T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
DO
OC
C
TO
T
OR
RA
AT
T
D
DE
E
L
L’
’U
UN
NI
IV
VE
E
RS
R
S
IT
I
TÉ
É
DE
D
E
T
TO
O
UL
U
L
OU
O
US
SE
E
Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Pharmacologie
JURY
Mme. Véronique BALDIN, HDR-CR1, CRBM-CNRS, Montpellier Rapporteur M. Bertrand FRIGUET, Professeur, Université Pierre et Marie Curie-Paris 6, Paris Rapporteur Mme. Sok Eng TEA, Professeur, Faculté de Pharmacie de Phnom Penh, Cambodge Examinateur M. Jean CROS, Professeur Emérite, Université Paul Sabatier, Toulouse Examinateur M. Jean Edouard GAIRIN, Professeur, Université Paul Sabatier, Toulouse Directeur de Thèse
Ecole doctorale : Biologie-Santé-Biotechnologies Unité de recherche : UMR2587,CRPS, CNRS-Pierre Fabre
Directeur(s) de Thèse : Pr. Jean Edouard GAIRIN
Présentée et soutenue par Rothmony EANG Le 17 décembre 2008
Titre : Mise au point d'outils de recherche et étude de modifications
REMERCIEMENTS
Ce travail de thèse a été effectué au sein de l’UMR2587, CRPS, ISTMT, CNRS-Pierre Fabre. Je tiens à remercier vivement Monsieur le Professeur Jean Edouard GAIRIN, Directeur de l’UMR, pour m’avoir chaleureusement accueilli au sein de son laboratoire, et de m’avoir prodigué de précieux conseils. Son soutien, sa disponibilité et la confiance qu’il m’a toujours témoignée m’ont permis de mener à bien ce travail et de préparer mon doctorat dans les meilleurs conditions.
J’adresse des remerciements sincères à Monsieur le Professeur Jean CROS, Professeur Emérite à Université Paul Sabatier, pour son aide, pour son soutien, pour la qualité des connaissances qu’il m’a transmise, pour le temps qu’il a consacré à suivre mon travail et pour la bienveillance qu’il m’a accordée. Je tiens à remercier également à Madame S. CROS qui m’apporté une aide efficace et indispensable.
Je tiens à remercier particulièrement Madame le Docteur Elisabeth
GIRBAL-NEUHAUSER et Madame le Docteur Nabila Jabrane-FERRAT pour leurs précieux
conseils qui ont été essentiels pour le démarrage de mon travail, pour le temps qu’elles ont consacré à la réalisation de la publication, pour la confiance qu’elles m’ont toujours accordée et pour l’expérience et les compétences qu’elles m’ont fait partager.
Je remercie infiniment Madame le Professeur Sok Eng TEA, Doyenne de la faculté de pharmacie de Phnom Penh, pour la connaissance qu’elle m’a prodiguée au cours de son enseignement, pour ses précieux conseils, pour l’attention qu’elle m’a témoignée, pour la bienveillance qu’elle m’accordée. Je suis très honoré qu’elle ait accepté de siéger dans mon Jury de thèse.
J’adresse également des remerciements particulièrement à Monsieur le Professeur
Bertrand FRUIGUET, Professeur à l’Université Pierre et Marie Curie-Paris 6, Responsable
de l’équipe 6 « Biologie Cellulaire du Vieillissement » de l’UMR7079, pour ses précieux conseils, et pour avoir accepté de siéger dans mon Jury et d’être rapporteur de ce travail.
Je tiens également à remercier vivement Madame Véronique BALDIN, Chargée de Recherche du CNRS au Centre de Recherche de Biochimie Macromoléculaire (Montpellier)
pour ses précieux conseils, pour avoir accepté de siéger dans mon Jury et d’être rapporteur de ce travail.
Je tiens à remercier sincèrement Monsieur le Docteur Hot BUN pour le temps qu’il m’a donné, afin que je puisse m’enrichir de nouvelles connaissances lors de son enseignement, des précieux conseils dont j’ai pu bénéficier, de son soutien sans faille, afin que je puisse mener ce travail dans les meilleures conditions.
Je tiens à remercier très vivement et infiniment Monsieur Pierre Fabre, Président de la Fondation Pierre Fabre, qui a subventionné mon travail de thèse en France. Qu’il trouve ici le témoignage de mon profond respect et de toute ma gratitude.
Je voudrais remercier publiquement la Fondation Pierre Fabre et, en particulier son Directeur Général, Monsieur Philippe BERNAGOU, pour avoir subventionné mon séjour en France pour que je puisse travailler dans les meilleures conditions. Qu’il trouve ici le témoignage de mon respect, de ma gratitude et également de l’attachement de tous les étudiants cambodgiens à la Fondation Pierre Fabre qu’il dirige.
Je remercie également Madame Véronique TESSIE, chargée de mission, pour sa gentillesse, sa disponibilité. Je n’oublierai jamais le soutien et l’aide sans cesse qu’elle m’a apportés ainsi qu’à l’ensemble des anciens thésards Khmers de la Fondation Pierre Fabre.
Je voudrais remercier aussi l’Association pour la Recherche sur le Cancer (ARC) pour avoir subventionné la quatrième année de ma thèse pour que je puisse mener à bien mon travail.
Je tiens à remercier à toute l’équipe de l’Amicale des Etudiants en pharmacie et des
Pharmaciens Khmers (AEPK) et particulièrement à Madame le Docteur Vary KHUN,
Présidente de l’AEPK, d’avoir subventionné la fin de ma thèse pour que je puisse terminer mon travail dans les meilleurs conditions. Qu’elle trouve ici le témoignage de mon respect et de ma gratitude.
Je remercie aussi Madame le Professeur Anne ROUSSIN, Madame le Docteur Isabelle
m’ont transmises, leurs accueils chaleureux et leurs précieux conseils tout le long de mon travail.
Je tiens à remercier sincèrement et infiniment l’ensemble des personnes, passées et présentes, de l’UMR : Martine KNIBIEHLER, Joëlle RION, Chantal ETIEVANT,
William RIQUET, Brigitte RAYNAUD-MESSIA, Laurence HAREN, Laurent
MAZZOLINI, Marie-Hélène REMY, Aude ESPIGAT-GEORGER, Vanessa
TILLEMENT, Bent RUBIN, Andreas MERDES, Anne Héry CONDON, Elisabeth SOULIE, Martine SOLER, Latifa BENAHMED pour leurs compétences, leurs
disponibilités, leurs conseils sans faille, leurs aides, leur soutien quotidien et leur bonne humeur tout au long de ma présence dans le laboratoire.
Je voudrais adresser mes remerciements les plus sincères à Anaïs BOUISSON, Marine
CHARTRAIN, Lucie KRZACZKOWSKI, Nicolas ROULLET, Christelle VEROLLET, Frédéric GIRARD, Delphine ROBERIOUX pour leur bonne humeur, leur soutien sans
faille, leurs encouragements quotidiens, leur disponibilité et surtout leur aide sans cesse lors de mon travail dans le laboratoire.
Je n’oublierai pas de remercier mes amis cambodgiens (de Toulouse et de Marseille) et toulousains, particulièrement à Sothea KIM, Labo CHHUN, Makara KHOV et la famille
CHHEM, avec qui j’ai partagé de nombreux souvenirs, des soirées inoubliables, des sorties
sportives, le toit et surtout leurs soutiens quotidiens inestimables sans cesse.
Enfin, je tiens à remercier vivement et infiniment ma famille, surtout mes parents pour leurs soutiens permanents et leurs conseils malgré notre éloignement. Je voudrais également remercier ma fiancée, Vichheka KHUON et ses parents pour leurs encouragements sans cesse et inestimables. C’est grâce à eux que j’ai pu réaliser mes rêves et avec qui je partage pour toute ma vie dans les moments heureux comme dans les instants difficiles.
Mise au poin d’outils de recherche et étude de modification par
phosphorylation du protéasome 20S humain
Table des matières page
RESUME --- 01
ABSTRACT --- 02
INTRODUCTION GENERALE --- 03
Chapitre I : Le protéasome --- 06
I.1. Systèmes protéolytiques intracellulaires «Système Ubiquitine-Protéasome» --- 06
I.2. Machinerie de polyubiquitination --- 06
I.3. Modes de reconnaissance des substrats à ubiquitiner par l’E3 --- 09
I.4. Cœur catalytique de l’Ub-P «le protéasome» --- 11
a. Structure du protéasome --- 11
b. Protéasome 20S--- 12
c. Biogenèse et assemblage du protéasome 20S--- 13
d. Maturation des sous-unités catalytiques en vue de complèter l’assemblage---- 14
e. Protéines accessoires dans la maturation du protéasome --- 15
I.5. Complexes régulateurs du protéasome 20S --- 16
a. Complexes régulateurs 19S (RP) --- 17
b. Complexes régulateurs 11S (PA28)--- 18
c. Complexes régulateurs PA200--- 18
I.6. Activité du protéasome --- 19
I.6.1. Etape de la dégradation des substrats ubiquitinés --- 19
I.6.2. Activités spécifiques du protéasome --- 22
a. Activité PGPH --- 22
b. Activité T-L --- 23
c. Activité CT-L --- 23
I.7. Localisation subcellulaire du protéasome --- 23
a. Protéasome cytoplasmique--- 23
b. Protéasome nucléaire --- 24
I.8. Protéasome et vieillissement --- 25
I.9. Protéasome, cible pharmacologique attractive--- 25
Chapitre II : Les inhibiteurs du protéasome --- 28
II.1. Principales classes d’inhibiteurs « catalytiques » du protéasome --- 28
II.1.1. Peptides aldéhydes--- 28
II.1.2. Peptides boronates --- 29
II.1.3. Lactacystine et le β-lactone--- 29
II.1.4. Peptides vinyl sulfones--- 29
II.1.5. Epoxykétones--- 30
II.2. Mécanismes d’induction de l’apoptose par les inhibiteurs du protéasome --- 30
II.2.1. Augmentation de l’activité p53 par inhibition du protéasome --- 30
II.2.2. Accumulation des inhibiteurs de croissance, la p27 --- 31
II.2.3. Accumulation de protéines pro-apoptose Bim et Bik--- 31
II.2.4. Activation de SAPK (stress-activated protein kinase) par l’inhibition du protéasome --- 31
II.2.5. Atténuation de l’effet anti-apoptose de NF-κB --- 32
II.2.7. Induction de l’activation de caspases par inhibition du protéasome --- 32
II.3. Inhibiteurs « on-catalytiques » du protéasome --- 32
Chapitre III : La phosphorylation du protéasome --- 34
III.1. Généralité sur des modifications post-traditionnellement des protéines --- 34
III.2. Modifications post-traductionnelles par la phosphorylation/déphosphorylation --- 34
III.3. Phosphorylation et le cancer--- 35
III.4. modifications post-traditionnelles du protéasome--- 37
III.4.1. O-β-N-acétylglucosamination --- 37
III.4.2. Acétylation N-terminale --- 37
III.4.3. Modification par phosphorylation du protéasome--- 38
a. Phosphorylation du protéasome 20S--- 38
b. Phosphorylation des complexes régulateurs 19S et PA28 --- 41
Chapitre IV : L’immunoprotéasome --- 43
IV.1. Structure de l’immunoprotéasome --- 43
IV.2. Immunoprotéasome et réponse immunitaire--- 43
IV.3. Immunoprotéasome et vieillissement --- 46
MATERIELS ET METHODES--- 48
Lignées Cellulaires--- 48
Réactifs et anticorps--- 48
Traitement la lignée HeLa par l’INFγ --- 48
Purification de l’anticorps monoclonal MCP21 --- 49
Couplage de MCP21 sur les billes de sépharose --- 49
Préparation des extraits cellulaires --- 49
Extraction des ARN et RT-PCP --- 50
Purification et quantification du protéasome 20S --- 50
Electrophorèse dénaturante mono- et bi-dimensionnelle--- 51
Western blot--- 51
Test d’activité du protéasome --- 52
Déphosphorylation du protéasome 20S par la phosphatase acide--- 54
Analyse de la séquence protéique --- 54
Construction de modèle 3D du protéasome--- 54
RESULTATS --- 55
Première partie : Mise au point d’outils pour l’étude des modifications Post-traditionnellement par phosphorylation du protéasome 20S--- 55
IV.I.1. Obtention du protéasome à partir d’érythrocytes humains --- 55
a-Purification de l’anticorps anti-α2 (MCP21) --- 55
b-Purification du protéasome 20S --- 56
c-Séparation des sous-unités du protéasome 20S par électrophorèse bi-dimensionnelle --- 57
d-Test d’activité du protéasome 20S purifié --- 58
IV.I.2. Détection des phosphoSérines sur les sous-unités β2 et β7 du protéasome 20S -- 59
IV.I.3. Analyse des sites potentiellement phosphorylées des sous-unités β2 et β7 du protéasome 20S --- 61
IV.I.4. Développement et validation des anticorps anti-phosphoSérines spécifiques --- 64
a-Conception du développement des anticorps anti-phosphoSérine --- 64
IV.I.5. Utilisation de l’anticorps validé pour détecter la nouvelle
isoforme phosphorylée de la sous-unité β7 --- 67 IV.I.6. Effet du traitement à la phosphatase acide
sur la forme phosphorylée de la sous-unité β7 --- 70 IV.I.7. Effet du traitement à la phosphatase acide sur l’activité PGPH --- 75 IV.I.8. Différence d’expression de la forme phosphorylée dans des lignées
normales et tumorales étudiées --- 77 IV.I.9. Différences d’expression d’activité PGPH entre la lignée HEK293 et U2OS --- 80 Deuxième partie : Mise au point d’outils permettant de quantifier
le taux relatif d’immunoprotéasome --- 82 IV.II.1. Synthèse de peptide PS et IP permettant une coupure différente
entre le protéasome standard et l’immunoprotéasome--- 82 IV.II.2. Caractérisation des modèles cellulaires--- 83 IV.II.3. Détermination de la quantité du protéasome nécessaire pour
la coupure du peptide PS --- 86 IV.II.4. Test d’activité des protéasomes d’extraits brut de HeLa, HeLa traité à l’INFγ, de HEK293 SP et de HEK293 IP avec les peptides PS et IP--- 87
DISCUSSION ET PERSPECTIVES --- 89 Références--- 99 Annexes : Article « Characterization and differential expression of a newly identified phosphorylated isoform of the human 20S proteasome β7 subunit in tumor vs. normal cell lines », Rothmony Eang, Elisabeth Girbal-Neuhauser, Bo Xu and
Liste des abréviations
AMM : Autorisation de Mise sur le Marché APC : Anaphase promoting complex CDK : cyclin-dependent kinase CKII : Caséine kinase II
CMH : Complexe Majeur d'Histocompatibilité CP : Complexe catalytique ou Protéasome 20S
CT-L : Chymotrypsine-like
DRiPs : Defective ribosomal products EDTA : Ethylène diamine tétracétique ER : Réticulum endoplasmique FDA : Food and Drug Administration
HECT : Homologous to E6-associated protein C-terminus INFγ : Interféron gamma
LMP2 : Low Molecular mass Protein-2
MECL-1 : Multicatalytic Endopeptidase Complex-like 1
NEDD8 : Neural precursor cell expressed, developmentally down-regulated 8 NF-κB : Nuclear Factor-kappa B
NLS : Signal de localisation nucléaire
PGPH : Peptidylglutamyl-peptide hydrolyzing PKA : Protéine kinase A
RE : Réticulum Endoplasmique RP : Complexe régulateur
Rpn : Regulatory particle non-ATPase
Rpt : Regulatory particle triple-A ATPase protein SCF : SKP1-Cullin-F box
SDS : Sodium Dodécyl Sulfate
TGFβ : Transforming Growth Factor beta
T-L : Trypsine-like
UBC domain : Ubiquitin-conjugating Domain UBL : Ubiquitin-like proteins
Mise au point d’outils de recherche et étude de modifications
post-traductionnelles du protéasome 20S humain
RESUME
Le protéasome 20S, cœur multi-catalytique du système Ubiquitine-Protéasome (Ub-P), joue un rôle majeur dans la régulation de la dégradation non-lysosomale des protéines importantes impliquées dans des processus cellulaires fondamentaux et donc, dans le cancer. Son inhibition constitue une des approches originales récentes utilisées dans le domaine de la recherche de médicaments anti-cancéreux innovants. Le Bortezomib ou VelcadeTM est le premier inhibiteur du protéasome ayant obtenu une AMM pour le traitement des myélomes multiples et récemment du lymphome du manteau. En revanche, son administration induit de nombreux effets indésirables. Différents programmes sont actuellement développés pour rechercher des molécules modulatrices de l’activité du protéasome ayant un mécanisme d’action différent afin de tenter de diminuer les effets secondaires. Dans le cadre de cette thèse, nous avons porté nos efforts sur la compréhension de deux mécanismes pouvant conduire à une modification de l’activité du protéasome : les modifications post-traductionnelles par phosphorylation d’une part et le changement des sous-unités catalytiques du protéasome conduisant à l’apparition de l’immunoprotéasome d’autre part.
Les modifications post-traductionnelles par phosphorylation pourraient jouer un rôle important dans la régulation de l’assemblage des sous-unités du protéasome et donc sur son activité. L’étude des mécanismes de phosphorylation/déphosphorylation pourrait donc contribuer à comprendre le mécanisme d’activation du protéasome. Actuellement, seules les phosphorylations des sous-unités α du protéasome 20S ont été caractérisées et identifiées. La présence de modification post-traductionnelle des sous-unités β est restée peu étudiée. Un des objectifs de mes travaux a consisté à développer des outils permettant d’identifier de nouvelles isoformes phosphorylées des sous-unités β du protéasome 20S humain. Les résultats obtenus ont montré, pour la première fois, que la sous-unité β7 du protéasome 20S, purifié à partir des érythrocytes humains, est phosphorylée en Sérine249 en utilisant un anticorps rationnellement développé. Cette forme peut être déphosphorylée par la phosphatase acide. L’activité PGPH est affectée par le traitement, alors que les activités CT-L et T-L restent inchangées. De plus, dans une série de lignées cellulaires tumorales étudiées nous montre que l’expression de cette forme phosphorylée diminue par rapport à celle dans les lignées cellulaires non-tumorales étudiées, suggérant son utilisation possible comme un biomarqueur.
L’activité catalytique du protéasome peut aussi être affectée par différents facteurs environnementaux et cellulaires conduisant à l’expression d’immunoprotéasome. Cette expression a pu être mise en évidence dans différentes lignées tumorales. L’objectif de mes travaux a consisté à mettre au point des outils de recherche permettant de différencier et de quantifier les activités du protéasome standard et de l’immunoprotéasome et de quantifier le taux relatif d’immunoprotéasome dans les cellules tumorale. Pour cela, deux substrats peptidiques fluorescents ont été synthétisés et analysés. L’utilisation de ces substrats a permis de mettre en évidence une activité différentielle des protéasomes et/ou immunoprotéasomes purifié à partir des lysats cellulaires mais n’a pas pu être étendue à l’étude d’extraits totaux.
2
ABSTRACT
The 20S proteasome, the multi-catalytic core particle of proteasome 26S, a major component of the Ub-P pathway playing a fundamental role in many basic cellular processes and in cancer since the majority of intracellular proteins, including many tumor suppressor proteins, are regulated by this pathway. Accordingly, it is very attractive for recent and innovative cancer treatment strategy. The proteasome inhibitor VelcadeTM/bortezomib has proven successfully anti-tumoral activity in vitro and in vivo with multiple tumor cell lines. Therefore, it was the first marketed proteasome inhibitor for the treatment of patients with multiple myeloma, and recently of mantle cell lymphoma. Nevertheless, adverse reactions have been found when using VelcadeTM and efforts are currently made to find modulatory molecules with different mechanism action in order to diminish side effects. During the thesis here, we focus our efforts on two mechanisms found to modify the proteasome activity: the post-translational modifications by phosphorylation and the change of catalytic subunits leading to immunoproteasome expression.
The modification post-translational by phosphorylation may regulate its association with the regulatory complex and then, the proteolytic activity of the proteasome. The study of phosphorylation/dephosphorylation mechanisms could help to comprehend the activation mechanism of the proteasome. At present, on the 20S proteasome, phosphorylated forms have only been characterized on α proteasome subunits and few evidences has been provided to prove the presence of phosphorylated form on β subunit. Objectif of my works here is to develop the research tools to investigate the phosphorylation modification of the β subunits of 20S human proteasome purified from human erythrocytes. We found that β7 subunit was phosphorylated on serine249 residues using a rationally developed antibody. The β7 phosphorylated form could be able to be dephosphorylated by acid phosphatase treatment and as a result, the PGPH activity was affected, whereas the other activities remained unchanged after the treatment. Moreover, the serine-phosphorylation on β7 subunit was observed in a panel of non-tumoral and tumoral cell lines and we found a decrease amount of phosphorylated serine on β7 subunits of the studied tumoral cell lines compared to the experimental non-tumoral cell lines, suggesting its possible use as a biomarker.
On the other hand, the proteasome catalytic activity could be also affected by different environmental and cellular factors, leading to immunoproteasome expression. The presence of the immunoproteasome was found in different tumoral cell lines. The other part of my works is to develop a research tools to differentiate and to quantify the activities of the two-type proteasomes in tumoral cells. Two fluorescent peptide substrates with different preferring cleavage site by “proteasome” and “immunoproteasome” were synthesized and analysed. The use of these two substrates allowed us to identify a differential activity of proteasomes and/or immunoproteasomes purified from cell lysates, but could not be extended to a study with cell total extracts.
INTRODUCTION GENERALE
L’homéostasie des protéines au niveau cellulaire est finement contrôlée par trois grands processus fondamentaux : la synthèse de novo, la modification post-traductionnelle, et la dégradation [1]. De plus, la machinerie de dégradation des protéines joue plusieurs rôles essentiels dans la cellule : 1- la destruction des protéines endommagées, mal-repliées, et des protéines en fin de vie, 2- l’activation par maturation des protéines « précurseurs » et 3- la dégradation des protéines « régulatrices » qui contrôlent divers processus cellulaires [1, 2]. Environ 80% des protéines intracellulaires sont dégradées par la voie Ubiquitine-Protéasome (Ub-P) [1, 2]. Certaines protéines associées au cancer, telle que les protéines « suppresseurs de tumeurs » p53 et p27, les récepteurs 1 (EGFR) et 2 (HEER2/neu) de « Epidermal Growth Factor (EGF), et des facteurs de transcription tels que NF-κB, sont dégradées par l’Ub-P [3]. De plus, des perturbations de ces mécanismes de dégradation ont été retrouvées associées à de nombreux cancers [4, 5]. Par conséquent, la voie Ub-P est devenue une des cibles attractives en thérapie anticancéreuse [3].
Le système Ub-P est principalement constitué par un groupe d’enzymes, liant la chaîne de poly-ubiquitine aux protéines à dégrader, et par le protéasome 26S [6]. Le protéasome 20S, doté des activités catalytiques, associé au(x) complexe(s) régulateur(s) 19S forment le protéasome 26S. Le protéasome 20S est un complexe multi-protéique et multi-catalytique de forme cylindrique. Il est composé de quatre anneaux accolés et constitués de sept sous-unités de type α (α1 à α7) et de type β (β1 à β7). Exprimé de façon ubiquitaire dans toutes les cellules, le protéasome est devenu, depuis quelques années, une cible pharmacologique sur laquelle de gros efforts de recherche sont entrepris [7]. En particulier, son inhibition est une des stratégies récentes dans le domaine de la recherche de médicaments anti-cancéreux innovants [8]. Son inhibition provoque le blocage du cycle cellulaire dans les cellules normales et induit l’apoptose dans les cellules tumorales [8]. Un des inhibiteurs du protéasome, le PS-341 ou le Bortézomib (VelcadeTM) a démontré une activité anti-tumorale in vitro et in vivo sur plusieurs lignées cellulaires tumorales [4]. Le Bortézomib est donc le premier inhibiteur du protéasome qui a obtenu une autorisation de mise sur le marché (AMM) pour le traitement des myélomes multiples par la Food and Drug Administration (FDA) en 2003 [9] et par l’Agence Européenne d’Evaluation des Produits Médicaux en 2006 [10]. Il a récemment été utilisé dans le traitement de « lymphome du manteau » [11]. Par ailleurs, au cours des dernières années de recherche sur les molécules thérapeutiques anti-cancéreuses,
4
l’émergence de molécules inhibitrices du protéasome, une des classes les plus prometteuses de médicaments anti-néoplasiques, était en quelque sorte inattendue. Cela montre que nos connaissances des propriétés des cellules tumorales sont encore loin d’être complètes. Toutefois, l’utilisation de l’inhibiteur du protéasome « Bortézomib » en thérapie induit de nombreux effets secondaires, y compris les risques d’insuffisance cardiaque, une neuropathie périphérique et la thrombocytopénie [12]. Des programmes sont actuellement en cours pour rechercher des molécules modulatrices de l’activité du protéasome ayant un mécanisme d’action différent afin de tenter de diminuer ces effets secondaires. Plusieurs mécanismes ont été démontrés comme régulant l’activité du protéasome. Dans le cadre de cette thèse, nous avons porté nos efforts sur deux mécanismes particuliers : les modifications post-traductionnelles par phosphorylation et le changement des sous-unités catalytiques du protéasome conduisant à l’expression de l’immunoprotéasome.
Les modifications post-traductionnelles par phosphorylation jouent des rôles importants dans la régulation de l’activité de certaines enzymes [13] et un taux anormal en protéines phosphorylées a été démontré comme la cause ou la conséquence de certaines maladies telles que le cancer [14]. La phosphorylation des protéines substrats a été impliquée dans la régulation de la dégradation des protéines par la voie Ub-P [1]. En outre, plusieurs composantes du protéasome et de ses complexes régulateurs ont été trouvés phosphorylées [15, 16], suggérant que cette modification post-traductionnelle peut jouer des fonctions variées [17]. La phosphorylation pourrait jouer en particulier un rôle important dans la régulation de l’assemblage des sous-unités, et l’activité du protéasome [18-20]. Par ailleurs, lors de l’étude de séquences des gènes codant les sous-unités du protéasome, de nombreux sites potentiellement phosphorylés en Sérine, Thréonine et Tyrosine ont été trouvés sur les deux types (α et β) de unités du protéasome [21-23]. Toutefois, excepté pour la sous-unité α7 où la phosphorylation a été localisée en spectrométrie de masse [18, 24], la présence de cette modification sur les autres sous-unités n’a pas été bien démontrée. La mise au point d’outils permettant d’identifier de nouvelles isoformes phosphorylées du protéasome constitue l’objectif de mes travaux de thèse. En outre, la recherche de la présence d’isoformes phosphorylées du protéasome dans des cellules tumorales sera effectuée dans le but de déterminer si le niveau d’expression de ces isoformes peut être un critère discriminant entre des cellules tumorales et des cellules non-tumorales.
En présence d’interféron γ (IFNγ), les sous-unités catalytiques du protéasome standard (β1, β2 et β5) sont remplacées par les sous-unités dites inductibles (β1i, β2i et β5i) et forment ainsi l’immunoprotéasome. Les sous-unités β1i, β2i et β5i possèdent les mêmes spécificités enzymatiques que leurs homologues respectives du protéasome standard mais les niveaux d’activité varient. Suite à ces modifications d’activité, les peptides générés par l’immunoprotéasome sont différents de ceux engendrés par le protéasome standard et les antigènes présentés à la surface des cellules du système immunitaire sont par conséquent différents. L’implication de l’immunoprotéasome dans la génération d’épitopes tumoraux a été étudiée. Son rôle reste toutefois mal compris et très controversé. D’une part, la présence de l’immunoprotéasome favorise la génération de peptides plus immunogènes que le protéasome standard [25]. D’autre part, certains travaux ont montré que l’expression d’immunoprotéasome était associée à une production décrue de certains peptides antigéniques à partir de précurseurs tumoraux. Ainsi, une diminution de la génération des antigènes tumoraux reconnus par les lymphocytes T cytotoxiques spécifiques des antigènes RU1 (protéine rénale ubiquitaire) et Melan-A (protéine humaine mélanocytaire) est observée après traitement des cellules présentatrices à l’IFNγ [26]. Dans ce cas, l’immunoprotéasome pourrait donc avoir un impact négatif sur la réponse immunitaire antitumorale. L’objectif de mes travaux était de tenter de mettre au point des outils de recherche permettant de différencier les activités du protéasome standard de celles de l’immunoprotéasome et de quantifier le taux relatif d’immunoprotéasome dans les cellules tumorales.
Dégradation des protéines intracellulaires
Système lysosomal Système Ubiquitine-Protéasome
Sélectivité : +/- +
Cibles : protéines exogènes et endogènes par
processus « hétérophagie et autophagie »
sont majoritairement DRiPs et aussi des protéines impliquées dans la régulation des processus cellulaires fondamentaux
Figure 1 : Système de dégradation des protéines intracellulaires. DriPs : Defective ribosomal products.
Chapitre I : Le protéasome
Le renouvellement en continu des protéines cellulaires ou « turnover protéique » est essentiel pour le maintien de l’homéostasie cellulaire et pour la régulation des multiples fonctions des cellules. L’idée d’une régulation des protéines par dégradation a été proposée il y a 60 ans, au moment où le concept protéolytique restait un domaine négligé et était considéré comme un processus cellulaire non spécifique. La découverte du lysosome dans les années 50 n’a pas sensiblement changé ce point de vue. Cet organelle était en effet connue principalement pour son implication dans la dégradation des protéines extracellulaires et les protéases lysosomales impliquées n’étaient pas spécifiques de leurs cibles [1]. L’avancée de la science nous a permis de découvrir d’autres machineries de dégradation protéique non-lysosomales dans les années 80 : le système Ubiquitine-Protéasome (figure 1). Ce système est impliqué dans le contrôle de nombreux processus cellulaires tels que la régulation du cycle cellulaire, de la division cellulaire, de l’apoptose, de la transcription, de la présentation de l’antigène, de la transduction des signaux, du contrôle de l’intégrité des protéines et encore du métabolisme [27]. A la vue de la multitude de substrats ciblés et de la myriade de processus cellulaires dans lesquels le système Ubiquitine-Protéasome est impliqué, il n’est pas surprenant que des aberrations de ce système soient impliquées dans les régulations observées dans de nombreuses maladies, notamment dans la tumeurogenèse et dans les processus de neurodégénérescence [1]. La dégradation protéique est donc devenue un domaine de recherche important de la biologie moderne. La découverte du complexe en cascade de la voie Ubiquitin-Protéasome a révolutionné ce domaine de recherche [27].
I.1. Systèmes protéolytiques intracellulaires «Système Ubiquitine-Protéasome»
Le système Ubiquitine-Protéasome (Ub-P) est un système protéolytique majeur extra-lysosomal des cellules eucaryotes [28]. Cette voie de dégradation est hautement complexe et joue un rôle majeur dans le maintien et la régulation des processus cellulaires très variés, tels que la différentiation, la prolifération, le cycle cellulaire, l’apoptose, la transcription des gènes, la transduction des signaux, la présentation de l’antigène et l’homéostasie cellulaire [29-31]. La dégradation des protéines via le système Ub-P peut être dissociée en deux étapes successives : un « pré-marquage » des substrats à dégrader par polyubiquitination, qui est une étape fréquente mais pas requise pour tous les substrats et le processus de dégradation proprement dit des substrats ou clivage protéolytique.
Ubiquitine Protéine K NH-CO G76 K48 G76 Ubiquitine NH -CO K NH-CO G76 K48
Lysine du substrat peptidique
Lysine 48 de l’Ubiquitine
Glycine 76 du C-terminal de l’Ubiquitine Liaison peptidique
Figure 2 : Chaîne bi-Ubiquine avec substrat peptidique.
Ubiquitine K48 K33 K29 K27 K11 K6 K63 -Dégradation des protéines -Régulation de l'activité de facteurs de transcription -Dégradation des protéines -Ubiquitination de Bagl ? -Réparation de l'ADN -Endocytose -Régulation de Kinases -Dégradation des protéines
Figure 3 : Rôles divers des 7 Lysines (K) de l’Ubiquitine permettant la conjugaison sur elle-même. Bagl : Bcl-2-associated athanogene.
L’ubiquitine (Ub) est une protéine composée de 76 acides aminés. La Glycine C-terminale (G76) est essentielle et nécessaire pour sa liaison avec les substrats ciblés ou pour sa liaison à d’autres molécules d’ubiquitines. L’Ub est généralement conjuguée sur ses substrats via une liaison isopeptidique entre l’extrémité carboxyterminale de son résidu glycine (G76) et la fonction amine en ε-NH2 de lysines acceptrices de ses substrats (figure 2). C’est le cas,
par exemple, de l’ubiquitination de la p53. Dans certains cas, mais très rare, l’Ub peut se conjuguer via une liaison peptidique classique entre son extrémité C-terminale et l’extrémité N-terminale de la protéine acceptrice. C’est le cas, par exemple de l’ubiquitination de l’oncoprotéine E7 (du papilloma virus humain HPV) [5]. L’Ub contient, elle-même, 7 résidus lysines (K6, K11, K27, K29, K33, K48, K63) utilisés pour la formation de chaînes polyubiquitines. Ces différents types de branchement confèrent une richesse topologique aux chaînes d’Ub, ce qui, vraisemblablement, permet l’adaptation du système à une grande variété de structures. De plus, ces différents types de chaînes ont été impliqués dans des processus divers, non liés nécessairement à la dégradation protéique [32-34]. Les chaînes K48 sont essentiellement impliquées dans le marquage des protéines à dégrader. Néanmoins, elles peuvent aussi être impliquées dans des fonctions non-protéolytiques, comme par exemple dans la régulation de l’activité de facteurs de transcription, soulignant la complexité de système Ub-P (figure 3) [5].
La polyubiquitination est accomplie en plusieurs étapes et en plusieurs cycles catalysés par trois classes différentes d’enzymes : les ubiquitine ligases E1, E2 et E3 (figue 4). La formation de la chaîne poly-ubiquitine peut aussi être catalysée directement par une autre enzyme E4 ou « Ubiquitin Chain assembly/elongator Factors » [28, 35]. Il existe une seule E1, des trentaines d’E2 et plusieurs centaines d’E3 différentes. L’E1 transfert l’Ub à toutes les E2s connues. La plupart des E2s interagissent avec plusieurs E3s et la plupart des E3s interagissent avec plusieurs protéines [1].
L’enzyme E1 ou « Ub-activating enzyme », est unique. Elle active l’Ub de manière ATP-dépendante pour former un complexe thio-ester E1-Ub. Chez la levure, l’inactivation du gène codant pour l’orthologue d’E1 : UBA1, est létale. De plus, cette protéine contient un signal de localisation nucléaire ou NLS et peut être phosphorylée, suggérant le rôle de cette modification post-traductionnelle dans la signalisation de déplacement dans le cycle cellulaire [36].
Les enzymes E2 ou « Ub-carrier/conjugating proteins » transfèrent l’Ub activée aux E3s. Dans certains cas, E2 peut catalyser l’attachement de l’Ub aux protéines cibles car certaines d’entre elles ont des activités spécifiques vis-à-vis de certaines protéines cibles [29].
E1 E2 E3 ATP ATP Présentation de l’antigène Dégradation par protéasome 26S peptides Protéine à dégrader Ubiquitination Réutilisation Acides aminés Déubiquitination : Ubiquitine
Les E2s possèdent un résidu cystéine nécessaire à la formation d’autres complexes thio-ester avec l’Ub et se distinguent par la présence de domaines UBC (ubiquitin-conjugating domain) requis à la liaison aux différentes E3s [37]. Plusieurs E2s ont été identifiées et sont impliquées dans de différents processus cellulaires : la dégradation protéique, mais aussi la réparation de l’ADN pour UBC2/RAD6 dans la levure. L’Ubc6 est une E2 trans-membranaire du réticulum endoplasmique (ER) qui est impliquée dans la dégradation des protéines ER [38].
Les enzymes E3s appartiennent à la famille Ubiquitin-proteines ligases, qui catalysent le transfert de l’Ub, à partir du complexe thio-ester E2s, sur le groupement NH2 de la protéine
cible ou sur celui d’une chaîne polyubiquitine déjà ancrée sur la protéine cible. E3 peut se lier directement aux protéines cibles ou indirectement à l’aide de protéines auxiliaires. Comme les protéines cibles se lient à E3 avant la conjugaison avec l’Ub, E3 devient la protéine clé dans la détermination de la spécificité du système. Même si les E3s sont hétérogènes, on peut les classer en deux grands groupes : les E3s à domaine HECT et les E3s à domaine « Ring finger ». Dans le cas des domaines HECT (homologous to E6-associated protein C-terminus), l’Ub activée est transférée à partir de E2 à la cystéine de E3 avant la conjugaison de cette Ub sur la protéine cible [1]. Les E3s à domaine HECT possèdent la séquence homologue au domaine C-terminal de la famille E6-AP. Ce domaine contient un résidu cystéine conservé recevant l’Ub transférée de l’E2. Le domaine N-terminal varie entre les différentes protéines à domaine HECT et est probablement impliqué dans la reconnaissance spécifique des substrats [39, 40]. La protéine Smurf1 est une autre E3 à domaine HECT qui interagit spécifiquement avec les protéines SMADs (Small Mothers Against Decapentaplegic), des effecteurs clés de la superfamille des facteur de croissance TGF-β. L’expression ectopique de Smurf1 affecte la croissance du Xénope [41]. Dans le cas des E3s à domaine Ring finger, E3 sert de support amenant E2 à la protéine cible et permettant le transfert direct de l’Ub de E2 à la protéine cible. A ce jour, deux types d’E3 à domaine Ring finger ont été identifiées : (i) des monomères ou homodimères qui contiennent à la fois le domaine à Ring finger et le site de reconnaissance des substrats dans la même molécule, et (ii) des complexes composés de plusieurs sous-unités (1). La protéine E3α et son homologue de levure UBR1 sont des E3s de type (i). Elles reconnaissent les résidus N-terminaux basiques ou hydrophobes des substrats selon la loi du résidu N-terminal (N-end rule). Cependant elles reconnaissent aussi les substrats sans N-end rule comme des protéines N-α-acétylées via un site qui n’est pas encore bien caractérisé [42]. La plupart des E3s restantes sont de type (ii) : comme le cyclosome ou
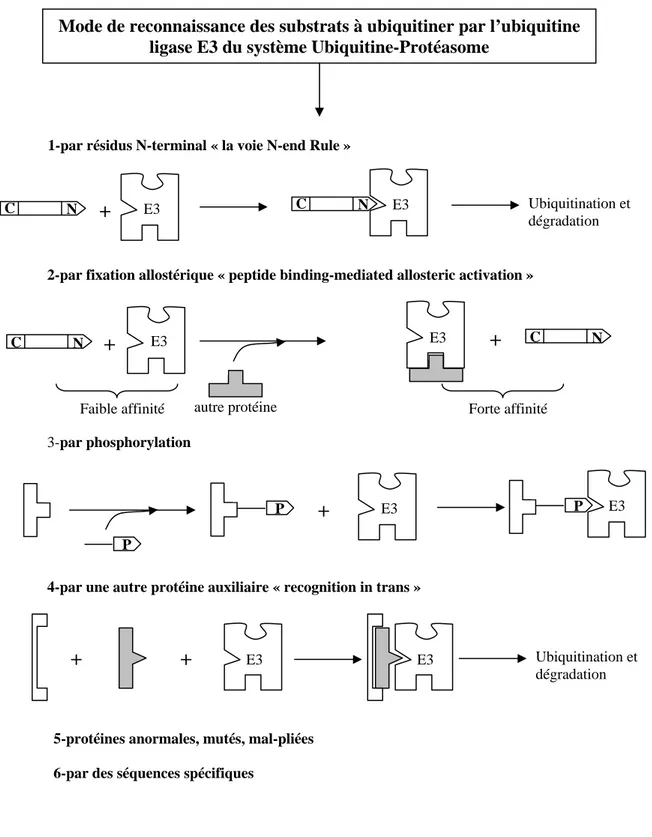
Mode de reconnaissance des substrats à ubiquitiner par l’ubiquitine ligase E3 du système Ubiquitine-Protéasome
1-par résidus N-terminal « la voie N-end Rule »
N C E3 E3 Ubiquitination et dégradation
+
N C2-par fixation allostérique « peptide binding-mediated allosteric activation »
E3
+
C N E3+
N C autre protéineFaible affinité Forte affinité
3-par phosphorylation
P E3 E3
P
+
P
4-par une autre protéine auxiliaire « recognition in trans »
+
+
E3 E3 Ubiquitination etdégradation
5-protéines anormales, mutés, mal-pliées
6-par des séquences spécifiques
: protéines à ubiquitinyler et à dégrader N
C
APC (anaphase promoting complex) responsable de la dégradation des régulateurs du cycle cellulaire comme CDK1 et la cycline B [1, 29].
Une autre famille d’enzymes désignées E4 ou « Ub-chain elongation factor » (appelé aussi U-box proteins) a été décrite pour sa capacité à rallonger la chaîne courte de polyubiquitine par transfert de molécules ubiquitine additionnelles sur la protéine cible [43]. L’Ufd2 (ubiquitin fusion degradation protein-2) appartient à cette famille et est impliquée dans la dégradation des substrats chimériques marqués par une molécule Ub à l’extrémité N-terminale [44]. Une autre exemple d’E4 est la protéine CHIP (C-terminal of Hsp-70-interacting protein) qui est impliquée dans la dégradation de CFTR (cystic fibrosis transmembrane conductance regulator) et des récepteurs des glucocorticoïdes [1]. Les protéines substrats des E4s sont liées par des chaperonnes, probablement à leur liaison avec les E4s [2].
I.3. Modes de reconnaissance des substrats à ubiquitiner par l’E3
Cibler les protéines substrats par la voie d’Ub-P implique la fixation spécifique entre les protéines ciblées et l’ubiquitine ligase E3 appropriée. En principe, la reconnaissance peut être faite par plusieurs mécanismes : soit le substrat est modifié pour être reconnu par les E3s, par exemple, le substrat doit être phosphorylé pour être reconnu par le complexe SCF (SKP1-Cullin-F box) ; soit c’est E3 elle-même qui peut être modifiée, par exemple, l’activité des E3s à domaine HECT dépend d’autres protéines auxiliaires faisant le pont entre l’enzyme et le substrat [1]. A ce jour, il existe plusieurs modes spécifiques de reconnaissance des substrats à ubiquitiner (figure 5) :
a. Reconnaissance par des résidus N-terminaux « la voie N-end Rule »
Lors de cette reconnaissance, les substrats se fixent directement sur la ligase via leurs résidus N-terminaux. Le E3α/UBR1 contient deux sites de reconnaissance par la voie N-end rule : site I pour les substrats avec des résidus N-terminaux basiques et site II pour les substrats avec des résidus N-terminaux hydrophobes [45, 46]. RGS4, un membre des protéines RGS (regulator of G protein signaling), modifié post-traductionnellemnent par arginylation au niveau des résidus N-terminaux, est ubiquitiné par UBR1 puis rapidement dégradé in vitro [47].
b. Reconnaissance par transition allostérique « peptide binding-mediated allosteric activation »
L’E3α/UBR1 possède aussi un troisième site de reconnaissance ciblant des motifs « body » non-identifiés en amont des résidus N-terminaux [45, 46]. Les protéines N-α
10
acétylées, protéine Gα de la levure (impliquée dans la régulation de la croissance par les phéromones), et le facteur de transcription Cup9 ont été démontrées comme interagissant avec cette ligase par l’intermédiaire du troisième site [1]. En fait, la fixation d’autres peptides sur le site I ou II active allostériquement l’UBR1 et augmente la vitesse d’ubiquitination puis la dégradation [47].
c. Reconnaissance par la phosphorylation
De nombreux substrats de la voie Ub-P sont modifiés par phosphorylation avant leur ubiquitination. Cette modification post-traductionnelle a été démontrée, dans certains cas, nécessaire pour que les substrats soient directement reconnus par les E3s appropriées [1]. L’activation du NF-κB est normalement achevée après la protéolyse de l’IκBα. La phosphorylation de l’IκBα sur les Sérine 32 (Ser32) et Ser36 (par IKKα et IKKβ) est nécessaire pour que ce facteur soit reconnu par le complexe SCF spécifique (SCFβ-Trcp) ubiquitiné et enfin dégradé [48]. D’autres protéines dont le temps de demi-vie est court contiennent une séquence riche en résidus PEST (Pro, Glu, Ser, Thr). Cette séquence est impliquée dans la dégradation de ces protéines à la suite de la phosphorylation sur les résidus Ser et Thr. Dans certains cas, au contraire, la phosphorylation inhibe l’ubiquitination [1]. L’addition de la protéine kinase A (PKA) aux fractions de cyclosome inhibe l’ubiquitination de la cyline B [49].
d. Reconnaissance par d’autres protéines auxiliaires « recognition in trans »
Dans certains cas, les protéines ciblées ne sont pas reconnues directement par la ligase, mais après la fixation de protéines auxiliaires [1]. Certains virus utilisent ce système pour leur propagation [39, 50]. Le prototype de cette interaction est l’interaction entre E6-AP contenant un domaine HECT et la protéine E6 des HPVs entraînant la dégradation de p53. La protéine E6 forme un pont entre E6-AP et p53. La formation de ce complexe entraîne l’ubiquitination de p53 [39]. La protéine Vpu des virus HIV-1 est, après modification par phosphorylation, reconnue par la F-box du complexe SCF. Cette protéine, va fixer additionnellement au récepteur CD4 des lymphocytes T. Cette fixation entre les trois protéines entraîne l’ubiquitination et la dégradation du CD4 par le complexe SCF [50].
e. Protéines anormales, mutées, mal-repliées
Les protéines dénaturées/mal-repliées à la suite de mutations ou de stress environnementaux post-traductionnels sont sélectionnées et dégradées efficacement par la voie Ub-P [51]. Les chaînes peptidiques, n’ayant pas encore atteint leurs structures définitives à cause d’erreurs lors de la traduction ou lors de processus post-traductionnels, sont aussi
Sous-unité β Sous-unité α Complexe 19S
protéasome 20S protéasome 26S
11
dégradées par cette voie [1]. La chaîne à dégrader est appelée DRiPs (defective ribosomal products). La nature des signaux et l’identité des ligases impliquées sont encore inconnus. Il est possible que les domaines hydrophobes soient utilisés comme signaux de reconnaissance de ces DRiPs [1].
f. Reconnaissance via des séquences spécifiques
L’ubiquitination des cyclines mitotiques peut être induite par le motif « small N-terminal » connu sous le nom de « destruction box ». Ce motif est composé de neuf résidus dont la séquence consensus est : R-A/T-A-L-G-X-I/V-G/T-N [52]. Le rôle de ce motif n’est pas très clair puisqu’il n’est pas phosphorylé ou ubiquitiné. Pourtant, il peut être utilisé comme site de fixation pour le complexe ligase APC [1].
I.4. Cœur catalytique de l’Ub-P : «le protéasome»
Dans la voie Ub-P, dans la majorité des cas, pour dégrader les protéines cibles, il existe deux étapes majeures successives : (i) l’ubiquitination des protéines cibles, assurée par la machinerie d’ubiquitination et (ii) la dégradation réelle des protéines cibles, assurée par le cœur catalytique de la voie : « le protéasome », un complexe catalytique et multi-protéique hautement conservé chez cellules procaryotes et eucaryotes [27, 53].
a. Structure du protéasome
Des particules de haut poids moléculaire dotées d’activités protéolytiques dénommées « particules cylindriques », « prosome », « protéases alcalines » ou bien « complexe protéase multi-catalytique » ont été découvertes dans les années 80. Toutes ces protéines ont des points communs : une forme cylindrique, une constante de sédimentation de 20S, un poids moléculaire environ 600 à 700 kDa et sont formées par l’assemblage de différentes sous-unités de poids moléculaires variables [53]. La constatation que ces particules cylindriques 20S sont identiques aux complexes protéases multi-catalytiques et a conduit à regrouper ces complexes sous le terme de protéasome [54]. Un complexe catalytique similaire à celui détecté chez la levure a été trouvé équivalent au protéasome chez la drosophile et les mammifères [55]. L’identification du complexe 26S contenant le complexe 20S comme cœur catalytique nous a permis d’élargir la connaissance de cette machinerie appelée actuellement «le protéasome 26S » dans de nombreux aspects fondamentaux cellulaires [53]. La capacité de dégrader spécifiquement et sélectivement des protéines par le protéasome est obtenue par l’association des deux composants du protéasome 26S : les particules régulatrices 19S et le cœur catalytique « protéasome 20S », en bref, le complexe 19S reconnaît sélectivement les substrats, le complexe catalytique 20S, quant à lui n’a pas de spécificité de reconnaissance et ne peut dégrader que les protéines une fois dépliées (figure 6) [1, 53].
b. Protéasome 20S
Le protéasome caractérisé par une structure hautement conservée, sous une forme ou une autre, a été trouvé dans tous les domaines de la vie [56]. Le protéasome 20S a une structure unique : il ressemble à un cylindre ou un baril avec des sites catalytiques protéolytiques résidant sur la face interne du cylindre. L’ancêtre des protéasomes des eucaryotes est celui des eubactéries et des archaebactéries [53]. La structure caractéristique de ce complexe est l’empilement de 4 anneaux composés de 7 sous-unités α ou β chacun : [α7β7β7α7] [53]. Seules les sous-unités β possèdent les sites catalytiques. Chez les bactéries,
chaque sous-unité β est active, le nombre de sites catalytiques s’élève à 2 x 7 sites actifs identiques. Les sous-unités α, dont le nombre est 2 x 7, sont aussi identiques chez le protéasome des eubactéries [57, 58].
Pour le protéasome chez les eucaryotes, les séquences des sous-unités α et β se sont diversifiées lors de l’évolution, conduisant à la naissance de 7 différents types de sous-unités α et β. Cette évolution permet la réduction du nombre de sites actifs des sous-unités β, cela permet au protéasome des eucaryotes d’avoir 2 x 3 sites protéolytiques [53]. Chez la levure, les sous-unités du protéasome sont codées par 14 gènes individuels et chez les cellules de mammifères, 3 gènes additionnels, inductibles par l’interféron gamma (INFγ), existent et codent d’autres variants des sous-unités β (LMP2/β1i, MECL-1/β2i et LMP7/β5i). L’incorporation de ces nouveaux types de sous-unités β conduit à la formation d’un sous-type de protéasome « l’immunoprotéasome » responsable de la genèse des peptides antigéniques présentés par le complexe majeur d’histocompatibilité (CMH) classe I [59, 60] et récemment décrit pour le complexe CMH de classe II [61].
Le protéasome 20S mesure 15 nm de long et 11 nm de diamètre. La structure [α7β7β7α7]
conduit à la formation de 3 cavités dans la particule : la cavité catalytique, appelée « chambre », d’environ 84 nm3 est formée par les anneaux β et 2 autres cavités, appelées « antichambres », sont formées par un anneau α et un anneau β et ont un volume d’environ 59 nm3 [57, 62]. La taille des antichambres indique qu’elles peuvent accueillir une quantité
importante de protéines ou de produits de dégradation des substrats [53]. Les sites catalytiques dans la cavité « chambre » active peuvent être atteints par deux pores axiaux d’un diamètre d’environ 2 nm formés par les sous-unités α à chaque extrémité du protéasome. Cette dimension de chaque pore assure que seuls les polypeptides dépliés peuvent entrer dans la cavité catalytique du protéasome. De plus, l’accès à l’intérieur est fermé et n’est possible
13
qu`après réarrangement des hélices N-terminales des sous-unités α. L’ouverture des pores est induite par le complexe régulateur 19S ou PA28/11S [30, 63, 64].
c. Biogenèse et assemblage du protéasome 20S
La structure du protéasome 20S a bien été caractérisée [57]. La question maintenant est de savoir comment cette structure est générée à partir de ses composantes. La forme moins complexe du protéasome 20S d’Archaea et d’Eubacteria nous a permis de récapituler les associations de l’assemblage des composantes du protéasome 20S.
Chez les Archaebactéries (par exemple, chez Thermoplasma acidophilum), l’expression des sous-unités α en absence de sous-unités β permet l’assemblage principalement en paire d’anneaux heptamèriques. En revanche, les sous-unités β restent sous forme monomèrique en absence d’expression des sous-unités α et elles sont inactives [65, 66]. Ces études nous suggèrent que la formation des intermédiaires lors de l’assemblage du protéasome d’Archaebactérie est due à l’assemblage des sous-unités α [67]. Cependant, les anneaux α n’ont pas encore été montrés in vivo comme l’intermédiaire de l’assemblage du protéasome d’archaebactérie [67].
Chez les Eubactéries, le premier protéasome étudié était celui de Rhodococcus sp. Ce protéasome est composé de 4 anneaux heptamèriques accolés des sous-unités α et β [67]. Cependant, il n’existe que 2 types différents de sous-unités α et de sous-unités β [68]. Contrairement au système du protéasome de Thermoplasma, aucun anneau n’est pas formé à partir de l’expression des sous-unités α seules ou β seules. La formation des structures en anneaux est trouvée seulement lors de l’expression simultanée des sous-unités α et β. Toutes les combinaisons d’hétérodimères (α1β1, α1β2, α2β1 et α2β2) donnent la naissance au protéasome final actif in vitro et in vivo [69].
Chez les eucaryotes, le protéasome 20S des cellules eucaryotes est plus complexe par rapport à son homologue bactérien puis qu’il est composé de plusieurs sous-unités différentes (7 différentes sous-unités α et 7 différentes sous-unités β), qui s’occupent les positions bien définies dans la particule 20S. 5 sous-unités parmi les sous-unités β (β1, β2, β5, β6 et β7) ont des propeptides N-terminaux qui sont clivés lors de leur maturation, et seulement 3 d’entre eux portent finalement les 3 sites catalytiques. Possédant plus de complexité, l’assemblage du protéasome des cellules eucaryotes est beaucoup plus compliqué que celui des procaryotes.
Les premières étapes de l’assemblage des sous-unités du protéaosme 20S chez les eucaryotes ne sont pas très bien connues. Les sous-unités α7 sont capables de former spontanément des doubles structures en anneaux. Quant à ses voisines, α6 et α1, elles sont
incapables d’en faire dans les mêmes conditions d’expériences. Les deux types de sous-unités α, en revanche, peuvent s’incorporer dans la structure d’anneaux lors de co-expression avec les sous-unités α7. Cette observation suggère que toutes les sous-unités α ne portent pas les informations pour le positionnement correct dans les anneaux des sous-unités du protéasome 20S, et elle suggère aussi que l’interaction avec les sous-unités β pourrait être nécessaire pour guider leur orientation de position dans les anneaux 20S [70, 71]. Il n’est pas très clair si l’assemblage du protéasome 20S des cellules eucaryotes est initié par un intermédiaire d’anneaux des sous-unités α ou s’il est initié par l’interaction des sous-unités α/β [67]. D’autres études de l’assemblage du protéasome 20S des levures et des mammifères ont révélé l’apparition d’autres intermédiaires plus complexes. Ces intermédiaires semblent être « un complexe demi-protéasome » composé d’un anneau des unités α, d’un anneau des sous-unités β inactives ainsi que le facteur de maturation du protéasome Ump1. Les analyses biochimiques ont montré que ces complexes sont à 13-15S, dont le poids moléculaire est 300-350 kDa en gel filtration [72, 73]. De plus, ces études ont montré aussi qu’il existe d’un autre complexe intermédiaire, qui est assez stable à être détecté, avant la formation de « demi-protéasome ».
d. Maturation des sous-unités catalytiques en vue de compléter l’assemblage
Pour le protéasome de Thermoplasma acidophilum, toutes les sous-unités β sont transcrites à partir d’un seul gène et s’expriment sous forme de précurseurs avec propeptides N-terminaux. Lors du processus de maturation du protéasome des archaebactéries, toutes les copies des sous-unités β sont activées par le processus post-traductionnel de maturation. Les propeptides N-terminaux sont clivés par le mécanisme d’autocatalyse intra-sous-unité rendant la forme mature des sous-unités β avec le résidu Thr à l’extrémité N-terminale qui attaque les liaisons peptidiques des substrats. Pour cette raison, on place le protéasome dans la famille « Ntn-hydrolases » (N-terminal nucleophile hydrolases) [74].
L’image est plus complexe pour le protéasome des eucaryotes puisqu’il contient 7 types différents des sous-unités β. Parmi les 7 sous-unités β, 5 sous-unités sont synthétisées avec des propeptides N-terminaux, qui diffèrent par leur séquence primaire et leur longueur . Ces sous-unités sont des sous-unités catalytiques (β1, β2 et β5) et 2 autres sous-unités sont non catalytiques (β6 et β7) [75]. La maturation des sous-unités non catalytiques avec propeptides N-terminaux (β6 et β7) n’est pas obtenue par le mécanisme intramoléculaire d’autocalyse. Les propeptides N-terminaux des sous-unités β non catalytiques sont clivés par les sous-unités catalytiques la plus proche. Chez la levure, les propeptides des sous-unités β6 et β7 sont
15
clivés par la sous-unité β2 qui est la plus proche sans l’aide de β1 ou de β5. Pourtant, les deux sous-unités non catalytiques peuvent atteindre leurs formes finales par le clivage des propeptides par les sous-unités β1 et β5 dans la levure contenant la β2 mutante inactive [76]. D’autres études sur la mutation des sites actifs ont montré un processus de maturation des sous-unités catalytiques (β1, β2 et β5) en 2 étapes successives. Très probablement, l’étape finale de la maturation des sous-unités catalytiques est le clivage autocatalytique au niveau des sites consensus. Cette étape est précédée par une réduction de la longueur des propeptides en longueurs uniformes favorisant le clivage autocatalytique [77].
Une autre question se pose sur les rôles des propeptides des sous-unités β dans le processus de maturation du protéasome. In vitro, les propeptides sont indispensables pour l’assemblage du protéasome de Thermoplasma acidophylum [75]. Chez la levure, les propeptides exercent des rôles inégaux reflétant une hiérarchie de fonction des sites actives : β5> β2 >β1 [78]. Parmi les trois sous-unités catalytiques, la présence du propeptide de la β5 est nécessaire pour la viabilité cellulaire puisqu’il est essentiel à l’exécution des fonctions de l’Ump1 dans l’assemblage et la maturation du protéasome (voir ci-dessous e. Protéines accessoires dans la maturation du protéasome). La déplétion de propeptide de la β1 a très peu d’effet sur la levure. Pourtant, la déplétion de propeptide de la β2 conduit à un ralentissement important de la croissance mais son rôle ne semble pas dépendant à l’Ump1. Concernant les sous-unités non catalytiques (β6 et β7), alors que la suppression complète du propeptide de la β7 n’a pas d’effet sur la croissance, la partie terminale du propeptide de la β6 semble être indispensable pour la viabilité cellulaire mais cette fonction a été trouvée seulement en présence de l’Ump1 [78]. Un autre rôle important des propeptides semble être de protéger le résidu Thr de l’extrémité N-terminal de l’inactivation par l’acétylation. D’autres études ont montré que les propeptides jouent un rôle important dans la coordination de l’assemblage des sous-unités β induites par l’INF γ conduisant la formation de l’immunoprotéasome. En bref, en plus de maintenir les sous-unités β en état dormante et protégée, les propeptides semblent avoir le rôle d’une chaperonne efficace dans le pliement et l’assemblage des sous-unités du protéasome en interaction avec d’autres facteurs de maturation [67].
e. Protéines accessoires dans la maturation du protéasome
La maturation du protéasome nécessite le support d’autres protéines accessoires qui sont importantes dans les étapes finales de maturation du protéasome. Un exemple est la protéine chaperonne hsc73. Cette protéine a été trouvée associer au complexe 16S des cellules mammifières. La hsc73 a pour rôle de maintenir partiellement le complexe en état non-plié
(pour permettre la maturation des sous-unités β), de corriger les pliements du complexe et d’incorporer d’autres sous-unités restantes du protéasome [75]. D’autres facteurs jouant un rôle crucial dans la maturation du protéasome est la protéine de levure Ump1 (underpin the maturation of the proteasome 1). L’Ump1 fait partie du complexe précurseur du protéasome et est dégradée lors de l’étape finale de maturation. L’Ump1 est nécessaire pour l’assemblage normal et la maturation du protéasome. Dans les cellules mutantes Ump1-∆, l’assemblage et la maturation du protéasome sont fortement affectées. La formation de la structure 20S à partir des précurseurs « demi-protéasome » est moins efficace et la maturation des trois sous-unités catalytiques β est considérablement réduite (la plupart des sous-sous-unités catalytiques retient encore des propeptides après l’assemblage en protéasome 20S), conduisant à la diminution totale des activités du protéasome et une altération de la croissance de levure [73, 79]. Il a été démontré précédemment que le propeptide de la β5 est essentiel pour la viabilité de la levure et que ce propeptide réagit comme un chaperon intramoléculaire nécessaire pour l’incorporation de la β5 dans le protéasome [78, 80]. Des résultats plus récents ont montré que le propeptide de la β5 n’est pas essentiel pour l’incorporation de la β5 dans le protéasome mais ce propeptide est nécessaire pour induire un changement de conformation ou de position de l’Ump1 lors de l’assemblage du protéasome [67, 73]. En absence de propeptide de la β5, l’Ump1 est dans la position ou dans la conformation qui n’est pas compatible avec l’étape suivante de maturation du protéasome ou avec la fonction du protéasome [67, 73]. En revanche, le propeptide de la β5 (qui est essentiel pour la viabilité de levure sauvage) n’affecte pas croissance dans les levures mutantes Ump1-∆ qui s’expriment ce propeptide [73]. Récemment, d’autres homologues de l’Ump1 ont été identifiés soit par co-purification avec le complexe 16S soit par la recherche dans les bases de données : POMP (proteasome maturation protein) [81], proteassembline [79], h/m UMP (human/mouse UMP) [82] ou PACs (proteasome assembling chaperones) [83]. Les analyses biochimiques des fractions lors des expériences ont montré que ces homologues ne sont associés qu’aux complexes précurseurs du protéasome et non aux protéasomes matures [79, 81-83].
I.5. Complexes régulateurs du protéasome 20S
Plusieurs études, utilisant la microscopie électronique, ont montré que le protéasome peut se présenter sous plusieurs formes dans la cellule : soit sous forme de protéasome 20S, soit sous forme de complexes « 20S-régulateurs » [58, 84]. Trois principaux complexes régulateurs du protéasome 20S ont été décrits. Le premier est le complexe régulateur 19S (RP) qui permet au protéasome 20S de dégrader les protéines polyubiquitinées de manière ATP-dépendante [84].
Lid Complexe 19S Base Rpn10 Rpt1 Rpt2 Rpt3 Rpt4 Rpt5 Rpt6 Rpn1 Rpn2 Rpn13 Rpn3 Rpn5 Rpn6 Rpn7 Rpn9 Rpn12 Rpn8 Rpn11 Rpn15
Figure 7: Représentation schématique du complexe 19S et rôles de ses sous-unités. Rpt : regulatory particle triple-A ATPase, Rpn : regulatory particle non-ATPase. Ouverture de l’orifice accédant à la particule catalytique Reconnaissance de la chaîne polyubiquitine Reconnaissance de la chaîne polyubiquitine Reconnaissance de la chaîne polyubiquitine Déubiquitination des substrats
Le deuxième complexe régulateur est le PA28 ou 11S REG qui stimule l’activité protéasique du complexe 20S par le changement de conformation des sous-unités du 20S, mais qui ne favorise pas la dégradation des protéines pliées. Le troisième complexe régulateur est le PA200, récemment identifié, qui se lie au protéasome 20S et stimule l’hydrolyse des petits peptides par le protéasome 20S [84].
a. Complexe régulateur 19S (RP)
Le RP est le complexe régulateur du protéasome 20S (CP) des cellules eucaryotes (217). Il a un poids moléculaire d’environ 700 kDa, et est constitué d’au moins 19 sous-unités (figure 7). Le RP peut se lier à l’une des extrémités du protéasome 20S ou les deux extrémités, formant ainsi le protéasome 26S [85]. Le RP lui-même peut se diviser en deux sous-complexes : le complexe «base» et le complexe «Lid-like» [86].
Le complexe « base » se localise à la proximité du CP et contient 6 sous-unités ATPases (Rpt 1 à 6 : Rpt= regulatory particle triple-A ATPase protein) et 4 sous-unités non-ATPases (Rpn1, Rpn2, Rpn10 et Rpn13 : Rpn= regulatory particle non-ATPase). Les six Rpts de la base forment un anneau et participent dépliement des protéines ubiquitinées à dégrader [87, 88]. Malgré la haute similarité de séquence de ces six Rpts de la base, ils n’ont pas de rôles équivalents [51]. Le Rpt2 jouerait un rôle dans l’ouverture de l’orifice permettant d’accéder au CP et donc de faciliter l’entrée du substrat à dégrader [89]. Le Rpt5 participe à la reconnaissance de la chaîne poly-ubiquitine [85]. Le Rpn10 est clairement un récepteur de l’ubiquitine [85]. Rpn1 et Rpn2 interagissent avec les protéines contenant le domaine UBL et ont des motifs riches en leucine connus pour être des domaines d’interaction protéine-protéine [85]. Donc, les Rpn1 et Rpn2 pourraient jouer un rôle dans la reconnaissance de la chaîne polyubiquitine et de protéines contenant le domaine UBL. De plus, il a été démontré que le Rpn13, comme le Rpn10, interagit avec le domaine UBL des protéines UBA (ubiquitin-associated domain). Le Rpn13 est capable de se lier à la mono- et di-ubiquitine, montrant que protéines monoubiquitinées sont aussi des cibles à dégrader par le protéasome [90].
Le complexe «Lid-like», formé par 8 sous-unités Rpns (Rpn3, Rpn5-7, Rpn8-9, Rpn11-12), se trouve à la partie distale du complexe «base». Ils ont une haute homologie avec deux autres complexes protéiques : le signalosome COP9 et le facteur d’initiation de traduction eIF3 [86, 91]. Les trois complexes sont caractérisés par deux motifs connus sous les noms MPN (Mov34, Pad N-terminal) et PCI (Proteasome, COP9, eIF3) [86, 91]. Les sous-unités contenant le domaine MPN sont Rpn11 et Rpn8. Les autres sous-unités possèdent le domaine PCI. Le domaine PCI n’est pas très bien connu mais le domaine MPN de Rpn11 possède l’activité « déubiquitine métallo-protease-like » qui enlève la chaîne Ub des substrats à
Protéasome 20S
PA200 PA28
Complexe 20S-PA28 Complexe 20S-PA200
Figure 8 : Représentation schématique des complexes régulateurs PA28 et PA200 avec le protéasome 20S.
dégrader [92]. Le domaine MPN de Rpn8 est très similaire à celui de Rpn11 mais il manque des résidus catalytiques [85]. Récemment un autre membre du complexe «lid-like», le Rpn15, a été identifié mais son rôle n’est pas encore bien connu [85].
Le complexe régulateur 19S a été seulement trouvé dans les cellules eucaryotes, mais chez certains archées, des homologues du complexe «base» ont été trouvés et partagent une similarité de structure et de fonction avec le complexe base du 19S [91].
b. Complexe régulateur 11S (PA28)
Le complexe régulateur PA28 a été identifié pour la première fois dans les cellules mammifères (figure 8) [93]. Sa présence n’a jamais été détectée dans les levures, les archées et les plantes [94]. Le complexe PA28 a été trouvé sous 2 formes le complexe PA28αβ constitué des sous-unités α et β (hétéroheptamère) et le complexe PA28γ, homoheptamère, constitué de sous-unité γ. Le complexe PA28 ne possède pas lui-même l’activité hydrolytique. Une fois lié au CP, il accélère la dégradation des peptides fluorescents de manière ATP-indépendante mais il ne stimule pas le CP à dégrader des protéines avec ou sans ubiquitine [58]. Très probablement, cette fonction est en relation avec l’implication du protéasome dans la présentation de l’antigène puisque ce complexe régulateur PA28 αβ est induit en même temps que l’immunoprotéasome par l’INFγ [58]. De plus, d’autres travaux basés sur les expériences avec protéine Tat VIH-1 ont proposé que le virus peut s’échapper de la surveillance immunitaire par l’empêchement de la formation du complexe 20S/11S [95]. Des conditions physiologiques ou pathologiques peuvent affecter le niveau d’expression du PA28αβ. La stimulation chronique du muscle squelettique du lapin augmente d’environ 70 fois le niveau d’expression de PA28αβ [96]. D’autres exemples, la carence en acides aminés due au stress nutritionnel et le vieillissement de la peau humaine, augmentent aussi le niveau d’expression de PA28αβ [97].
Les sous-unités PA28 α et β ont été détectées dans plusieurs organes, mais sont particulièrement abondantes dans les tissus immunitaires [94]. De manière étrange, le PA28β n’est pas présent dans le cerveau alors que l’expression de PA28 α et γ y est élevée [98]. Au niveau intracellulaire, le PA28αβ est principalement cytoplasmique, alors que le PA28γ est nucléaire [94]. De plus, le PA28αβ est inductible par l’INFγ et par l’infection, alors que le PA28γ n’est pas affecté par l’INFγ et il est diminué lors de l’infection [99].
c. Complexe régulateur PA200
Un autre complexe régulateur du protéasome 20S, le PA200, a été récemment identifié (figure 9) [84]. C’est une protéine de poids moléculaire d’environ 200 kDa se liant à une ou aux