The function of the pseudokinase domain of BUBR1 in
mitosis.
Thèse
Luciano Gama Braga
Doctorat en biologie cellulaire et moléculaire
Philosophiæ doctor (Ph. D.)
The function of the pseudokinase domain of BUBR1 in
mitosis
Thèse
Doctorat en biologie cellulaire et moléculaire - Philosophiæ Doctor (Ph. D.)
Luciano Gama Braga
Sous la direction de :
Résumé
La mitose est un point critique de la division cellulaire, où la distribution précise du matériel génétique garantit la viabilité de la descendance. En conséquence, la ségrégation correcte des chromosomes pendant la mitose dépend de la capacité du point de contrôle d'assemblage du fuseau mitotique (SAC) à détecter l’interaction des chromosomes avec les microtubules. Ainsi, la voie de signalisation du SAC est responsable d’inhiber la séparation des chromatides-sœurs jusqu’à l’attachement correct de tous les chromosomes aux microtubules provenant des pôles opposés du fuseau mitotique. Une fois que les chromosomes sont fixés, le signal du SAC est éteint.
L'extinction du SAC dépend d'une réaction rapide aux attachements, orchestrée par deux forces qui s’opposent au niveau des centromères : les activités kinases et phosphatases. Lorsque tous les chromosomes sont correctement attachés, l'activité phosphatase augmente et éteint le signal du SAC. Notamment, la protéine pseudokinase BUBR1 est cruciale pour la génération de l’activité phosphatase au niveau d’un grand complexe protéique établi aux centromères, le kinétochore. En bref, la voie de contrôle mitotique conduit à la phosphorylation de la protéine KNL1, un des principaux centres d’échafaudage du kinétochore, provoquant une accumulation de BUBR1 et d'autres protéines impliquées dans le SAC. La phosphorylation de BUBR1 à son domaine KARD crée un motif de liaison pour la sous-unité B56 de la phosphatase PP2A. Par conséquent, le complexe BURR1-PP2AB56 est essentiel pour la dephosphorylation de plusieurs sites aux kinétochores et éteindre le SAC. Pour cette raison, comprendre comment le chemin évolutif de la pseudokinase BUBR1 l'a amenée à promouvoir la déphosphorylation est une question intrigante.
D'un point de vue évolutif, les gènes de la famille Bub, incluant le gène codant pour BUBR1, ont évolué à partir d'un seul gène ancestral appelé Madbub. Le gène
Madbub a subi des événements de duplication de gènes distincts au cours de
indispensable pour le SAC appelé KEN box, mais a conservé un autre domaine crucial, le domaine kinase. Cependant, l'autre copie MAD3 a conservé le domaine KEN box et a perdu le domaine kinase. Remarquablement, la copie de la protéine MAD3 chez un certain nombre d'insectes et de vertébrés, appelé BUBR1, a conservé le domaine kinase malgré une dégénérescence résultant un domaine kinase inactif, ou pseudokinase. Il existe, notamment, des exceptions comme la Drosophila, qui présente une kinase active. En tous cas, l'avantage évolutif conféré par le maintien de ce domaine serait la stabilité qu’il confère à la protéine entière. Les mutations dans le gène codant pour BUBR1 qui causent la déstabilisation de la protéine sont associées à l’aneuploïdie variée en mosaïque, une maladie sévère qui entraîne le développement du cancer chez l’enfant. Également, des mutations au niveau de plusieurs résidus situés au niveau du domaine pseudokinase de BUBR1 déstabilisent la protéine entière. Toutefois, étant donné que BUBR1 tronqué au niveau de son domaine kinase est en fait plus stable que le type sauvage et que l'homologue BUBR1 dépourvu de kinase, MAD3, est présent dans la majorité des organismes inférieurs, cela soulève la question de savoir si le domaine pseudokinase confère un autre attribut à la fonction ou régulation de BUBR1, en particulier chez les organismes plus complexes.
La présente étude vise à préciser si le domaine pseudokinase de BUBR1 régule la fonction du domaine KARD pendant la mitose. Nous présentons un aperçu d'un domaine de pseudokinase dans le contrôle de la liaison d'une phosphatase, car nos données confirment que le domaine pseudokinase régule l'affinité du KARD pour la phosphatase PP2AB56. Finalement, nous avons dévoilé un nouveau rôle du domaine pseudokinase de BUBR1 qui est crucial pour certaines fonctions mitotiques et qui aide à expliquer le chemin évolutif particulier subit par son gène.
Abstract
Mitosis is a critical point of cell division, where the accurate distribution of genetic material guarantees the viability of the progeny. Proper chromosome segregation during mitosis relies on the capacity of the spindle assembly checkpoint (SAC) to sense the attachment of chromosomes to microtubules. The SAC pathway is responsible for halting sister chromatid separation until all chromosomes are correctly attached to microtubules originating from opposing poles of the mitotic spindle. After the chromosomes are successfully attached, the SAC signal is extinguished.
SAC extinction depends on a swift response to microtubule attachments to sister-chromatids, which is orchestrated by the tug-of-war between two opposing sides: kinase and phosphatase activities. Each sister-chromatid possesses a kinetochore, a great protein complex that serves as interface between chromosomes and microtubules. At kinetochores unattached to microtubules, kinase activity dominates and turns the signalling on. Once all kinetochores are properly attached, phosphatase activity increases and silences the signalling. Importantly, the protein pseudokinase BUBR1 is crucial for the generation of phosphatase activity at kinetochores.
When active, the mitotic checkpoint leads to the phosphorylation of MELT motifs in the kinetochore protein KNL1, causing BUBR1 and other SAC protein accumulation at kinetochores. In turn, BUBR1 phosphorylation at its kinetochore-microtubule attachment domain (KARD) creates a binding motif for the B56 subunit of the phosphatase PP2A. Surprisingly, the pseudokinase BUBR1, in complex with PP2AB56, acts as an important phosphatase of the mitotic checkpoint by counteracting the SAC-activating kinases AURORA B and MPS1. How the evolutionary path of a protein kinase ultimately led it to promote dephosphorylation, the opposite role from its original kinase domain, is an intriguing question.
the kinase and the KEN box domain. Accordingly, Madbub undertook distinct gene duplication events throughout evolution leading to a parallel subfunctionalization of the protein yielding two different copies. One of the copies, BUB1, lost the KEN box domain but retained the kinase domain. However, the other copy, MAD3, retained the KEN box and lost the kinase domain. Barring a few exceptions, the MAD3 copy in a number of insects and vertebrates, called BUBR1, retained the kinase domain albeit severely degenerated, yielding an inactive kinase domain, or pseudokinase. Retaining this catalytically inactive domain is believed to be an evolutionary advantage since it confers stability to the whole protein. Indeed, mutations in the BUBR1 pseudokinase domain are associated with the disease Mosaic Variegated Aneuploidy, a severe condition that causes the development of cancer in children due to a reduction in BUBR1 levels. Nevertheless, since BUBR1 truncated at its kinase domain is in fact more stable than wild-type and the kinase-lacking BUBR1-homolog MAD3 is present in the majority of lower eukaryotes s raise questions concerning whether the pseudokinase domain provides another attribute to BUBR1 function or regulation, especially in more complex organisms.
The present study aims to clarify whether the pseudokinase domain of BUBR1 regulates KARD function in mitosis. We present the first insight of a pseudokinase domain controlling its binding to a phosphatase, as our data supports that the pseudokinase regulates KARD affinity to PP2AB56. Here we demonstrate that, besides its role in AURORA B centromeric recruitment, BUB1 has also a role in opposing AURORA B activity by promoting PP2AB56 tethering to BUBR1. Collectively, we unraveled a new role of the BUBR1 pseudokinase domain that is crucial for proper mitotic functions and helps explain the characteristic evolutionary path undertaken by the Bub1b gene.
Table of contents
Résumé ... ii Abstract ... iv Table of contents ... vi List of figures ... ix List of abbreviations ... x Acknowledgement ... xv Foreword ... xvi Introduction ... 1 Historical context ... 1The cell cycle ... 4
The CYCLINs and CYCLIN-dependant kinases ... 6
Cell cycle checkpoints ... 8
Mitosis ... 9
The mitotic spindle ... 10
The kinetochore ... 14
Chromosome Congression ... 23
The Spindle Assembly Checkpoint ... 28
The Anaphase-promoting complex (APC/C) ... 30
The Mitotic Checkpoint complex (MCC) ... 31
The canonical SAC pathway for the formation of C-MAD2 ... 36
The non-canonical MCC formation pathway ... 38
The SAC silencing signalling pathway ... 41
BUBR1 and SAC-related proteins in cancer ... 47
CIN and aneuploidy in tumor development ... 48
The relationship between mitosis and cancer treatment ... 49
BUBR1 and cancer ... 50
Pseudokinases... 54
Introduction on kinases ... 54
Hypothesis for the function of the pseudokinase domain of BUBR1 ... 57
BUBR1 ... 59
Historical Background ... 59
Structural diversity of MAD3/BUBR1 orthologs ... 61
Functions of the distinct domains of hBUBR1 ... 63
Post-translational modifications - PLK1 is a major regulatory kinase of BUBR1 ... 66
Functions of BUBR1 during mitosis ... 67
Spindle Assembly Checkpoint activation and MCC formation ... 68
Regulation of K-MT attachments and SAC silencing ... 69
Mitotic timer... 70
Non-mitotic functions of BUBR1 ... 71
Summary ... 71
REFERENCES ... 71
Recent developments in our understanding of BUBR1 ... 80
1. The BUBR1 pseudokinase domain promotes efficient kinetochore PP2A-B56 recruitment to regulate spindle checkpoint silencing and chromosome alignment ... 82
1.1 Résumé ... 82
1.2 Abstract ... 83
1.3 Introduction ... 83
1.4 Results ... 86
Co-evolution of the KARD and pseudokinase domains of BUBR1. ... 86
The pseudokinase domain is essential for BUBR1 stability and phosphorylation ... 88
The BUBR1 pseudokinase domain is required for PP2A-B56 kinetochore recruitment and downstream phosphatase signalling ... 92
BUBR1 pseudokinase mutants undermine PP2A-B56 functions during mitosis ... 94
The pseudokinase is required in cis for KARD phosphorylation ... 97
BUB1 kinase-BUBR1 pseudokinase heterodimerization drives KARD phosphorylation ... 100
1.5 Discussion... 103
1.6 Material and Methods ... 106
Evolutionary analysis ... 106
Cell culture and transfection... 108
Antibodies and dye ... 109
Immunofluorescence ... 110
Immunoprecipitation and Immunoblotting ... 110
Microscopy ... 110 Image analysis ... 111 Statistical Analysis ... 111 1.7 Acknowledgements: ... 112 1.8 References... 112 1.9 Supplemental Material ... 118
Perspectives and conclusion ... 127
Which sites targeted by AURORA B are dephosphorylated by PP2AB56? ... 127
Can PP1-CENP-E dephosphorylate AURORA B targets? ... 128
AURORA B and MPS1, which kinase responds first to biorientation? ... 128
Is there a correlation between BUBR1 levels and chromosome alignment? ... 129
Which proteins are responsible for CENP-E kinetochore recruitment? ... 130
What are the proteins recruited to the outer kinetochore in distinct mitotic kinetochore-microtubule attachment conditions? ... 130
Is there an organism with a functional BUB1 KARD? ... 131
Did the hypothetical protein MADBUB formed homodimers?... 132
Did the BUB1 B3BD followed KN1 evolutionary divergence in order for BUB1 to retain its ability to bind to KNL1? ... 133
Conclusion ... 134
List of figures
Figure 1 Cell cycle phases ... 5
Figure 2 The cell cycle plasticity ... 6
Figure 3 Representation of the mitotic stages ... 9
Figure 4 The mitotic spindle ... 11
Figure 5 Scheme of the kinetochore assembly factors and their interactions ... 15
Figure 6 Outer kinetochore ... 17
Figure 7 Aurora B tension mechanism ... 21
Figure 8 Congression model ... 26
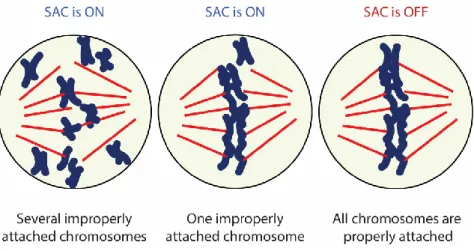
Figure 9 The SAC sensing mechanism ... 29
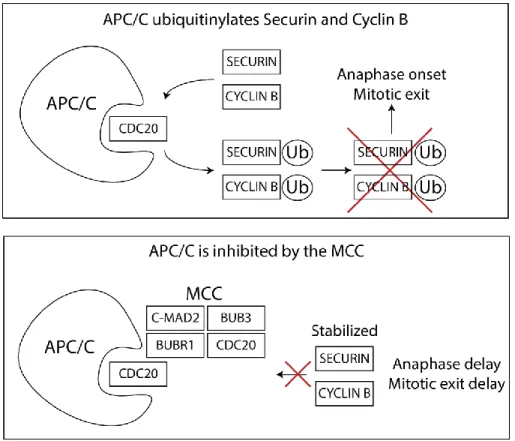
Figure 10 APC/C is inhibited by the MCC ... 31
Figure 11 Domain structure of SAC proteins ... 32
Figure 12 Template model for the formation of C-MAD2 ... 34
Figure 13 Integration of several pathways during prometaphase ... 40
Figure 14 Negative feedback at the kinetochore ensures a rapid response to attachment formation 44 Figure 15 SAC and tumor development ... 49
Figure 16 CIN levels correlate with SAC strength ... 52
Figure 17 Phosphorylation mechanism ... 55
Figure 18 Domain architecture of BUBR1 ... 62
Figure 19 The Function of BUBR1 in the SAC ... 68
Figure 20 BUBR1-mediated priming of SAC silencing and stabilization of K-MT attachments ... 70
Figure 1.1 Co-evolution of the BUBR1 pseudokinase and KARD domains... 87
Figure 1.2 BUBR1 KARD Phosphorylation is attenuated in BUBR1 pseudokinase domain mutants . 91 Figure 1.3 Signalling events downstream of KARD phosphorylation are attenuated in BUBR1 pseudokinase domain mutants ... 93
Figure 1.4 BUBR1 pseudokinase mutants undermine PP2A-B56 functions during mitosis ... 96
Figure 1.5 KARD phosphorylation requires the BUBR1 pseudokinase domain to be present in cis. . 99
Figure 1.6 BUBR1 and BUB1 C-termini allosterically regulate each other. ... 102
Figure 1.7 Suppl. Fig. 1.1: Conservation between BUB1 and BUBR1 motifs across evolution ... 119
Figure 1.8 Suppl. Fig. 1.2: BUBR1 is a pseudokinase domain that lacks phosphotransfer activity. . 120
Figure 1.9 Suppl. Fig. 1.3: BUBR1 and B56 expression in the Hela T-Rex stable cell lines. ... 121
Figure 1.10 Suppl. Fig. 1.4: Mitotic phenotypes associated with BUBR1 pseudokinase domain mutations. ... 122
Figure 1.11 Suppl. Fig 1.5: Effect of CENP-E depletion on KARD phosphorylation. ... 123
List of abbreviations
AAA+
ATPases Associated with diverse cellular Activities ... 47 APC/C Anaphase-promoting Complex... 30 ATP Adenosine triphosphate ... 54 B3BD BUB3-binding domain ... 36 BBC BUBR1-BUB3-CDC20 ... 35 BUB
Budding Uninhibited by Benzimidazole ... 3 BUB1
Budding Uninhibited by Benzimidazoles 1 ... 19 BUB3
Budding Uninhibited by Benzimidazoles 3 ... 19 BUBR1
BUB1-related kinase 1 ... 19 CCAN
Constitutive Centromere-Associated Network ... 14 CD1
Conserved domain 1 ... 39 CDC
Cell Division Cycle ... 3 CDH1 CDC20 homolog 1 ... 30 CDK CYCLIN-dependent kinase ... 3 CDK1 CYCLIN-dependent kinase 1 ... 3 CENP Centromere protein ... 15 CENP-A
Histone H3-like centromeric protein A ... 2 CENP-F Centromere protein F ... 2 CH Calponin Homology ... 18 CIN Chromosomal Instability ... 48 CPC
D box Destruction box ... 30 DNA Deoxyribonucleic acid ... 3 DSN1 Dosage Suppressor of NNF1 1 ... 16 EB End-binding protein ... 12 EG5 Kinesin-5 ... 13 GEF
Guanine nucleotide exchange factors ... 13 GLEBS
Gle2-binding-sequence ... 61 GTP
Guanosine-5'-triphosphate ... 12 INCENP
Inner Centromere Protein ... 16 KARD
Kinetochore Attachment Regulatory Domain ... 42 KEN
Lysine (K) – Aspartic Acid (E) – Asparagine (N) ... 30 KI
Lysine (K) – Isoleucine (I) ... 20 KID
Kinesin-like DNA-binding protein ... 25 KMN KNL1C-MIS12C-NDC80C ... 16 KNL1 Kinetochore Scaffold 1 ... 16 KNL1C KNL1 Complex ... 20 MAD
Mitotic Arrest Deficient ... 3 MAD1 Mitotic-arrest deficient 1 ... 33 MAD2 Mitotic-arrest deficient 2 ... 31 MAP Microtubule-associated Proteins ... 12 MCAK
Mitotic Centromere-associated Kinesin ... 21 MCC
Mitotic Checkpoint Complex... 30 MELT
Methionine (M) – Glutamate (E) – Leucine (L) – Threonine (T) ... 19 MIS12
Minichromosome Instability 12 ... 16 MIS12C MIS12 Complex ... 16 MPF Maturation-promoting factor ... 3 MPS1 Monopolar Spindle 1 ... 3 MVA
Mosaic Variegated Aneuploidy... 50 NDC80
Nuclear Division Cycle 80 ... 16 NDC80C
NDC80 Complex ... 17 NNF1
Necessary for Nuclear Function 1 ... 16 NSL1
NNF1 Synthetic Lethal 1 ... 16 NUF2
NUclear Filament-containing protein 2 ... 17 PCS
Premature Chromatid Separation ... 50 PMF1
Polyamine Modulated Factor 1 ... 16 PP1
Protein Phosphatase 1 ... 19 PP2A
Protein Phosphatase 2A ... 42 RAN
RAS-related Nuclear Protein ... 13 RANGAP1
RAN GTPase-activating protein 1 ... 13 RCC1
Regulator of chromosome condensation 1... 13 ROD1
Regulator of differentiation 1 ... 38 ROR
Receptor tyrosine kinase-like Orphan Receptor ... 55 RVSF
Arginine (R) – Valine (V) – Serine (S) – Phenylalanine (F) ... 19 RWD
RING finger - WD repeat containing proteins - DEAD-like helicases ... 18 RYK
Receptor-like Tyrosine Kinase ... 55 RZZ
ROD-ZW10-ZWILCH ... 38 SAC
SGO2
Shugoshin 2 ... 42 SILK
Serine (S) – Isoleucine (I) – Leucine (L) – Lysine (K) ... 19 SKA
Spindle and Kinetochore-associated protein ... 22 SPC24
Spindle Pole Component 24 ... 17 SPC25
Spindle Pole Component 25 ... 17 TIP
Microtubule plus-end tracking proteins ... 12 TPR
TetratricoPeptide Repeat ... 20 TPX2
Targeting Protein for Xklp2 ... 12 TRIP13
Thyroid Hormone Receptor Interactor 13 ... 47 γTuRCs
« These facts show that mitosis is due to the co-ordinate play of an extremely complex system of forces which are as yet scarcely comprehended. Its purpose is, however, as obvious as its physiological explanation is difficult. It is the end of mitosis to divide every part of the chromatin of the mother-cell equally between the daughter-nuclei. All the other operations are tributary to this. We may therefore regard the mitotic figure as essentially an apparatus for the distribution of the hereditary substance, and in this sense as the especial instrument of inheritance. »
— Edmund Beecher Wilson, The Cell in Development and Inheritance (1896)
Acknowledgement
I would like to express my deepest appreciation to Sabine Elowe, who never wavered in her support for my growth as a scientist.
I am also grateful to my colleagues Michelle Mathieu, Philippe Thebault, Guillaume Combes, Adeel Asghar, Chantal Garand, Diogjena Prifti, Katheryn Ouellet-Boutin, Alexsandro Dos Santos, Romain Devillers, Pawan Kumar Saini, Angel F. Cisneros, Christian R. Landry, Maxime Clerc, Pauline Garcia, Baptiste Lottin, and Joanie Patenaude.
Finally, I gratefully acknowledge all Canadian taxpayers for supporting this endeavour.
Foreword
The research presented in this thesis comprises the experimental investigative work performed in Dr. Elowe laboratory on the subject of the pseudokinase domain of BUBR1. In a nutshell, our goal was to understand the role of this domain during mitosis. Dr. Elowe’s previous research led to several advances in the field, such as the discovery of key phosphorylation sites in this protein, notably at serine 676. Subsequent work by other groups demonstrated the importance and consequences of this phosphorylation site in mitosis. The present study aims to understand the regulation of this phosphoresidue by the BUBR1 pseudokinase domain. Since there was no consensus in the field about the function of this pseudokinase domain, the present study offers valuable information for the advance of our current molecular model of mitosis. The result of our work was synthesized in an article submitted to Nature Cell Biology on the 13th of August 2019 (Paper #:
NCB-E40671), which can be found at
“https://www.biorxiv.org/content/10.1101/733378v1”.
The introduction of this thesis provides a brief historical perspective of mitosis, followed by an introduction on the cell cycle. Mitosis is subsequently described in more depth, where key mitotic structures pertinent to the understanding of this work, the mitotic spindle and the kinetochore, are highlighted. The following chapter describes the SAC (Spindle Assembly Checkpoint) signalling and extinction. The first part of the introduction closes on a section on the implication of SAC disfunction on diseases, notably cancer.
The second part of the introduction describes pseudokinases and our hypothesis concerning the pseudokinase domain of BUBR1. Finally, the section is closed with a chapter published in the second edition of the “Encyclopedia of Signaling Molecules”, Springer 2016 (DOI 10.1007/978-1-4614-6438-9_101975-1, ISBN: 978-1-4614-6438-9,). I am the first author of this chapter and Dr. Sabine Elowe is the last author. The chapter outlines the gene Bub1b and its encoding protein, BUBR1. Since the chapter was published in 2016, there were a few
after the encyclopedia entry. In this manner, the introduction gradually focuses the spotlight on the molecular mechanism of study, from generalities of the cell cycle to BUBR1-dependant microtubule attachment regulation.
The work performed by Dr. Elowe laboratory on BUBR1 is found at the center of this thesis. I am the first author of the article, which concentrated virtually all my efforts during my Ph.D. studies. All the figures and experiments were performed by me with the exception of: Fig. 2A-F (Michelle Mathieu), Suppl. Fig. 1D (Sabine Elowe), Suppl. Fig. 2A (Sabine Elowe), Suppl. Fig. 11C (Philippe Thebault), and the co-evolution analysis. The project was initiated and partially developed by Michelle Mathieu during her M.Sc. studies. Her work is seen in Figure 2A-F. Angel F. Cisneros and Christian R. Landry were responsible for the evolutionary analysis present in the article. Philippe Thebault was responsible for creating a few cell lines used throughout the project. Moreover, Chantal Garand oversaw the development of several plasmids, while the interns Maxime Clerc, Pauline Garcia, and Baptiste Lottin helped me with experiments throughout the 5 years I worked on this project.
Finally, the thesis closes with a section of prospective research discussion and conclusion. In this section I enumerated several questions and possible experiments that may further elucidate our understanding of the subjects discussed along the thesis.
Introduction
Historical context
Mitosis, from the Greek word “μίτος” for “thread”, is the process by which one cell becomes two cells. The term was coined by the German histologist Walther Flemming in 1878, who also described the stages comprising the mitotic phenomenon (Flemming, 1880). Flemming observed a structure he called chromosome (Greek for colored bodies) migrate to the middle zone inside the cytoplasm, and then move to opposing sides in the epidermal cells from a salamander larva. The chromosomes from these cells are relatively larger in size, which allowed Flemming to see them more clearly than in other cells.
In 1825, it was first noted that all cells originate from previous cells (“Omnis
cellula e cellula” or “every cell from a cell”) by the French scientist Raspail (Bechtel,
2006; Harris, 2000; Wright and Poulsom, 2012). However, the link between mitosis and inheritance was made later, by Weismann in 1885, who proposed that chromosomes are responsible for storing the genetic material (McIntosh and Hays, 2016; Weismann, 1885). Arguably, the founder of our modern concept of mitosis came from the German biologist Theodor Boveri, who was essential in establishing the connection between mitosis and genetics. After studying cells from sea urchins, a model used due their characteristic fast division, Boveri remarkably reached to three fundamental conclusions for the study of cell division (Boveri, 1904; Yanagida, 2014):
1. Chromosomes are individual bodies in the cell nucleus 2. Different chromosomes carry different genetic material
3. Abnormal mitosis generates abnormal gene content, which might be the cause of carcinogenesis.
occur biochemically, ensuing the discovery of several mitotic-related structures and proteins. The mitotic spindle, for instance, was proposed by Flemming (Flemming, 1878, 1880; Müller-Reichert et al., 2018), but its existence was subject of intense debate, since it was thought that the spindle fibers were artifacts caused by the fixation and staining of the cells. Eventually progress in microscopy and other technologies in the twentieth century permitted further development in our understanding of mitosis.
In the 1950s, Inoue used polarization microscopy to confirm the existence of the mitotic spindle (Inoué, 1951, 1953; Müller-Reichert et al., 2018). Later on, Edwin Taylor used radioactive colchicine, a spindle poison, to isolate the main component of the spindle; the protein TUBULIN (Weisenberg et al., 1968). Electron microscopy was then used by Brinkley and Stubblefield in 1966 to see the polymerized fibrils of TUBULIN, called microtubules, attach at special paired places at the chromosome middle-region (Brinkley and Stubblefield, 1966). These loci received the name “kinetochores”, as they were believed to be responsible for chromosomal movement (“kineto” means “of movement” and “khoros”, place).
After the discovery of the kinetochore, substantial work was done to identify its core components. To accomplish this, researchers exploited the fact that a few human auto-immune syndromes are caused by the production of antibodies against kinetochore targets. Notably, in 1984 Bill Earnshaw and Don Cleveland used these antibodies to identify several centromeric associated proteins, called CENP-A, to CENP-F (CENP for centromeric protein) (Earnshaw et al., 1984; McIntosh and Hays, 2016). The discovery of these proteins had a major impact on our understanding of kinetochores. For instance, CENP-A is the histone responsible for marking the centromere region where kinetochores will assemble during mitosis, while other CENP proteins have different functions, such as microtubule binding. While the components at the interface between the chromosome and microtubules were being identified, other groups focused on the cytoplasmic environment during mitosis.
cytoplasmic profile (Johnson and Rao, 1970; Rao and Johnson, 1970). They discovered that there were factors in the mitotic cytoplasm capable of turning interphasic cells (non-mitotic cells) into mitotic cells. Later, these factors were identified as Maturation-promoting factor (MPF) and Cyclin-dependent kinases (CDKs) (Yanagida, 2014). Furthermore, Leland Hartwell continued the quest for mitotic genes with an elegant experiment where he isolated S. cerevisiae strains unable to form colonies at 36°C (Hartwell et al., 1970; McIntosh and Hays, 2016). After investigating the specific effect of each mutation, as DNA synthesis or cell division, his group identified a series of genes related to specific stages of the cell-cycle. The yeast strains were named cdc for cell-division cycle, later identified as the gene encoding for CDC28, a subunit of CDK1, a major mitotic kinase. Similarly, groups led by Hoyt and Murray devised an experiment where mutated yeast cells are challenged with a microtubule poison that halts cell cycle during mitosis. In this manner, by isolating the mutated genes in colonies unable to arrest the division, these groups identified an array of essential mitotic genes, such as the bubs (Budding Uninhibited by Benzimidazole) (Hoyt et al., 1991), mads (Mitotic Arrest Deficient) (Li and Murray, 1991), and mps1 (Monopolar Spindle 1) (Winey et al., 1991). By the 1990s, Schimke and Sherwood proposed the ultimate function of these proteins in defining the mammalian spindle assembly checkpoint (SAC) as a mechanism to “couple cell cycle progression to the completion of certain karyokinetic events” (Kung et al., 1990). Thus, the identification of the key mitotic genes that started in the 60s reached its conclusion in the 90s with the start of the description of the SAC signalling pathway.
Still in the 1990s, Paul Nurse demonstrated that the aforementioned MPF and CDK are conserved throughout evolution, from yeast to mammals (Nurse, 1990). Likewise, in 1996 Lane and Nigg showed the conservation of Plk (Polo-like kinase) and Aurora from fungi to higher eukaryotes (Lane and Nigg, 1996; Yanagida, 2014). Importantly, these genes encode for kinase enzymes that regulate a plethora of mitotic functions, including spindle dynamics and SAC signalling. Similarly, prokaryotic counterparts from many mitotic key players have been identified in
studies combined the knowledge from different mitotic models into a conserved biochemical background for all organisms. Despite the divergence in certain structures, such as the kinetochore (Vleugel et al., 2012), the evidence gathered demonstrates similarities concerning molecular components, the regulatory principles, and the overall organization of the cell cycle (Margolin and Bernander, 2004). The divergence observed in the case of kinetochores and other structures likely reflects its specialization in different cells and tissues. For these reasons, the universality of mitosis places this mechanism at the base of the tree of life.
In the last 20 years, research in mitosis has focused on understanding the progressively complex interplay between key mitotic proteins relative to their networks (Saurin, 2018), as discussed in the next chapters. Indeed, it will be a formidable task for future generations to translate the complexity of the network into comprehensible concepts.
As every cell originates from previous cells, our cells are descendants from the first primordial cell, which arose 750 million years after the Earth was formed (Cooper, 2000). It is indeed remarkable that mitosis is an ongoing process lasting 3.8 billion years, lying as the core mechanism of life inheritance.
The cell cycle
The cell cycle is defined as “the series of events that leads to the reproduction of the cell” (Morgan, 2006). For unicellular organisms, the reproduction of the cell guarantees the continuation of the species. In contrast, multicellular organisms use cell division not only for the creation of new organisms, but also to replenish damaged or lost cells.
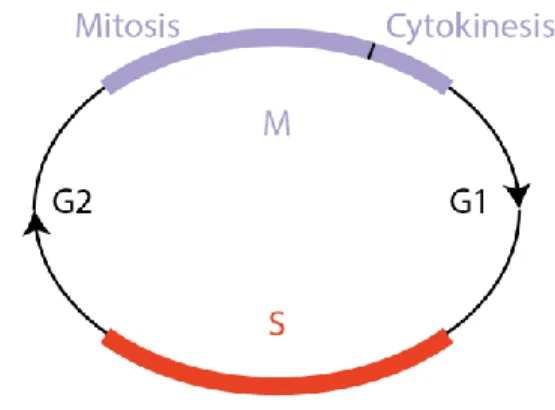
The complex architecture of cell division is organized in distinct steps, which are repeated in each generation, thus the use of the word cycle. Broadly speaking, the cell cycle is divided in two distinct phases: Interphase (composed of G1, S phase, and G2) and M phase (composed of mitosis and cytokinesis). In a human somatic cell, the order of phases is illustrated chronologically in Figure 1. After the gap phase
G1, the cell enters the S phase, when the chromosomes are duplicated, and sister-chromatids are formed (S for DNA synthesis). Subsequently, the cell enters a gap period (G2), which is followed by mitosis, when the DNA content is equally separated into each new cell. When mitosis is concluded, the cytoplasm is divided by the cell membrane, physically separating the two cells. This event is called cytokinesis (Murray and Kirschner, 1989).
Figure 1 Cell cycle phases
The order of each phase is outlined in the scheme. Interphase is composed of G1, S, and G2, whereas the M phase is subdivided in mitosis and cytokinesis.
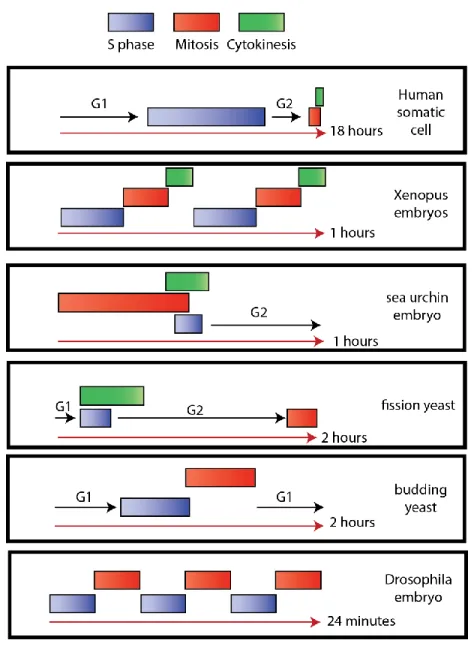
Interestingly, the timing of the cell cycle, as well as the allocated time for each phase, varies vastly (Figure 2). In a human somatic cell, the duration of G1, S, G2, and M phases is about 18 hours. S phase is the longest, lasting about 8 hours, while the M phase is the shortest, lasting only 1 hour. Strikingly, the Xenopus laevis embryo does not enter gap phases and divides every 30 minutes, making it an excellent model for mitotic studies. The sea urchins observed by Boveri present also a short cycle of only one hour, where cytokinesis, mitosis and S phase overlap. As part of a syncytium, nuclei of Drosophila embryoscan divide every 8 minutes without cytokinesis nor gap phases (Morgan, 2006). The plasticity of the cell cycle also characterizes different cells from the same organism. Normal human epidermis displays a cell cycle duration of 311 hours (Weinstein et al., 1984), while precursor B cells divide every 90.5 hours (Cooperman et al., 2004). Thus, the cell adapts its
This cyclic nature of the cell is, in turn, dictated by a specific set of proteins called CYCLINs.
Figure 2 The cell cycle plasticity
Scheme depicting the varying order and duration of the cell cycle in different species. Based on (Morgan, 2006).
The CYCLINs and CYCLIN-dependant kinases
In order to achieve this clockwork-like control of the cell cycle, the cell requires a timing mechanism able to alert and push forward the new cell cycle phase. The
cell thus takes advantage of an on/off biochemical switch composed by CYCLIN-dependent kinases (CDKs) and a stimulus ligand necessary for their activation, which in this case comprises the CYCLINs (Morgan, 1997). The CDK-CYCLIN complexes phosphorylate a myriad of target proteins, leading to transcriptomic changes, as well as alterations at the level of organelle structure and cell morphology in each cell cycle phase. The irreversible transition of cell phases is linked to each CDK’s rapidly rising activity.
As the ligand binds to the enzyme and activates it, the process creates a hyperbolic response curve (also known as Michaelis-Menten curve), in which activity increases with ligand binding until arriving to a plateau. On the other hand, CDKs-CYCLIN activity curves follow a more complex case of the hyperbolic curve, the sigmoidal response curve, in which small amounts of the ligand do not interfere greatly with the enzyme activity until a threshold is breached. Once the ligand reaches this limit, the enzyme activity increases dramatically. Subsequent increases in ligand concentration does not enhance the enzyme activity significantly. In this manner, the concentration of each CYCLIN during the cell peaks according to the cell cycle stage, which in turn activates its corresponding CDK (Lim and Kaldis, 2013).
In essence, during the start of the cell cycle (end G1, before the start of S phase), G1/S CYCLINs (CYCLIN D and E) concentration increases and activates G1/S CDKs (CDK4/6) (Lim and Kaldis, 2013). Their activity is reduced during S phase, when S CYCLINs (CYCLIN A) concentration rises, causing the activation of its corresponding CDK (CDK2). At the G2/M transition, M CYCLINS (CYCLIN B) peak and activate the M phase CDK (CDK1). Towards the anaphase transition and late mitosis, M CYCLINS concentration declines, which resets the system at its initial state. For these reasons, CDK-CYCLINs are the master switches of the cell cycle. However, the existence of different CYCLINS does not explain how their activities are timely regulated. To achieve this control, the cell takes advantage of the cell cycle checkpoints (Barnum and O’Connell, 2014; Hartwell and Weinert, 1989).
Cell cycle checkpoints
The goal of the cell cycle is to allow cell division to occur in an orderly fashion, lest the cell divides improperly. While the CYCLINS dictate the phase of the cell cycle, the orderly regulation of phase duration is controlled by the checkpoints. Hence, the cell cycle checkpoints are a series of dependent signalling pathways where the onset of subsequent events relies on the fulfilment of early events. To accomplish this goal, the checkpoints have three main components: a sensor , a transducer, and an effector mechanism (Bradshaw and Dennis, 2009). The sensor mechanism is used by the cell to evaluate if it should advance through the cell cycle or not. Whenever the cell senses that the cell cycle should not advance, the pathway is activated, which will relay the signal to a transducer. In turn, the role of the transducer is to activate the effector mechanism, which will enforce the cell cycle arrest. In this manner, the cell progresses adequately through several phases of the cell cycle in an orderly fashion, where the whole cell works as a unit (Hartwell and Weinert, 1989).
The governing points of the cell cycle are mainly three checkpoints: Start checkpoint, G2/M checkpoint, and the metaphase-anaphase or mitotic checkpoint. The Start checkpoint occurs between G1 and S phases. Briefly, it is a checkpoint that, when satisfied, imposes a commitment to enter a new cell cycle (Pardee, 1989). In this fashion, the cell only commits to cell division if specific conditions are met to avoid unproductive divisions. This checkpoint is governed by cell growth factors or mitogens that guarantee the proper rate of cell proliferation, thus ensuring the proper size of the cell or organ. The second checkpoint, the G2/M checkpoint, is responsible for ensuring that the DNA content in a cell was properly copied during the previous S phase, thus guaranteeing a faithful copy of DNA in the daughter cells (Melo and Toczyski, 2002). Whenever DNA damage is sensed by this mechanism in S phase or G2, mitotic entry is prevented. Once the G2/M checkpoint is silenced, the cell enters mitosis.
Mitosis
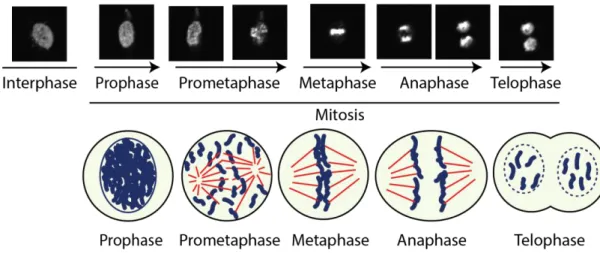
To better understand the mitotic phenomenon in humans, researchers have subdivided mitosis in 5 stages: prophase, prometaphase, metaphase, anaphase, and telophase (Figure 3). Prophase starts with chromosome condensation, followed by migration of centrosomes to opposing poles of the cell. At this stage, the chromatin is compacted inside the nucleus in the form of sister-chromatids, each possessing a copy of each chromosome. As soon as the nuclear envelope breakdown occurs, the mitotic spindle is formed, and prometaphase begins (Clift and Schuh, 2015; Güttinger et al., 2009). During prometaphase the chromosomes migrate to the equatorial plane of the cell. Chromosomal positioning and alignment in a bioriented manner at the middle of the cell is called metaphase. Next, each sister-chromatid is pulled apart in a drastic movement. The chromosomes thus migrate to opposing poles of the mitotic spindle in anaphase. Finally, during telophase the spindle is disassembled, and the chromosomes are engulfed in a new nucleus, while a contractile ring pinches the axis of the cell in two. After mitosis, cytokinesis ensues with the division of the cell in two separated bodies (Morgan, 2006).
Figure 3 Representation of the mitotic stages
Scheme and live-cell imaging experiment performed by the author with human cells (HeLa)
expressing GFP (green fluorescent protein)-tagged Histone H2B on an Olympus IX80 spinning disk confocal microscope system (20X objective). Note the chromatin configurational and positional
changes at each mitotic stage. In the representation below, each mitotic stage is depicted with chromatin in blue and microtubules in red.
Mitosis is a critical point of cell division, where its goal is to evenly distribute the hereditary components of the cell to its progeny. The focus of this work is on prometaphase and the metaphase-anaphase transition events. Specifically, we will address how the interaction between chromosomes and microtubules from the mitotic spindle is regulated, how chromosomes migrate to the metaphase plate, and how the SAC ensures that anaphase onset takes place only when all chromosomes are properly attached to the spindle.
The mitotic phase is characterized by several distinctive features, but two are crucial for the understanding of subsequent concepts in this work: the mitotic spindle and the kinetochore.
The mitotic spindle
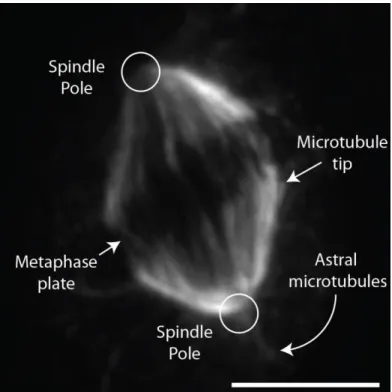
The mitotic spindle distinctive fusiform shape possesses a mirror symmetry along the equator of the cell (Figure 4), which ensures that each sister chromatid faces each pole of the spindle. It displays a large architecture, measuring up to 60 μm in length in X. laevis (Wühr et al., 2008), and is composed of hundreds of thousands of microtubules and around a thousand other proteins (Brugués et al., 2012; Sauer et al., 2005). Importantly, the mitotic spindle is the macromolecular machine responsible for separating sister-chromatids to two daughter cells (Walczak and Heald, 2008).
Figure 4 The mitotic spindle
Immunofluorescence of HeLaS3 cells performed by the author. The cell was fixed and stained with an anti-α-TUBULIN antibody. Each structure is defined by arrows. The poles are localized at the extremities of the spindle, while the metaphase plate lies at the center. The end of microtubules can be seen close to the metaphase plate. Astral microtubules are microtubules polymerized outside the spindle. As observed in this figure, their concentration is less important than spindle microtubules. The white scale represents 5 µm. Acquisition performed with an Olympus IX80 microscope system equipped with a Yokogawa spinning disk (100X objective).
Microtubule nucleation and flux
The dynamic nature of the spindle structure arises from the nucleation of microtubules, which have a half-life of 60 to 90 seconds (Inoué and Salmon, 1995; Sawin and Mitchison, 1991). Microtubules are composed of a 6S GTP-binding protein termed TUBULIN, more precisely by dimers of α and β-TUBULIN (Brinkley, 1997). The nucleation process begins at γ-TUBULIN ring complexes (γTuRCs) (Kollman et al., 2011), which serve as a templating scaffold. The γTuRCs present a 13-fold symmetry, where 13 protofilaments of TUBULIN are assembled in an
end-the microtubule directly interacts with end-the γTuRC (Zheng et al., 1995) and defines the minus end, while the β- TUBULIN side of the microtubule corresponds to its plus end. Concentrations of TUBULIN above a certain threshold in the presence of GTP (Guanosine-5'-triphosphate) lead to the nucleation event and microtubule formation. However, the reaction is reversible, causing a dynamic instability in the process, and is characterized by random growing and shrinking phases. Interestingly, microtubules can nucleate, grow and shrink in solutions containing solely TUBULIN and GTP (Mitchison and Kirschner, 1984).
The control of microtubule nucleation is accomplished by microtubule-associated proteins (MAPs). There are roughly one thousand MAPs (Goshima et al., 2007; Petry, 2016), which are divided in different classes depending on their activity, such as in microtubule nucleation, dynamics, transport, and cross-linking. Importantly, the dynamic nature of the spindle is regulated by microtubule dynamic MAPs responsible for stabilizing the straight conformation of TUBULIN dimers or causing microtubule catastrophe by supporting curvatures of the dimers. The regulation of MAP at microtubule ends is performed by microtubule plus-end tracking proteins (+TIPs), as end-binding proteins (EB). For instance, EB1 is a microtubule maturation factor that catalyses GTP-TUBULIN conversion to GDP-TUBULIN, which enhances microtubule growth velocity (Maurer et al., 2014). Likewise, TPX2 (Targeting Protein for XKLP2) nucleates microtubules and generates branching of microtubules (Alfaro-Aco et al., 2017; Petry, 2016). Furthermore, another interesting property of microtubules is flux. Microtubules continuously slide toward the pole in a minus-end movement characterized by the disassembly of microtubules at the minus end (at the poles) and assembly towards the plus ends (at the spindle equator) (Buster et al., 2007). However, microtubule nucleation and dynamics do not dictate the full structure of the mitotic spindle.
Spindle architecture
Spindle architecture is promoted by molecular motors (kinesins) and microtubule crosslinking proteins. Kinesins are proteins comprised of a motor
domain, which binds a specific molecule (Welburn, 2013). Thus, microtubules can be transported as cargo along microtubules. The kinesins DYNEIN (Tanenbaum et al., 2013) and EG5 (kinesin-5) (Kapitein et al., 2005) have two motor domains, enabling these kinesins to slide microtubules along the spindle axis in an anti-parallel manner. The cross-linking of microtubules is achieved by the tethering of kinesins to parallel microtubules, which dictates the spindle architecture. DYNEIN is thought to focus the pole flux of microtubule by cross-linking the different bundles of microtubule at the pole (Ito and Goshima, 2015; Petry et al., 2013).
Mitotic spindle formation
In human cells, nucleation of microtubules, which leads to the spindle formation, originates from centrosomes or alternatively from chromosome-mediated pathways (Prosser and Pelletier, 2017). In the centrosome mediated pathway, upon mitotic entry, the centrosomes migrate to opposing sides of the cell. Subsequently, they activate microtubule nucleation after nuclear envelope breakdown occurs. Several centrosome-localized proteins recruit γ-TuRC, which nucleates the microtubules (Choi et al., 2010; Haren et al., 2006; Liu and Wiese, 2008; Lüders et al., 2006; Prosser and Pelletier, 2017). Interestingly, despite experiments demonstrating the capacity of spindle formation in the absence of chromosomes (Gruss and Vernos, 2004), the chromosomes are also key players in the formation of microtubules.
In the chromosome-mediated microtubule nucleation pathway, which has been mostly studied in Xenopus egg extracts, chromatin produces a localized biochemical environment that promotes nucleation and microtubule growth. The RAS-related nuclear protein (RAN) in its GTP-bound state notably concentrates around the chromatin. Its GEF (Guanine nucleotide exchange factor) , Regulator of chromosome condensation 1 (RCC1), is associated with chromatin, while the RAN GTPase-activating protein 1 (RANGAP1) is present at the cytosol (Kaláb et al., 2006). Consequentially, close to the chromatin RAN is present at its GTP-bound state, which binds to IMPORTIN-β (Gruss et al., 2001; Prosser and Pelletier, 2017;
negative regulator of spindle formation factors, as it sequesters nucleation factors, such as TPX2 (Askjaer et al., 2002; Gruss et al., 2001; Nachury et al., 2001). In this manner, chromatin and centrosomes are able to support mitotic spindle formation. Nevertheless, to better comprehend the role of chromatin in spindle formation we need to understand the focal point of spindle regulation at the chromosome, the kinetochore.
The kinetochore
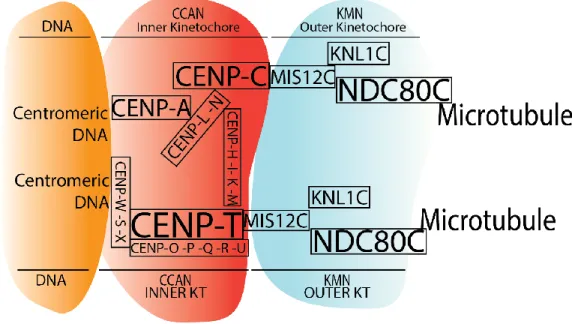
The kinetochore is a large protein complex assembled at the centromeres of chromosomes that serves as the anchoring point at which the mitotic spindle tethers to each sister chromatid during mitotic division. The vertebrate kinetochore comprises over 100 proteins divided in sub-complexes (Pesenti et al., 2016), and it also serves as signalling hub for a myriad of proteins. Importantly, the kinetochore is composed of a trilaminar structure: the inner kinetochore, the outer kinetochore and the fibrous corona. The inner kinetochore is the region closest to the centromere of chromosomes, while the outer kinetochore is formed closer to the mitotic spindle. The fibrous corona, on the other hand, is formed externally at the outer plate in the absence of microtubule attachment (Musacchio and Desai, 2017).
Inner kinetochore
The inner kinetochore presents two structures pertinent to this study: the CCAN (Constitutive Centromere-Associated Network) and the CPC (Chromosome Passenger Complex).
The CCAN is constitutively recruited to the centromeres of chromosomes (Hara and Fukagawa, 2017), where a characteristic set of epigenetic marks targets its formation. An exception to this rule is shown in S. cerevisiae, in which kinetochore tethering is based on DNA sequence rather than epigenetics (Westhorpe and Straight, 2015). The histone 3 variant CENP-A is responsible for CCAN tethering in higher eukaryotes, as it promotes the formation of a centromere-specific nucleosome complemented by histone H4 and H2A/B (Fukagawa and Earnshaw, 2014; McKinley and Cheeseman, 2016; Westhorpe and Straight, 2015) (Figure 5). Furthermore,
acetylation of histone H4 K5 and K12 in the CENP-A pre-nucleosome and monomethylation of histone H4 K20 in the CENP-A nucleosome induce CCAN assembly (Hori et al., 2014; Shang et al., 2016).
Figure 5 Scheme of the kinetochore assembly factors and their interactions
Centromeric DNA is shown in orange. The inner kinetochore is represented in red, whereas the outer kinetochore is shown in blue. The intricate array of proteins at the inner kinetochore serve to anchor the base of the kinetochore firmly to the DNA. The outer kinetochore is fixed to the inner kinetochore and its goal is to capture microtubules. Scheme loosely based on (Hara and Fukagawa, 2018).
Importantly, the CCAN complex is also sub-divided in 4 complexes (Musacchio and Desai, 2017): the CENP-OPQRU complex (De Wulf et al., 2003; Hori et al., 2008; Schmitzberger and Harrison, 2012), the CENP-LN complex (Hinshaw and Harrison, 2013; McKinley et al., 2015; Pot et al., 2003; Weir et al., 2016), the CENP-HIKM complex (Basilico et al., 2014; De Wulf et al., 2003, 2003; Klare et al., 2015; Measday et al., 2002; Okada et al., 2009; Pekgöz Altunkaya et al., 2016; Weir et al., 2016), and the CENP-TWSX complex (Amano et al., 2009; Hori et al., 2008). As CENP-A docks to the centromere, CENP-C and CENP-N binds to CENP-A directly (Carroll et al., 2010; Guo et al., 2017; Kato et al., 2013; Weir et al., 2016), while other CCAN proteins are recruited in series. CENP-T is able to bind
chromatin directly (Hori et al., 2008; Nishino et al., 2012; Takeuchi et al., 2014) and recruits CENP-OPQRU and bridge CENP-A through CENP-HIKM (Fukagawa and Earnshaw, 2014; Hara and Fukagawa, 2017; McKinley and Cheeseman, 2016; Pesenti et al., 2016; Westhorpe and Straight, 2015). Interestingly, despite the existence of organisms lacking some of its constituents, the CCAN proteins are fairly conserved throughout evolution (Drinnenberg et al., 2016; Hara and Fukagawa, 2018; Hooff et al., 2017).
Unlike the CCAN, the CPC is composed of only 4 proteins: the kinase AURORA B, BOREALIN, SURVIVIN, and INCENP (Inner Centromere Protein). At mitotic entry, this complex is found along chromosome arms, but as mitosis progresses, it focuses at the inner centromere, in the space in-between sister-chromatids and kinetochores (Hindriksen et al., 2017). The CPC is further divided in two functional modules connected by INCENP. The first module is composed of BOREALIN and SURVIVIN, which bind to the N-terminus of INCENP and have a role in the localization of CPC. Indeed, phosphorylation of histone H3 at T3 (Wang et al., 2010) provides the reader epitope for SURVIVIN, thus leading CPC recruitment to the centromeres. The second module is composed of AURORA B and the C-terminal region of INCENP, which recruits AURORA B. Notably, CPC has two important functions: the regulation of kinetochore-microtubule attachments (discussed in a subsequent chapter) and promoting the localization of other complexes to the outer kinetochore region (Hindriksen et al., 2017).
Outer kinetochore
In the outer kinetochore region lies the KMN, which is an aggregate of 3 complexes: KNL1 (Kinetochore Scaffold 1), MIS12 (Minichromosome Instability 12), and NDC80 (Nuclear Division Cycle 80) complexes (Hara and Fukagawa, 2018) (Figure 6). Its localization depends on a series of steps. First, AURORA B is responsible for phosphorylating the protein DSN1 (Dosage Suppressor of NNF1 1) at S100 and S109 in humans (Petrovic et al., 2016). The phosphorylation of these sites leads to allosteric changes that allow its interaction with CENP-C. Notably,
recruited at interphase (Obuse et al., 2004). Thus, the MIS12C is formed at the outer kinetochore region. Secondly, NDC80C (NDC80, NUF2, SPC24, and SPC25) localizes to the centromere through MIS12C interaction during nuclear envelope breakdown, as it is prevented from entering the nucleus. Interestingly, NDC80C binds to CENP-T via CDK1-CYCLIN B-dependant phosphorylation of CENP-T in human cells, which plays a role also in CENP-T-MIS12C interaction. Despite the importance of CENP-T in NDC80C kinetochore localization, NDC80C recruitment to CENP-C requires MIS12C-CENP-C interaction (Rago et al., 2015). Based on the current model, MIS12C acts as an interface between the inner and the outer kinetochore, while NDC80C acts as the interface between kinetochores and microtubules.
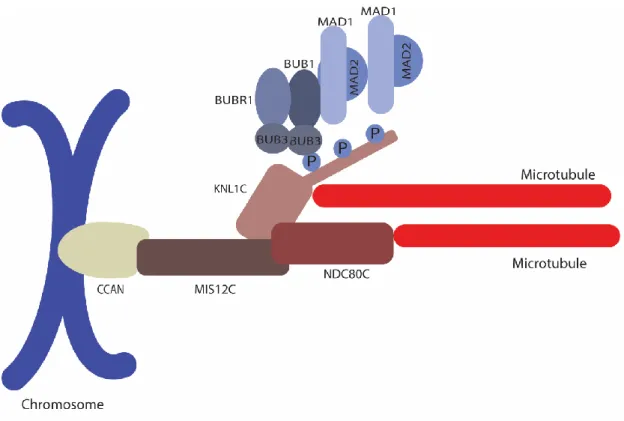
Figure 6 Outer kinetochore
The outer kinetochore includes MIS12C, NDC80C and KNL1C. MIS12C anchors the outer kinetochore to the CCAN. Microtubule interaction is mediated through NDC80C and KNL1C. The KNL1C serves as platform for the localization of several SAC proteins, such as BUB3, BUB1,
BUBR1, MAD1, and MAD2. SAC proteins recruitment to the kinetochore is regulated by phosphorylation of the KNL1C (Hara and Fukagawa, 2018).
The function of the kinetochore in microtubule attachment is illustrated by the direct interaction between NDC80C and microtubules. NDC80C interacts with microtubules at its globular heads located at the N-terminus of NDC80 and NUF2, whereas its kinetochore tethering occurs at its other extremity at SPC24 and SPC25 along its 55 to 60 nm axis (Ciferri et al., 2005; Musacchio and Desai, 2017; Wei et al., 2005). The microtubule interface comprises calponin-homology (CH) domains, which directly interacts with the lateral lattice of both TUBULIN monomers in microtubules (Alushin et al., 2010, 2012; Valverde et al., 2016). Apart from the CH domains, it has been demonstrated that human NDC80 can interact with microtubules through its disorganized N-terminal tail. Strikingly, phosphorylation by AURORA B in two different sites of the NDC80 tail regulates its affinity to microtubules (Alushin et al., 2012). In this fashion, AURORA B creates a negative charge at the NDC80 tail which destabilizes its optimum interaction with microtubules (Tooley and Stukenberg, 2011).
The last subunit of the KMN network, KNL1, is its largest (Musacchio and Desai, 2017). Composed of 2316 residues in humans, this protein is recruited to kinetochores through its RWD domain (discussed subsequently) interaction with circa 20 residues from the C-terminus of the MIS12C subunit NSL1 (Petrovic et al., 2010, 2014). KNL1 has the distinctive characteristic of having several regions of low complexity, which is also present in several hub proteins, as it allows the flexibility to form interactions with distinct molecules (Ghongane et al., 2014). Therefore, KNL1 is a scaffolding platform for several proteins involved in the SAC and kinetochore-microtubule attachment. Interestingly, KNL1 can also directly contribute to microtubule tethering to kinetochores (Espeut et al., 2012).
As a center stage for the molecular mechanisms in this work, the protein interactions at KNL1 will be dissected for each of its motifs.
KNL1 RWD motif
At its C-terminus, the RWD motif in KNL1 is a conserved region organized in a compact globular domain (Petrovic et al., 2010, 2014) composed of 3 domains: C-terminal RING finger, WD repeat, and DEAD-like helicases domains. The RWD interacts with MIS12 and ZWINT, which respectively have roles in KNL1-kinetochore recruitment and SAC signalling.
KNL1 RVSF/SILK motif
Localized at KNL1 N-terminal regions, the RVSF (RVXF) and SILK ([SG]ILK) motifs interact with the Protein Phosphatase 1 (PP1) in humans and yeast (Liu et al., 2010; Rosenberg et al., 2011). This phosphatase is responsible for opposing AURORA B activity at the kinetochore and, consequently, stabilizing kinetochore-microtubule attachments. Notably, AURORA B phosphorylation of KNL1 inhibits PP1 docking, leading to the formation of a negative feedback loop that culminates in maximum phosphatase activity at metaphase and anaphase onset (Espeut et al., 2012; Lesage et al., 2011). This mechanism will be thoroughly described later.
KNL1 MELT motifs
The MELT motifs consist of a sequence consensus of (M/I/L/V)-(E/D)-(L/M/I/V)-(T/S). The phosphorylation of the threonine in position 4 by the mitotic kinases MPS1 and PLK1 triggers the direct tethering of BUB3 to KNL1 (Primorac et al., 2013) in humans, which then serves as scaffold for BUB1 (Budding Uninhibited by Benzimidazoles 1) and BUBR1 (BUB1-related kinase 1) kinetochore recruitment (Ikeda and Tanaka, 2017; London et al., 2012; Overlack et al., 2015, 2017; Primorac et al., 2013; von Schubert et al., 2015; Shepperd et al., 2012; Yamagishi et al., 2012), ultimately leading to SAC activation and the regulation of kinetochore-microtubule interaction. The MELT motifs are in fact part of a larger set of repeated motifs in KNL1, comprising a TΩ motif (TxxΩ; Ω is an aromatic residue), and a SHT at the C-terminal extremity from the MELT motif (Krenn et al., 2014; Tromer et al., 2015; Vleugel et al., 2013, 2015). Interestingly, the number of MELT motifs varies in
different species (Cheeseman et al., 2004; Vleugel et al., 2012, 2013). Despite the fact that human KNL1 possesses 20 MELT repeats, it was demonstrated that only 6 repeats are necessary to recruit the human BUBs to kinetochores. It was suggested that duplication of the gene portion that creates the repeats occurred dynamically to mirror the required amount of BUB proteins on a kinetochore to support SAC signalling. A higher number of BUB proteins tethering to MELT motifs resulted in increased efficiency in microtubule binding corrections (Tromer et al., 2015).
KNL1 KI motifs
The two N-terminal KI motifs (sequence KI(D/N)FxxF(L/I)xRL) are found only in vertebrates and directly interact with the tetratricopeptide repeat (TPR) motif found in BUB1 and BUBR1 (Bolanos-Garcia and Blundell, 2011; Kiyomitsu et al., 2011; Krenn et al., 2012; Tromer et al., 2015). Despite not being essential for BUB1 and BUBR1 kinetochore recruitment, the KI motifs enhance and stabilize BUB1 and BUBR1 binding to MELT motifs (Krenn et al., 2014; Vleugel et al., 2013).
Kinetochore-microtubule attachment
Regulation in kinetochore-microtubule attachments concerning AURORA B and the spindle and kinetochore associated (SKA) complex will be discussed in this section.
The role of AURORA B in kinetochore-microtubule attachment formation
As previously observed, microtubules attach to kinetochores primarily at the NDC80C (Musacchio and Desai, 2017), and are subject to regulation by AURORA B. Indeed, AURORA B phosphorylates several targets in the outer kinetochore region, such as NDC80, MIS12C, and KNL1C, leading to the weakening of the microtubule-kinetochore interaction (Carmena et al., 2012; Cheeseman et al., 2006; Ciferri et al., 2008; DeLuca et al., 2011; Lampson et al., 2004; Welburn et al., 2010; Zhou et al., 2017). This is considered the fundamental mechanism for the correction of erroneously attached microtubules. Moreover, AURORA B also promotes kinetochore-microtubule detachment indirectly through the microtubule
depolymerase MCAK (Mitotic Centromere-associated Kinesin) (Knowlton et al., 2006), and other microtubule binding proteins (Iimori et al., 2016) (Figure 7).
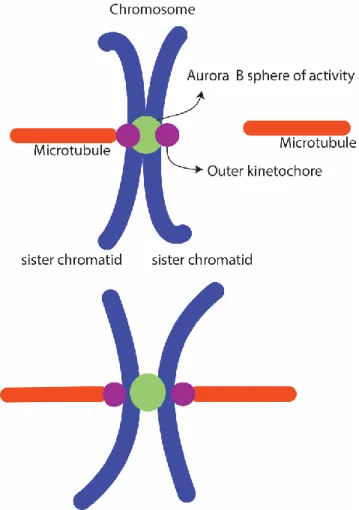
Figure 7 Aurora B tension mechanism
From the centromere, the Aurora B sphere of activity reaches the outer kinetochore. Phosphorylation of Aurora B targets decreases kinetochore affinity for microtubules. When biorientation occurs (each sister chromatid is attached to microtubules originating from opposing poles of the mitotic spindle) it creates tension, which increases the distance between the centromere and outer kinetochore, undermining Aurora B capacity to reach its outer kinetochore substrates (Krenn and Musacchio, 2015).
To better understand the role of AURORA B in the formation of stable kinetochore-microtubule attachments, each step in the process will be presented separately:
• Unattached kinetochores display high AURORA B activity (Welburn et al., 2010). In this state, microtubules are still able to interact with kinetochores. • Upon microtubule attachment, the microtubules are believed to move the
outer kinetochore away from the centromere of chromosomes, separating AURORA B from its substrates in the outer kinetochore (Krenn and Musacchio, 2015). Microtubules can still detach from the kinetochore due to AURORA B phosphorylation, since AURORA B is still within reach of the outer kinetochore.
• After several turns of attachment and detachment, biorientation finally occurs, when each sister-chromatid is attached to opposing poles of the mitotic spindle. This results in a significant increase in the tension between sister-chromatids, which in turn results in increased distance between AURORA B and its substrates (Cheerambathur and Desai, 2014; Lampson and Grishchuk, 2017; Sarangapani and Asbury, 2014). AURORA B can no longer phosphorylate its substrates, and the kinetochore-microtubule attachment is thus stabilized.
The SKA complex is important for bridging microtubules and kinetochores
Despite its importance, the KMN is not sufficient to establish stable kinetochore-microtubule attachments. Another important complex for the formation of such attachments is the human SKA complex, which consists of SKA1, SKA2 and SKA3. This complex localizes to kinetochores and to microtubules, and its main two functions are to promote kinetochore-microtubule attachment and to silence the SAC signalling pathway (Zhang et al., 2018b). The role of the SKA complex in attachment will be discussed in this section, while its role in SAC silencing will be addressed in the “SAC silencing” section.
SKA recruitment to kinetochores depends on NDC80C (Zhang et al., 2017b) and on AURORA B (Chan et al., 2012). SKA3 directly binds to NDC80, while AURORA B phosphorylates the SKA complex inhibiting the NDC80 interaction. Therefore, in prometaphase the SKA complex is closer to AURORA B at unattached kinetochores, which results in SKA phosphorylation and reduction of its accumulation at kinetochores. When AURORA B activity decreases following bi-orientation, SKA complexes accumulate at kinetochores and promote the stabilization of kinetochore-microtubule attachment. Importantly, while bridging the microtubules to kinetochores, the SKA complex has also major role in translating microtubule forces to chromosomes in order to generate movement during chromosome congression.
Chromosome Congression
As prometaphase occurs and the nuclear membrane breakdowns, chromosomes are required to be transported to the metaphase plate. This process is called chromosome congression, which ensures that all chromosomes start their anaphase poleward movement at equal positions relative to the spindle pole (Maiato et al., 2017; Matos et al., 2009). Furthermore, congression also increases the probability of chromosome capture by opposing sides of the spindle at the cell equator (Joglekar, 2016), leading to the required tension at the kinetochores, which the cell uses to sense biorientation (King and Nicklas, 2000). Models for chromosome congression date as far 1895, in which migration is thought to be achieved through chromosome-pulling and ejection forces, which are balanced and reach a net zero movement at the metaphase plate (Drüner, 1895). Current models aim to explain the biochemical mechanism behind these forces.
The presence of forces exerted at the chromosome arms and at the kinetochore level were elegantly demonstrated by laser microsurgery experiments. Chromosomes presenting laser-damaged kinetochores were pushed away from the spindle pole, while armless chromosomes (isolated kinetochore fragments from chromosomes) were pulled toward the spindle pole (Rieder et al., 1986). These
ejection forces are exerted onto chromosome arms, and poleward force is exerted upon the kinetochores.
In order to facilitate the study of chromosome congression, the origin of the forces acting on chromosomes are broadly divided in two categories: microtubule flux and motor-driven forces. Both categories can exert polar ejection forces (chromosome movement away from the pole) or pulling forces (chromosome movement in direction to the pole) (Maiato et al., 2017).
The generation of chromosome movement through microtubule flux
Microtubule flux generates both polar ejection and kinetochore pulling forces. Polar ejection forces occurs upon microtubule contact at chromosomes arms, which exerts pressure on the chromosome due to the polymerization of microtubules (Ault et al., 1991). In contrast, kinetochore pulling forces are generated through induced microtubule depolymerization (Auckland and McAinsh, 2015). While the kinetochore remains attached to the depolymerizing microtubule, it generates a poleward force on the chromosome (Inoué and Salmon, 1995). Microtubule depolymerization can be achieved through MCAK activity in the outer kinetochore region (Honnappa et al., 2009; Kline-Smith et al., 2004), thus promoting poleward movement (Armond et al., 2015). Additionally, it has been demonstrated that the binding of curved protofilaments to kinetochores also causes the depolymerization of microtubules (McIntosh et al., 2008). Notably, the SKA complex is able to follow depolymerizing microtubules plus-ends, and harness the resulting force, as demonstrated in experiments using polystyrene beads in vitro (Schmidt et al., 2012; Welburn et al., 2009). The SKA complex is thus thought to act as interface between depolymerizing microtubules and kinetochores, where it is recruited to kinetochores upon microtubule binding (Gaitanos et al., 2009; Hanisch et al., 2006; Raaijmakers et al., 2009; Schmidt et al., 2012; Theis et al., 2009; Welburn et al., 2009). Interestingly, the protein CENP-F was also shown to bind curved protofilaments and transduce the generated force to beads (Volkov et al., 2015), which is believed to simulate its biological role in transporting chromosomes through microtubule flux. In support of
congression (Bomont et al., 2005; Feng et al., 2006; Holt et al., 2005; Yang et al., 2005). Recent reports support that BUB1 is the anchoring factor at the kinetochore for CENP-F (Ciossani et al., 2018), where CENP-F also promotes the kinetochore recruitment of kinesins CENP-E and DYNEIN (Bomont et al., 2005; Vergnolle and Taylor, 2007; Yang et al., 2005).
The generation of chromosome movement through chromokinesins
The motor-driven forces of chromosome congression are associated with chromokinesins, which are motor proteins capable of binding to chromosomes and moving along the microtubule lattice. Despite the role of poleward flux in congression, motor-driven congression is thought to be the main source of force for congression, as 80% reduction in poleward flux has no impact in human chromosome congression (Ganem et al., 2005), and only a minor fraction of polymerizing microtubules on chromosome arms has an effect on congression (Barisic et al., 2014). On the contrary, chromokinesins are the main source for polar ejection forces (Brouhard and Hunt, 2005). These motor proteins present plus-end directed motility, such as KID (Kinesin-like DNA-binding protein) and CENP-E (Kim et al., 2008; Yajima et al., 2003), thus they are thought to carry chromosomes away from the pole (Figure 8). On the other hand, minus-end motor proteins, such as DYNEIN, oppose polar ejection forces and transport chromosomes poleward (Barisic et al., 2014; Pfarr et al., 1990; Steuer et al., 1990; Vergnolle and Taylor, 2007).
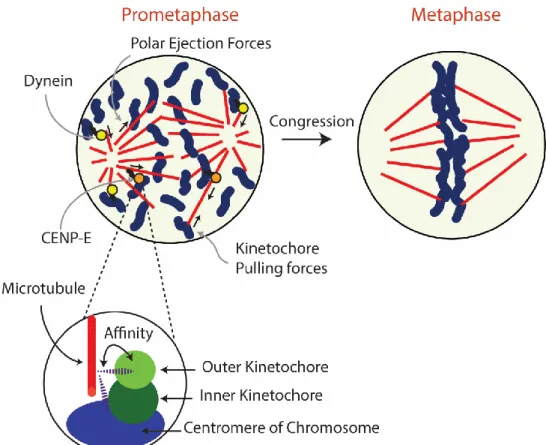
Figure 8 Congression model
Motor-driven chromosome congression and polar ejection forces occur simultaneously and depend on each other to promote faithful chromosome segregation. Dynein and kinetochore pulling forces bring chromosomes to the mitotic spindle pole, while CENP-E and polar ejection forces drive chromosomes to the metaphase plate (Maiato et al., 2017). Chromosome-microtubule interaction occurs at kinetochores, which can promote or destabilize the kinetochore-microtubule attachment. This regulation is crucial for proper chromosome migration and alignment at the metaphase plate.
To illustrate the current congression model, each step will be presented chronologically:
• During early prometaphase, DYNEIN is localized at unattached kinetochores. If the chromosome lies outside the mitotic spindle, DYNEIN is responsible for transporting the chromosome to the poles (Auckland and McAinsh, 2015). In support of this, it has been observed that chromosomes tend to firstly migrate toward the poles (Maiato et al., 2017).