Université de Sherbrooke
ETUDE DE LA PROTEASE ADENOVIRALE: CLONAGE, SEQUENCAGE, EXPRESSION DANS E. COLI ET PROPRIETES DE L'ENZYME RECOMBINANTE
Par Alain Houde
Département de microbiologie
Thèse présentée à la Faculté de médecine en vue de l'obtention du grade de
philosophiae doctor (Ph.D.) Copyright 30 mars 1990
Canada to microfilm this thesis and to lend or aell copies of the film.
The author (copyright owner) bas reserved other
publication rights, and neither the thesis nor extensive extracts from it may be printed or otherwise reproduced without hie/ber written permission.
du Canada de aicrof ilmer cette thèse et de prêter ou de vendre des exemplaires du
film.
L'auteur (titulaire du droit d'auteur) se réserve les autres droits de publication: ni la thèse ni de longs extraits de celle-ci ne doivent être imprimés ou autrement reproduits sans son autorisation écrite.
Un grand sage a dit: "Il ne faut pas forcer la Nature
àcoïncider avec nos théories, mais plutôt la laisser venir
à
nous. Ce qui est apparemment fantastique et
irration-nel change alors d'aspect ... "
A ma fille Lison,
A ceux qui ont cru en moi,
A ceux qui se reconnaissent .•.
Table des matières Table des matières
Liste des figures Liste des tableaux Liste des abréviations Résumé
Introduction
A - Généralités sur les protéases B - Protéase adénovirale
c -
Projet Matériel et méthodes A - Manipulations de virus i iii iv V vii 1 1 6 12 14 141 - Cellules et infection virale 14
B
-2 - Préparation du substrat 14
3 - Purification d'ADN viral 15
4 - Isolement de noyaux de cellules infectées 15 5 - Essais de la protéase nucléaire in vitro 17
Manipulation de l'ADN 18
6 - Enzymes de restriction, de modification
d'ADN et ligase T4 18
7 - Bactéries et plasmides 18
8 - Electroélution d'ADN 19
9 - Préparation des cellules compétentes 21
10 - Transformation d'ADN double brin 22
11 - Fabrication d'un stock de phages 23 12 - Minipréparation de plasmides
ou de formes réplicatives 24
13 - Amplification de plasmide 24
14 - Transfert d'ADN sur
membrane (Southern blot) 24
16 - Isolement de l'ADN simple brin et
réactions de séquence
17 - Oligonucléotides
18 - Gel de polyacrylamide pour le
séquençage d'ADN
ii
25
25
26
c -
Expression
27
19 - système d'expression
27
20 - Induction de l'expression
27
21 - Purification de la protéine fusion
29
22 - Transfert de protéines et iodination
30
23 - Séquençage N-terminal de protéines
30
24 - Marquage des protéines induites
31
25 - Enrichissement des corps d'inclusion
31
Résultats
33
A - Complémentation in vitro des protéases de
divers sérotypes
33
B - Séquençage du gène de la protéase de Ad3 et Adl2
40
c -
Expression
51
- Expression de la protéase dans
~coli
52
- Clonage de la protéase dans le vecteur
inductible pRIT2T
55
- Expression de la protéase par le
vecteur pRIT2T
57
- Purification et activité protéolytique
60
- Spécificité de clivage
65
- Optimisation de l'expression et
enrichissement de l'enzyme
71
- Expression de la protéase par le
vecteur tronqué
74
D - Propriétés générales de l'enzyme
81
Discussion
87
- Conclusions
108
Remerciements
110
Bibliographie
112
Liste des f iqures
1 - Montage utilisé pour l'électroélution
d'un fragment d'ADN 20
2 - Carte génétique du plasmide d'expression pRIT2T 28 3 - Clivage in vitro des précurseurs viraux marqués
à la méthionine- [ 35S
J
3 74 - Localisation des segments d'ADN séquencés par rapport aux gènes des protéases de Adl2
(a) et de Ad3 (b) 42
5 - Séquence nucléotidique (brin 1) du gène de la protéase de Adl2 (souche Pereira 1131) et
la séquence déduite d'acides aminés 44
6 - Séquence nucléotidique (brin 1) du gène de la protéase de Ad3 et la séquence déduite
d'acides aminés 46
7 - Comparaison de la séquence en acides aminés
des protéases adénovirales 50
8 - Construction du vecteur d'expression 56
9 - Mise en évidence de l'expression d'une
protéine recombinante après induction 59
10 - Purification de protéines par immune-affinité 62
11 - Autoclivage de la protéase recombinante 64
12 - Clivage spécifique des précurseurs de
H2tsl-[35S] par la protéase recombinante 66 13 - Clivage en trans de la protéase recombinante 68 14 - Profil d'inhibition et clivage de l'ovalbumine
de poule par la protéase recombinante 70 15 - Marquage à la méthionine-[35S] des protéines
induites à partir de différents
vec-teurs. 75
16 - Immuno-blots avec des sérums polyclonaux
anti-protéase 78
iv
Liste des tableaux
1 - Classification des protéases
3
2 - Protéases virales
5
3 - Classification des adénovirus humains
7
4 - Clivage intersérotypique des précurseurs
chez les virus recombinants
35
5 - Clivage in vitro du précurseur préVII de H2tsl
39
6 - Composition en acides aminés des protéases
48
7 - Profil d'inhibition de la protéase virale
83
8 - Comparaison des sites potentiels de clivage
relevés dans !'adénovirus avec les sites
déjà caractérisés in vivo
85
9 - Alignement des sites de clivage de protéases
de différentes origines
86
10 - Comparaison des signaux de régulation de la
Ad:
ADN:
ADNc:
ARN:
ARN-m:
BSA:
CPE:
DFP:
DMEM:
D. 0. :EDTA:
FMDV:
g:HIV-1:
H2tsl:
IgG:
IPTG:
k: kb: kd:LB:
LBA:
mA:
mci:
MCS:
mg:
MIM:
ml:
M.M.:
mm:
Liste des abréviations
Adénovirus
Acide déoxyribonucléique
ADN complémentaire ou copie
Acide ribonucléique
ARN messager
Albumine sérique de boeuf
Effet cytopathique
Diisopropylfluorure
Milieu Eagle modifié par Dulbecco
Densité optique
Ethylènediamine tétraacétate
Foot-and-mouth disease virus
Unité de force centrifuge
Virus de l'immunodéficience humaine
Mutant tsl de l'Adénovirus de type 2 humain
Immunoglobuline de classe G
Isopropyl-B-thio-galactoside
Kilodalton
Kilobase
Kilodalton
Milieu Luria-Bertani
Milieu Luria-Bertani
+ampicilline
Milliampère
Millicurie
Site de clonage multiple (multiple cloning site)
Milligramme
Milieu d'induction maximale (Maximum Induction
medium)
Millilitre
Masse moléculaire
Millimètre
MOI:
NEM:
ng:
nm:
NP-40:
N. S. :Multiplicité
infection)
N-éthylmaléimide
nanogramme
nanomètre
Nonidet P-40
Non synthétisé
vi
d'infection
(multiplicity
of
PAGE-SDS: Gel de polyacrylamide-sulfate dodécyl de sodium
p .b.:
PCMB:
PFU:PI:
PMSF:
pTP:
P/V:
SBTI:
SDS:
TB:
TBE:
TE:
TP:
TPCK:
ts:
TST:
µCi:
µg:
µl:vs:
V/V:
X-Gal:
Paire de bases
p-chloromercuribenzoate
Unité de formation de plage (Plaque forming unit)
Point isoélectrique
Phénylméthylsulfonylfluorure
Précurseur de la protéine terminale liée
àl'ADN
Poids/volume
Inhibiteur de la trypsine de la fève de soya
Sulfate dodécyl de sodium
Bouillon terrif ique (Terrif ic Broth)
Tampon Tris-HCl 50
mMpH 8,0, Borate 50
mMEDTA 1
mMTampon Tris-HCl 10
mM,EDTA 1
mMProtéine terminale (Terminal protein)
L-l-Tosylamine-2-phényléthylchlorométhylcétone
Thermosensible
Tampon Tris-Hel 50
mMpH 7,6, NaCl 150
mM,Tween 80 0,5%
Microcurie
Microgramme
Microlitre
Versus
Volume/Volume
5-bromo-4-chloro-3-indolyl-B-galactoside
Résumé
ces travaux sur la protéase adénovirale ont permis de démontrer que les enzymes de Ad2 et Ad5 sont interchangeables in vivo par analyse de recombinants intersérotypiques. Des tests de clivage in vitro sur les précurseurs de H2tsl par des membres du sous-groupe c (Adl, 2, 5 et 6), du sous-groupe B (Ad3 et 7), du sous-groupe E (Ad4) et du sous-groupe D( Ad9) ont confirmé la présence de cette enzyme chez 3 nouveaux sous-groupes et ont montré que tous ces sérotypes peuvent cliver préVII aussi efficacement que leur niveau d'infection le permet. Des expériences de séquençage de Ad3 (sous-groupe B) et de Adl2 (sous-groupe A) ont permis de comparer la séquence de la protéine avec celles de Ad2 et Ad5 (sous-groupe C), de Ad4 (sous-(sous-groupe E), de Ad40 (sous-(sous-groupe F) et de Ad41 (sous-groupe G) afin de toucher à tous les adénovirus humains. Ces protéines montrent une homologie brute de 58,8% qui atteint 72, 0% en tenant compte des changements semi-conservatifs. L'analyse de séquences réglant la traduction de la protéase chez ces sérotypes semble établir une corréla-tion directe avec l'efficacité de produccorréla-tion de particules infectieuses. Tous ces résultats suggèrent que la protéase adénovirale représente une cible de choix pour une chimiothé-rapie contre tous les adénovirus par l'utilisation d'une seule drogue spécifique. Cette protéase peut être produite par~ coli mais s'avère toxique lorsqu•exprimée d'un vecteur faiblement contrôlé. L'enzyme mature par une autocatalyse en trans et maintient sa spécificité de clivage pour ses subs-trats naturels. Ainsi, la présence de protéines virales, de protéines cellulaires ou encore de modifications post-traduc-tionnelles spécifiques aux cellules d'eucaryotes ou même une phosphorylation ne sont pas absolument essentielles. Des protéines autres que les substrats naturels peuvent aussi être hydrolysées spécifiquement. Un système d'expression,
viii d'enrichissement de l'enzyme et de criblage par anticorps a été mis au point et permet d'envisager la purification à grande échelle afin de poursuivre l'étude de cette protéine. Le site actif usuel des sérine-protéases GDSGG est absent et la protéine n'a présentement aucun homologue par hybridation d 'ADN ou par analyse de banques de séquences et pourrait ainsi représenter une nouvelle classe d'enzyme. Son profil d'inhibition non-classique l'associe aux cystéine-protéases de type chymotrypsine qui représentent un intermédiaire évolutif et structural entre les cystéine-protéases et les sérine-protéases.
INTRODUCTION
A - Généralités sur les protéases
Il n'y a pas si longtemps, les deux seuls rôles attribués aux protéases étaient de digérer les protéines nutritives et de participer au renouvellement des protéines cellulaires. La défini tien de ces fonctions provenaient de 1 'étude des protéases les plus connues comme la trypsine, la chymotryp-sine, la pepsine et les enzymes lysosomales, cathepsine B et D. Plus récemment, il a été démontré que la protéolyse limitée, par opposition à la dégradation, a un rôle-clé dans divers processus cellulaires (Holzer et Heinrich, 1980; Holzer et Tschensche, 1979; Bond et Butler, 1987). L'habi-leté des enzymes protéolytiques à faire des modifications sélectives de protéines par des clivages limités signifie que les protéases ont aussi une fonction de régulation comme dans les cas de coagulation sanguine, de l'activation d'hormone, de l'activation du complément, de la fertilisation et de la
fibrinolyse (Kay et Dunn, 1990) . La protéolyse restreinte est aussi un processus important pour diverses voies biosynthéti-ques et métabolibiosynthéti-ques d'une cellule dans le transport post-traductionnel de polypeptides sécréteurs au travers des membranes microsomales, dans le transport post-traductionnel de polypeptides à l'intérieur des organelles, dans l'élimination des protéines anormales ou non fonctionnelles,
2
dans la rapidité d'adaptation des compléments de protéines en fonction des besoins physiologiques de la cellule (Schoe-nheimer, 1942; Pine, 1980; Kreil, 1981). L'activité protéasique est ubiquite et a été détectée dans plus de 150 genres de bactéries, de protozoaires, de moisissures, de levures et de cellules d'eucaryotes supérieurs (North, 1982; Jones, 1984; Bond et Butler, 1987). Les protéases sont cataloguées en 4 grands groupes: les aspartate-protéases, les métallo-protéases, les cystéine-protéases et les sérine-protéases. Certains inhibiteurs-type ou inhibiteurs diagnos-tiques permettent d'associer une protéase à la famille à
laquelle elle appartient (cf. tableau 1) . Par exemple, l'inhibition d'une protéase par le diisopropylfluorure (DFP) ou encore, quoique moins efficacement, par le phénylméthyl-sulfonylfluorure (PMSF) catalogue celle-ci dans la classe des sérine-protéases.
Plus récemment, la protéolyse limitée a aussi été mise en évidence au cours du cycle infectieux d'une multitude de virus (3 revues Wellink et van Kammen, 1988; Krausslich et Wimmer, 1988; Kay et Dunn, 1990) . Cette activité a été associée à certains segments du matériel génétique viral qui ont été clonés dans plusieurs cas en vue de caractériser l'enzyme. Les protéases virales sont généralement impliquées dans le clivage de polyprotéines afin de produire les composants structuraux et non structuraux matures essentiels
Type of. Examples Diagnostic inhihitors active site
Aspartic pcpsin, gastricsin,
renin, cathepsin D, pepstatin. cathcpsin E.
Cysteine papain, cathepsin L, E-64; cystatins, cathepsin B, leupeptin,
cathcpsin H. p-chloromercuribcnzoate. Met allo collagenascs, meprin; EDTA, phcnanthroline,
thermolysin, EC 24.11 phosphoramidon. Serine trypsin-like, DFP, lcupcptin,
chymotrypsin-like, DFP, chymostatin, clastase-Jike. DFP, c1astatinal.
Tableau 1. Classification des protéases en 4 groupes selon
des inhibiteurs caractéristiques. Tiré de Kay et
Dunn, 1990.
4
à la réplication et l'assemblage des particules virales. Elles sont aussi responsables de la régulation du cycle infectieux en produisant précisément dans le temps la bonne proportion des protéines nécessaires à la poursuite de 11 infection. La haute spécificité de ces enzymes permet
d'espérer le développement d'une chimiothérapie contre les virus pathogènes, de les utiliser comme outil dans la tech-nologie de manipulation de protéines et d'étudier un nouveau mécanisme de régulation post-traductionnelle de l'expression. Les protéases virales les mieux caractérisées sont regroupées au tableau 2. Il est intéressant de noter que cette activité n'est pas seulement liée aux virus à acide ribonucléique (ARN) et que les protéases virales sont réparties dans tous les groupes exception faite des métallo-protéases. On retrouve les aspartate-protéases chez les rétrovirus, les caulimovirus (virus de plante) et les hepadnavirus, les cystéines-protéases chez les picornavirus, les comovirus (plantes) et les poty-virus (plantes) et les sérine-protéases chez les flavipoty-virus, les togavirus et les adénovirus. Les adénovirus ont été jusqu'à tout récemment les seuls virus eucaryotiques à acide déoxyribonucléique (ADN) à posséder une protéase. Cette activité a ensuite été rapportée chez les poxvirus (Yang et al., 1988; Yang et Bauer, 1988) et les papovavirus (Bowen et al., 1984). Etant donné que les virus à ADN produisent des ARN messagers (ARN-m) monocistroniques par épissage, donc
Virus Genetie Name/ Proteinase material protein type Adeno DNA Adeno 23K serine Toga RNA Sindbis nsP2 serine (Alpha) RNA Semliki forcst serine (Pesti) RNA Bovine Viral
Diarrhoca serine Flavi RNA Yellow Fever NS3 serine? Pi coma RNA Polio 2A cysteine
(~ntero/ RNA Rhino2A cysteine
Rhino) RNA Polio 3C cysteine RNA Rhino 3C cysteine RNA Hepatitis A 3C cysteine (Cardio) RNA EMCV3C cysteine (Aphtho) RNA FMDV LProtein cysteine?
FMDV3C cysteine Corno RNA Cowpea Mosaic 24K cysteine Poty RNA Tobacco Etch Nia cysteine RNA Plum Pox Nia cysteine Retro RNA HIV-1 PR aspartic RNA HIV-2 PR aspartic RNA MoMLVPR aspartic RNA HTLV-1 PR aspartic RNA BLVPR as partie
RNA RSVPR aspartic
RNA MAVPR aspartic
Caulimo DNA Cauliflower Mosaic aspartic? Hepadna DNA Hepatitis B as partie?
Tableau 2. Liste des protéases virales les mieux
caractéri-sées. Tiré de Kay et Dunn, 1990.
6 n'expriment aucune polyprotéine, la protéase codée par ces virus pourrait jouer un rôle différent de ceux déjà men-tionnés.
B - Protéase adénovirale
Les adénovirus (revue Philipson, 1984; Horwitz, 1985) sont des virus oncogènes à ADN double brin linéaire d'un poids moléculaire de 2
o
à 2 3 X 106 daltons et qui contiennentenviron 35000 à 36000 paires de base (p. b.) (Green et al., 1967; Van der Eb et al., 1969). L'extrémité 5 1 de chaque
brin du génome viral est liée de façon covalente à un polypep-tide viral, la protéine terminale (TP) • L'expression du matériel génétique de ce virus pendant son cycle infectieux est divisée en deux phases, précoce et tardive, et les gènes viraux sont transcrits à partir des deux brins d'ADN (Tooze,
1981). Les adénovirus humains ont été classifiés en 7 sous-groupes (cf. tableau 3). Les critères de classification sont le pourcentage en G
+
C et les homologies d'ADN, les groupes d'hémagglutination avec les érythrocytes de rat et de singe et leur potentiel oncogénique.Les adénovirus humains infectent différentes cellules du système respiratoire, du système gastro-intestinal et des yeux. Les infections adénovirales ont été associées à des
Oncogenic potential
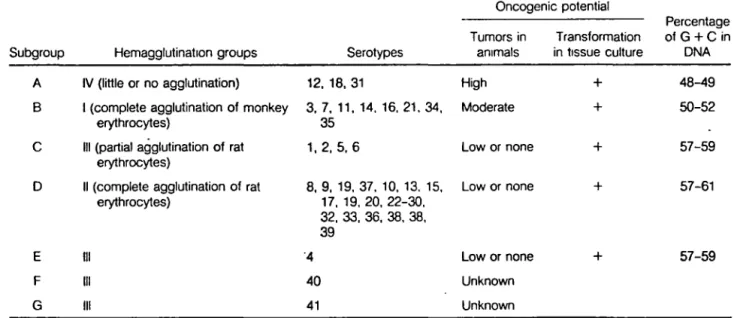
Percentage Tumors in Transformation of G+C in Subgroup Hemagglutinat1on groups Serotypes animais in tissue culture DNA
A N (little or no agglutination) 12, 18, 31 High + 48-49
8 1 (complete agglutination of monkey 3, 7, 11, 14, 16, 21. 34, Moderate + 50-52 erythrocytes) 35
c
Ill (partial agglutination of rat 1,2, 5,6 Low or none + 57-59 erythrocytes)D li (complete agglutination of rat 8, 9, 19, 37. 10, 13. 15, Low or none + 57-61 erythrocytes) 17, 19. 20, 22-30.
32, 33, 36, 38. 38, 39
E Ill ·4 Low or none + 57-59
F Ill 40 Unknown
G Ill 41 Unknown
Tableau 3. Classification en sous-groupes des adénovirus
humains selon Horwitz, M.
s.
dans Fields, B. N.et al. Virology. Raven Press. New York. 1985.
8
amygdalites, des pharyngites, des pneumonies, des maladies
respiratoires aiguës (acute respiratory disease) , des
gastro-entérites aiguës, des conjonctivites, des
kérato-conjonctivi-tes et
àla fièvre pharyngo-conjonctivale principalement chez
les jeunes enfants et les recrues militaires (Horwitz, 1985).
Le cycle infectieux de ces virus est lytique et permet de
libérer les pentons non utilisés qui sont toxiques pour les
cellules.
Des lésions dans un tissu peuvent ainsi être
occasionnées. Dans les cas de complications, les cellules
nerveuses peuvent être atteintes provoquant ainsi une
méningo-encéphalite. Certaines de ces maladies peuvent être létales
pour les jeunes enfants ou encore causer de nombreux
désagré-ments
àtous les âges.
En 1975, un mutant thermosensible de Ad2 déficient dans
une fonction tardive fut mis en évidence (Bégin et Weber,
1975; Weber et al., 1975) Ce mutant, appelé H2tsl ou Ad2tsl,.
est défectif dans le clivage des peptides précurseurs pVI et
pVIII qui deviennent, après clivage, les protéines de capsides
VI et VIII (Bhatti et Weber, 1979), et des précurseurs pVII,
87k et llk qui deviennent les protéines liées
àl'ADN, VII,
55k (TP) et X ou
µ(Walter et Maizel, 1974; Weber, 1976;
Weber et al., 1977; Lishwe et Sung, 1977; Challberg et al.,
1980; Lewis et Mathews, 1980; stillman et al., 1981; Weber
et Anderson, 1988; Anderson et al., 1989). Les particules
virales, produites
à39°C, sont non infectieuses et réfèrent
aux virions immatures déjà observés pendant le cycle infectieux de l' adénovirus (Ishibashi et Maizel, 1974; Weber, 1976). Il y a aussi de fortes évidences suggérant que la protéolyse survient entre une glycine et une alanine ou après le dipeptide glycine-glycine. Mais tous ces liens ne sont pas nécessairement hydrolysés car on observe une seule protéine mature par précurseur (Akusjarvi et Persson, 1981; Tremblay et al., 1983; Weber et Anderson, 1988; Anderson et al., 1989) et aucun clivage des autres protéines du système in vitro (Tremblay et al., 1983).
Les ARN-m codants pour les polypeptides structuraux tardifs fonctionnels sont transcrits à partir du brin r du génome viral et peuvent être groupés en 5 familles (Ll à L5) de séquence (Ziff et Fraser, 1978). Chaque famille d'ARN-m a en commun la position de leur site de polyadénylation à l'extrémité 3 ' de 1 'uni té de transcription et la même séquence de tête (leader) en trois parties à l'extrémité 51 • La
séquence de tête est obtenue par un épissage qui conserve les séquences positionnées à 16, 3, 19, 7 et 26, 5 en unités de génome (Chow et al., 1977; Berget et al., 1977). La mutation tsl a été localisée par des expériences de recombinaison et de séquençage à la position 61,1 dans l'unité de transcription L3 . Cette mutation ponctuelle correspond à la transi tien d'un C en T (Yeh-Kai et al., 1983) changeant une proline en leucine et est située dans un cadre de lecture de 612 nucléotides
10 découvert précédemment chez Ad2 et Ad5 (Kruijer et al., 1980; Akusjarvi et al., 1981) codant pour une protéine de 23k. La famille L3 est comprise entre les coordonnées 50,08 (Akusjarvi et Persson, 1981) et 62,45 (Akusjarvi et al., 1981; Le Moullec et al., 1983) en unités génomiques et code pour 3 polypeptides, pVI de 50,09 à 52,17 (Sung et al., 1983), II (hexon) de 52,41 à 60,5 (Jornvall et al., 1981; Akusjarvi et al., 1981) et 23k de 60.0 à 61,7 (Akusjarvi et al., 1981).
L'activité protéasique a été détectée dans les virions matures, dans les noyaux de cellules infectées (Bhatti et Weber, 1979a) et associée aux matrices nucléaires et à la chromatine virale (Everitt et Ingelman, 1984; Khittoo et al., 1986). La fonction de cette enzyme est de convertir les jeunes virions non infectieux en virions matures infectieux par le clivage des précurseurs. L'analyse de 17 révertants spontanés de Ad2tsl obtenus de 3 expériences indépendantes et produisant un patron protéique normal a révélé que tous les révertants retrouvent leur activité protéasique par une réversion vraie suggérant que la transition C/T est la seule cause du phénotype mutant et n'est pas suppressible (Weber et Houde, 1987) .
La protéase adénovirale de type sauvage a été soumise à divers tests biochimiques. L'activité maximale est située entre les pH 6,5 et 7,0 et est stable jusqu'à 45°C. L'enzyme
reconnaît son substrat même dénaturé et la synthèse de
nouvelles protéines n'est pas nécessaire pour que le clivage
se poursuive
àla même vitesse car la cycloheximide n'a aucun
effet sur elle (Weber, 1976).
L'activité enzymatique est
inhibée par le phénylméthylsulfonylfluorure (PMSF) , par le
L-l-tosylamine-2-phényléthylchlorométhylcétone (TPCK) et le
diisopropylfluorophosphate (DFP) signifiant alors une activité
protéolytique de type chymotrypsine d'une sérine-protéase mais
ce n'est pas une métallo-enzyme car l'éthylènediamine
tétra-acétate (EDTA) n'a aucun effet sur elle (Bhatti et Weber,
1978; Tremblay et al., 1983). La présence physique de la
protéine n'a pas encore été démontrée étant donné la rareté
de son ARN-m (environ 1000 fois moins que l'hexon).
Larsen et son groupe (1979) a rapporté qu'il existe
seulement 2 régions distinctes d'homologie entre les génomes
de Ad2 et l 'adénovirus murin FL. Celles-ci sont situées entre
les coordonnées 12
à18% et 51
à62% en unités génomiques de
Ad2. Cette donnée est intéressante car elle signifie que la
région de la protéase (60,0
à61,7) est homologue du point de
vue ADN entre un adénovirus humain et murin. La comparaison
en acides aminés des adénovirus humains 2, 5 (sous-groupe C)
et 4 (sous-groupe E) montre une homologie brute de 72,1% qui
atteint 81,4% en tenant compte des changements conservatifs
12
C - PROJET
Les études sur le mutant H2tsl ont démontré que la protéase responsable de la maturation des adénovirus est codée dans l'unité de transcription L3 et qu'elle est à l'origine d'une fonction essentielle du cycle infectieux. Cette enzyme, très spécifique dans le choix et le clivage des protéines, semble agir comme les enzymes de restriction sur 1 'ADN et représente un intérêt pour la manipulation des protéines. L'importance de la caractérisation de la protéase adénovirale devient alors évidente.
En vue d'évaluer le potentiel de cette enzyme à servir de cible pour un traitement de chimiothérapie, divers séroty-pes seront testés in vitro sur les précurseurs de H2tsl. Ces expériences ont pour but de vérifier si les protéases adénovirales ont une spécificité différente pour chaque sérotype, pour chaque sous-groupe ou si cette caractéristique est commune et universelle. Pour compléter cette étude, les gènes de la protéase des sérotypes 3 (sous-groupe B) et 12 (sous-groupe A) seront séquencés, traduits en acides aminés et comparés avec les séquences publiées de Ad2 et Ad5 (sous-groupe C), Ad 4 (sous-(sous-groupe E), Ad40 (sous-(sous-groupe F) et Ad41
(sous-groupe G) (Kruijer et al, 1980; Houde et Weber, 1987; Akusjarvi et al, 1981; Vos et al., 1988).
Toutes les études portant sur l'activité biologique de la protéase adénovirale ont été faites avec des lysats cellulaires, des noyaux de cellules infectées ou le virus lui-même, et personne n'a vraiment encore démontré la présence physique de cette protéine. La puissante technologie de 1 'ADN recombinant sera utilisée afin de mettre au point un système d'expression. Etant donné la nature eucaryotique de la protéine, nous avons cru au départ devoir utiliser un vecteur d'expression eucaryote mais la détection de l'activité protéasique adénovirale dans un intermédiaire de clonage procaryotique a réorienté le projet. Afin de contourner le problème de toxicité de la protéase dans les bactéries, un vecteur inductible, produisant une protéine fusionnée à la protéine A de Staphylococcus aureus et permettant une purif i-cation par immune-affinité sera utilisé. Les caractéristiques de 1 'enzyme recombinante seront comparées à celles de 1 'enzyme virale. Après avoir acquis la certitude que la protéase adénovirale peut être produite adéquatement dans le système, la queue protéine A du vecteur sera délétée. Ce nouveau plasmide permettra une expression plus élevée d'une protéine qui ressemble davantage à l'enzyme virale et qui peut être enrichie sommairement par isolement de corps d'inclusion. L'utilisation du vecteur tronqué permettra aussi de tester les sérums polyclonaux produits par Lyne Delorme contre des peptides synthétiques provenant des régions hydrophiles de la protéase.
14
MATERIEL ET METHODES
A - Manipulations de virus
1 - Cellules et infection virale
Des cellules de type Hep-2 (Moore et al., 1955) sont cultivées dans des pétris de culture cellulaire (100 mm) dans du milieu de Eagle modifié par Dulbecco (DMEM) (Dulbecco et Freeman, 1959) supplémenté avec 10% V/V de sérum de veau. La couche monocellulaire est infectée lorsqu'elle atteint 90 à 95% de confluence avec une multiplicité d'infection (MOI) de 10 unités de formation de plage/cellule (PFU, plaque forming unit), à moins que ce ne soit décrit autrement, et la concentration de sérum est ensuite abaissée à 2,5% V/V.
2 - Préparation du substrat
Des cellules Hep-2 sont infectées tel que décrit précédemment avec 1 'adénovirus de type 2 mutant tsl (H2tsl ou Ad2tsl) (Weber, 1976). Les cellules infectées sont incubées à 39°C pendant 20 heures, puis les protéi-nes virales sont marquées avec de la méthionine- [ 358
J
millitre (µCi/ml) pendant 3 heures. Après le marquage, 8 ml de milieu DMEM sont ajoutés à chaque pétri afin de combler les besoins cellulaires jusqu'à l'obtention de l'effet cytopathique {CPE), habituellement 48 heures après l'infection. Les cellules sont ensuite récoltées et lysées par 6 cycles de gel-dégel. Les particules virales présentes dans le surnageant sont concentrées sur gradient de chlorure de césium {CsCl) préformé (1,2-1,5 g/ml) par une ultracentrifugation à 180000 g pendant lj heure à 4°C (Weber, 1976; Khittoo et Weber, 1977). La bande virale à 1,345 g/ml est récoltée par aspiration, remise sur un gradient autoformé de CsCl (1,4 g/ml) et ultracentrifugée à 100000 g pendant 18 heures à 4°C. Les particules virales isolées sont ensuite dialysées contre un tampon de Tris-Hel 10 mM pH 8,0 et EDTA 1 mM (TE lX) à 4°C afin d'éliminer le sel. Les capsides sont éclatées par une traitement à la pyridine 10% V/V pendant 30 minutes. Après une autre dialyse contre du TE lX, le
substrat de H2tsl radioactif est prêt pour les tests in vitro.
16
3 - Purification d'ADN viral
Les virus, purifiés de la même façon que Ad2tsl (cf.
section ci-dessus), sont dilués dans un tampon Tris-Hel
10 mM pH 7,5, KCl 15 mM et sulfate dodécyl de sodium
(SDS) 0,5% P/V et traités avec de la protéinase K (500
µg/ml)
à37°C pendant 1 heure. L'ADN viral est extrait
2 fois avec du phénol, saturé avec du Tris-HCl 100 mM pH
7, 5, 2 autres fois avec du chloroforme-isoamylalcool
(24:1) et dialysé contre du TE lX.
4 - Isolement de noyaux de cellules infectées
Des cellules Hep-2 sont infectées comme précédemment
décrit avec Adl, Ad2, Ad3, Ad4, Ad5, Ad6, Ad7 et Ad9
à50 PFU/cellule pendant 20 heures
à39°C. Les cellules
sont lysées dans un tampon Tris-HCl 10 mM pH 7,8/KCl 5
mM/MgC1
2O, 5 mM/B-mercaptoéthanol lmM et Nonidet-P40
(NP40) 0,1% V/V
à4°C par agitation au vortex jusqu'à ce
que 95% de lyse soit observée par microscopie
àcontraste
de phase (2
à5 minutes). Les noyaux sont centrifugés
à 1000 g, lavés et resuspendus dans le tampon de lyse
sans NP40 (120 µl/pétri). Afin de mesurer la qualité de
1
1infection, des extraits cellulaires de chacune des
infections sont préparés en ajoutant 1 ml de solution de
lyse-SDS (Tris-HCl 0,05 M pH 6,8/SDS 1% P/V/
B-mercaptoéthanol 1 mM/glycérol 10% V/V). Ces échantil-lons sont récoltés, bouillis pendant 3 minutes et prêts pour l'électrophorèse sur gel de polyacrylamide-SDS (SDS-PAGE) (Maizel, 1969).
5 - Essais de la protéase nucléaire in vitro
Les noyaux isolés de cellules infectées et non infectées (mock) sont soniqués pendant 5 secondes à 40 cycles (Sonic Dismembrator-Artek Co.) et incubés pendant 16 heures à 37°C avec le substrat (H2tsl-35S) (Tremblay et
al., 1983). Les réactions sont arrêtées par addition de la solution de lyse-SDS et bouillies pendant 3 minutes. L'activité protéasique est détectée en mesurant la conversion de la protéine préVII en VII sur gel de SDS-PAGE (Maizel, 1969).
B - Manipulations de l'ADN
6 - Enzymes de restriction, de modification d'ADN et ligase T4
18
Les enzymes de restriction, de modification d'ADN et la ligase T4, achetées chez Pharmacia et Amersham, ont été utilisées respectivement tel que recommandées par leur fabricant.
7 - Bactéries et plasmides
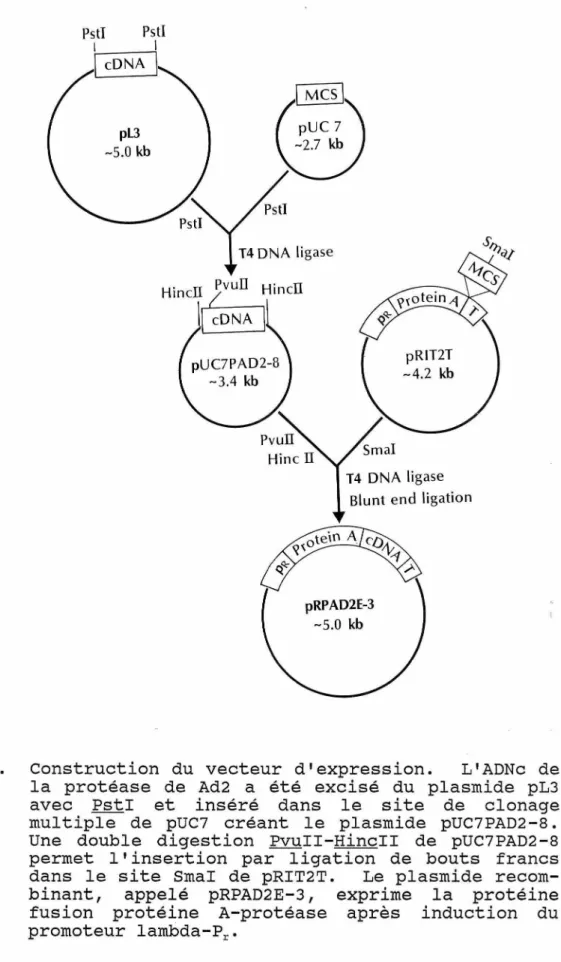
Le plasmide pL3, décrit par Yeh-Kai et son groupe en 1983, contient l'ADN complémentaire (ADNc) du gène de la protéase de Ad2 inséré au site PstI de pBR322. Ce plasmide a été utilisé pour le clonage de la protéase dans un vecteur d'expression et comme source d'ADN pour la préparation de sondes spécifiques à la protéase. Le plasmide pBR3 2 2 a servi de vecteur de clonage pour enrichir le fragment HindIIIC de Ad3 d'environ 4, 5 kilobases (kb) dans les expériences de séquençage. Pour le sous-groupe A, le fragment Adl2 BamHIC de la souche Pereira 1131 inséré au site BamHI de pBR322 nous a été
fourni par le docteur Stanley Mak. Tous ces plasmides sont contenus dans ~ coli HBlOl décrit par Bolivar et
Backman en 1979.
Le plasmide pUC7 nous a été fourni par le docteur J. Messing (Messing et al., 1981) . Les formes réplicati-ves des phages Ml3mpl8 et Ml3mpl9 de même que l'hôte, ~
coli JM109, ont été achetés chez Pharmacia.
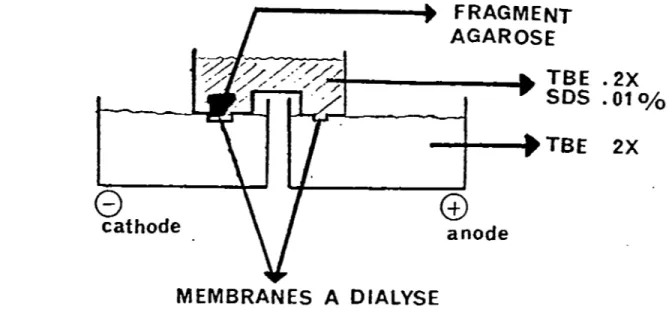
8 - Electroélution d'ADN
Après digestion de l'ADN par une enzyme de restric-tion, le produit réactionnel est déposé sur un gel d'agarose à base de TBE (Tris-HCl 50 mM pH 8,0, Borate 50 mM et EDTA 1 mM) (Maniatis et al., p. 150-162). La bande d'agarose correspondant au fragment d'ADN désiré est découpée. Des membranes à dialyse d'une porosité de 12 kilodaltons (kDa) sont fixées sur les 2 extrémités de la microcuvette (Iseo #68-1754-010) (cf. figure 1) en s'assurant qu'il n'y a pas de fuites. La cuvette est ensuite remplie avec 10 ml de TBE 0,2X, SDS 0,05% P/V et la bande d'agarose-ADN est déposée dans le gros puits correspondant à l'extrémité cathodique. Les bassins extérieurs sont remplis de TBE 2X et le courant ajusté à 10 milliampères (mA) par cuvette d' électroélution.
FRAGMENT
AGAR OSE
~---_.TSE.2X
SOS .
010;0
-+----TSE 2X
8
cathode
0
anode
MEMBRANES A DIALYSE
Figure 1. Montage utilisé pour 11 électroélution d'un fragment d'ADN dans une microcuvette.
Après 45 minutes, la polarité du courant est inversée 30
secondes afin de décoller l 'ADN de la membrane, puis
200µ1 sont recueillis dans le fond de la cuvette à
1
1extrémité anodique.
Cette étape est répétée deux
autres fois et l'ADN est précipité avec 1 volume
d'iso-propanol.
9 - Préparation de cellules compétentes
Une colonie bactérienne est ensemencée dans 5 ml de
milieu Luria-Bertani (LB) (Maniatis et al., p. 68) et
incubée avec agitation à 3 7°C jusqu'à une densité optique
(D.O.) à 550 nanomètres (nm) de 0,3. La culture est
alors diluée 20 fois et incubée de la même façon jusqu'à
une D.O. de 0,48. La suspension est centrifugée à 5000
g pendant 5 minutes à 4°C et le culot est repris dans 40
ml de tampon TfbI ( Acétate de potassium 30
mM,RbC1
2100
mM,CaC1
210
mM,MnC1
250
mM,glycérol 15% V/V, pH 5,8)
et gardé à 4 °C pendant 5 minutes.
Après une autre
centrifugation dans les mêmes conditions, le culot est
repris dans 4 ml de TfbII (MOPS 10
mM,CaC1
275
mM,RbC1
210
mM,glycérol 15% V/V, pH 6,5). Cette suspension est
gardée sur glace 15 minutes, aliquotée par échantillons
de 200 µl, gelée dans l'azote liquide et conservée à
-70°C jusqu'à utilisation.
22 10 - Transformation d'ADN double brin
Les bactéries compétentes sont dégelées et gardées sur glace pendant 10 minutes. L'ADN double brin, soit sous forme de plasmide ou de formes réplicatives du phage Ml3, ne doit pas excéder 100 ng et/ou un volume de 40 µl pour 200 µl de cellules. L'ADN est ajouté à la suspen-sion qui est gardée sur glace pendant 30 minutes. Les bactéries sont alors soumises à un choc thermique à 42°C pendant 90 secondes et remises sur glace 2 minutes. Quatre volumes de LB sont ajoutés aux transformants et la suspension est incubée à 3 7°C pendant une heure. Après centrifugation, le culot bactérien est repris dans 200 µl de milieu LB et prêt pour l'étalement.
L'étalement diffère selon la transformation de plasmide ou de formes réplicatives du phage Ml3. Dans le cas de plasmides, les cellules transformées sont étalées sur un pétri de milieu LB supplémenté de l'anti-biotique approprié pour la sélection. L' étalement de bactéries transformées par les formes réplicatives du phage Ml3 nécessite la préparation de 3 ml d'agar mou pour chaque pétri. A chacun de ces tubes fondus au micro-onde et tempérés à 45°C sont ajoutés 10 µl d'iso-propyl B-thio-galactoside (IPTG) 10 mM, 40 µl de 5-bromo-4-chloro-3 indolyl-B-galactoside (X-Gal) 2% (P/V), 200
µl de ~ coli JM109 en phase logarithmique de croissance et 40 µl de bactéries transformées. Les tubes sont bien mélangés puis vidés rapidement sur pétri. Une fois l'agar de surface solidifié, les pétris sont incubés à
37°C pendant la nuit.
11 - Fabrication d'un stock de phages
Une colonie de ~ coli JM109 provenant d'un pétri de milieu minimum est ensemencée dans 2 ml de milieu et incubée à 3 7°C avec agitation pour la nuit. Le len-demain, la culture est diluée 100 fois et réincubée pendant 5 à 6 heures. Ensuite, 1 ml de bactéries est infecté avec une plage prélevée à l'aide d'une pipette pasteur stérile sur le pétri d'étalement. La suspension est incubée à 37°C pendant 1 heure pour favoriser l'adsorption et la pénétration des phages. Après l'incubation, 8 ml de milieu LB sont ajoutés aux bacté-ries infectées et cette culture est incubée à 37°C avec agitation pendant la nuit. La suspension est centrifugée
à 5000 g pendant 10 minutes à 4°C. Le culot de bactéries peut être utilisé pour la mini-préparation de formes réplicatives afin de déterminer les orientations des recombinants tel que décrit ci-dessous.
24
12 - Minipréparation de plasmides ou de formes réplicatives
La technique utilisée pour la mini-préparation de
plasmide est décrite par Maniatis et al., aux pages
368-369 avec certaines modifications. Les 5 ml de bactéries
sont centrifugées et utilisés pour la lyse plutôt que 1, 5
ml et 1
1étape d'extraction au phénol/chloroforme est
remplacée par une extraction au phénol sui vie d'une
extraction au chloroforme.
13 - Amplification de plasmide
L'amplification des plasmides sélectionnés avec du
chloramphénicol est décri te par Maniatis et al. aux pages
86-94. La méthode utilisée pour éclater les bactéries
est celle de la lyse alcaline décrite
àla page 90.
14 - Transfert d'ADN sur membrane (Southern blot)
La technique de transfert d 'ADN sur membrane de
Nylon (Hybond N d'Amersham) est la même que celle décrite
par Maniatis et al. aux pages 382-389.
15 - Préparation d'une sonde d'ADN
L'ADN utilisé comme sonde est rendu radioactif par
le système de marquage
àamorces multiples (Amersham,
multiprime DNA labelling system, RPN 1601). Ce système
est utilisé tel que recommandé par la compagnie Amersham.
16 - Isolement de l'ADN simple brin et réactions de séquence
Une fois les phages recombinants identifiés et leur
orientation déterminée, l'ADN simple brin pour le
séquençage par la méthode de Sanger Ml3/didéoxy est
purifié selon la méthode décrite par "United States
Biochemical Corporation" (USB) dans le livret explicatif
de la trousse "Sequenase". Ce même livret explicatif
décrit toutes les étapes de l'hybridation de l'amorce et
de l'élaboration des réactions de séquençage et fut suivi
intégralement.
17 - Oligonucléotides
Dans plusieurs réactions de séquençage, l'amorce
universelle du phage Ml3 de 17 nucléotides a été utilisée
mais pour compléter la séquence des deux brins d'ADN, 4
26 oligonucléotides de 15 bases ont été commandés au laboratoire de synthèse d'oligonucléotides du Dr. Ken Deugau à Kingston. Ces amorces et leurs utilisations sont décrites ci-dessous:
amorce #423 51-TGTAGCATACAATTA-3 1 Adl2 brin r
amorce #424 5'-GCAGGAGCCCCTCGT-31 Ad3 brin r
amorce #485 51-TGTACAAGGACCGTT-3 1 Adl2 brin 1
amorce #486 51-TACCCAAAGCGTGCA-31 Ad3 brin 1
18 - Gels de polyacrylamide pour le séguencage d'ADN
Le matériel utilisé, le montage, le coulage et les conditions de migration du gel sont décrits dans le livret explicatif du système de séquençage Macrophor 2010 vendu par LKB. Les gels permettant de lire la séquence de O à 250 paires de base ont une épaisseur de 0,2 mm, une concentration en polyacrylamide de 8% P/V et ont migré à 60°C pendant 3 heures à puissance constante de 25 watts alors que ceux permettant de lire entre 200 et 450 paires de base ont la même épaisseur, une concentration d' acrylamide de 6% P/V et ont migré à 55°C pendant 9 heures à la même puissance.
C - Expression
19 - Système d'expression
Le vecteur d'expression pRIT2T (vendu par Pharmacia) permet d'insérer un gène dans le si te de clonage multiple
(MCS) dans le même cadre de lecture que la protéine A et de sélectionner pour la résistance à l'ampicilline (cf. figure 2). La protéine chimérique est sous le contrôle du promoteur lambda PR. Les plasmides recombinants sont d'abord sélectionnés dans ~ coli N99CI+ (Rosenberg et al., 1983), qui possède le répresseur sauvage, afin d'éviter le problème de toxicité de la protéase et sont ensuite transférés dans~ coli N4830 (Gottesman et al.,
1980), qui produit le répresseur lambda thermosensible CI857, en vue de l'expression.
20 - Induction de l'expression
Le plasmide est inséré dans ~ coli N4830 par les techniques décrites précédemment. Une colonie est ensemencée dans 5 ml de milieu LB
+
ampicilline (50 µg/ml) et incubée 5 heures à 3 0°C. La suspension est transférée dans 500 ml de milieu LBA (LB+
ampicilline) préchauffé et incubée toute la nuit à 30°C sansFigure 2.
pro git un ••• •r11 1•1 ••' ,.1 ••P .. ,,. gin
CCG GGG AAT TCC CGG GGA TCC GTC GAC CTG CAG
IDfSJ) ~)15rn1i' ~1mHIS.1l1Mintll 1 ,.,, 1
map unit or.gin
Cl·0.52 PUCB 1631·26Hf1·2l41 Cl·0.13 pEMILI 0~-0153 Phage 11 11407·10411 0 93-0.16 APA, ri ·m 111n1 (1:11 eodonal m1p unh DH·Dl3 D 13·0 I• o .... o 13 0 13·1,[I W .. ~ pBR322 Orl Hoe li (0.10) Prolein /.. 13~!1-1103! pUCI '°IYl'"le1 Pr01ein A 11660·11201 P'rolam A J -noncodrng
Carte génétique du plasmide d'expression pRIT2T
(lambda
PR;promoteur droit du phage lambda; Ampr,
agitation. Lorsque la densité optique
à600 nm atteint
0,8
à1,0 unité, un volume égal de milieu LBA préchauffé
à
54°C est ajouté
àla suspension qui est incubée
à42°C
avec agitation pendant 2
à4 heures.
21 - Purification de la protéine fusion
Les bactéries induites sont centrifugées
à5000 g
pendant 5 minutes
à4°C, lavées avec 20 ml de Tris 50
mMpH 8,1 et resuspendues dans 9 volumes de TE lX par gramme
de bactéries. Les cellules sont lysées par 6 cycles de
gel-dégel suivis de 3 sonications de 30 secondes
à60
cycles.
Le lysat est centrifugé
à12000 g pendant 5
minutes et le surnageant est déposé sur une colonne
d'immune-affinité appelée "IgG-Sépharose 6 FF"
(Phar-macia). Les protéines non adsorbées sont lavées avec 20
ml de TST (Tris-Hel 50
mMpH 7,6, NaCl 150
mM,Tween 80
O, 5% V /V) .
La colonne est ensui te rincée avec 10 ml
d'acétate d'ammonium 5
mMet les protéines sont éluées
avec de l'acide acétique 0,5 M pH 3,4. Le pH de chaque
fraction de 0,5 ml est neutralisé avec 1 volume d'acétate
d'ammonium
o,
5M. Les fractions sélectionnées par le test
de Bradford (Bradford, 1976) sont rassemblées et
dialy-sées pendant la nuit
à4°C contre du TE lX.
30
22 - Transfert de protéines et iodination
Après une migration sur gel de SDS-PAGE, les protéines sont transférées sur filtre de nitrocellulose (Hybond C d'Amersham) par le système d'électro-transfert polyblot de la compagnie ABN tel que décrit par le fabricant. La préhybridation, l'hybridation et le lavage des filtres sont décrits par Towbin et al. (1979) exception faite de l'albumine sérique de boeuf qui est utilisée comme agent bloquant au lieu du lait en poudre. L' iodination de 1 'anticorps ou de la protéine A avec 125I par la méthode à la chloramine T est décrite par McConahey et Dixon (1980).
23 - Séquençage N-terminal de protéines
Les extrémités N-terminales de peptides ont été séquencées à partir de transfert sur membrane immobilon PVDF (Millipore) à Montréal au 11Shriner's Hospital" par Michel van der Rest.
24 - Marquage des protéines induites
Les bactéries hôtes N4830 sont induites par une
augmentation de la température
à42°C pendant 30 minutes.
De la méthionine-[
355] est ajoutée
àraison de 100 µCi/ml
à
la suspension bactérienne qui est réincubée
à42°C
pendant une autre demi-heure.
Les bactéries sont
récoltées par centrifugation, lavées avec Tris-HCl 50
mMpH 8,1 et resuspendues dans du TE-lX.
25 - Enrichissement des corps d'inclusion
Après l'induction, les bactéries sont centrifugées
à
5000g, lavées avec du Tris-HCl 50
mMpH 8,1,
recentri-fugées et le culot humide est pesé. Pour chaque gramme
de bactéries, 3 ml de tampon de lyse, composé de
Tris-HCl 50
mMpH 8,1, EDTA 1
mMet NaCl 100
mM,sont ajoutés
de même que 80 µl d'une solution de lysozyme fraîchement
préparée
à10 mg/ml. Le tube est brassé
occasionnelle-ment pendant 20 minutes. Après cette étape, 4 mg de
déoxycholate sont ajoutés par gramme de bactéries, et le
tube est agité continuellement pendant 30 minutes
à4°C.
La solution devient très visqueuse et les acides
nucléi-ques sont détruits par 3 sonications de 5 secondes
à40
cycles.
Le lysat bactérien est centrifugé
à12000 g
32 pendant 5 minutes dans une micro-centrifugeuse. Le traitement au déoxycholate est répété une seconde fois. Le culot est lavé avec du TE lX pour éliminer le déter-gent puis resuspendu dans 9 volumes de tampon de lyse contenant O, 5% P/V de sulfobétaïne, homogénéisé au vortex et centrifugé à 12000 g. Cette étape est répétée une seconde fois mais la suspension est d'abord centrifugée à 120 g pendant 5 minutes. Le surnageant est transféré dans un tube propre et centrifugé à 12000 g pendant 5 minutes. Le culot contenant les corps d'inclusion est resuspendu dans 3 volumes de TE lX.
RESULTATS
Les protéases virales occupent une position stratégique
dans le cycle infectieux des virus où elles ont été mises en
évidence (Butterworth, 1977; Wellink et van Kammen, 1988;
Krausslich et Wimmer, 1988; Kay et Dunn, 1990). Celle de
!'adénovirus de type 2 fut l'une des premières
àêtre
démon-trée chez les virus eucaryotiques. Dans ce travail, l'étude
de cette enzyme a été menée sur trois fronts. Premièrement,
1
1activité enzymatique existe-t-elle chez les autres sérotypes
et, dans l'affirmative, la spécificité est-elle conservée?
Deuxièmement, la structure de l'enzyme est-elle similaire chez
d'autres sous-groupes? Et finalement, est-ce que l'enzyme
peut être produite en grande quantité afin de prouver
physi-quement sa présence et de pouvoir poursuivre son étude plus
aisément?
A - Complémentation in vitro des protéases de divers sérotypes
sur le précurseur pré VII de H2tsl
Il y a une dizaine d'années, l'isolement de recombinants
génétiques après un croisement
à33°C entre les mutants
ther-mosensibles Ad2tsl, Ad2ts3, Ad2ts4, Ad2ts48 et Ad5tsl, Ad5ts2,
Ad5ts22, Ad5ts36 a permis de localiser les coordonnées
physiques de ces mutations (Hassel! et Weber, 1978; Weber et
34
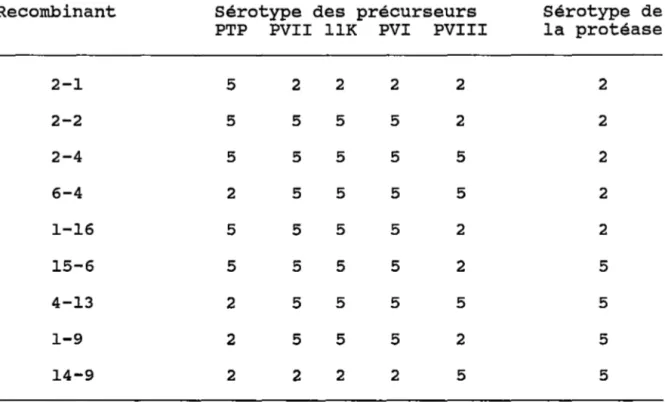
Hassell, 1979). Chacun de ces recombinants, purifié à 39°, était viable, infectieux et avait un patron protéique normal démontrant ainsi gue la protéase remplissait adéquatement son rôle. La comparaison de la cartographie génétique, établie par enzymes de restriction à 1 'époque pour chaque recombinant, avec les séquences de Ad2 publiées plus récemment a permis de déterminer précisément l'origine sérotypigue des précurseurs et de la protéase de chacun de ces recombinants. Ceux d'entre eux exprimant des caractères intersérotypigues face à ces gènes ont été sélectionnés et sont rapportés au tableau 4. Ces résultats montrent clairement gue chacun des 5 précurseurs d'un sérotype peut être clivé in vivo par la protéase de l'autre. Ainsi, les protéases de Ad2 et de Ad5 sont inter-changeables. Les séquences en acides aminés de ces enzymes ont été déduites des séquences d'ADN publiées (Akusjarvi et al., 1981; Kruijer et al., 1980). Ces deux virus appartien-nent au même sous-groupe (C) et leurs protéases ne diffèrent gue par un seul acide aminé, soit l'arginine 63 de Ad2 gui est remplacée par une histidine chez Ad5. Cependant, seules quelques portions du génome de Ad5 ont été séquencées et aucune d'entre elles n'impliquent la séquence codante d'un précurseur. Les seules autres données disponibles montrent des différences dans les cartes de restriction et dans la mobilité de quelques protéines virales des deux sérotypes. La
chez les virus recombinants
Recombinant Sérotype des précurseurs Sérotype de PTP PVII llK PVI PVIII la protéase
2-1 5 2 2 2 2 2 2-2 5 5 5 5 2 2 2-4 5 5 5 5 5 2 6-4 2 5 5 5 5 2 1-16 5 5 5 5 2 2 15-6 5 5 5 5 2 5 4-13 2 5 5 5 5 5 1-9 2 5 5 5 2 5 14-9 2 2 2 2 5 5
Tableau 4. Virus recombinants sélectionnés par comparaison de cartographie génétique (Weber et Hassell, 1979) avec les séquences de Ad2 publiées plus récemment démontrant la possibilité de clivage intersérotypique.
36
complémentation a donc été testée avec des membres d'autres
sous-groupes humains par le biais d'un test in vitro sur le
précurseur préVII de H2tsl.
Des cellules Hep-2 ont été infectées avec des adénovirus
de type 1, 2, 3, 4, 5, 6, 7 et 9. Les noyaux ont été isolés
20 heures plus tard comme source d'enzyme. Etant donné que
la protéase est exprimée
àun très faible niveau, sa
produc-tion est directement proporproduc-tionnelle
àla qualité de
l'infec-tion et doit être standardisée entre chacun des sérotypes.
La séquence codante de l'hexon précède celle de la protéase
dans l'unité de transcription L3 et cette protéine est
clairement visible dans les extraits cellulaires déposés sur
un gel PAGE-SDS coloré au bleu de coomassie. cette protéine
a donc été choisie pour standardiser les niveaux d'infection
de chaque virus par rapport
àAd2 par lecture densi tométrique.
L'infection des cellules Hep-2 par les membres du sous-groupe
c (Adl, 2, 5 et 6) est très efficace alors que celle par les
autres groupes s'est avérée plus laborieuse (cf. tableau 5).
Les noyaux isolés de ces différentes infections, après une
préincubation
à4°C pendant 24 heures, ont été mis en présence
des précurseurs marqués
àla méthionine-[
35S] fournis par H2tsl
à
température non permissive, incubés
à37°C pendant 16 heures
et analysés sur PAGE-SDS. Un exemple de résultat de ce type
d'expérience est montré
àla figure 3 (piste c).
seur préVII est la protéine immature la plus
Le
précur-f acile
àFigure 3.
VIII
IX
a
b
c
d e
<JPVll
Clivage in vitro des précurseurs viraux de H2tsl marqués à la méthionine- [ 358] . La piste a contient
le virus de type sauvage comme marqueur de poids moléculaire. Les pistes b à e contiennent en plus du substrat, des noyaux de cellules Hep-2 non infectées (b), des noyaux de cellules Hep-2 infectées avec Adl (c), Ad3 (d) et Ad9 (e) incubés à 37°C pendant 16 heures. Les triangles pleins montrent le clivage de l'hexon en 2 fragments.
38
localiser et sa conversion en VII est le test de clivage le plus approprié pour détecter l'activité de protéases adénovi-rales. Le taux de clivage a été déterminé par densitométrie sur les autoradiogrammes exposés adéquatement. La possibilité d'un clivage par activité endogène de H2tsl ou encore par des protéases cellulaires a été écartée par une incubation du substrat avec du tampon, en remplacement de noyaux (résultats non montrés), et avec des noyaux de cellul es non infectées (figure 3, piste b). Dans ces 2 cas, aucune activité protéa-sique n'a été détectée. Les résultats de ces expériences pour chacun des sérotypes ont été normalisés par rapport à leur niveau d'infection et sont rapportés au tableau 5. Tous les adénovirus testés ont été capables de cliver le précurseur préVII de H2tsl aussi efficacement que leur niveau d'infection le permettait. Certaines expériences ont permis d'observer occasionnellement, en plus du clivage de préVII en VII, le clivage spécifique du précurseur préVI en VI et de l'hexon en deux fragments de 95 et 15k (figure 3, pistes d, e) par la plupart des sérotypes. Ces deux protéolyses additionnelles, bien que présentes à des degrés variables, appuient plus fortement l'idée de propriétés communes de cette enzyme pour tous les sérotypes humains notamment pour la spécificité du clivage. La digestion de l'hexon en 2 fragments spécifiques révèle aussi la présence d'un si te cryptique pouvant être occasionnellement hydrolysé par les différents sérotypes humains.
Sérotype de la protéasea Adl Ad2 Ad3 Ad4 Ad5 Ad6 Ad7 Ad9 Sous-groupe de l'adénovirus
c
c
B Ec
c
B D Efficacité de 1 ' inf ectionb (%) 100 100 50 50 100 100 40 30 Clivage de pVII norma-lisée 100 100 100 100 100 100 100 100a - Noyaux isolés de cellules Hep-2, _20 heures après infection par le virus indiqué.
b - Efficacité estimée à partir de la quantité d'hexon par rapport à Ad2 sur PAGE-SOS colorés avec du bleu de coomassie.
c - Données normalisées par rapport à l'efficacité d'infection.
Tableau 5. Clivage in vitro du précurseur préVII de H2tsl par les protéases de différents sérotypes provenant de noyaux de cellules infectées.
40 B - Séquençage du gène de la protéase de Ad3 et Adl2
Les expériences précédentes suggèrent l'existence d'une similarité structurale de 1 'enzyme qui se traduit par une activité biologique homologue in vitro. L'étude des réver-tants du mutant H2tsl a permis d'associer un lien direct entre l'activité protéasique et le gène 23k de la région L3 (Weber et Houde, 1987). Ce gène a déjà été séquencé pour les séroty-pes Ad5 (Kruijer et al., 1980), Ad2 (Akusjarvi et al., 1981), H2tsl (Yeh-Kai et al., 1983) et Ad4 (Houde et Weber, 1987). Des informations intéressantes sur les domaines importants de l'enzyme, notamment le site actif, pourraient être obtenues par comparaison de ces résultats avec la séquence de sérotypes membres d'autres sous-groupes.
En séquençant le gène 72k de Adl2 (souche Huie), Kruijer et al. (1983) ont publié la séquence nucléotidique des derniers résidus de la protéase de ce sérotype, membre du sous-groupe A. L'homologie stricte en acides aminés avec les séquences de Ad2 et Ad4 est de 60,2%. Le docteur Stanley Mak a cloné tous les fragments BamHI de l'adénovirus de type 12, souche Pereira 1131, dans pBR322 et nous a gentiment fourni le plasmide contenant la région localisée entre 59,6 et 73.0% du génome, soit le fragment BamHIC d'environ 4, 5 kb. Le plasmide a été amplifié au chloramphénicol en présence d'ampicilline et purifié sur gradient de chlorure de césium.
En se basant sur la carte de restriction de Adl2 souche Huie (Tooze, J., 1981) et sa portion de séquence disponible, le gène de la protéase a été localisé entre les sites de restric-tions BamHI (59,6%) et HindIII (62,1%) (cf. figure 4a). Le fragment, par clonage forcé, a été inséré dans les deux orientations en utilisant le site inversé de clonage multiple des phages Ml3mpl8 et Ml3mpl9. Le gène a aussi été sous-cloné par double digestion avec BglII (60,0%) et HindIII (62,1%) dans Ml3mpl9 de même que BglII (60,0%) et BamHI (59,6%) dans Ml3mpl8 pour faciliter le séquençage. L'utilisation d'un autre site situé à 60,9% (KpnI) du génome de la souche Huie s'est avérée impossible. Tous ces sous-clones ont été criblés par transfert de Southern en utilisant comme sonde l'ADNc de la protéase de Ad2. L'intensité du signal s'est avérée très faible mais suffisante pour sélectionner les phag,es recom-binants positifs. Etant donné l'importance de la longueur du
fragment BglII-HindIII (environ 7 50 p. b.) , 2 oligonucléotides de 15 bases, dont la composition fut déterminée après séquen-çage d'une première portion du segment, ont été utilisés comme amorces pour compléter l'analyse des 2 brins. L'amorce #485 a permis de compléter le brin 1 et l'amorce #423, le brin r. La stratégie de séquençage et la localisation des sites de restriction sont montrées à la figure 4a. Après avoir complété ce travail, 1 'analyse de la séquence d' ADN par ordinateur a révélé que le site KpnI situé à 60, 9% de la souche Huie est inexistant dans la souche Pereira 1131, ce
a) Adl2 Protéase 1485 B Bl H 165 809 1423 b) Ad 3 Protéase 1486 K B Sm Sp Bl 0 193 476 852 1273 1424
Figure 4. Localisation des segments d'ADN séquencés par rapport aux gènes des protéases de Ad12 (a) et de Ad3 (b) . Les si tes de restrictions sont représentés par des lettres (BarnHI (B), BglII (Bl), HindIII (H), KpnI(K), SmaI (Sm) et SphI (Sp)).
a) Le séquençage du gène de la protéase de Ad12 a nécessité le sous-clonage des fragments BarnHI-BglII et BarnHI-HindIII dans M13mpl9 , celui des fragments HindIII-BarnHI et BglII-BarnHI dans M13mpl8 et l'utilisation des amorces #423 et #425, différentes de l'amorce universelle est indiquée au-dessus de la portion d'ADN qu'elles ont permis de séquen-cer.
b) Le séquençage du gène de la protéase de Ad3 a nécessité le sous-clonage des fragments KpnI-BglII, BamHI-SphI et SmaI-BglII dans M13mpl9, celui des fragments SmaI-BglII-KpnI, SphI-BglII, SphI-BarnHI, SmaI-KpnI et BarnHI-KpnI dans Ml3mpl8 et 1 'utilisation des amorces #424 et #486, différentes de l'amorce universelle est indiquée au..;.dessus du segment d'ADN qu'elles ont permis de séquencer.
qui explique l'impossibilité que nous avons rencontrée en tentant d'utiliser cette enzyme dans le sous-clonage. La séquence en ADN de 809 paires de base (cf. figure 5) comprend un cadre de lecture ouvert de 206 acides aminés codant pour une protéine de 23419 daltons. La comparaison de l'acide nucléique avec la région correspondante pour Ad2 et Ad4 a permis d'identifier le site accepteur d' épissage pour la protéase après la base 5, le codon de terminaison de l'hexon (98), le signal de polyadénylation de la région L3 (771-775) et l'inverse de celui de la protéine 72k (787-782) située sur le brin r. La séquence en acides aminés de la protéine a été déduite par ordinateur à partir de la séquence nucléotidique grâce à la propriété de conservation du code génétique. Le site accepteur d'épissage pour Ad2 a été déterminé par essai à la nucléase Sl (Akusjarvi et al., 1981).
Le sous-groupe B comprend les adénovirus de type 3, 7, 11, 14, 16, 21, 34 et 35 (cf. tableau 3). Les gènes adénovi-raux sont colinéaires, aussi la comparaison des cartes de restriction de Ad3 avec Ad2 et Ad5 (D'Halluin et al., 1983; Niel et D'Halluin, 1984) a permis de localiser facilement la région codant pour la protéase. Le fragment HindIIIC d'envi-ron 4,5 kb situé entre les cordonnées 50,4 et 63,8% a été cloné dans le site HindIII de pBR322. Le gène de la protéase a été mis en évidence, par transfert de Southern, entre les sites KpnI (59,9%) et BglII (62,9%) (cf. figure 4b). Ce