DÉVELOPPEMENT DE NOUVELLES MÉTHODOLOGIES PAR SPECTROMÉTRIE DE MASSE A TRÈS HAUTE RÉSOLUTION POUR L'ANALYSE STRUCTURALE DE COMPLEXES PROTÉIQUES
237
0
0
Texte intégral
Figure

+7



Outline
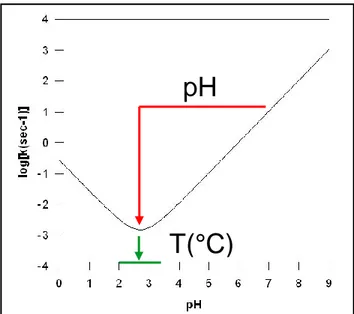
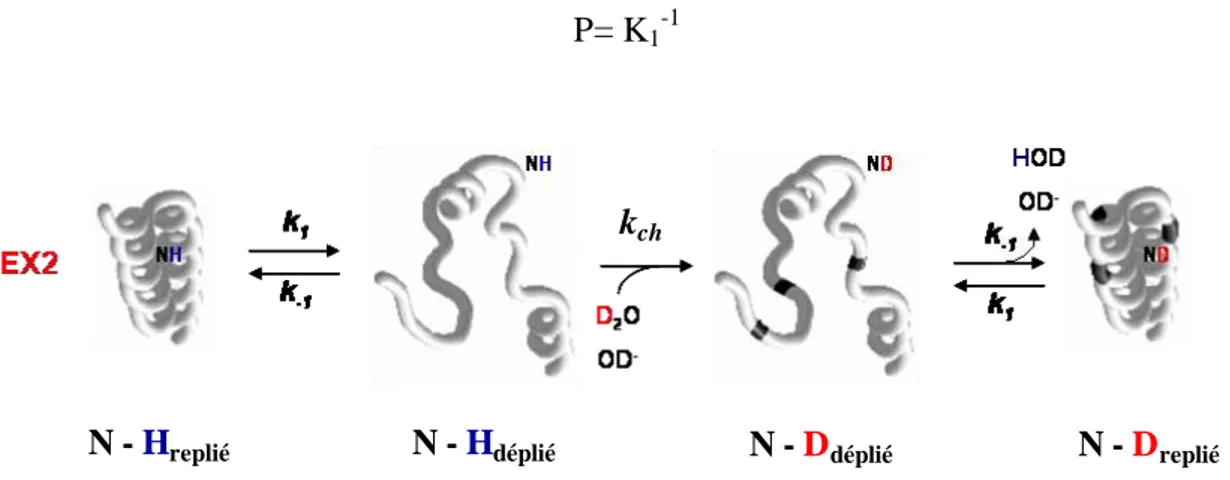
Méthode associant les échanges hydrogène/deutérium à la spectrométrie de
2.3.2.1 Digestion protéolytique et analyse par LC-MS
2.3.2.2 Fragmentation en phase gazeuse des peptides protéolytiques
Conclusion sur l’APEX III
Autres instruments FT-ICR
Analyse de la sous-unité aIF2α3
Analyse de la protéine aIF2α3 avec une étiquette poly-histidines
Détermination du taux maximal de deutération : mise en évidence du
Qu’est-ce que la « cryosource » ?
Echange inverse
Documents relatifs
