T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Biologie Cellulaire
JURY
M. CHAP Hugues - Professeur Université Paul Sabatier Toulouse M. CRISTOL Jean-Paul - Professeur Université Montpellier 1 Mme. MAZIERE Cécile - Professeur Université de Picardie Amiens
M. NEMOZ Georges - Chargé de Recherche INSERM U870 M. SALVAYRE Robert - Professeur Université Paul Sabatier Toulouse
Mme. VINDIS Cécile - Chargée de Recherche INSERM U858
Ecole doctorale : Biologie Santé Biotechnologie Unité de recherche : Inserm U858, équipe 10
Directeur(s) de Thèse : Pr. SALVAYRE Robert et Dr. VINDIS Cécile Rapporteurs : Pr. MAZIERE Cécile et Dr. NEMOZ Georges
Présentée et soutenue par
INGUENEAU Cécile
Le 15 février 2010
Titre : Régulation de la signalisation calcique dans l'apoptose
induite par les lipoprotéines oxydées. Implication dans l'athérosclérose.
Ce travail a été réalisé au sein de l’équipe 10 de l’unité INSERM 858 dirigée par le Docteur Anne Nègre-Salvayre.
Je tiens tout d’abord à remercier les membres du jury, Madame le Professeur Cécile Mazière, Monsieur le Docteur Georges Nemoz et Monsieur le Professeur Jean-Paul Cristol, qui m’ont fait l’honneur de juger ce travail. Permettez moi de vous adresser ma reconnaissance la plus sincère.
Je remercie Monsieur le Professeur Hugues Chap, d’avoir accepter la présidence de ce jury de thèse. Merci pour votre disponibilité, votre soutien et vos conseils avisés dont vous m’avez fait part tout au long de mon cursus hospitalo-universitaire. Veuillez trouver ici l’expression de ma profonde gratitude et de mon respect le plus sincère.
Merci à Madame Docteur Anne Nègre-Salvayre de m’avoir accueilli dans son équipe de recherche il y a cinq ans pour mon master-2-recherche, et de m’avoir donnée l’opportunité de poursuivre mes travaux de recherche dans le cadre de ma thèse. Merci pour tous vos conseils et le partage de votre expérience scientifique. C’est avec gand plaisir que je continuerai à travailler au sein de votre équipe pendant de nombreuses années je l’espère.
Je remercie Monsieur le Professeur Robert Salvayre d’avoir dirigé cette thèse. Merci pour l’intérêt permanent que vous avez porté à ce travail, pour vos idées scientifiques et remarques toujours pertinentes ainsi que pour votre disponibilité. Je vous suis également très recconnaissante de m’avoir donner la possibilité de réaliser mon souhait de carrière hospitalo-universitaire. Vous m’avez accueillie tout d’abord comme Assistante-Hospitalo-Universitaire dans votre service, puis vous m’avez soutenue jusqu’à l’obtention d’un poste de Maître de Conférences Universitaire-Praticien-Hospitalier. Merci pour la confiance que vous m’avez témoignée.
Merci également à Madame le Docteur Cécile Vindis qui a encadré mes travaux de recherche de mon master-2-recherche jusqu’à l’aboutissement de cette thèse. Cécile, je te remercie pour ta rigueur et tes compétences scientifiques, ton énergie, ton envie d’innover, ton investissement dans ce travail et ta disponibilité. Merci de me communiquer ta passion pour la recherche et de m’aider à y croire dans les moments difficiles que traverse, je l’imagine, tout étudiant en thèse. Je suis ravie de poursuivre ce travail de recherche avec toi et j’espère continuer à bénéficier de tes grandes qualités de chercheur dans les années à venir.
La réalisation de ce travail n’aurait pu se faire sans la présence, la participation et la collaboration d’un grand nombre de personnes que je tiens également à remercier.
Merci à tout le personnel de l’équipe 10 : les chercheurs, Nathalie et Françoise ; le personnel technique, Jean-Claude, Elodie, Marie-Hélène, Corinne, Stéphanie, Christophe ; les étudiants, Aurélie, Marie, Sylvain, Cindy, Raphael, Carole, Caroline, Magalie, Christel, Pauline.
Merci aux secrétaires Danièle, Eve et Chantal.
Merci également à tout le personnel du laboratoire de Biochimie de Rangueil, biologistes, techniciens et secrétaires.
Et enfin, je remercie tout mon entourage personnel, famille et amis, pour m’avoir soutenue dans la réalisation de ce travail.
LISTES DES ABREVIATIONS
……….. 1RESUME
……….. 4LISTE DES FIGURES ET TABLEAUX
……… 5I- REVUE GENERALE
……….. 61. ATHEROSCLEROSE ………. 7
1.1. Définition et description anatomo-pathologique……… 7
1.2. Manifestations cliniques………... 9
1.3. Epidemiologie……… 10
1.4. Facteurs de risque………. 10
1.5. Physiopathologie……… 11
1.5.1. Physiologie……….. 12
1.5.1.1. Structure d’une artère normale……… 12
1.5.1.2. Rôle physiologique de l’endothélium………. 12
1.5.2. Athérogénèse……….. 13
1.5.2.1. La dysfonction endothéliale……… 13
1.5.2.2. Pénétration et oxydation des LDL dans l’intima……… 14
1.5.2.3. Recrutement des leucocytes……… 15
1.5.2.4. Formation des cellules spumeuses……….. 16
1.5.2.5. Migration et prolifération des cellules musculaires lisses….. 17
1.5.3 Evolution de la plaque ……… 18
2. LES LDL OXYDEES……… 20
2.1. Les lipoprotéines……… 20
2.1.1. Structure et rôle des lipoprotéines………20
2.1.2. Métabolisme général des lipoprotéines……… 22
2.2. Métabolisme des LDL……… 22
2.2.1. Origine des LDL……….. 22
2.2.2. Catabolisme des LDL……….. 22
2.3. Oxydation des LDL……… 26
2.3.1. Mécanisme de la peroxydaion lipidique……….. 26
2.3.2. Oxydabilité des LDL………28
2.3.3. Les produits d’oxydation lipidiques……… 28
2.3.4. Oxydation des LDL in vivo... 30
2.4. Métabolisme des LDL oxydées……… 31
2.4.1. Captation des LDL oxydées par les récepteurs scavenger…………. 31
2.4.2. Dégradation des LDL oxydées……… 34
2.5. Effets biologiques des LDL oxydées……… 34
2.5.1. Cibles moléculaires des LDL oxydées……… 34
3. APOPTOSE……… 36
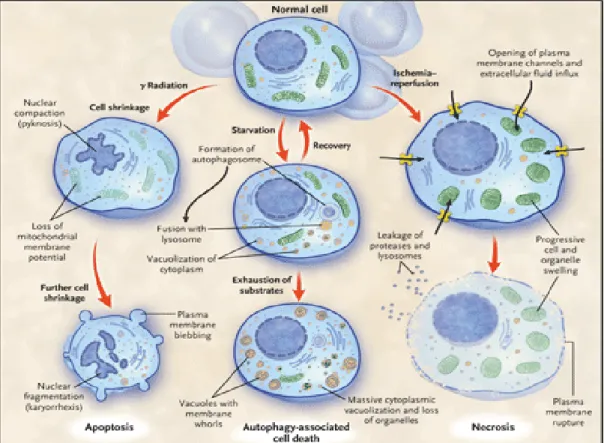
3.1. La mort cellulaire……….. 36
3.2. L’apoptose dans les lésions d’athérosclérose………. 39
3.3. Les mécanismes apoptotiques……….. 42
3.3.1. Principales voies de signalisation apoptotique……… 42
3.3.1.1. La voie extrinsèque………. 43
3.3.1.2. La voie intrinsèque mitochondriale……… 44
3.3.1.3. La voie du réticulum endoplasmique……….. 45
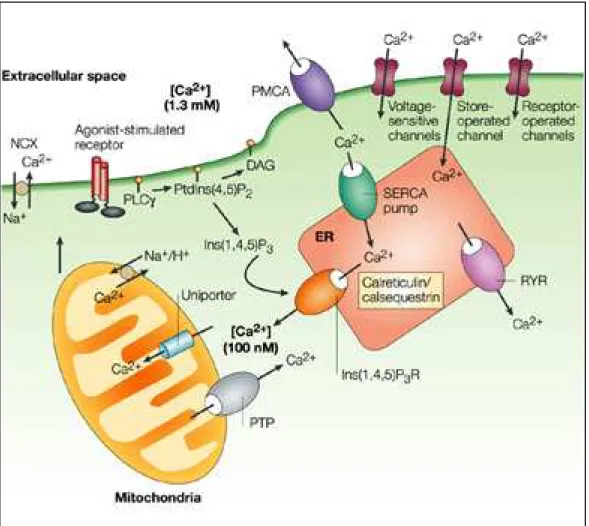
3.3.2. Rôle du calcium dans la signalisation apoptotique………. 46
3.4. Signalisation apoptotique induite par les LDL oxydées………. 47
4. LES CAVEOLES ET LA CAVEOLINE-1………. 51
4.1. Les cavéoles……… 51
4.1.1. Définition et morphologie des cavéoles……… 51
4.1.2. Spécificité tissulaire des cavéoles……… 52
4.1.3. Composition et propriétés biochimiques des cavéoles……… 52
4.1.4. Biogénèse des cavéoles……….. 53
4.2. La cavéoline-1……….. 54
4.2.1. Isoformes α et β de la cavéoline-1………. 54
4.2.2. Structure de la cavéoline-1……… 55
4.3. Roles des structures cavéoles/cavéoline-1……….. 57
4.3.1. Rôle dans le transport vésiculaire……… 57
4.3.2. Rôle dans la régulation de l’homéostasie du cholestérol……… 59
4.3.3. Rôle dans la transduction du signal……… 60
4.3.3.1. Régulation des récepteurs couplés aux protéines G…………. 61
4.3.3.2. Régulation des récepteurs tyrosine kinase………. 62
4.3.3.3. Régulation de la signalisation calcique……….. 63
4.3.4. Rôle de la cavéoline-1 localisée en dehors des cavéoles……… 64
4.4. Souris kO-cavéoline-1……… 64
4.5. Cavéoline-1 et athérosclérose……… 65
5. LES CANAUX TRPC ……….. 69
5.1. La superfamille des Transient Receptor Potential (TRP)………. 69
5.2. Les TRPC……… 71
5.2.1. Structure des TRPC………..71
5.2.2. Mécanismes d’activation des TRPC………. 77
5.2.2.1. Activation par les produits de la phospholipase C …………. 78
5.2.2.2. Activation par translocation à la membrane……… 79
5.2.2.3. Activation mécanique……….. 80
II-
MATERIEL et METHODES
………. 821. MATERIEL et REACTIFS UTILISES……….. 83
2. CULTURE CELLULAIRE……….. 86
2.1. Types cellulaires……….. 86
2.1.1. Cellules musculaires lisses……….. 86
2.1.2. Cellules endothéliales………. 87
2.2. Conditions de culture………. 87
2.3. Transfection par des siRNA……….. 88
3. PREPARATION ET OXYDATION DES LDL……… 88
3.1. Préparation des LDL……….. 88
3.2. Oxydation des LDL………. 88
4. DETERMINATION DE LA CAPTATION DES LDL……….. 89
5. IMMUNODETECTION (WESTERN BLOT)……… 89
5.1. Extraction et dosage des protéines cellulaires……….. 89
5.2. Biotinylation des protéines de surface………90
5.3. Electrophorèse des protéines en gel de polyacrylamide (SDS-PAGE) et western blot (WB)……… 90
6. IMMUNOCYTOCHIMIE……… 91
7. IMMUNOHISTOCHIMIE……….. 91
8. MICROSCOPIE ELECTRONIQUE……….. 91
9. MESURE DU CALCIUM INTRACELLULAIRE……… 92
10. MESURE DE L’INCORPORATION DE THYMIDINE TRITIEE………….. 92
11. EVALUATION DE LA CYTOTOXICITE ………. 92
11.1. Test de viabilité cellulaire au MTT……… 92
11.2. Dosage des LDL……….. 93
11.3. Marquage des cellules au syto13/Iodure de Propidium……….. 93
11.4. Marquage fluorescent des cellules apoptotiques par le test Polycaspases……… 93
11.5. Mise en évidence des cellules apoptotiques par la technique Apostain®……… 93
12. ANALYSE DU CHOLESTEROL ET DU 7-CETOCHOLESTEROL……….. 94
III-
OBJECTIFS et RESULTATS
……….. 951. OBJECTIFS ……….. 96
2. RESULTATS………. 97
2.1. Rôle des structures cavéoles/cavéoline-1 dans la signalisation apoptotique des LDL oxydées……….. 97
2.1.1. Introduction………. 97
2.1.2. Caveolin-1 sensitizes vascular smooth muscle cells to mildly oxidized LDL-induced apoptosis. ……….. 97
2.1.3. Discussion………... 98
2.2. Mise en évidence de l’implication et de la régulation du canal Calcique TRPC1 dans l’apoptose des cellules vasculaires induite par les LDL oxydées……… 100
2.2.1. Introduction……… 100
2.2.2. TRPC1 is regulated by caveolin-1 and is involved in oxidized LDL-induce apoptosis of vascular smooth muscle cells………… 100
2.2.3. Discussion……….. 101
2.3. Rôle de la protéine chaperonne du réticulum endoplasmique ORP150 dans la signalisation calcique et l’apoptose induites par les LDL oxydées………. 104
2.3.1. Introduction……… 104
2.3.2. Oxygen-Regulated-protein-150 prevents calcium homeostasis deregulation and apoptosis induced by oxidized LDL in vascular cells………. 104
2.3.3. Discussion………... 105
IV- CONCLUSION et PERSPECTIVES
……….. 107V- BIBLIOGRAPHIE
……….. 110VI- ANNEXE
……… 13513-HPODE : Acide 13-HydroPeroxyde OctaDEcanoïque 4-HNE : 4-HydroxyNonEnal
9-HODE : Acide 9-Hydroxy OctaDEcanoïque ACAT : AcylcoA-Cholesterol-Acyl-Transferase ADN : Acide DésoxyriboNucléique
AGPI : Acides Gras Poly-Insaturés AIF : Apoptosis Inducing Factor
Apaf-1 : Apoptotic Proteases Activating Factor 1 Apo: Apoprotéine
ARN : Acide Ribo Nucléique
ARNm : Acide Ribo Nucléique messager ATF6 : Activating Transcription Factor 6 CAD : Caspase Activated Desoxyribonuclease Caspase : Cystéinyl Aspartate Specific ProteASE CCE : Entrée Capacitative de Calcium
CC-N et CC-C : Coiled Coil domain N et C terminaux CE : Cellule Endothéliale
CETP : Cholesteryl Esterase Transfert Protein
CHOP : CCAAT/enhancer binding protein Homologous Protein CIRB : Calmodulin IP3R Binding
CML : Cellule Musculaire Lisse COX : Cyclo-OXygénase
CSD : Caveolin-1 Scaffolding Domain DAG : DiAcyl Glycérol
DD : Death Domain
DED : Death Effector Domain
DIABLO : Direct Inhibitor of Apoptosis Binding Protein with LOw pI EGFR : EGF Récepteur
eNOS : endothelial NO Synthase
EROs : Espèces Réactives de l’Oxygène FADD : Fas Associated Death Domain FAK : Focal Adhesion Kinase
FLICE : FADD-homologous ICE-Like protease FLIP : FLICE Inhibitory Protein
GPI : GlycosylPhosphatidylInositol HDL : High Density Lipoprotein
HMG-CoA : HydroxyMethylGlutaryl-Coenzyme A IC : Immuno-Cytochimie
ICAD : Inhibitor of CAD
ICAM-1 : Inter Cellular Adhesion Molecule 1 IDL : Intermediate Density Lipoprotein IGFR : Insulin Growth Factor Receptor IH : Immuno-Histochimie
IL : InterLeukine
IP3 : Inositol (1, 4, 5) triPhosphate
IP3R : Inositol (1, 4, 5) triPhosphate Receptor IRE1: Inositol Requiring 1
JNK : c-Jun N-terminal Kinase KO : Knock Down
LDL : Low Density Lipoprotein LH : Lipase Hépatique
LOX : Lipo-OXygénase
LOX-1 : Lectin-like OXidized low density receptor LPC: LysoPhosphatidylCholine
LPL : Lipoproteine Lipase
MCP-1 : Monocyte Chemoattractant Protein 1 M-CSF : Macrophages Colony Stimulating Factor MDA : MalonDiAldéhyde
MscCa : Mecanosensible Cation channel NCX : Antiport Na+/Ca2+
NHERF : Na+/H+ Exchanger Regulatory Factor NO : Oxyde Nitrique
ORP150 : Oxygen Regulatory Protein 150 kDa PDGF : Platelet Derived Growth Factor
PDGFR : PDGF Récepteur
PERK : PKR like Endoplasmic Reticulum Kinase PIP2 : Phosphatidyl Inositol diPhosphate
PKC : Proteine Kinase C PLC : PhosphoLipase C
PMCA : Plasma Membrane Calcium ATPase PS : Phosphatidyl Sérine
PTP : Permeability Transition Pore
PTRF : Polymérase I and transcript Release Factor RCPG : Récepteur Couplé à la Protéine G
RE : Réticulum Endoplasmique RS : Récepteur Scavenger
RTK : Récepteur Tyrosine Kinase RyR : Ryanodine Receptor
SERCA : Sarcoplasmique Endoplasmic Reticulum Calcium ATPase SMAC : Second Mitochondrial derived Activator of Caspase
SMC : Smooth Muscle Cell SOC : Store Operated Channel
SREBP : Sterol Regulatory Element Binding Protein TBARS: ThioBarbituric Acid Reactive Substance TG : TriGlycérides
TGFβ : Transforming Growth Factor TNFα : Tumor Necrosis Factor α
TRADD : TNF Receptor Associated Death Domain TRAF2 : TNF-Receptor Associated Factor
TRP : Transient Receptor Potential
TRPC : Transient Receptor Potential Canonical UPR : Unfolding Protein Response
VCAM-1 : Vascular Cell Adhesion Molecule 1 VLDL : Very Low Density Lipoprotein
L’athérosclérose et ses complications cardiovasculaires représentent une des principales causes de morbi-mortalité dans les pays développés et posent un problème majeur de santé publique. Parmi les facteurs proathérogènes, il est couramment admis que les LDL oxydées jouent un rôle prépondérant dans la génèse et la progression des lésions d’athérosclérose. Les effets proapoptotiques des LDL oxydées sur les cellules vasculaires, participent à la formation du centre nécrotique, et pourraient contribuer à l’érosion et à l’instabilité des plaques d’athérosclérose.
L’objectif de ce travail a été d’étudier la régulation de la signalisation pro-apoptotique induite par les LDL oxydées dans les cellules vasculaires. Nous nous sommes plus particulièrement intéressés aux mécanismes moléculaires à l’origine d’un influx calcique impliqué dans l’activation des voies de signalisation apoptotique.
Dans une première partie de nos travaux, nous avons étudié le rôle des cavéoles, microdomaines lipidiques membranaires, dans la signalisation apoptotique et calcique des LDL oxydées. Nous avons montré que la cavéoline-1, protéine structurante des cavéoles, sensibilisait les cellules musculaires lisses vasculaires à l’apoptose induite par les LDL oxydées en potentialisant l’activation des voies de signalisation apoptotique dépendantes du calcium.
Dans une deuxième partie, nous avons montré que l’activation par les LDL oxydées du canal TRPC1 (Transient Receptor Potential Cannonical de sous type 1) conduisait à un influx calcique à l’origine de l’apoptose des cellules musculaires lisses vasculaires. L’influx calcique induit par les LDL oxydées nécessite la translocation de TRPC1 d’un compartiment intracellulaire vers les cavéoles.
Enfin, nous avons étudié le rôle de la protéine ORP150 (Oxygen-Regulated Protein), une protéine chaperonne du réticulum endoplasmique, dans l’apoptose induite par les LDL oxydées. Nous avons montré que ORP150 protégeait les cellules endothéliales vasculaires de l’apoptose induite par les LDL oxydées en bloquant la signalisation calcique.
Les résultats de ce travail mettent en évidence l’importance des structures cavéoles/cavéoline-1 dans la régulation d’une signalisation apoptotique dépendante du calcium et impliquant TRPC1. La compréhension des mécanismes moléculaires conduisant à l’apotose des cellules vasculaires représente un enjeu majeur pour prévenir les phénomènes de rupture de plaque à l’origine des complications athérothrombogènes de la maladie.
Atherosclerosis and its cardiovascular complications represent a main cause of human morbidity and mortality in the developed countries and raise a major problem of public health. Among pro-atherogenic factors, oxidized LDL are thought to play an important role in atherogenesis and progression of atherosclerotic lesions. The pro-apoptotic effects of oxidized LDL are thought to be involved in necrotic core formation and in plaque rupture or erosion, that may lead finally to athero-thrombotic events.
The aim of this work was to study the regulation of the oxidized LDL-induced apoptotic signalling pathways in vascular cells. We were more particularly interested in the molecular mechanisms leading to a calcium entry, upstream from the activation of the apoptotic pathway.
In the first part of our works, we studied the role of caveolae, lipid microdomains of the plasma membrane, in the oxidized LDL-induced apoptotic and calcium signaling. We reported that caveolin-1, a structural protein of caveolae, increases the sensitivity of the vascular smooth muscle cells to the oxidized LDL-induced apoptosis by potentiating the calcium-dependent mitochondrial apoptotic pathway induced by oxidized LDL.
In the second part, we showed that the oxidized LDL-induced activation of the TRPC1 channel (Transient Receptor Potential Cannonical 1) led to a calcium entry that triggers the apoptosis of vascular smooth muscle cells. The calcium entry requires the translocation of TRPC1 from internal compartment to caveolae.
Finally, we investigated the role of ORP150 (Oxygen-Regulated Protein), a chaperon protein located in the reticulum endoplasmic, in oxidized LDL-induced apoptosis. We showed that ORP150 protected vascular endothelial cells against the oxidized LDL-induced apoptosis by blocking the calcium signalling pathway.
The results of this work point out the importance of caveolae and caveolin-1 in the regulation of calcium-dependent apoptosis and the critical role of TRPC1. The understanding of the molecular mechanisms leading to the vascular cells apoptosis is of importance to prevent the plaque rupture and erosion which are involved in the trigger of atherothrombotic events.
LISTE DES FIGURES :
Figure 1: Structure de la paroi d’une artère, évolution de la plaque d’athérome
et conséquences pathophysiologiques ……… 9
Figure 2 : Structure générale d’une lipoprotéine ……… 20
Figure 3 : Origine des LDL : voie endogène du métabolisme lipidique………. 23
Figure 4 : Captation et dégradation cellulaires des LDL ……… 25
Figure 5 : Représentation simplifiée de la peroxydation lipidique ………. 27
Figure 6 : Principaux récepteurs scavenger impliqués dans la captation des LDL Oxydées ……….. 32
Figure 7 : Caractéristiques morphologiques des trois principaux types de mort cellulaire : apoptose, autophagie et nécrose……… 37
Figure 8 : Voies apoptotiques extrinsèque et intrinsèque ……….. 43
Figure 9 : Régulation de l’homéostasie calcique ……… 47
Figure 10 : Voies de signalisation apoptotiques induites par les LDL oxydées ………… 48
Figure 11 : Morphologie des cavéoles ……….. 51
Figure 12 : Structure de la cavéoline-1 ……….. 55
Figure 13 : Transport vésiculaire impliquant les cavéoles : transcytose, endocytose et potocytose……… 58
Figure 14 : Arbre phylogénétique de la superfamille des TRP……….. 70
Figure 15 : Structure des TRPC ……….. 71
Figure 16 : Principaux mécanismes d’activation des TRPC ……….. 77
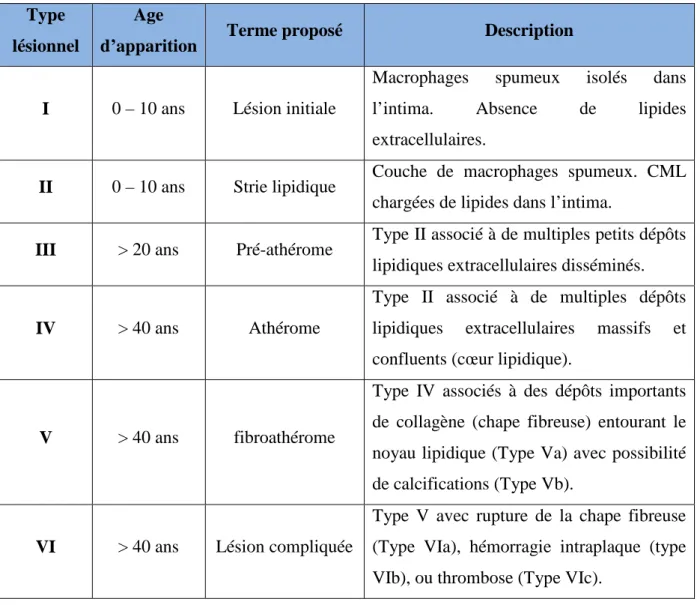
LISTE DES TABLEAUX : Tableau 1 : Classification des lésions d’athérosclérose ………. 8
Tableau 2 : Facteurs de risque génétiques et environnementaux associés à l’athérosclérose ……… 11
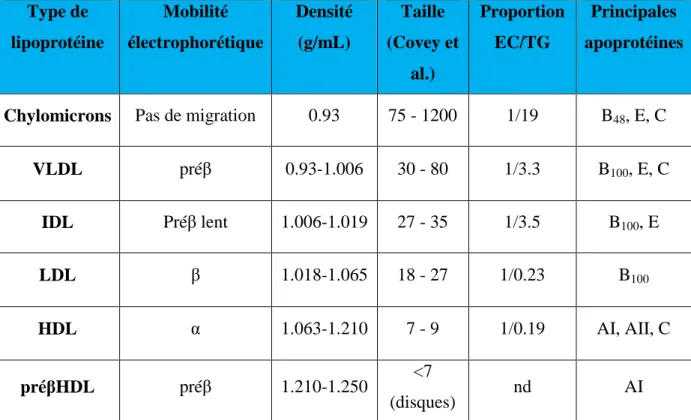
Tableau 3 : Caractéristiques physico-chimiques des lipoprotéines plasmatiques humaines……….. 21
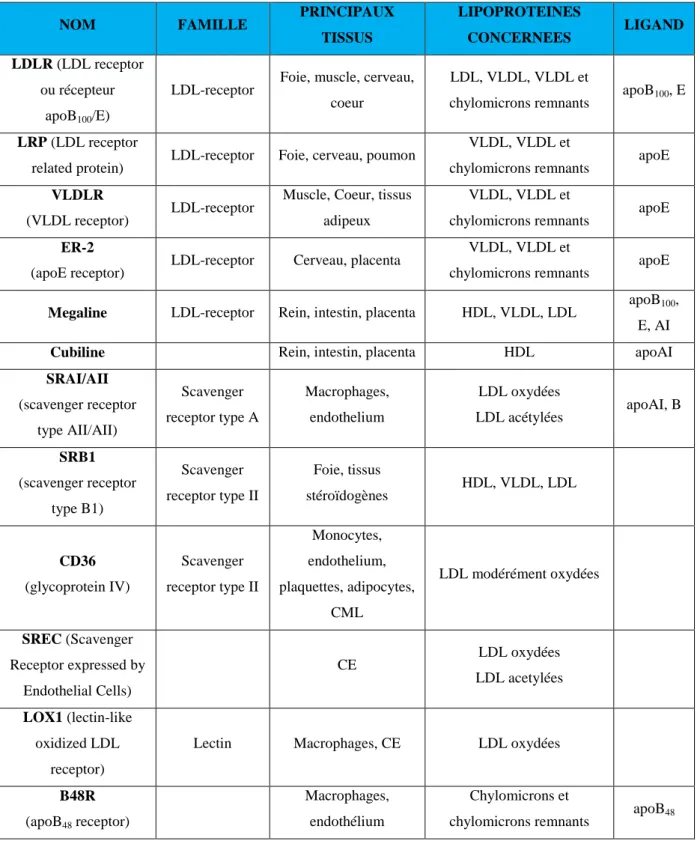
Tableau 4 : Récepteurs cellulaires des lipoprotéines ……….. 24
Tableau 5 : Principales propriétés des TRPC ………. 76
1. L’ ATHEROSCLEROSE
L’athérosclérose est une maladie chronique de la paroi artérielle, d’évolution lente, à l’origine d’évènements ischémiques aigus tels que l’infarctus du myocarde, l’accident vasculaire cérébral ou l’artériopathie oblitérante des membres inférieurs.
A l’origine d’une très forte morbi-mortalité et d’un coût socio-économique particulièrement lourd, l’athérosclérose et ses conséquences font l’objet d’une attention soutenue de la part de la communauté scientifique.
1.1. Définition et description anatomo-pathologique
L’athérosclérose est définie selon l’Organisation Mondiale de la Santé comme « une association variable de remaniements de l'intima des artères de gros et moyen calibre, consistant en une accumulation locale de lipides, de glucides complexes, de sang et de produits sanguins, de tissu fibreux et de dépôt calcaires ; le tout s'accompagnant de modifications de la media ». En fait, l’athérosclérose aboutit à la formation de plaques au niveau de la paroi des artères. Ces plaques sont composées de dépôts lipidiques riches en cholestérol (athérome) enveloppés dans une gangue fibreuse (sclérose) (Lusis, 2000).
La description anatomo-pathologique simplifiée de l’athérosclérose retient trois stades évolutifs : la strie lipidique, la lésion fibrolipidique, et enfin la lésion compliquée. Une classification plus détaillée définit six stades dont chacun est l’évolution du précédent (Stary et al., 1995). Le tableau 1 décrit ses six stades.
Les lésions précoces (types I, II, III et IV de Stary) correspondent à une accumulation de cellules (principalement des macrophages) surchargées en cholestérol appelées cellules spumeuses. Ces lésions appelées stries lipidiques, ne sont pas cliniquement symptomatiques mais sont les précurseurs des lésions plus avancées correspondant aux lésions fibrolipidiques (type V de Stary). Les lésions fibrolipidiques sont composées d’un centre nécrotico-lipidique isolé de la lumière artérielle par une chape fibreuse, constituée de cellules musculaires lisses et de matrice extracellulaire. La plaque d’athérosclérose compliquée (type VI de Stary) survient habituellement après 40 ans à la suite de phénomènes hémorragiques intra-plaque, ou thrombotiques. Le processus de cicatrisation permet l’organisation de la thrombose ou de l’hémorragie au sein de la plaque, contribuant ainsi à la progression de la sténose.
Tableau 1 : Classification des lésions d’athérosclérose (d’après Stary et al., 1995) Type
lésionnel
Age
d’apparition Terme proposé Description
I 0 – 10 ans Lésion initiale
Macrophages spumeux isolés dans
l’intima. Absence de lipides
extracellulaires.
II 0 – 10 ans Strie lipidique Couche de macrophages spumeux. CML chargées de lipides dans l’intima.
III > 20 ans Pré-athérome Type II associé à de multiples petits dépôts lipidiques extracellulaires disséminés.
IV > 40 ans Athérome
Type II associé à de multiples dépôts lipidiques extracellulaires massifs et confluents (cœur lipidique).
V > 40 ans fibroathérome
Type IV associés à des dépôts importants de collagène (chape fibreuse) entourant le noyau lipidique (Type Va) avec possibilité de calcifications (Type Vb).
VI > 40 ans Lésion compliquée
Type V avec rupture de la chape fibreuse (Type VIa), hémorragie intraplaque (type VIb), ou thrombose (Type VIc).
La figure 1 schématise l’évolution de la plaque d’athérosclérose et ses conséquences physiopathologiques.
Figure 1 : Structure de la paroi d’une artère, évolution de la plaque d’athérome et conséquences pathophysiologiques. (D’après Libby, 2002).
1.2. Manifestations cliniques
Le processus athéromateux débute dès l’enfance et évolue pendant longtemps de façon infraclinique. Les premières complications de l’athérosclérose sont liées à l’épaississement de la paroi qui accompagne la progression de la plaque. Le diamètre artériel longtemps maintenu normal, peut se réduire localement dans les lésions avancées sténosantes. La diminution du débit sanguin en aval de la lésion est à l’origine de douleurs (angines de poitrine) correspondant à une souffrance du muscle cardiaque en manque de nutriments et d’oxygène. Les angines de poitrine sont souvent un avertissement à l’apparition d’un phénomène plus
grave qu’est la survenue d’un infarctus, provoqué lors de l’oblitération d’un vaisseau par un thrombus, qui se forme suite à l’érosion ou à la rupture de la plaque.
La mortalité et la morbidité associées à la maladie athéroscléreuse sont essentiellement liées aux complications thromboemboliques de la rupture de plaque, c'est-à-dire aux épisodes d’occlusion artérielle, dont les principales manifestations sont les syndromes coronaires aigus (infarctus du myocarde, angor instable, mort subite), les accidents vasculaires cérébraux ischémiques et les ischémies aiguës des membres inférieurs.
1.3. Epidémiologie
En 2005, le rapport de l’OMS estimait à 17,5 millions le nombre d’individus décédés de maladies cardiovasculaires, ce qui représentait 30% de l’ensemble des décès. Parmi ces 17,5 millions, 45% succombaient à un accident vasculaire cérébral, et 30% à un infarctus. Ces dernières années, les données ont été modifiées en France car la mortalité d'origine cardiovasculaire a baissé de 50 % en vingt-cinq ans, alors que celle liée au cancer n'a que peu diminué: 30% des décès sont attribués au cancer contre 29% pour les maladies cardiovasculaires. Des régions du monde comme l’Afrique, l’Inde et l’Asie, jusqu’à présent épargnées par les maladies cardiovasculaires s’y trouvent aujourd’hui confrontées en raison de l’évolution de leur mode de vie se rapprochant du modèle occidental (Ranjith et al., 2005). Ces nouvelles données ont amené à prendre en compte les déterminants sociologiques et économiques comme facteurs de risque des maladies cardiovasculaires (80% d’entre elles concernent les populations au revenu faible ou moyen).
1.4. Facteurs de risques
L’athérosclérose est une pathologie multifactorielle qui résulte de la combinaison entre des facteurs de risque non-modifiables que sont l’âge, le sexe masculin et les antécédents familiaux, et des facteurs environnementaux modifiables tels qu’une alimentation riche en graisses et en sucres, l’inactivité et le tabagisme. De telles conditions de vie favorisent l’installation d’une hypertension artérielle, d’un diabète de type II, de dyslipidémies et de l’obésité, pathologies qui accélèrent la formation des plaques et leurs complications (Lusis, 2000). Il est maintenant clairement établi que l’hypercholestérolémie avec une augmentation du cholestérol liée aux lipoprotéines de faible densité (LDL) est le risque cardiovasculaire principal. Le tableau 2 résume les principaux facteurs de risque génétiques et
Tableau 2 : Facteurs de risque génétiques et environnementaux associés à l’athérosclérose (d’après Lusis, 2000).
Facteurs de risque à composante
génétique Facteurs de risque environnementaux
Taux plasmatiques élevés de LDL/VLDL Alimentation riche en graisses et sucres
Taux plasmatiques réduits de HDL Tabac
Taux plasmatiques élevés de lipoprotéine(a) Sédentarité, manque d’activité physique
Hyperhomocystéinémie Agents infectieux (chlamydia pneumoniae)
Antécédants familiaux de maladies
cardiovasculaires Faibles taux d’anti-oxydants
Diabètes et obésité
Taux plasmatiques élevés de facteurs hémostatiques (fibrinogène, inhibiteur de l’activateur du plasminogène de type I, hyper réactivité plaquettaire)
Sexe masculin
Marqueurs de l’inflammation plasmatiques élevés (CRP)
Syndrome métabolique
1.5. Physiopathologie
Il y a une trentaine d’années, la théorie de l’athérogenèse était dominée par le rôle des lipides étant données les fortes corrélations qui existaient entre l’hypercholestérolémie et l’apparition des plaques d’athérosclérose (Ross and Harker, 1976). L’implication des facteurs de croissance et la prolifération des cellules musculaires lisses à l’origine de la sténose ont ensuite pris le relais dans l’intérêt porté aux mécanismes de l’athérogenèse (Pietila and Nikkari, 1983). A la fin des années 80, la plaque était donc visualisée comme un amas de débris lipidiques enrobés dans une capsule de cellules musculaires lisses en prolifération. La meilleure connaissance des cellules immunitaires depuis ces 20 dernières années a laissé, petit à petit, la place à une théorie inflammatoire de l’athérogenèse (Luscinskas et al., 1991). Le système immunitaire participerait activement à la formation de la plaque et également à sa progression.
Les études expérimentales les plus récentes, associées aux observations anatomo-pathologiques de plaques humaines, permettent d’affirmer aujourd’hui que l’athérosclérose est une maladie inflammatoire chronique des grosses et moyennes artères, à localisation intimale (Ross, 1999) (Shi et al., 2000). Les LDL modifiées, notamment par oxydation, sont des agents d’agression majeurs à l’origine de la réaction inflammatoire (Lusis, 2000; Steinberg et al., 1989b).
1.5.1. Physiologie
1.5.1.1. Structure d’une artère normale :
La figure 1 montre l’organisation d’une artère normale en trois tuniques concentriques. De la lumière du vaisseau vers l’extérieur, on distingue l’intima, la média puis l’adventice.
- l’intima ou tunique interne : dans la paroi artérielle saine, l’intima est fine, à peine visible en microscopie optique et constituée par une monocouche continue de cellules endothéliales reposant sur la membrane basale et plus en profondeur sur la couche sous endothéliale constituée de macromolécules de la matrice extracellulaire. La couche sous endothéliale est le site préférentiel de développement des lésions d’athérosclérose.
- la média ou tunique moyenne : responsable de l’essentiel de l’épaisseur de la paroi artérielle à l’état normal, la média est constituée de cellules musculaires lisses entourées de macromolécules de matrice extracellulaire et est limitée par des fibres élastiques. Les cellules musculaires lisses de la média sont de phénotype contractile.
- l’adventice ou tunique externe : constituée de collagène fibrillaire, d’amas de cellules musculaires lisses et de microvaisseaux appelés vasa vasorum.
1.5.1.2. Rôle physiologique de l’endothélium
L’endothélium vasculaire est constitué d’une monocouche de cellules endothéliales qui recouvrent la surface interne des vaisseaux sanguins. Pendant de nombreuses années, l’endothélium n’a été considéré que comme une simple barrière physique séparant le flux sanguin des tissus sous-jacents. Il est désormais considéré comme une véritable glande endocrine capable de sécréter des molécules impliquées dans la régulation du tonus vasculaire, dans la réponse inflammatoire et dans la coagulation sanguine. L’endothélium joue un véritable rôle dans le maintien de l’homéostasie vasculaire, en modulant des réponses physiologiques résultant de l’écoulement sanguin (forces de cisaillement) et de la synthèse de
contractants, procoagulants et anticoagulants, inflammatoires et anti-inflammatoires, fibrinolytiques et antifibrinolytiques, oxydants et anti-oxydants (Arnal et al., 2003).
L’endothélium sécrète de nombreuses substances vasoactives dont la principale est l’oxyde nitrique (NO). Le NO est synthétisé de façon constitutive par la NO synthase endothéliale (eNOS) dont l’activité est stimulée par les forces de cisaillement résultant de l’écoulement sanguin sur la paroi endothéliale (Raij, 2006). Le NO est un facteur relaxant clé, synthétisé par l’endothélium, qui joue un rôle pivot dans le maintien du tonus vasculaire et sa réactivité. Le NO induit différents effets :
- il exerce un fort pouvoir vasodilatateur
- il permet le maintien d’une surface anti-thrombotique en inhibant l’adhésion plaquettaire
- il inhibe l’expression de molécules d’adhésion ou de chémokines
- il favorise la prolifération des cellules endothéliales permettant ainsi la réparation de l’endothélium.
- il exerce un fort pouvoir anti-oxydant en éliminant les espèces réactives de l’oxygène dont l’anion superoxyde (O2●– ) qui endommage fortement l’endothélium.
- il a une action sur les cellules musculaires lisses de la media d’une part en inhibant leur prolifération, et d’autre part en ayant un effet relaxant sur ces cellules.
L’effet vasodilatateur du NO est équilibré par la sécrétion de peptides vasoconstricteurs, tels que l’endothéline et l’angiotensine II. La balance des effets de ces substances dicte le tonus du vaisseau qui lui-même conditionne l’écoulement du flux sanguin.
1.5.2. Athérogénèse
1.5.2.1. La dysfonction endothéliale
Lors de son passage, le flux sanguin applique des forces de frottement contre la paroi vasculaire. Ces forces de frottement imposées directement sur l’endothélium, modulent sa structure et ses fonctions par des mécanismes de mécano-transduction. Lorsque ces forces de frottement sont constantes, cela confère à l’endothélium un statut inflammatoire, anti-oxydant et anti-thrombotique (Cunningham and Gotlieb, 2005).
Certaines zones de l’arbre circulatoire telles que les bifurcations ou les courbures artérielles, imposent une contrainte physique à l’écoulement sanguin, rendant les forces de frottement faibles voire négatives. Cet événement perturbe l’homéostasie de l’endothélium
vasculaire et est à l’origine de la dysfonction endothéliale (Topper and Gimbrone, 1999). Ceci explique le développement préférentiel des plaques d’athérome au niveau des bifurcations artérielles (Gimbrone, 1999). Ce dysfonctionnement en partie causé par une diminution de la biodisponibilité du NO, est aggravé par la présence des facteurs de risques précédemment décrits : hypercholestérolémie, diabète, hypertension, tabagisme, ainsi que certains agents infectieux ou toxines (Tedgui and Chapman, 2003).
Lorsque cette dysfonction endothéliale existe on observe : - une vasoconstriction
- une augmentation de la perméabilité endothéliale facilitant la pénétration des lipoprotéines athérogènes présentes en plus ou moins grande quantité
- une expression d’intégrines ou de protéines d’adhésion telles que VCAM-1 (Vascular Cell Adhesion Molecule) ou ICAM-1 (Inter Cellular Adhesion Molecule) à la surface de l’endothélium, participant à la réaction inflammatoire en favorisant l’adhésion puis la pénétration dans l’espace sous-intimal de cellules inflammatoires comme les monocytes.
- une activation plaquettaire et une coagulation imparfaite favorisant les thromboses - l’induction d’un stress oxydatif qui altère de nombreuses fonctions de l’endothélium
dont le tonus vasomoteur. Ce phénomène biologique complexe est également induit par les effets de divers facteurs de risque (hypertension, hypercholestérolémie, diabète et tabac).
La dysfonction endothéliale contribue à la phase d’initiation de l’athérosclérose, mais il est maintenant clairement établi qu’elle participe également à la progression et l’évolution des plaques conduisant aux complications de la maladie. La présence d’une dysfonction endothéliale mesurable par différentes techniques d’imagerie serait un marqueur prédictif du risque de complications de l’athérosclérose (Roquer et al., 2009)
1.5.2.2. Pénétration et oxydation des LDL dans l’intima
L’importance du cholestérol et plus particulièrement des LDL dans l’athérogénèse n’est plus contestée, depuis que les essais cliniques de prévention primaire et secondaire chez les sujets hypercholestérolémiques ont démontré qu’il était possible de réduire le risque cardiovasculaire en diminuant le cholestérol-LDL à l’aide de statines (Steinberg et al., 1989b).
Les forces de cisaillement conditionnent la morphologie et la perméabilité des cellules endothéliales (Chatzizisis et al., 2007). Dans les zones artérielles où le flux laminaire est perturbé, la diminution des forces de cisaillement augmente la perméabilité de l’endothélium et facilite l’infiltration des LDL dans l’espace sous-endothélial (Gimbrone et al., 2000). De plus, la diminution des forces de cisaillement entraîne une activation endothéliale des SREBP (Sterol Regulatory Element Binding Protein) qui vont réguler positivement l’expression des gènes codant pour le récepteur des LDL et la cholestérol synthase (Liu et al., 2002b). Dans un contexte d’hyperlipidémie, ceci contribue à une augmentation de la captation et de la synthèse des LDL par les cellules endothéliales à l’origine d’une accumulation sous-endothéliale de LDL. Le passage des LDL à travers l’endothélium est facilité par une concentration circulante élevée et par la petite taille des LDL : plus elles sont petites, plus elles sont athérogènes (Carmena et al., 2004).
Une fois l’endothélium traversé, ces LDL se retrouvent dans l’espace sous-endothélial où elles restent piégées en raison d’interactions qui s’établissent entre des constituants de l’apoprotéine B100 (apoB100) et les protéoglycanes de la matrice (Boren et al., 1998). La survenue préalable d’une dysfonction endothéliale conduit à une production d’espèces réactives de l’oxygène (Dietrich et al.), qui vont attaquer la partie lipidique et protéique des LDL emprisonnées dans l’intima. Ce processus aboutit à l’oxydation des LDL et à la formation de produits dérivés (cf : 2. Les LDL oxydées) qui induisent l’expression de molécules d’adhésion par les cellules endothéliales permettant le recrutement des leucocytes sur le site de la lésion (Napoli et al., 1997).
1.5.2.3. Recrutement des leucocytes
L’invasion de la paroi artérielle par les leucocytes est un des évènements précoces de l’athérogénèse. L’adhérence des monocytes et des lymphocytes T à l’endothélium implique leur liaison à des molécules de structure exprimées à la surface des cellules endothéliales. Les molécules d’adhésion spécifiques (sélectines, intégrines, superfamille des immunoglobulines) contribuent aux processus de ralentissement, de « roulement » et d’extravasation des leucocytes. L’attachement et le « roulement » des monocytes à la surface des cellules endothéliales sont médiés par les sélectines (L-, P- et E- sélectines). L’interaction entre, d’une part les intégrines (β1 et β2 intégrines) et le ligand VLA-4 exprimées à la surface des leucocytes, et d’autre part, les molécules d’adhésion de la superfamille des immunoglobulines ICAM-1 et VCAM-1, exprimées à la surface des cellules endothéliales, permet de renforcer
l’attachement des leucocytes sur l’endothélium et leur extravasation dans la paroi artérielle (Quehenberger, 2005).
Les sélectines et les intégrines sont fortement exprimées par l’endothélium activé à proximité des lésions athéroscléreuses (Cybulsky and Gimbrone, 1991). De plus, la présence de VCAM-1 et d’ICAM-1 est associée avec une augmentation de l’accumulation intimale de leucocytes (O'Brien et al., 1993). L’expression de ces molécules d’adhésion peut être induite par les LDL oxydées (Takei et al., 2001), par les cytokines pro-inflammatoires secondairement synthétisées par les cellules de la plaque (Tumor Necrosis Factor α (TNF α), Interleukine 1 (IL-1)), et également sous l’influence de la perturbation du flux sanguin (Topper and Gimbrone, 1999).
Le monocyte adhérent pénètre dans l’intima à travers les jonctions inter-endothéliales sous l’effet de facteurs chimiotactiques, dont le MCP-1 (Monocyte Chemoattractant Protein-1) et l’IL-8 (interleukine-8) qui sont retrouvés dans la plaque d’athérosclérose humaine abondamment exprimés par les macrophages et les cellules musculaires lisses (Libby, 2002).
Les monocytes/macrophages dans la plaque ont la capacité de se multiplier. Le Macrophages Colony Stimulating Factor (M-CSF), facteur hématopoïétique de différenciation et de prolifération des monocytes, est produit localement par les cellules endothéliales et les cellules musculaires lisses de la plaque d’athérosclérose humaine, puis par le macrophage lui même. La multiplication et la différenciation des monocytes en macrophages dans la plaque est d’une importance capitale dans le processus d’athérogénèse, comme en témoigne l’absence quasi-totale de lésions athéroscléreuses chez les souris Knock Down pour l’apoprotéine E (KO apoE) et déficientes en M-CSF (Smith et al., 1995). Les monocytes qui pénètrent dans la paroi artérielle se différencient en macrophages. Leur rôle d’épuration du cholestérol de l’intima est plutôt bénéfique mais par contre ils participent au processus de l’athérogénèse en activant les cellules endothéliales et en augmentant la perméabilité aux LDL par le biais de la production de cytokines pro-inflammatoires. De plus, en se chargeant en lipides, ils se transforment en cellules spumeuses dont la formation correspond à la première étape de l’athérogénèse.
1.5.2.4. Formation des cellules spumeuses
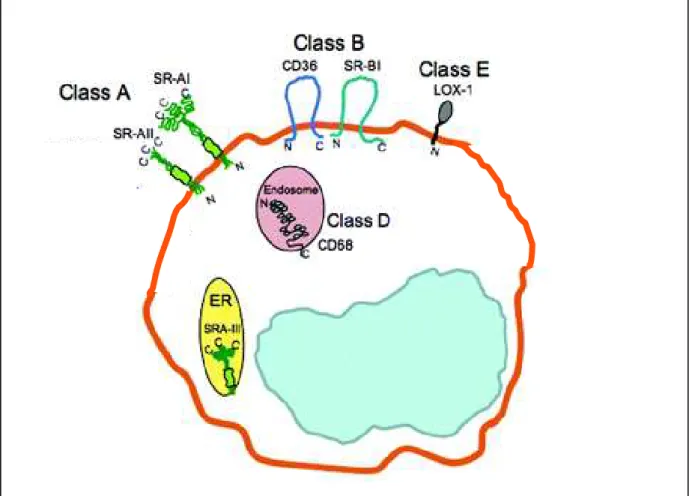
Pour se transformer en cellules spumeuses, les macrophages internalisent des LDL modifiées (et/ou oxydées) par l’intermédiaire de récepteurs dits scavenger. A l’inverse du
le contrôle inhibiteur du contenu intracellulaire en cholestérol (De Winther et al., 2000), ce qui permet une captation de grandes quantités de LDL oxydées. Les macrophages se surchargent en cholestérol et se transforment en cellules spumeuses qui vont s’accumuler dans la paroi artérielle (Tontonoz et al., 1998) pour former les stries graisseuses caractéristiques des lésions précoces de l’athérosclérose. La majorité des cellules spumeuses retrouvées dans les lésions athéroscléreuses proviennent des macrophages, mais les cellules musculaires lisses peuvent également se surcharger en lipides provenant de la captation des LDL modifiées (et/ou oxydées) par l’intermédiaire des récepteurs scavengers (Stary et al., 1994).
Dans les lésions précoces (stries lipidiques), les lipides sont essentiellement intracellulaires puisqu’ils sont stockés par les macrophages. Puis, dans les lésions avancées, lorsque la capacité épuratrice de ces cellules est dépassée, les lipides se regroupent au centre de la plaque pour former un amas appelé cœur lipidique composé de cholestérol et d’esters de cholestérol, mais également de débris cellulaires (cf 3. Apoptose).
1.5.2.5. Migration et prolifération des cellules musculaires lisses
Sous l’influence de divers stimuli athérogènes, les cellules musculaires lisses vont migrer de la média vers l’intima où elles vont proliférer. On observe un changement de phénotype des cellules musculaires lisses qui passent d’un phénotype contractile à un phénotype sécrétoire (Doran et al., 2008). Les cellules musculaires lisses retrouvées dans la média expriment majoritairement des molécules impliquées dans les fonctions contractiles comme la chaîne légère de la myosine, ou de l’α-actine. A l’inverse, les cellules musculaires lisses retrouvées dans l’intima expriment peu ces protéines contractiles, ont un indice prolifératif plus élevé et une plus grande capacité à synthétiser des molécules de la matrice extracellulaire, des protéases et des cytokines (Owens et al., 2004). Ces événements se produisent en réponse à :
- la levée d’inhibition de prolifération des cellules endothéliales sur les cellules musculaires lisses (Fingerle et al., 1990)
- la sécrétion du CD40 ligand par les lymphocytes T qui participe à la migration des cellules musculaires lisses (Schonbeck et al., 2000).
- la production d’EROs (Coatrieux et al., 2007) et de cytokines (TNF-α) (Tellier et al., 2007) par les cellules de la paroi vasculaire (Ross, 1993).
- aux LDL oxydées, qui à faible concentration stimulent la migration, la prolifération des cellules musculaires lisses en culture et la synthèse de collagène à la fois
directement et indirectement par l’intermédiaire de l’expression de facteurs de croissance et de cytokines (Schonbeck et al., 2000)
- la production de facteurs de croissance, dont le PDGF (Platelet Derived Growth Factor) et le TGFβ (Transforming Growth Factor) libérés par les macrophages, les cellules musculaires lisses et les cellules endothéliales (Ross, 1993; Stiko-Rahm et al., 1992).
La migration, la prolifération, et la synthèse de matrice extracellulaire par les cellules musculaires lisses permettent la formation d’une chape fibreuse qui va recouvrir le cœur lipidique de la plaque (Raines and Ross, 1993).
1.5.3. Evolution de la plaque
La plaque se développe longtemps sans altérer le calibre vasculaire, car le vaisseau s'adapte par un élargissement compensatoire appelé remodelage vasculaire excentrique. Lorsque la masse intimale excède 40 % de la surface totale de la paroi, le remodelage de la paroi n'est plus suffisant pour contenir la plaque. Son développement se fait alors au détriment de la lumière artérielle et conduit à une sténose progressive. La migration, la prolifération et la synthèse de matrice extracellulaire par les cellules musculaires lisses participent à l’action sténosante des plaques.
Cependant, les manifestations cliniques graves de la maladie athéromateuse sont peu en rapport avec la taille de la plaque mais sont essentiellement dues à son instabilité qui augmente les risques de thrombose (Mann and Davies, 1996). Il est largement admis que l’évènement initial entraînant la formation d’un thrombus au niveau d’une plaque athéroscléreuse est la rupture (60% des cas) ou l’érosion (40% des cas) de la plaque, abolissant l’interface endothéliale et mettant en contact le matériel thrombogène contenu dans la plaque et le sang circulant (Taubman et al., 1997). Le thrombus formé peut, par son volume ou sa localisation, entraîner une occlusion partielle ou complète de la lumière artérielle, se morceler ou migrer dans un autre territoire artériel, ou être incorporé à la plaque par un processus de cicatrisation qui participe à la croissance de la plaque (Tedgui and Mallat, 1999).
La stabilité de la plaque dépend de plusieurs facteurs :
- la présence d’un cœur lipidique important (plus de 40 % de son volume) diminue la résistance physique de la plaque.
- l’épaisseur et la composition de la chape fibreuse : plus la chape fibreuse est épaisse, plus la plaque est stable. Ainsi, sa richesse en cellules musculaires lisses qui synthétisent la matrice extracellulaire contribue à la formation d’une chape fibreuse épaisse et solide, augmentant la stabilité de la plaque (Burke et al., 2002). Au contraire, sa richesse en cellules inflammatoires synthétisant des cytokines proinflammatoires contribue à un amincissement de la chape fibreuse. En effet les cytokines proinflammatoires stimulent l’activité des métalloprotéinases (MMP2, MMP9, MMP3) qui vont dégrader la matrice extracellulaire. De plus, l’IFN γ inhibe la production de collagène par les cellules musculaires lisses (Kolodgie et al., 2001).
- Par ailleurs, l’apoptose des cellules vasculaires (cellules endothéliales, cellules musculaires lisses, macrophages) augmente la vulnérabilité et la thrombogénicité des plaques. L’apoptose des cellules endothéliales contribue à l’érosion de l’endothélium, et est à l’origine de la formation de micro-thrombi en raison des propriétés pro-adhésives et pro–coagulantes des cellules endothéliales en apoptose (Sugiyama et al., 2004). L’apoptose des cellules musculaires lisses fragilise la chape fibreuse en diminuant sa cellularité et la synthèse de matrice extracellulaire. De plus, la plaque d’athérome est riche en facteur tissulaire, dont l’activation est dépendante de la présence de micro-particules libérées par les cellules en apoptose (principalement les macrophages) responsables de la thrombogénicité de la plaque (Leroyer et al., 2007).
La compréhension des mécanismes responsables d’une instabilité de la plaque pouvant aboutir à sa rupture ou son érosion, est donc primordiale dans la prévention des complications thrombogènes gravissimes de l’athérosclérose.
Bien que les travaux récents sur l’athérogénèse soient principalement centrés sur les aspects inflammatoires, le rôle des LDL reste primordial dans la formation et l’évolution de la lésion. Nous allons voir dans le chapitre suivant la structure et les fonctions de ces molécules dont les effets sur les cellules vasculaires sont très nombreux.
2. LES LDL OXYDEES :
2.1. Les lipoprotéines
2.1.1. Structure et rôle des lipoprotéines
Les lipides sont des molécules hydrophobes, insolubles dans les milieux biologiques aqueux. Pour être véhiculés à travers les différents compartiments extracellulaires de l’organisme, les principaux composés lipidiques (cholestérol non estérifié, esters de cholestérol, triglycérides et phospholipides) s’assemblent avec des protéines (apoprotéines) en complexes macromoléculaires nommés lipoprotéines (Macheboeuf, 1928). Les lipoprotéines permettent le transport des lipides d’origines endogène et exogène, des sites d’absorption ou de production, vers les tissus d’utilisation, de stockage ou de transformation.
La figure 2 schématise la structure générale d’une lipoprotéine.
Figure 2 : Structure générale d’une lipoprotéine.
Si l’on considère une lipoprotéine de faible densité (LDL), son cœur contient en moyenne 1600 molécules d’esters de cholestérol, 100 molécules de triglycérides et 1 molécule de caroténe. Sur la couche externe, on trouve 600 molécules de cholestérol, 700 molécules de phospholipides (dont les acides gras polyinsaturés représentent 25 à 50%) et 6 molécules de tocophérol (Salvayre et al., 2002).
Les triglycérides et les esters de cholestérol forment le cœur des lipoprotéines. Celui-ci est recouvert d’une enveloppe amphiphile constituée de phospholipides et de cholestérol non estérifié et dans laquelle les apoprotéines viennent s’insérer. Les apoprotéines jouent non seulement un rôle structural et stabilisateur essentiel, mais elles sont également responsables des propriétés fonctionnelles et du devenir métabolique des lipoprotéines. Ainsi les apoprotéines permettent l’assemblage et la sécrétion des lipoprotéines, leur interaction avec des récepteurs cellulaires spécifiques, ou encore l’activation ou l’inhibition d’enzymes impliquées dans leur métabolisme intravasculaire. Les lipoprotéines subissent des remaniements constants durant leur transit dans l’espace intra-vasculaire, jouant sur leur composition lipidique et en apoprotéines.
Les lipoprotéines sont subdivisées en plusieurs catégories selon leurs caractéristiques physico-chimiques (densité, taille, compositions lipidique et protéique) (cf tableau 3). On distingue aujourd’hui cinq populations plasmatiques de lipoprotéines. Ainsi par ordre croissant de leur densité on a : les chylomicrons, les VLDL (Very Low Density Lipoprotein), les IDL (Intermediate Density Lipoprotein), les LDL (Low Density Lipoprotein), les HDL (High Density Lipoprotein) et les préβ-HDL (Gofman et al., 1949).
Tableau 3 : Caractéristiques physico-chimiques des lipoprotéines plasmatiques humaines. (d’après Lagrost et al., 2004)
Type de lipoprotéine Mobilité électrophorétique Densité (g/mL) Taille (Covey et al.) Proportion EC/TG Principales apoprotéines
Chylomicrons Pas de migration 0.93 75 - 1200 1/19 B48, E, C
VLDL préβ 0.93-1.006 30 - 80 1/3.3 B100, E, C IDL Préβ lent 1.006-1.019 27 - 35 1/3.5 B100, E LDL β 1.018-1.065 18 - 27 1/0.23 B100 HDL α 1.063-1.210 7 - 9 1/0.19 AI, AII, C préβHDL préβ 1.210-1.250 <7 (disques) nd AI
EC : esters de cholestérol ; TG : triglycérides ; nd : non détectable
Les lipoprotéines les plus légères (chylomicrons et VLDL) transportent majoritairement dans leur cœur lipidique des triglycérides et les lipoprotéines de densité plus élevée (LDL et HDL) transportent essentiellement du cholestérol estérifié. En ce qui concerne les apoprotéines, les apo A sont principalement associées aux HDL, les apo B aux LDL (apoB100) et chylomicrons (apo B48) et les apo C aux VLDL, chylomicrons et HDL (Gustafson et al., 1966) (Lagrost et al., 2004).
2.1.2. Métabolisme général des lipoprotéines
Lorsque l’on considère le métabolisme des lipoprotéines, il est classique de distinguer trois types de tissus : l’intestin, le foie, et les tissus périphériques. L’intestin permet l’absorption des lipides alimentaires et leur intégration dans des lipoprotéines de grande taille et riches en triglycérides, néosynthétisées au sein de l’entérocyte: les chylomicrons. Ces chylomicrons vont contribuer au transport des lipides d’origine alimentaire vers les tissus et le foie. Les triglycérides contenu dans le cœur des chylomicrons seront hydrolysés et captés par les tissus périphériques pour y être stockés (tissus adipeux), ou dégradés à des fins énergétiques (muscles striés). Le foie constitue l’organe central de gestion du métabolisme et du transport des lipides dans l’organisme. Il prend en charge les lipides résiduels d’origine intestinale et les intègre dans de nouvelles lipoprotéines afin de les redistribuer aux tissus périphériques. Cette voie centrifuge consiste en une cascade impliquant les VLDL, les IDL et les LDL. Enfin les tissus périphériques captent les lipides (principalement cholestérol et acides gras libres non estérifiés) par endocytose et hydrolyse des lipoprotéines d’origine hépatique et intestinale. La plupart des tissus périphériques ne peuvent pas métaboliser le cholestérol, et ont donc recours, via les HDL, à une voie de transport centripète vers le foie, seul organe capable de l’éliminer par voie biliaire.
2.2. Métabolisme des LDL
2.2.1. Origine des LDL
Les LDL sont issues de la transformation des VLDL qui subissent l’action d’enzymes dans la circulation sanguine. La figure 3 schématise la voie endogène du métabolisme des lipoprotéines à l’origine de la formation des LDL.
Figure 3 : Origine des LDL : voie endogène du métabolisme lipidique.
PL : PhosphoLipides ; CL : Cholestérol Libre ; CE : Cholestérol Estérifié ; TG : TriGlycérides ; LPL : LipoProtéine Lipase ; LH : Lipase Hépatique ; CETP : Cholestéryl Estérase Transfert Protein ; LCAT : Lecithine Cholesterol Acyl Transferase.
Les VLDL, synthétisées et sécrétées par le foie, initient une voie métabolique qui permet de transporter les lipides endogènes du foie vers les tissus périphériques. Après sécrétion dans le compartiment intravasculaire, les VLDL hépatiques subissent l’action de la LipoProtéine Lipase endothéliale (LPL) puis de la Lipase Hépatique (LH), qui hydrolysent leurs triglycérides. La perte des triglycérides, entraînant un enrichissement relatif du cœur hydrophobe en esters de cholestérol, s’accompagne d’un transfert de phospholipides et d’apoprotéine C aux HDL. De plus, la Cholestéryl Estérase Transfert Protein (CETP) permet un échange de triglycérides et d’esters de cholestérol entre les HDL et les VLDL contribuant également à un enrichissement de la VLDL en esters de cholestérol. In fine, et après formation transitoire de particules IDL intermédiaires, la combinaison des activités LPL (sur les VLDL), LH (sur les IDL) et CETP conduit à la formation des LDL. Les LDL contiennent à leur surface uniquement l’apoprotéine B100 après perte des apoprotéines C et E contenues sur les VLDL et IDL (Lagrost et al., 2004).
CETP CE
TG
2.2.2. Catabolisme des LDL
Tout au long de la cascade VLDL-IDL-LDL, les lipoprotéines peuvent interagir avec des récepteurs cellulaires spécifiques localisés au niveau des hépatocytes ou au niveau des tissus périphériques. Le tableau 4 regroupe les principaux récepteurs des lipoprotéines.
Tableau 4 : Récepteurs cellulaires des lipoprotéines (d’après Lagrost et al., 2004))
NOM FAMILLE PRINCIPAUX
TISSUS LIPOPROTEINES CONCERNEES LIGAND LDLR (LDL receptor ou récepteur apoB100/E)
LDL-receptor Foie, muscle, cerveau, coeur
LDL, VLDL, VLDL et
chylomicrons remnants apoB100, E
LRP (LDL receptor
related protein) LDL-receptor Foie, cerveau, poumon
VLDL, VLDL et
chylomicrons remnants apoE
VLDLR
(VLDL receptor) LDL-receptor
Muscle, Coeur, tissus adipeux
VLDL, VLDL et
chylomicrons remnants apoE
ER-2
(apoE receptor) LDL-receptor Cerveau, placenta
VLDL, VLDL et
chylomicrons remnants apoE
Megaline LDL-receptor Rein, intestin, placenta HDL, VLDL, LDL apoB100, E, AI
Cubiline Rein, intestin, placenta HDL apoAI
SRAI/AII (scavenger receptor type AII/AII) Scavenger receptor type A Macrophages, endothelium LDL oxydées LDL acétylées apoAI, B SRB1 (scavenger receptor type B1) Scavenger receptor type II Foie, tissus stéroïdogènes HDL, VLDL, LDL CD36 (glycoprotein IV) Scavenger receptor type II Monocytes, endothelium, plaquettes, adipocytes, CML LDL modérément oxydées SREC (Scavenger Receptor expressed by Endothelial Cells) CE LDL oxydées LDL acetylées LOX1 (lectin-like oxidized LDL receptor)
Lectin Macrophages, CE LDL oxydées
B48R
(apoB receptor)
Macrophages, endothélium
Chylomicrons et
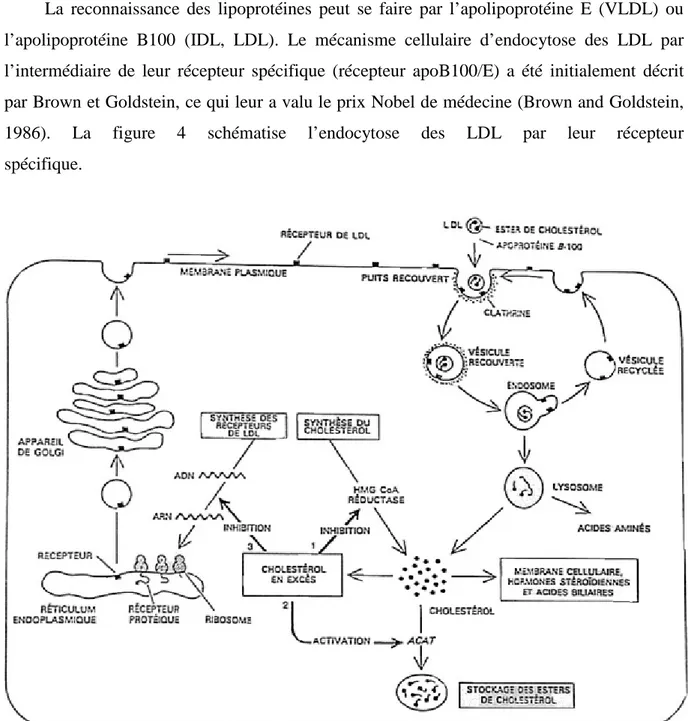
La reconnaissance des lipoprotéines peut se faire par l’apolipoprotéine E (VLDL) ou l’apolipoprotéine B100 (IDL, LDL). Le mécanisme cellulaire d’endocytose des LDL par l’intermédiaire de leur récepteur spécifique (récepteur apoB100/E) a été initialement décrit par Brown et Goldstein, ce qui leur a valu le prix Nobel de médecine (Brown and Goldstein, 1986). La figure 4 schématise l’endocytose des LDL par leur récepteur spécifique.
Figure 4 : Captation et dégradation cellulaire des LDL (d’après Brown and Goldstein, 1986) HMGCoA réductase : HydroxyMethylGlutaryl-Coenzyme A réductase ; ACAT : AcylcoA-Cholesterol-Acyl-Transferase
Les LDL circulantes se fixent sur leur récepteur localisé dans des puits recouverts de clathrine au niveau de la membrane plasmique des cellules. Les LDL sont ensuite internalisées par endocytose dans des vésicules lysosomales où tous les constituants lipidiques et protéiques sont dégradés. Ce mécanisme assure à la cellule un approvisionnement substantiel en cholestérol qui va exercer un rétrocontrôle inhibiteur à trois niveaux :
- inhibition de la synthèse endogène du cholestérol par inhibition de l’HydroxyMéthylGlutaryl-Coenzyme A (HMG-CoA) réductase.
- augmentation de l’activité d’estérification, et donc du stockage du cholestérol via l’ AcylCoA-Cholestérol-Acyl-Transférase (ACAT)
- inhibition de l’expression des récepteurs des LDL à la surface des cellules, bloquant ainsi la voie principale d’entrée du cholestérol dans les cellules.
Les VLDL, IDL et LDL peuvent également se fixer sur les récepteurs scavenger SR-B1 permettant la captation sélective des esters de cholestérol (Williams et al., 1999).
2.3. Oxydation des LDL
L’oxydation des LDL intervient majoritairement dans l’intima des artères après leur passage à travers l’endothélium (Steinberg and Gotto, 1999). L’oxydation des LDL est un processus progressif qui conduit à la génération de LDL oxydées à divers degrés (faiblement à fortement oxydées). Elle se produit en réponse au stress oxydant généré par les cellules de la paroi artérielle (cellules endothéliales, cellules musculaires lisses) et les cellules inflammatoires (macrophages et lymphocytes T) présentes sur le site de la lésion. Les LDL peuvent être oxydées in vitro en présence de métaux de transition ou par des cellules en cultures qui vont produire des radicaux oxydants (Salvayre, 2004).
2.3.1. Mécanisme de la peroxydation lipidique
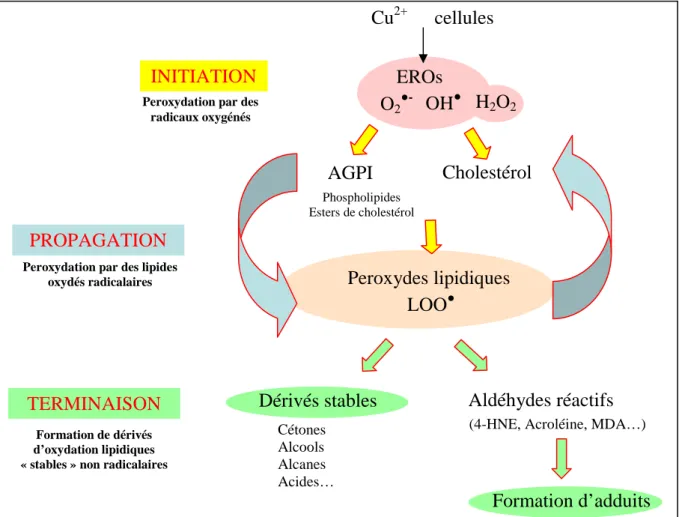
L’oxydation des LDL par voie chimique implique des oxydants et des ions de métaux de transition. La peroxydation chimique des lipides résulte de l’attaque de substrats oxydables (les Acides Gras Poly-Insaturés (AGPI) étant plus oxydables que les acides gras saturés) par des radicaux oxydants. La dégradation en chaîne des acides gras, conduit à la formation d'hydroperoxydes instables qui vont également attaquer la composante protéique de la lipoprotéine (Witztum and Steinberg, 1991). Selon le modèle d’oxydation par le cuivre (Esterbauer et al., 1990) (Jurgens et al., 1987), l’oxydation des LDL se déroule en plusieurs étapes (résumées figure 5) :
- la phase de latence permet la consommation des antioxydants par les EROs (Terentis et al., 2002).
- la phase d'initiation correspond à l’attaque des AGPI. Ils sont particulièrement vulnérables du fait de leurs doubles liaisons. Cette initiation de la peroxydation induit
la formation de diènes conjugués et d’un radical L●H qui peut fixer O
2 pour former des
radicaux peroxyles (LOO●) et d’autres dérivés oxygénés (radicaux alkoxyles, endoperoxydes, époxydes).
- les radicaux peroxyles vont à leur tour réagir avec une molécule d’O2 pour induire une
réaction d’oxydation en chaîne à l’origine de la propagation de la peroxydation lipidique.
- la phase de terminaison de la réaction d’oxydation survient soit par épuisement du substrat, soit par formation de dérivés stables (alkanes, alcools, cétones, aldéhydes, acides carboxyliques).
Figure 5 : Représentation simplifiée de la peroxydation lipidique
EROs : Espèces réactives de l’oxygène ; AGPI : Acides Gras Poly Insaturés ; HNE : 4-HydroxyNonEnal ;MDA : MalonDiAldéhyde
AGPI Cu2+ cellules O2●- OH● H2O2 Peroxydes lipidiques LOO● Cétones Alcools Alcanes Acides…
(4-HNE, Acroléine, MDA…)
Cholestérol Formation d’adduits Phospholipides Esters de cholestérol INITIATION PROPAGATION TERMINAISON EROs
Peroxydation par des radicaux oxygénés
Peroxydation par des lipides oxydés radicalaires
Formation de dérivés d’oxydation lipidiques « stables » non radicalaires