, making them very hard to compare with one another. The great majority of such studies present evaluations of NF-κB status by immunohistochemistry (IHC). In this technique, the positivity is defined by nuclear accumulation of the protein. One of the main limitation of this method is that nuclear localization does not necessarily correspond to the activation of NF-κB family members (102), neither the amount of protein accumulation is proportional to the activation (as observed in our cell lines, discussed ahead). Another issue is that it is not known how significant levels of activity correlate to protein levels detectable by IHC. There is no consensus about the criteria of IHC positivity either.
Electrophoretic mobility shift assay (EMSA) remains the gold standard assay to evaluate NF-κB activity (103). This technique allows the qualitative and quantitative determination of direct DNA binding of NF-κB subunits. In addition to being extremely sensitive, with aid of supershift with specific antibodies, it is possible to precisely define the dimers involved. It also allows the appreciation of
49 the relative intensities of each subunit as they are all seen at the same time in each case. The main disadvantages are the need of radioactive isotopes and that the technique requires a good level of expertise both for execution and interpretation. Further details are presented in the section “Materials and Methods”.
EMSA has never been largely used in the study of NF-κB status in DLBCL patients and our study is the first to analyze the NF-κB DNA binding status using EMSA combined with supershift in a cohort of de novo DLBCL patients.
I.3.1 - Alternative pathway and RelB
There is growing interest in the alternative NF-κB pathway, whether it is activated and its role in pathogenesis of DLBCL (65,70,93–96,104). However, in spite of being demonstrated to have crucial roles in other B-cell lymphomas (25,105–107), the importance of the alternative pathway of NF-κB in DLBCL remains largely unknown.
Ramachadiran et al. have associated the alternative NF-κB pathway to the control of chromosome stability and DNA repair in p100 knockdown models of DLBCL (104). Recently, Zhang et al. (65) have shown that enforced alternative NF-κB pathway activity pushes the B-cell towards plasma cell differentiation. When associated with Bcl-6 dysfunction this differentiation is blocked resulting in ABC DLBCL-like tumors in mice. They suggest that frequent TRAF3 mutations described before, for abolishing the negative regulation of NIK, might result in constitutive activation of the alternative pathway of NF-κB in DLBCL.
50 Studies concerning RelB in DLBCL face the same technical and methodological problems described in the previous section. Perhaps because of the lack of convincing evidence of RelB constitutive activation in DLBCL so far, its role in this disease remains poorly understood. The status per se of RelB DNA-binding activity in DLBCL patients and cell lines remains an open question.
I.3.2 - NF-κB as therapeutic target
Given the central importance of NF-κB in DLBCL pathogenesis and maintenance, it is not surprising that it is one of the main candidates for development of target therapy. All alterations described downstream the BCR are potential targets for indirect anti-NF-κB therapy. Due to the mentioned evidence pointing to ABC DLBCL depend on BCR signaling, up to this point most of the trials with new drugs targeting upstream of NF-κB have been performed exclusively in non-GCB DLBCL patients.
Proteasome inhibitors, such as bortezomib, block the degradation of classical NF-κB inhibitors. Bortezomib is ineffective alone, but in a phase II trial has shown benefits in non-GCB patients (31). Nevertheless, a large scale randomized phase III trial recently published showed no benefit in the progression free survival in DLBCL patients, both GCB and ABC subgroups, receiving bortezomib in addition to R-CHOP (108)
Lenalidomide is an oral immunomodulatory drug that acts through multiple mechanisms and it is believed to selectively kill ABC DLBCL cells (92). In a study with 40 refractory relapse patients it has shown efficacy (109). However, phase III trials showed no event free survival benefit of lenalidomide in combination with
R-
51 CHOP in ABC type DLBCL (ROBUST trial, Celgene, press release, April 25, 2019) and no overall survival benefit as maintenance therapy in DLBCL patients (110)
Ibrutinib is an inhibitor of the BTK complex, which is immediately downstream to the BCR. Patients bearing CARD11 mutations did not respond to ibrutinib and the status of CD79B did not seem to interfere. MYD88 mutated patients bearing a wild type CD79B showed no response to the drug, however
MYD88 and CD79B double mutated patients were the group with best response.
This indicates that ibrutinib requires specific selecting of patients, which depends on the mutational profile of their tumors (31,92,111). However, in a recent phase III trial (Phoenix), ibrutinib did not improve the event free survival of patients with non-GCB DLBCL. Additionally, in elderly patients (>60 year old), and added in serious adverse events, hindering them from receiving complete R-CHOP therapy (112).
Inhibitors of other BCR associated kinases, such as PI3K, SRC and SYK have been tested, especially in combination with ibrutinib in order to prevent and overcome resistance (113–115). Most studies haven’t reached clinical testing yet. Observation in vitro and using patient-derived xenografts in mice suggest they might be an effective alternative. However, a phase II clinical trial for the use of SYK inhibitor fostamatinib showed poor response in DLBCL patients (116).
SMAC mimetics are IAP antagonists that prevent activation of canonical NF-κB pathway. However, IAPs also control negatively NF-κB alternative pathway. Because of ABC DLBCL addiction to BCR signaling, SMAC mimetic birinapant showed to be beneficial in ABC, but not GCB, DLBCL cell lines.
52 However, authors conclude care has to be taken in the use of the drug in patients with constitutive activation of NF-κB alternative pathway (117). The use of this class of drugs still lacks further studying and robust clinical trials.
Other drugs targeting NF-κB are facing preliminary tests and new ones are being developed. The main concerns involving this modality are toxicity, side effects in long-term administration and clinical criteria for selecting patients (31,118). The results so far showed that, at least for the moment, addition of
targeted therapy has not been successful in demonstrating superiority to R-chop
alone in any phase III DLBCL trial. The design of diagnostic biomarkers is
53
OBJECTIVE
We aimed to define the activation status of the RelB NF-κB subunit in DLBCL and evaluate its prevalence and impact. To do so, we used two different strategies: human-derived DLBCL lines and DLBCL patient samples.
In cell lines we planned first to determine the activation status of NF-κB subunits. With aid of RNA interference, we aimed to knockdown RelB protein expression on cell lines with constitutive activation of RelB to study its importance on DLBCL cell survival and resistance to treatment.
In patients, our aim was to determine the NF-κB subunits activation status by EMSA in a group of newly diagnosed DLBCL patients included in clinical trials and determine the frequency of RelB constitutive activation. Following, we wished to establish, the impact of RelB activation in patient outcome. In the next step, we defined a gene expression signature linked to RelB activation status to study larger groups of patients.
54
MATERIAL AND METHODS
M.1 - Patient selection and biopsies
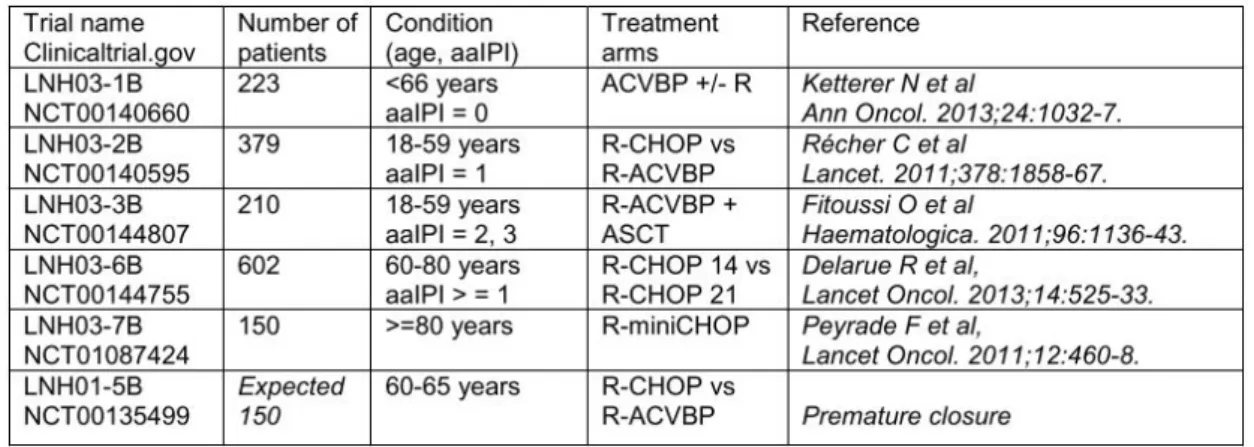
Patients were selected from the GHEDI (Deciphering the Genetic Heterogeneity of Diffuse large B-cell lymphoma in the rituximab era) study program of the LYSA group, previously published and described (119). Patients were enrolled in previous trials summarized on table M1, with available frozen tumor samples, centralized histopathological review, adequate DNA/RNA quality and complete clinical information (120). COO molecular classification was obtained with HGU133+2.0 Affymetrix GeneChip arrays (Affymetrix), grouping patients into ABC, GCB and “unclassified”. We had access to 70 frozen samples from de novo DLBCL patients, as well as complete clinical and transcriptomic data of 202 patients. Patients included in each part of the study are listed on table M2.
For all the analyses patients who received R-CHOP 14, R-CHOP 21 and
mini-CHOP were grouped as “CHOP”. Patients who received