ROLE OF 17β-HYDROXYSTEROID DEHYDROGENASE TYPE 5
IN BREAST CANCER STUDIED BY INTRACRINOLOGY
Thèse DAN XU Doctorat en physiologie-endocrinologie Philosophiae Doctor (Ph.D.) Québec Canada © Dan Xu, 2015
RÉSUMÉ COURT
Le cancer du sein (BC) est la deuxième cause de décès relié au cancer chez les femmes. Son incidence continue d'augmenter en particulier chez les femmes post-ménopausées. L’exploration de la pathogenèse et la recherche de nouveaux traitements restent des points d’intérêt. L'expression et le rôle de la 17β-HSD5 (AKR1C3) sont controversés. Ici, nous avons répondu à la question si la 17β-HSD5 est une cible potentielle pour le traitement du BC et nous avons comparé son expression chez des échantillons de tumeurs mammaires et des tissus normaux. De plus, nous avons proposé qu’une plus faible expression d’AKR1C3 puisse être utilisée comme biomarqueur pour un pronostic du BC. Nous suggérons de fournir du DHEA comme source d'hormone intracrinologique et de comparer le rôle des enzymes en utilisant la DHEA et leurs substrats directs. La provision de DHEA est un bon choix pour mimer un état post-ménopausique dans le métabolisme cellulairedes stéroïdes.
SHORT SUMMARY
Breast cancer (BC) is the second leading cause of cancer death in western women. Its incidence continues to increase, especially in post-menopausal women. Exploring the pathogenesis and seeking new treatments remain hotspots. In spite of the increasing number of studies on 17β-hydroxysteroid dehydrogenases (17β-HSDs), the expression and role of 17β-HSD5 (AKR1C3) remain controversial. Here we answered the question whether 17β-HSD5 is a possible target for BC therapy and made the comparison of AKR1C3 expression in normal breast and tumor samples. In addition, we propose that the lower expression of AKR1C3 is a biomarker for poor prognostic in breast cancer. We suggest to provide DHEA as intracrinological hormone source and to compare the role of steroid-converting enzymes using DHEA and their direct substrates. We demonstrated that provision of DHEA was a good choice to mimic postmenopausal condition in steroid metabolism in cell culture.
RÉSUMÉ LONG
Dans cette thèse, je présente une étude (1) du rôle de la 17β-HSD5 dans la modulation des taux d'hormones et dans la prolifération, et l'impact de l'expression de la 17β-HSD5 sur d’autres protéines de BC cellules; (2) une étude comparative sur trois enzymes (17β-HSD1, 17β-HSD7 et 3α-HSD3) avec la provision de DHEA et ses substrats directes soit l’E1 ou la DHT. Les principaux résultats obtenus dans cette étude sont les suivants: (1) en utilisant l'ARN d’interférence de la 17β-HSD5, des immunodosages enzymatiques et des tests de prolifération de cellules démontrent que l'expression de la 17β-HSD5 est positivement corrélée à un niveau de T et de DHT dans les BCC, mais négativement corrélée pour l’E2 et la prolifération des cellules de BC (2) les analyses quantitatives de PCR en temps réel et de Western blot ont démontré que l’inhibition de l’expression de la 17β-HSD5 régule à la hausse l'expression de l'aromatase dans les cellules MCF-7. (3) L’analyse d’ELISA de la prostaglandine E2 a vérifié que
l'expression accrue de l'aromatase a été modulée par des niveaux élevés de PGE2 après l’inactivation de l’expression du gène de la 17β-HSD5. (4) Le test de cicatrisation a montré que l’inactivation de l’expression du gène de la 17β-HSD5 favorise l’augmentation de la migration cellulaire. (5) L'expression du gène 17β-HSD5 dans des échantillons cliniques, à partir de l'analyse de base de données ONCOMINE, a montré que sa plus faible expression a été corrélée avec le statut de l’HER-2 et de la métastase de la tumeur. (6) Les données protéomiques révèlent également que des protéines impliquées dans les voies métaboliques sont fortement exprimées dans les cellules MCF-7 après l’inactivation de l’expression du gène de la 17β-HSD5. (7) L’étude n'a démontré aucune différence dans la fonction biologique de la 17β-HSD1 et de la 17β-HSD7 lorsqu'elles sont cultivées avec différentes stéroïdes: tel que les niveaux de stéroides, la prolifération cellulaire et les protéines régulées. (8) Toutefois, la supplémentation du milieu de culture se révèle avoir un impact marqué sur l'étude de la 3α-HSD3. (9). Nous avons proposé que l'utilisation de la DHEA comme source d'hormone puisse entraîner une meilleure imitation des conditions physiologiques
LONG SUMMARY
Human 17β-hydroxysteroid dehydrogenase type 5 (17β-HSD5) mainly synthesizes the activate androgen testosterone (T) from △4-androstenedione (4-dione), then 4-dione and T aromatazion to estrone (E1) and estradiol (E2) by the action of aromatase. 17β-HSD1 and 7 catalyze the formation of E2 from E1 and inactivate androgen dihydrotestosterone (DHT). In this thesis, I present the study of (1) the roles of 17β-HSD5 in the modulation of hormone levels and in the proliferation. and the proteomic study of the impact of the 17β-HSD5 knock down in BCC; (2) a comparative study of three enzymes (17β-HSD1,7 and 3α-HSD3) with the provision of DHEA and the direct substrates, E1 or DHT. The main results obtained in this study are as follow: (1) Using RNA interference of 17β-HSD5, enzyme immunoassays, and cell proliferation assays demonstrate that 17β-HSD5 expression is positively correlated with T and DHT levels in BCC, but negatively correlated with E2 levels, and BCC proliferation. (2) Quantitative real-time PCR analyzes and western blot showed that 17β-HSD5 knockdown up-regulates aromatase expression in MCF-7 cells. (3) Prostaglandin E2 ELISA assay verified that aromatase expression increase was modulated by elevated PGE2 levels after 17β-HSD5 knockdown. (4) Wound healing assay showed that with the knockdown of 17β-HSD5 expression, cell migration increased. (5)17β-HSD5 gene expression in clinical samples from ONCOMINE analysis showed its lower expression was correlated with HER-2 status and tumor metastasis. (6) The proteomic data also reveal that proteins involved in metabolic pathways are highly expressed in 17β-HSD5 knockdown MCF-7 cells. (7)Cell biology study showed no difference in biological function for 17β-HSD1 and 17β-HSD7 when cultured with different steroids cell proliferation and estradiol levels decreased, whereas DHT accumulated; cyclin D1, PCNA, and pS2 were down-regulated after knocking down these two enzymes. (8) The culture medium supplementation was found to have a marked impact on the study of 3α-HSD3. (9) We first proposed that
using DHEA as hormone source may result in better mimicking of the physiological conditions of post-menopausal in cell culture according intracrinology.
TABLE OF CONTENTS
RÉSUMÉ COURT ... III SHORT SUMMARY ... V RÉSUMÉ LONG ... VII LONG SUMMARY ... IX List of Publication ... XV Foreword ... XVII ACKNOWLEDGEMENTS ... XIX List of Tables ... XXI List of Figures ... XXIII List of Abbreviations ... XXV
CHAPTER Ⅰ ... 1
GENERAL INTRODUCTION ... 1
1.1 Breast cancer emergence and impact factor ... 3
1.2. Origins of estradiol ... 8
1.3. Enzymes involved in the synthesis of estradiol ... 11
1.3.1. Aromatase ... 11 1.3.2. Steroid sulfatase ... 11 1.3.3. 17β-HSDs ... 13 1.3.3.1. 17β-HSD1 ... 13 1.3.3.2. 17β-HSD7 ... 14 1.3.3.3. 17β-HSD5 ... 14 1.3.4 3α-HSD3 ... 15
1.4. Anti-estrogen approaches for breast cancer endocrine therapy and prevention ... 16
1.4.1 Selective estrogen receptor modulators ... 16
1.4.2 Aromatase Inhibitors (AIs) ... 18
1.4.3. Selective estrogen receptor down regulator (fulvestrant) ... 21
1.4.4. Steroid sulfatase inhibitors ... 23
1.5. Working hypothesis and Research Objectives ... 23
1.5.1. Hypothesis ... 23
1.5.2. Objectives ... 25
Chapter Ⅱ ... 27
Hypo-expression of AKR1C3 in breast tumors undergoes further reduction with metastasis: the enzyme knockdown up-regulates aromatase by increasing PGE2 ... 27
2.1 Résumé en français ... 29
2.2. Summary ... 31
Article 1 Hypo-expression of AKR1C3 in breast tumors undergoes further reduction with metastasis: the enzyme knockdown up-regulates aromatase by increasing PGE2 .. 33
Chapter Ⅲ ... 65
Proteomic study reveals that the knockdown of 17beta-hydroxysteroid dehydrogenase type 5 in MCF-7 cells up-regulates proteins such as GRP78 and enhances breast cancer cell development ... 65
3.1 Résumé en français ... 67
3.2. Summary ... 69
Article 2 Proteomic study reveals that 17beta-hydroxysteroid dehydrogenase type 5 knockdown in MCF-7 cells up-regulates proteins such as GRP78 and enhances breast cancer cell development ... 71
Chapter Ⅳ ... 107
Mimicking postmenopausal steroid metabolism in breast cancer cell culture: differences in response to DHEA or other steroids as hormone sources ... 107
4.1. Résumé en français ... 109
4.2. Summary ... 111 Article 3
DISCUSSION AND GENERAL CONCLUSION ... 145 REFERENCES ... 153
List of Publication
1. Dan Xu and Sheng-Xiang Lin. Mimicking postmenopausal steroid metabolism in breast cancer cell culture: differences in response to DHEA or other steroids as hormone sources. J. Steroid Biochem. Mol. Biol. 2015 Jul 19. pii: S0960-0760(15)30022-4. doi: 10.1016/j.jsbmb.2015.07.009.
2. Dan Xu, Tang li, Xiaoqiang Wang, Sheng-Xiang Lin. Hypo-expression of AKR1C3 in breast tumors undergoes further reduction with metastasis: the enzyme knockdown up-regulates aromatase by increasing PGE2. (article under
submission)
3. Dan Xu and Sheng-Xiang Lin. Proteomic study reveals that the knockdown of 17beta-hydroxysteroid dehydrogenase type 5 in MCF-7 cells up-regulated proteins such as GRP78 and enhance breast cancer cell development. (article under
Foreword
This thesis is submitted to the “Faculté des études supérieures de l'Université Laval” for the requirement of a doctor’s degree in science. Except for the short and long summaries and the abstract of each article, which are in French, the thesis is written in English in the form of three scientific manuscripts. One article has been accepted by J. Steroid Biochem. Mol. Biol. and is now under revision. The other two are being submitted for publication or in preparation.
In chapter I, the general introductory section, the breast cancer incidence, emergence and impact factor were reviewed. The estradiol synthesis and the enzymes involved in such synthesis, especially 17β-HSD 1, 17β-HSD 5 and 17β-HSD 7, are summarized. Anti-estrogen approaches for breast cancer endocrine therapy and prevention now used are also discussed. Working hypothesis and research objectives are described at the end of this chapter.
Chapter II contains an article in preparation, presenting the use of DHEA as hormone source and the use of 17β-HSD5 specific small interfering RNAs (siRNAs) to silence 17β-HSD5 gene transcription selectively. It is demonstrated that expression of 17β-HSD5 is reduced in breast tumor cells, and undergoes further reduction with metastasis. The enzyme knockdown up-regulates aromatase by PGE2 increasing cell proliferation, significantly increases estradiol levels, and induce cell cycle progression. Chapter III contains an article in preparation presenting proteomic study results, which reveal that proteins involved in metabolic pathways are highly expressed in breast cancer cells with 17β-HSD5 knockdown. Especially, the expression of glucose-regulated protein (GRP78) and phosphoglycerate kinase 1 (PGK1) were significantly elevated. These enzymes can be potent therapeutic targets for breast
cancer. In chapter IV we used three target human enzymes, 17β-HSD 1, 17β-HSD 7 and 3α-HSD3 to analyze their functional differences by providing DHEA and other steroids as hormone source. We demonstrated that provision of different steroids as substrates or hormone sources may promote modified biological effects. In cell culture, provision of DHEA is a proper choice to mimic postmenopausal condition in steroid metabolism. The conclusion is summarized in chapter V.
All experimental work in each publication was my individual contribution, except that Tang Li analyzed and processed the clinical raw data from ONCOMINE database in the chapter II.
The references of chapters I and V are listed at the end. References of publications are listed after the text of each article.
ACKNOWLEDGEMENTS
I would like to express my immeasurable gratitude to my director of research, professor Sheng-Xiang Lin, for accepting me as a PhD candidate. My educational background is internal medicine and major in hematology and oncology. I was not familiar with research. During my period of study for this degree, it was his meticulous guidance, enlightening discussions and continual encouragement that opened my interests, established confidence in research that enabled me to present this thesis. I have learned a lot from him: not only on fundamental theories of breast cancer endocrinology, new targets for therapies and on approaches to scientific research, but also on the importance of hard work and his tireless insistence on scientific research and independently thinking. I believe that the attitudes and skills I have acquired will be beneficial for my entire life and future career.
I was fortunate to have Dr. Sylvie Mader, Dr. Jacques Huot and Dr. Donald Poirier as the reviewers of my thesis.
I would like to thank all the professors and professional staff of our research center, and all the graduate students and postdoctoral fellows, who kindly helped me when I had questions, doubts or problems. Thank Jean-Francois Theriault for helping of my French writing and thank Alessander Laurentino and Preyesh Stephen for my English revision.
I also thank all the members of my research group: Ming Zhou, Dao-Wei Zhu for their help in familiar with the surroundings; thank you Mouna Zerradi, Xiao-Qiang Wang and Jian Song for giving me some suggestions on my research. I also shall remember friendship with them and with Hui Han, Rui-Xuan Wang and Xin-Xia Liang.
Four years of financial support from China Scholarship Council (CSC) were of unimaginable value. It would have been impossible for me to study abroad without their financial support.
Finally, I am very grateful to my family, especially to my parents in China, who always showed their love, encouragement and support to me during my studies.
List of Tables
Table 3. 1 Sequences of 17β-HSD5 and GRP78 specific siRNA ... 103 Table 3.2 Mass spectrometry identification of proteins spot upregulated in MCF-7-17β-HSD5 siRNA as compare to MCF-7 control siRNA * .... 105
Table 4. 1 Sequences of 17β-HSD1 and 17β-HSD7 specific siRNAs ... 135 Table 4. 2 Primers used in RT-PCR ... 135
List of Figures
Figure 1. 1 The percentage of all estimated new cancer cases and death in women in 2015. ... 4 Figure 1. 2 Average number of new cases per year and age-specific incidence rates
per 100,000 population, Females, UK. ... 5 Figure 1. 3 The biological effects of estardiol (E2) are mediated through at least
four ER pathway. ... 7 Figure 1. 4 Schematic representation of DHEA as the unique source of sex steroid
in women after menopause. ... 9 Figure 1. 5 Human steroidogenic and steroid-inactivating enzymes in peripheral
intracrine tissues. ... 10 Figure 1. 6 Proposed regulation of aromatase gene expression in breast adipose
tissue. ... 12 Figure 1. 7 A model for the action of SERMs (such as tamoxifen) through estrogen
response element (ERE)-dependent and non-ERE-dependent (e.g.
API-tethered) pathways in target tissues. ... 17 Figure 1. 8 Mechanism of action of aromatase inhibitors and tamoxifen. ... 20 Figure 1. 9 Mechanism of action of fulvestrant. ... 22 Figure 2. 1 AKR1C3 expression and siRNA knockdowm. . ... 55 Figure 2. 2 Modulation of cell cycle, cell proliferation migration. . ... 56 Figure 2. 3 Relationship between the expression of AKR1C3 and aromatase. .. 58 Figure 2. 4 Schematic representation of AKR1C3 knockdown. . ... 60 Figure 2. 5 AKR1C3 expression levels in breast cancer tissues. . ... 61
Figure 3. 1 Representative 2-D gel images for MCF-7 cells and 17β-HSD5
knock-down MCF-7 cells showing some differentially expressed spots. . ... 93 Figure 3. 2 Functions of the proteins differentially expressed in 17β-HSD5 knock
Figure 3. 3 The first network: ... 95 Figure 3. 4 The second interaction network ... 96 Figure 3. 5 Protein ubiquitination pathway generated by the ingenuity pathway
analysis (IPA) software. ... 97 Figure 3. 6 Negative crosstalk between expression of 17β-HSD5 and GRP78. ... 98 Figure 3. 7 MCF-7 cell growth and E2 production. . ... 99 Figure 3. 8 The expression of PGK1 was up-regulated in 17β-HSD5 knockdown
MCF-7 cells. ... 100
Figure 4. 1 Simplified pathway showing human steroidogenic and
steroid-inactivating enzyme involved in the steroid metabolism pathway in peripheral intracrine tissues. ... 137 Figure 4. 2 Expression of 17β-HSD1 and 17β-HSD7 and knockdown effect by
siRNA. ... 138 Figure 4. 3 Cell proliferation after transfection with siRNA. ... 139 Figure 4. 4 Effect of 17β-HSD1 and 17β-HSD7 knockdown on E2 and DHT
production. ... 140 Figure 4. 5 Cell cycle was evaluated by flow cytometry. . ... 141 Figure 4. 6 Cyclin D1, PCNA and pS2 expression in 17β-HSD1- or
17β-HSD7-knockdown cells compared with normal MCF-7 cells in response to 1 μM DHEA, 0.5 nM E1S, or 0.1 nM E1 as hormone sources in the culture medium. ... 142 Figure 4. 7 Apoptosis-regualted proteins and cell viability. ... 143
List of Abbreviations
△4-dione △4-androstenedione
17β-HSD 17β-hydroxysteroid dehydrogenase 3β-diol 5α-androstane-3β,17β-diol
5-diol androst-5-ene-3β,17β-diol
AIs aromatase inhibitors
AKR aldo-ketoreductase
BC breast cancer
Bcl-2 B-cell lymphoma 2
CHOLS cholesterol sulfate
CoA co-activator molecules
DCIS ductal carcinoma in situ
DHEA dehydroepiandrosterone
DHEAS dehydroepiandrosterone sulfate
DHT dihydrotestosterone
E1S estrogen sulfate
E2S estradiol sulfate
ER estrogen receptor
ERBB2/HER2 human epidermal growth factor receptor 2 EREs estrogen responsive elements
ERα estrogen receptor α
Estradiol E2
Estrone E1
FBS fetal bovine serum
GRP78 glucose-regulated protein 78
H hour
IC50 half maximal inhibitory concentration IDC infiltrating ductal carcinoma
ILC infiltrating lobular carcinoma LCIS lobular carcinoma in situ
M metastasis
Min minute
NAD+ nicotinamide adenine dinucleotide
NADPH nicotinamide adenine dinucleotide phosphate
nm nanometer
nM nanomolar
NME1 nucleoside diphosphate kinase
OD optical density
PAGE polyacrylamide gel electro phoresis
PGE2 prostaglandin E2
PGK1 phosphoglycerate kinase
PI propidium iodide
PRAP prolactin receptor-associated protein PREGS pregnenolone sulfate
Q-RT-PCR quantitative real-time PCR
RT-PCR real-time PCR
S second
SDR short chain dehydrogenase/reductase
SDS sodium dodecyl sulfate
SERDs selective estrogen receptor down regulators SERMs selective estrogen receptor down modulators
siRNA small interfering RNA
STS steroid sulfatase
ml microliter
CHAPTER Ⅰ
1.1 Breast cancer emergence and impact factor
Breast cancer (BC) is the most commom cancer among Canadian women. It is the second leading cause of cancer death in women. BC can also occur in men, but it is not common [1]. It is estimated that 25,000 women will be diagnosted BC in 2015 [2].
Figure 1.1 showed the percentage of all estimated new cancer cases and deaths in
women in 2015 [2]. Women diagnosted with BC often after age of 45, they most after menopause [3]. Figure 1.2 showed Average number of new cases per year and age-specific incidence rates per 100,000 Population, Females, UK [3]. BC emerges by a multistep process including the transformation of normal cells through hyperplasia, precancerous lesions, in situ carcinoma, progression of primary breast cancer and metastasis formation. In this developmental process, the hormones estrogen and hormones, acting through their receptors, stimulate cellular proliferation by receptor-mediated signaling pathways as well as the accumulation of various genetic alternation [4].
Estradiol (E2), the most potent estrogen, plays a critical role in tumor cellular proliferation and cancer development [5], stimulating cellular proliferation through nuclear receptor-mediated signaling pathways [6, 7]. E2, a small hydrophobic ligand that can diffuse through the cell membrane, distributes throughout the cell and binds to estrogen receptor α (ERα), which is located in the nucleus bound to heat shock protein 90 (hsp90). The binding of E2 to ERα induces a conformational change in the receptor, which results in the dissociation of hsp90 and formation of the ER homodimer. Subsequent interaction of the ER homodimer with estrogen responsive elements (EREs) in the E2-responsive gene promoter leads to binding of co-activator molecules (CoA), upregulating gene transcription and leading to cellular proliferation increase [6, 7] (ER signaling pathway. Four distinct pathways of estrogen signaling through ER are shown in Figure 1.3). Therefore, in the field of endocrine research, much emphasis has been placed on functional studies of estrogen and estrogen receptor.
Figure 1. 1 The percentage of all estimated new cancer cases and death in women in 2015. 25,000 women will be diagnosed with breast cancer. This represents 26% of
all new cancer cases in women in 2015. 5,000 women will die from breast cancer. This represents 14% of all cancer deaths in women in 2015.
Figure 1. 2 Average number of new cases per year and age-specific incidence rates per 100,000 population, Females, UK.
Tumorigenesis involves amplification of oncogenes and mutation or loss of tumor suppressor genes by cytogenetic and molecular genetic analysis. For example, the modification of predisposing genes (BRCA1, BRCA2, P53), oncogenes (c-myc, erbB2) and growth factors (EGF, TGFa) is one of these mechanisms. These defets result in atypical cellular proliferation and further progress to ductal carcinoma in situ, lobular carcinoma in situ, infiltrating ductal carcinoma, infiltrating lobular carcinoma in a multistep process [4].
Figure 1. 3 The biological effects of estardiol (E2) are mediated through at least four ER pathway. Path 1 is called the classical genomic pathway; E2 bounds ER,
leading to its dimerization of the later. After a conformational change, ER binds to EREs in regulatory regions to activate the transcription of target genes. Path 2 is the ligand-independent pathway. In the E2-independent pathway, ER is activated through phosphorylation induced by growth factors (GF). Path 3 describes the ERE-independent pathway, in which E2-ER complexes alter transcription of genes containing alternative response elements. Path 4 highlights the nongenomic pathway, which involves a small pool of ER located close to the membrane that, through recruitment of protein kinases (e.g. Scr and PI3K, not shown), activates signaling cascades.
1.2. Origins of estradiol
Estrogens in women are produced from two sources: 1) direct secretion from the ovary; 2) conversion from the adrenal precursors dehydroepiandrosterone-sulfate (DHEA-S), dehydroepiandrosterone (DHEA) and androstenedione in the peripheral tissues [8, 9]. In premenopausal women, DHEA-S is produced exclusively by the adrenal gland [10], and while 50% of DHEA is produced in the adrenal gland, 20% of DHEA production occurs in the ovary and the other 30% by peripheral conversion of DHEA-S by sulfatase [11]. Half of androstenedione is produced by the ovary and the other half by the adrenals [8]. Interestingly, half of testosterone (T) in women is produced via the peripheral conversion of androsterone [11]. In post-menopausal women, the ovaries become atrophied and almost cease function, such that nearly all E2 is produced in peripheral target tissues from precursor steroids of the adrenal glands. Therefore, the aromatase and 17β-hydroxysteroid dehydrogenases (17β-HSDs) are critical for E2 formation in these sites, particularly for postmenopausal estrogen-dependent breast cancer [9]. These steroidogenic enzymes responsible for the synthesis of all steroid hormones and are expressed in many peripheral tissues, providing the basis for a new area in the study of hormone action, namely intracrinology [9]. Intracrinology is a term coined in 1988, which describes the local formation, action and inactivation of sex steroids from the inactive sex precursor DHEA [10]. The best estimate of the intracrine formation of estrogens in peripheral tissue of women is about 75% before menopausal and almost 100% after menopause [9, 13] Figure 1. 4 is a schematic representation of DHEA as the unique source of sex steroid in women after menopause [13]. High amounts of DHEA secreted by the adrenals, serving as precursor, are converted to various estrogens and androgens by the action of steroid-converting enzymes expressed in peripheral tissue in the manner of intracrinology (Figure 1.5 Human steroidogenic and steroid-inactivating enzyme in peripheral intracrine tissue) [13]. Intracrine activity describes the formation of active
enzymes and their encoding genes may represent primary targets of novel therapies for hormone-dependent cancers, especially breast cancer [9, 14].
Figure 1. 4 Schematic representation of DHEA as the unique source of sex steroid in women after menopause. In pre-menopausal women about 20% is circulating
DHEA that released from the ovary and approximately 80% is of adrenal origin. After the menopause, the ovaries atrophy and lose their secretory function. All estrogens and androgens are then made locally from DHEA in peripheral target tissues. Thus, DHEA becomes the unique source of estrogens after menopause. GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone; CRH, corticotropin-releasing hormone; ACTH, an adrenocorticotropic hormone. (Labrie F and Labrie C. 2013)
Figure 1. 5 Human steroidogenic and steroid-inactivating enzymes in peripheral intracrine tissues. 4-dione, androstenedione; A-dione, 5α-androstane-3,17-dione; ADT,
androsterone; epi-ADT, epiandrosterone; E1, estrone; E1-S, estrone sulfate; E2,
17β-estradiol; E2-S, estradiol sulfate; 5-diol, androst-5-ene-3β, 17β-diol; 5-diol-FA,
5-diol fatty acid; 5-diol-S, 5-diol sulfate; HSD, hydroxysteroid dehydrogenase; testo, testosterone; RoDH-1, Ro dehydrogenase 1; ER, estrogen receptor; AR, androgen receptor; UGT2B28, uridine glucuronosyl transferase 2B28; Sult2B1, sulfotransferase 2B1; UGT1A1, uridine glucuronosyl transferase 1A1 (Labrie F and Labrie C, 2013)
1.3. Enzymes involved in the synthesis of estradiol
1.3.1. Aromatase
Aromatase is an enzyme belonging to the cytochrome P-450 superfamily. Aromatase is expressed from the CYP19 gene [15]. In humans, aromatase is expressed in cells such as the ovarian granulosa cells, the placental syncytiotrophoblast, the testicular Leydig cells, and the brain and skin fibroblasts [16]. Additionally, aromatase is expressed in the human adipose tissue. However, the highest levels of aromatase are in the ovarian granulosa cells in pre-menopausal women, the adipose tissue becoming the predominant site expressing aromatase after the menopause [17-18]. The expression of aromatase is regulated in a tissue-specific manner by promoters that are in turn controlled by hormones, cytokines, and other factors [19–21]. Estrogen-dependent BC uses four promoters (Ⅱ, Ⅰ.3, Ⅰ.7, andⅠ.4) to regulate the expression of aromatase [22]. PromoterⅠ.4 regulates aromatase gene expression by cytokines. Malignant breast epithelial cells secrete factors that induce aromatase expression in adipose fibroblasts and in fibroblasts of the tumor itself via promoter Ⅱ [23].One possible factor is prostaglandin E2 (PGE2), which is produced by malignant breast epithelium as well as by macrophages recruited to the tumor site [23]. (Figure 1.6 proposes regulation of aromatase gene expression in breast adipose tissue from cancer-negative and -positive subjects). The enzyme aromatase is responsible for synthesis of estrogens estrone (E1) from the preferred substrate androstenedione and of estradiol from testosterone. Aromatase inhibitors work by inhibiting the action of the enzyme in this process [24].
1.3.2. Steroid sulfatase
Steroid sulfatase (STS) is widely distributed throughout the body and it involved in physiological processes and pathological conditions [25]. STS is a member of the mammalian sulfatase superfamily. The substrates of STS include cholesterol sulfate
(CHOLS), pregnenolone sulfate (PREGS), dehydroepiandrosterone sulfate (DHEAS) and estrogen sulfate (E1S) [26-28]. It was revealed that the expression of STS in the breast is significantly higher than in normal tissues, which suggests that the STS might be a potent target for breast cancer treatment [29].
Figure 1. 6 Proposed regulation of aromatase gene expression in breast adipose tissue.In cancer-free individuals, the expression is stimulated primarily by class I
cytokines and TNFα produced locally in the presence of systemic glucocorticoids. Promoter I.4–specific transcripts of aromatase predominate. In breast cancer, PGE2 produced by the tumorous epithelium, tumor-derived fibroblasts, and/or macrophages recruited to the tumor site becomes the major factor stimulating aromatase expression, indicated by the predominance of promoter II and I.3–specific transcripts of aromatase. (Simpson ER, et al. Annu.Rev.Physiol.2002.64:93-129)
1.3.3. 17β-HSDs
17β-Hydroxysteroid dehydrogenases (17β-HSDs) are NADPH/NAD+-dependent
oxidoreductases that play a significant role in estrogen and androgen metabolism [30]. Until now, fourteen different isoforms of 17β-HSDs have been identified in mammals [31-33]. Exceptionally, 17β-HSD5 belongs to the aldo-ketoreductase (AKR), while all other 17β-HSDs belong to the short chain dehydrogenase/reductase (SDR) protein family [34-35]. The participation of several types of 17β-HSDs in the pathophysiology of human diseases has been suggested, and some of them were reported to play roles in breast cancer. 17β-HSD types 1, 5, 7 are the most important reductive enzymes in the synthesis of estrogens.
1.3.3.1. 17β-HSD1
17β-HSD1 catalyzes the activation of estrone (E1) to the most potent estrogen 17β-estradiol (E2) (Fig.1.3) [36]. E2 is known to have a critical role in the development of estrogen-dependent diseases. 17β-HSD1 mRNA expression levels and its induced E2/E1 ratio were significantly higher in postmenopausal than in premenopausal breast cancer [37], 17β-HSD1 catalysis also the inactivation of the potent androgen dihydrotestosterone (DHT) into 5α-androstane-3α/β, 17β-diol (3α/β-diol). This demonstrates its importance in BC cells due to the anti-proliferation effect of DHT that has been found in estrogen-dependent breast cancer cell lines (MCF-7 and ZR-75-1) [38-40]. 17β-HSD1 up-regulates BC cell proliferation by a dual action on E2 synthesis and DHT inactivation, and 17β-HSD1 can enhance the expression of estrogen responsiveness gene pS2 [41]. High levels of 17β-HSD1 activity facilitate the E2 synthesis and DHT inactivation in BC cells. Thus, a potent inhibitor of 17β-HSD1 could open new possibilities in clinical endocrine therapy.
1.3.3.2. 17β-HSD7
17β-HSD7 is a membrane-associated reductive enzyme. It is another recently found member of the family that can synthesize E2 and it was detected in humans in 1999 [42]. 17β-HSD7 was formerly named the prolactin receptor-associated protein (PRAP). It was later established as a novel 17β-HSD, and named 17β-HSD7 [43-45]. This enzyme has specificity for E1 conversion to E2 [44]. Recently, 17β-HSD7 was reported to possess dual enzymatic activity; it also participates in cholesterol biosynthesis [45-46]. Another interesting finding is the stimulation of 17β-HSD7 expression by E2 in MCF-7 cells [47]. Moreover, expression of the 17β-HSD7 gene is positively correlated with tumor E2 levels in postmenopausal women [48]. Together, these data provided convincing evidence that 17β-HSD7 is an essential enzyme for E2 biosynthesis in BC cells. Thus, 17β-HSD7 should be a potent target for breast cancer treatment.
1.3.3.3. 17β-HSD5
17β-HSD5 is an essential enzyme in androgen conversion with roles in BC cells. 17β-HSD5 belongs to the aldo-keto reductase (AKR) superfamily [49] and is widely expressed in human tissues including the prostate, endometrium and mammary gland [50]. 17β-HSD5 synthesizes 5-diol from DHEA and catalyzes 4-Dione reduction to T, which, when followed by aromatization by CYP19 aromatase, provides a route for E2 biosynthesis independent of 17β-HSD1. However, the expression of 17β-HSD5 and its relationship with the E2 level is controversial. Some researchers found that 17β-HSD5 was detected in mammary glands of premenopausal women [51]. Vihko and co-workers found that 17β-HSD5 mRNA expression was detected in the epithelial cells of normal and malignant breast tissue specimens from both pre- and postmenopausal
Some researchers found that high 17β-HSD5 was related to the significantly higher risk of late relapse in ER-positive BC patients [53]. Penning’s group found that over-expression of 17β-HSD5 (AKR1C3) increases cellular proliferation in MCF-7 cells and also reduces the anti-proliferative effects of prostaglandins, particularly PGD2, such that cellular proliferation can be inhibited by non-steroidal anti-inflammatory drugs [54, 55]. However, other researchers held the opposite opinions: using immunocytochemistry they studied the expression of 17β-HSD5 in 50 specimens of breast carcinoma and adjacent non-malignant tissues. They found that 17β-HSD5 was expressed in 56% of BC samples, indicating that the enzyme was significantly lower in BC than in normal adjacent tissues [56]. Ben P. and co-workers found that the expression of 17β-HSD5 was significantly lower in ER+ tumors compared with normal tissues [57]. Therefore, the expression and the role of 17β-HSD5 in ER+ BC are not entirely clear. Thus, further research is necessary to determine if 17β-HSD5 is a target for BC treatment.
1.3.4 3α-HSD3
3α-Hydroxysteroid dehydrogenase type 3 (3α-HSD3) is a member of the aldo-keto reductase (AKR) family, also named AKR1C2. The best known activity of 3α-HSD3 is the transformation of the most potent natural androgen dihydrotestosterone (DHT) into its much less active form, 5α-androstan-3α, 17β-diol (3α-diol) [58–59]. Because of the importance of 3α-HSD3 in prostate growth, 3α-HSD3 was mostly studied in prostate cancer cells. However, DHT also plays an important role in inhibiting BC cell proliferation, as mentioned above. Therefore, 3α-HSD3 may be critical in BC progression.
1.4. Anti-estrogen approaches for breast cancer endocrine therapy and prevention
1.4.1 Selective estrogen receptor modulators
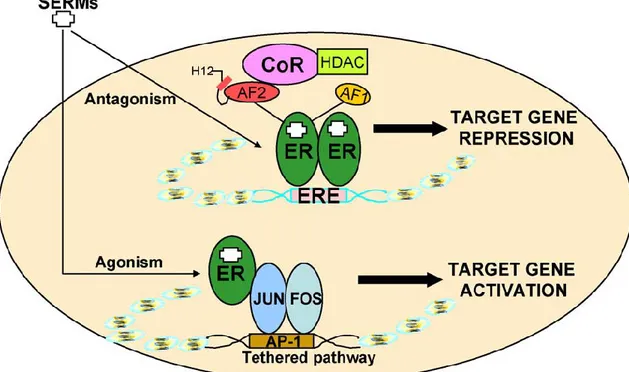
Selective estrogen receptor modulators (SERMs) are therapeutic agents available for the treatment of estrogen receptor-positive breast cancer. Three of the most known SERMs are tamoxifen (Nolvadex), raloxifene (Evista) and toremifene (Fareston). Figure 1.7 shows the model for the action of SERMs (tamoxifen and raloxifene) [60]. SERMs bind to ER and induce different conformational states that facilitate the interaction of corepressors (CoR) to the ERE-driven promoter, which inhibits the recruitment of the basal transcription machinery and thus inhibits transcription [60].
Tamoxifen, whose generic name is Nolvadex, is the oldest and most-prescribed SERM. It is an E2 competing antagonist that binds to the estrogen receptors in breast cells. Thus it prevents binding of estradiol onto its receptor and inhibits transcriptional activity of the receptor. Tamoxifen is approved by the U.S. Food and Drug Administration (FDA) to treat diagnosed hormone-receptor-positive early-stage BC after surgery (and/or possibly chemotherapy and radiation) [61].
Figure 1. 7 A model for the action of SERMs (such as tamoxifen) through estrogen response element (ERE)-dependent and non-ERE-dependent (e.g. API-tethered) pathways in target tissues. Tamoxifen and raloxifene work as estrogen antagonists via
the ERE, but estrogen agonist via the AP-1 tethered pathway. (J.S. Lewis, V.C. Jordan. Mutation Research 591.247-263.2005)
Tamoxifen, the first clinically relevant SERM, is an antagonist of ER in the breast and has been used for more than 30 years to treat ER-positive breast cancer. It has been firmly established that tamoxifen is used for both premenopausal and postmenopausal BC adjuvant endocrine therapy. In women with ER BC, oral tamoxifen adjuvant therapy can decrease the annual odds of recurrence by 39% and the annual odds of death by 31%, regardless of the use of chemotherapy, patient age, menopausal status, or axiliary lymph node status [62]. However, long-term oral tamoxifen has several side effects, one of the worst being the increased risk of endometrial cancer because SERMs are agonists in some tissues such as bone and liver, but are antagonists in other tissues such as brain and breast. In fact, they have the double function of agonist/antagonist in the uterus [60]. Tamoxifen has also other side effects: hot flushes and sweats, change in menstrual periods, vaginal discharge, nausea and indigestion, weight gain, and thrombosis, among others[63].
1.4.2 Aromatase Inhibitors (AIs)
To date, three generations of aromatase inhibitors have been developed as representative drugs. Figure 1.8 showed the mechanism of action of aromatase inhibitors and tamoxifen [64]. One of the most important developments in BC therapy has shown that letrozole is superior to tamoxifen as the first-line treatment for advanced BC [65-67], thus estrogen levels decreased and the growth of breast cancer cells was slowed. It has been indicated that aromatase inhibitors (AIs) can effectively treat the hormone-sensitive breast cancer in postmenopausal women. Overall trails indicated that AIs are superior to tamoxifen in postmenopausal ER+BC women, and also decreased recurrence and caused a reduction in undesirable side effects, typically endometrial cancer and venous thrombosis. The disadvantage of aromatase inhibitors is the higher incidence of osteoporosis that subsequent increases the risk of bone fracture
used in ER+ postmenopausal BC women. They are not to be used in premenopausal women because the reduction of estrogen activates the hypothalamus and pituitary axis causing an increased secretion of gonadotropin, the later in turn not only stimulates the ovary to increase the production of androgen, but also the heightened gonadotropin levels up-regulate the aromatase promoter, consequently increasing aromatase production with increased androgen substrate. As a consequence, the estrogen is increased when AIs are used in premenopausal women [68].
Figure 1. 8 Mechanism of action of aromatase inhibitors and tamoxifen. Aromatase
inhibitors inhibit the synthesis of estradiol by blocking the androstenedione conversion to estrone and of testosterone to estradiol. Tamoxifen inhibits the growth of breast tumors by competitive antagonism of estrogen at its receptor site. (Smith IE, Dowsett. Aromatase inhibitors in breast cancer. M. N Engl J Med. 2003. 348:2431-2442.)
1.4.3. Selective estrogen receptor down regulator (fulvestrant)
Fulvestrant, whose generic name is Faslodex, belongs to the first selective estrogen receptor down regulators (SERDs). It is an estrogen receptor (ER) antagonist that competitively binds to the ER with a much greater affinity than that of tamoxifen [69]. The fulvestrant down-regulates ER, and it has no estrogen agonist effects. The absence of estrogen agonist effects for fulvestrant is different from tamoxifen and the other selective ER modulator (SERMs) [70]. Fulvestrant-ER binding impairs receptor dimerization and affects the energy-dependent nucleo-cytoplasmic shutting, thus blocking nuclear localisation of the receptor. Also, the fulvestrant-ER complex enters the nucleus which cannot activate the transcription because the AF1 and AF2 stop working. In the end, the fulvestrant-ER complex is erratic and causes accelerated degradation of ER. Down regulation of ER proteins occurs without the decrease of mRNA in ER. Therefore, the fulvestrant binding to ER leads to complete inhibition of estrogen signaling pathway [71]. Figure 1.9. Represent the Mechanism of action of fulvestrant. Until now, it has been demonstrated that this new type of endocrine therapy is not only agonism-free in estrogen but also a lack of cross-resistance between the SERMs. Thus, fulvestrant will be useful in patients with tamoxifen-resistant disease [69, 71].
Figure 1. 9 Mechanism of action of fulvestrant. ERE = estrogen response element;
ER = estrogen receptor; F= fulvestrant. (CK Osborne, A Wakeling and RI Nicholson. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. British Journal of Cancer 90 (Suppl 1), S2 – S6, 2004.)
1.4.4. Steroid sulfatase inhibitors
The first reports on STS inhibition started in the 1970s. Maltais & Poirier have recently published a comprehensive review covering the steroid sulfatase inhibitors regarding the promising 2000-2010 decades [72]. This section intends to discuss clinical trials. 667 COUMATE was the first STS inhibitor to enter the phase I clinical trial in postmenopausal women with hormone-dependent breast cancer [73-74]. Phases I/II clinical trials on 667 COUMATE now have given it the generic name Irosustat. In a phase II endometrial cancer trial Irosustat did not reach the endpoint. The use of STS inhibitors as a treatment for estrogen or androgen-dependent cancers remains to be studied [75].
1.4.5. 17β-HSDs inhibitors
Inhibitors of 17β-HSD isoforms are worth studying, as they may block the formation of hydroxysteroids that stimulate estrogen-sensitive cancers. Several families of 17β-HSD inhibitors were reported to block the synthesis of estradiol. Poirier D gives a description of novel inhibitors of 17β-HSDs from 2003 to 2010 [76-77]. Inhibitors of 17β-HSD1 are suitable in the treatment of estrogen-dependent BC, but the role of types 5, 7 and 12 is still controversial [77].
1.5. Working hypothesis and Research Objectives
1.5.1. Hypothesis
Based on the above introduction, it is known that estradiol (E2) plays a critical role in breast cancer (BC). Endocrine therapy of BC decreases E2 activity or block its synthesis. Letrozole, tamoxifen and fulvestrant have been shown to be effective in BC treatment
in clinical or pre-clinical trials, but because of the side effects, limited use and resistance, further development of drug targets and enzyme inhibitors are necessary.
First, the role of 17β-HSD5 (AKR1C3) is controversial. We thus proposed to study
if 17β-HSD5 is a target for ER+BC therapy. We first provided DHEA as hormone source to mimic the postmenopausal conditions in women. Following 17β-HSD5 depletion by siRNA in ER+BC cells, DHEA cannot be converted to 5-diol, nor Δ 4-dione into testosterone. As showed steroidogenic pathway in the peripheral tissues in
Figure 1.3. Testosterone is a direct precursor steroid used for the synthesis of E2. Thus,
it is hypothesized that E2 level is reduced and cellular proliferation decreases. If this is the case, the inhibitor of 17β-HSD5 may be used independently or in combination with the inhibitors of 17β-HSD 1 and /or 17β-HSD 7 for breast cancer therapy.
Second, most studies of 17β-HSD1 and 17β-HSD7 inhibitots in vivo and in vitro
used estrone (E1) as substrate, while studies of 3α-HSD3 used DHT as substrate. These studies all used the direct substrate as hormone source. However, E2 synthesis involves multiple pathway, conversion from E1 to E2 being the last step, so providing E1 cannot fully mimic the physiological condition, As well as reflect the difference in the origins of estrogen before and after menopause. After the menopause, close to 100% estrogens are synthesized in peripheral target tissues from precursor steroids of adrenal origin. We are suggesting to provide DHEA as intracrine hormone source and to compare the role of steroid-converting enzymes using DHEA and their direct substrates when an extensive understanding of the mechanism is necessary. Thus, we hypothesized that providing different hormone sources may have similar or different effects on 17β-HSD1, 17β-HSD7 or 3α-HSD3 knockdown cells. Providing DHEA is mimicking the postmenopausal condition in steroid metabolism in BC cell culture.
1.5.2. Objectives
Objective one: To evaluate of the biological function of 17β-HSD5 in MCF-7 BC
cells. To achieve this, we will employ siRNA or 17β-HSD5 inhibitor (such as EM1404) in the MCF-7 cell line, followed by assaying cellular proliferation and cell cycle changes. At the same time, androgens and estrogens related to 17β-HSD5 (such as DHEA, 5-diol, 4-dione, T, DHT, E1 and E2) and cell migration will be determined.
Objective two: To identify mechanisms responsible for cell proliferation and
steroidal changes after 17β-HSD5 depletion.
Objective three: To use clinical sample information in ONCOMINE data base to
determine the 17β-HSD5 expression in BC and normal tissues.
Objective four: To compare the differences of using DHEA or E1S as substrates
with E1 as substrate in the previous experiments and to demonstrate that BC cell biology changes after knockdown the expression of 17β-HSD1 or 17β-HSD7. To compare the differences of providing DHEA as hormone sources and providing DHT to show the function of 3α-HSD3 in BC cells.
Chapter Ⅱ
Hypo-expression of AKR1C3 in breast tumors undergoes further reduction with
2.1 Résumé en français
La sous-expression d’AKR1C3 dans les tumeurs du sein subit est augmentée lors de la formation de métastases: l’inhibition de l’expression du gène de l’enzyme
régule à la hausse l'aromatase en augmentant la PGE2 .
Des rapports sur l’expression de l’AKR1C3 (17β-HSD5) dans des échantillons de cancer du sein (BC) et son utilité comme cible potentielle pour le cancer du sein sont controversés. En utilisant les informations d’échantillons cliniques dans la base de données Oncomine, nous avons démontré l'expression significativement inférieure de l’AKRIC3 dans des cancers du sein positifs pour le récepteur des œstrogènes par rapport aux échantillons de tissu de sein normal. En outre, le gène AKR1C3 est moins exprimé chez les patients avec des métastases. Nous avons fourni du DHEA comme source d'hormone pour imiter les conditions post-ménopausiques ales chez les femmes. Des petits ARN interférents ont été utilisés pour éteindre la transcription du gène AKR1C3 dans les cellules MCF-7 ce qui a produit une diminution de la quantité des androgènes actifs (T, DHT), ainsi qu’une augmentation simultanée inattendue du niveau d’E2 (1,8 fois) et de prolifération cellulaire (46%). L’inhibition du gène AKR1C3 a également stimulé la migration des cellules MCF-7 de 12%. Par ailleurs, nous démontrons que l’inhibition de l’expression du gène AKR1C3 a stimulé l'expression du gène de l'aromatase de 31% en raison de l'augmentation de la prostaglandine E2 (PGE2). Par conséquent, l’expression moindre de l’AKR1C3 dans le
cancer du sein conduit à la régulation positive de l’aromatase, à la prolifération cellulaire et à la métastase. Une telle expression plus faible peut être utilisée pour prédire un pronostic lors du suivi clinique.
2.2. Summary
Human 17β-hydroxysteroid dehydrogenase type 5 (17β-HSD5, AKR1C3) belongs to the aldo-keto reductase (AKR) superfamily. This enzyme synthesizes androst-5-ene-3β , 17 β -diol (5-diol) from dehydroepiandrosterone (DHEA) and catalyzes △4-androstenedione (△4-dione) reduction to testosterone (T), which is further converted to estradiol (E2) by aromatase in breast cancer cells (BCC). Therefore, 17β-HSD5 must play a critical role in estrogen receptor (ER)-positive breast cancer (ER+BC). However, its function in BCC development remains unclear and its expression in BC tumor samples is controversial. We provided DHEA as hormone source to mimic the postmenopausal conditions in women. Small interfering RNAs were used to silence AKR1C3 gene transcription in MCF-7 cells. Cell proliferation was assayed by CyQUANT cell proliferation kit, steroidal hormone levels were determined by ELISA kit and cell migration was measured by wound healing assay. Aromatase and AKR1C3 regulation were determined by western blot and Q-RT-PCR. We used clinical sample information available on Oncomine data base to compare the expression of AKR1C3 in different conditions and tissues. The silencing of AKR1C3 gene transcription produced a decrease in active androgens (T and DHT), an unexpected simultaneous increase of E2 levels (1.8 fold) and MCF-7 cell proliferation (46%). Inhibition of AKR1C3 also stimulated MCF-7 cells migration by 12%. AKR1C3 knockdown stimulated expression of the aromatase gene by 31% due to the increase of prostaglandin E2 (PGE2). PGE2 increases the binding activity of the Jun and ATF groups of transcription factors of the aromatase promoter I.3/II region. AKR1C3 expression was significantly lower in estrogen-receptor BC cells (p=2.88E-06) than in normal breast tissue. It was also significantly lower in HER-2 positive patients than in HER-2 negative cases (P=0.0003). In ductal breast carcinoma, AKR1C3 gene is less expressed in patients with metastasis than without it (P=0.008). The lower expression of AKR1C3 in BC leads to aromatase up-regulation, MCF-7 cell proliferation and metastasis. These findings are important as they propose a new mechanism to explain reduced expression of AKR1C3 in BC compared to normal breast tissue. This reduced
expression of AKR1C3 in BC may serve as a poor prognostic factor, which is strongly supported by clinical sample statistics, bur remains to be further validated in clinical tests and observation.
Hypo-expression of AKR1C3 in breast tumors undergoes further reduction with metastasis: the enzyme knockdown up-regulates aromatase by increasing PGE2
Dan Xu1, Tang li1, Xiaoqiang Wang1, Sheng-Xiang Lin1
1Centre de recherche du Centre hospitalier universitaire de Québec – Laval University,
Quebec City, Canada G1V 4G2
Authors’ e-mail addresses:
Dan Xu: Dan.Xu@crchuq.ulaval.ca Tang Li: tang.li.1@ulaval.ca
Xiaoqiang Wang: xiaoerqd@gmail.com
Corresponding author: Sheng-Xiang Lin, PhD CHUL Research Center Laval University
2705 Blvd. Laurier, Quebec City, Quebec, G1V 4G2, Canada. Phone: +1 418 525 4444 ext 46367;
Abstract
Introduction AKR1C3 (HSD17B5) is widely expressed in human tissues including
mammary gland. AKR1C3 synthesizes Δ5-androstene-3β,17β-diol (5-DIOL) from dehydroepiandrosterone (DHEA) and catalyzes the reduction of Δ4-androstenedione (4-DIONE) into testosterone (T). Subsequent T aromatization by CYP19 aromatase provides a route for E2 biosynthesis independent of HSD17B1. However, reports of its expression in BC tumor samples, and whether AKR1C3 is a potent target for BC treatment are controversial. A limited number of conflicting studies have investigated the importance of AKR1C3 in BC. Methods We provided DHEA as hormone source to mimic the postmenopausal conditions in women to study the enzyme role in BC. Small interfering RNAs were used to silence AKR1C3 gene transcription in MCF-7 cells, cell proliferation was assayed by CyQUANT cell proliferation kit, steroidal hormone levels were determined by ELISA kit and cell migration were measured by wound healing assay. Aromatase and AKR1C3 regulation were determined by western blot and Q-RT-PCR. We used clinical sample information available on Oncomine data base to compare the expression of AKR1C3 in different conditions and tissues. Results The silencing of AKR1C3 gene transcription produced a decrease in active androgens (T and DHT), an unexpected simultaneous increased in E2 levels (1.8 fold) and MCF-7 cell proliferation (46%). Inhibition of AKR1C3 also stimulated MCF-7 cells migration by 12%. Meanwhile, AKR1C3 knockdown stimulated expression of aromatase gene by 31% due to the increase of prostaglandin E2 (PGE2). PGE2 increases the binding activity of the Jun and ATF groups of transcription factors to the aromatase promoter I.3/II region. AKR1C3 expression was significantly lower in estrogen-receptor BC cells (p=2.88E-06) than in normal breast. In HER-2 positive patients it was also significantly lower than in HER-2 negative cases (P=0.0003). In ductal breast carcinoma, AKR1C3 gene is less expressed in patients with metastasis than without it (P=0.008). Conclusion The lower expression of AKR1C3 in BC leads to aromatase up-regulation, MCF-7 cell proliferation and metastasis. Such lower expression could be used as a biomarker for poor prognosis after further clinical follow-up.
Introduction
Breast cancer (BC) is the most common cancer in women regardless of race and socioeconomic factors, and is the second leading cause of cancer death in women in North America [1–2]. Estradiol (E2), the most potent estrogen, plays a critical role in tumor cellular proliferation and cancer development by stimulating cell proliferation via estrogen receptor (ER) activation [3–4]. Interestingly, several studies suggest that androgens may lead to resistance to the proliferative effects of estrogens and progesterone in the mammary gland as androgens have been shown to exert anti-proliferative effects [5–8].
The balance between the stimulatory effects of estrogens versus the inhibitory effects of androgens is one critical factor that regulates mammary cell proliferation in both normal and carcinomatous tissues [9]. AKR1C3 (HSD17B5) is widely expressed in human tissues including the prostate, endometrium and mammary gland [10–11]. AKR1C3 synthesizes 5-androstene-3β,17β-diol (5-DIOL) from dehydroepiandrosterone (DHEA) and catalyzes the reduction of 4-androstenedione (4-DIONE) into testosterone (T). Subsequent T aromatization by CYP19 aromatase provides a route for E2 biosynthesis independent of HSD17B1 [12]. The structure of AKR1C3 was reported by Qiu et al. 2004, which provided a foundation for inhibitor development [13]. However, reports of its expression in BC tumor samples, and of its usefulness as a potential target for BC treatment are controversial. A limited number of conflicting studies have investigated the importance of AKR1C3 in BC: Oduwole and colleagues [14] found that AKR1C3 mRNA was detected in 65% of breast cancer specimens and was significantly higher in breast tumors than in normal tissue. The prognosis for patients with a high level of expression of tumoral AKR1C3 was worse than for those with low or no expression. Agneta et al. [15] found that a high level of AKR1C3 was related to a significantly higher risk of late relapse in ER+ patients compared with tumors
expressing low and intermediate levels of the enzyme. Nevertheless, Han et al [16] found that lower levels of AKR1C3 were expressed in tumor tissue than in adjacent
normal tissue. Ben et al [17] found that the expression of AKR1C3 was significantly lowered (4.3-fold) in ER+ tumors compared with normal tissue. It is currently unclear
whether AKR1C3 is expressed more highly in normal tissue or in tumor tissue. Therefore, the correlation between AKR1C3 expression and BC is also unclear. In this study, we analyzed a large number of clinical samples from the Oncomine database [18], and provide statistical evidence that lower expression of AKR1C3 correlates with ER+, ERBB2+ and ductal breast carcinoma metastasis. Small interfering RNAs were
used to silence AKR1C3 gene transcription in MCF-7 cells. We demonstrate that AKR1C3 knockdown stimulated expression of the aromatase gene by 31% due to an increase in prostaglandin E2 (PGE2) expression levels.
Materials and methods
Cell culture
MCF-7 cells were from the American Type Culture Collection (ATCC). MCF-7 cells were derived from a 69 years female mammary gland metastatic site. MCF-7 cells were maintained in phenol red-free, low glucose DMEM supplemented with 10% fetal bovine serum (FBS). Cells were cultured at 37℃ in a humidified atmosphere of 95% air and 5% CO2. Cells were plated in charcoal-treated medium to eliminate exogenous
hormones and to permit application of different concentrations of DHEA in order to simulate the physiological conditions of postmenopausal women. A stable expression of aromatase, ER positive MCF-7 cell line, named MCF-7-aro, was generated by aromatase cDNA transfection and G418 selection in the laboratory of Shiuan Chen [19, 20]. The culture method used in MCF-7 was used in MCF-7-aro.
siRNA synthesis and transfections
Lipofectamine 2000 (Invitrogen) and a 100nM mixture of the three siRNA duplexes. Control siRNA were used as a negative control in the transfection experiments.
Western blot
Total proteins were extracted from cells with RIPA buffer (Invitrogen) supplemented with a 1% protease inhibitor cocktail (EMD Chemicals, Gibbstown, NJ, 100:1 v/v). The Bradford method was used to quantify proteins: 40μg total proteins from each sample were separated on a 12% SDS-polyacrylamide gel and then electro-blotted overnight onto a nitrocellulose membrane. The membranes were blocked with 5% skimmed milk in TBS-Tween 20. The membranes were hybridized to a polyclonal antibody directed against rabbit AKR1C3 or aromatase (Abcam) at dilutions of 1:1,000. The membranes were subsequently incubated with a goat anti-rabbit IgG peroxidase conjugated secondary antibody (Santa Cruz Biotechnology) at a dilution of 1:2,000. A 1:5,000 dilution of monoclonal anti-β-actin antibody produced in mouse (Sigma) was used as a loading control. Membranes were washed with TBST and proteins were visualized using Western Lighting™ Plus ECL (Perkin Elmer), followed by exposure of the membranes to X-ray films. The radiographic films were scanned and the Image program (Molecular Dynamics, Sunnyvale, CA) was used to quantify band density.
Steroid quantification in MCF-7 culture medium
Cells were seeded into 24-well plates at a density of 5×104cells/well in 500μl
hormone-free culture medium. Cells were transfected with 100nM siRNA or control siRNA as negative control after 24h. Each condition was performed in duplicate. The culture medium was replaced with hormone-free medium, hormone-free medium containing 8, 20, 100nM or 1μM DHEA five hours after transfection. The medium was collected from wells 4 days after transfection and immediately frozen at −80℃ until analysis. The levels of DHEA (Eagle Biosciences), 4-Dione (Eagle Biosciences), T (Cayman Chemical), DHT (Alpha Diagnostic), E1 (Abnova), and E2 (Cayman
Chemical) in the medium were determined by commercial ELISA kit. The levels of E2 and prostaglandin E2 (PGE2) in MCF-7 BC cell line supernatants were determined using a commercial E2 enzyme immunoassay kit and PGE2 EIA Kit (Cayman Chemical). All assays were performed according to the supplier’s protocols.
Cell cycle assay
MCF-7 cells were seeded in 6-well plates at a density of 5×104 cells per well in
hormone-free medium and were transfected with 100nMAKR1C3-specific siRNA(mixed siRNA1, siRNA2 and siRNA3) or control siRNA using lipofectamine 2000. 5h after transfection the medium was replaced with medium containing 1μM DHEA. Cell incubation was continued for 4 days after which the cells were washed and collected, fixed at −20℃ in 70% ethanol, stained with PI, and read by flow cytometry.
Quantitative real-time PCR
MCF-7 cells were seeded in 6-well plates at a density of 2.5×105 cells/well in
hormone-free medium and transfected with 100nM AKR1C3-specific siRNA or control siRNA using lipofectamine 2000. 5h after transfection the medium was replaced with medium containing 1μM DHEA. Cell incubation was continued for another 4 days, after which total RNA was extracted from cells using Trizol reagent and then sent to the Q-RTPCR Platform service (Research Center of the Laval University Hospital Center, Quebec, Canada) for quantification of ARK1C3 and CYP19A mRNA by Quantitative real-time PCR. The quantity of total RNA was measured using a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and total RNA quality was assayed on an Agilent BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA). Oligo primer pairs were designed by GeneTool 2.0 software (BiotoolsInc, Edmonton, AB, CA) and their specificity was verified by
(CYP19A1 primers 5’-CGACAGGCTGGTACCGCATGCTC/AAGAGGCAATAATAAAGGAAATCCA
GAC-3’, AKR1C3 primers
5’-CAACCAGGTAGAATGTCATCCGTAT/ACCCATCGTTTGTCTCGTTGA-3’).
Cell proliferation assay
Cell proliferation was determined by CyQuant cell proliferation kit. MCF-7 cells (3×103) were plated into 96-well plates containing 100μl hormone-free culture medium.
After 24h cells were transfected with 100nM siRNA and the culture medium was replaced with medium containing different concentrations of DHEA. Cells were cultured for 4 days, the culture medium was removed. Cells were washed with PBS, and frozen overnight in 96-well plates at −80℃. The plates were thawed at room temperature for 15 min then 200μl of CyQuant GR dye/cell-lysis buffer were added. The sample was protected from light and incubated for 2–5 min at room temperature. Sample fluorescence was measured using a fluorescence micro plate reader at 480nm excitation and 520nm emission.
Cell migration assay
Cell migration was carried out using a wound-healing assay. Cells were plated at a density of 5×105 cells in 24-well plates in E2-free medium containing 5% fetal bovine
serum (FBS). After 24h the wound was created by manually scraping the cell monolayer using a 20μl tip. The cells were washed 3 times, then the cells were treated with 6.4nM (2-fold IC50) AKR1C3 inhibitor (EM1404) [21]. Scratched cells were removed and a photograph was taken (0h). Cells were incubated in E2-free medium supplemented with 5% FBS. Photographs were taken at 24h intervals. All experiments were performed in quadruplicate. The scratch widths were measured at specific time points using Image J software.
Clinical data from the Oncomine data base
The expression of AKR1C3 in normal breast and breast carcinoma was analyzed by mRNA level using 4 different probes. We retrieved the raw data from 593 cases from the TCGA cohort from the Oncomine platform. The data had been previously processed and normalized [18]. Seventeen normal cases were filtered from 61 normal postmenopausal cases, and 160 tumor cases were filtered from 524 postmenopausal, ER+ tumor cases. We then employed differential expression analysis using the non-parametric Mann–Whitney U test as a measure of significance, and fold change as a method of differences evaluation. Since all the equal variances were assumed the t-statistic was used as the method of measurement for the differential fold change analysis of different ER status, BC type, ERBB2 status, and M stage.
Results
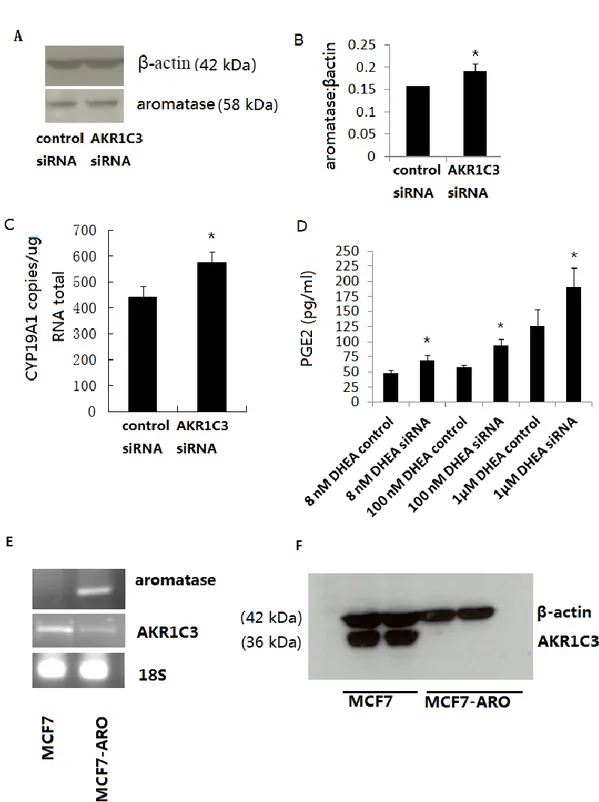
Regulation of principal androgens and estrogens with AKR1C3 knockdown
AKR1C3 mRNA levels were analyzed by quantitative real-time PCR (Q-RT-PCR) 4 days after transfection with control of AKR1C3 targeted siRNAs: this revealed 863,000 copies of mRNA/μg total RNA in control siRNA, and 207,000 copies mRNA/μg total RNA after transfection with AKR1C3 siRNA. Thus AKR1C3 is expressed in MCF-7 cells, and the siRNAs specifically silence approximately 80% of AKR1C3 gene expression (Figure 2.1A). In comparison with control siRNA, expression of the AKR1C3 protein in MCF-7 cells was decreased by 60% 48h after transfection with AKR1C3 siRNA (100nM) (Figure 2.1A). Therefore, these siRNAs were used in following experiments at a concentration of 100nM.