Évaluation du rôle neuroprotecteur de la petite GTPase
Rin pour le traitement de la maladie de Parkinson
Mémoire
Anne-Marie Castonguay
Maîtrise en neurobiologie - avec mémoire
Maître ès sciences (M. Sc.)
Évaluation du rôle neuroprotecteur de la petite
GTPase Rin pour le traitement de la maladie de
Parkinson
Mémoire
Anne-Marie Castonguay
Sous la direction de :
Résumé
La maladie de Parkinson (MP) est caractérisée par l'accumulation d'alpha-synucléine (aSyn) mal repliée dans la substantia nigra pars compacta (SNpc), entraînant la mort des neurones dopaminergiques (DA). Les mécanismes qui sous-tendent la toxicité de l’aSyn sont encore peu connus, mais on suppose qu'ils impliquent des défauts dans l'autophagie (ALP). Les mutations dans LRRK2 sont fréquentes dans la MP familiale et sporadique. L'inhibition pharmacologique de l'activité de la kinase LRRK2 réduit les déficits dans l’ALP et les inclusions d’aSyn phosphorylée (paSyn), ce qui suggère que ces phénotypes dépendent de l'hyperactivation de LRRK2. Nous avons observé une diminution de l’expression du gène RIT2 dans des cellules mutantes LRRK2 (G2019S). RIT2 encode la petite GTPase Rin, qui est enrichie dans les neurones DA, et moins abondante dans la SNpc des cerveaux des patients atteints de la MP. Notre objectif est d'évaluer si Rin peut moduler l'activité de LRRK2 pour contrecarrer les altérations dans l'autophagie et promouvoir la clairance de l’aSyn. Nous avons utilisé des cellules de neuroblastome exprimant LRRK2-G2019S ou LRRK2-sauvage (WT) et évalué les déficits dans l’ALP et la toxicité de l’aSyn avec ou sans surexpression de Rin. Nous avons ensuite testé notre approche in vivo en utilisant des vecteurs viraux encodant aSyn et/ou Rin afin de surexprimer ces gènes chez la souris. Nous avons évalué la déficience locomotrice et fait une analyse histopathologique de coupes de cerveau. La surexpression de Rin dans les cellules LRRK2-G2019S a permis de renverser les altérations dans l'ALP et de diminuer les inclusions d'aSyn. Dans notre modèle de souris, la surexpression de Rin a empêché les déficits moteurs induits par l'injection d'AAV-aSyn. La surexpression de Rin a également protégé contre la perte d'axones dopaminergiques dans le striatum et la dégénérescence neuronale. Nos données indiquent que Rin inhibe LRRK2 pour compenser le déficit ALP et contrecarrer l'agrégation d'aSyn et les déficits connexes. Cela suggère que de cibler la signalisation par Rin pourrait représenter une nouvelle stratégie pour combattre la MP familiale et sporadique.
Abstract
Parkinson's disease (PD) is characterized by the accumulation of misfolded alpha-synuclein (aSyn) in the substantia nigra (SNc), leading to the death of dopaminergic (DA) neurons. The mechanisms underlying aSyn pathology are still unclear but hypothesized to involve autophagy and endosome-lysosome pathways (ALP). LRRK2 mutations are a major cause of familial and sporadic PD. Pharmacological inhibition of LRRK2 kinase activity ameliorates ALP deficits and reduces paSyn inclusions, indicating that these phenotypes depend on LRRK2 hyperactivation. We observed selective downregulation of the novel PD risk factor RIT2 in LRRK2 mutant cells (G2019S). RIT2 encodes the small GTPase Rin, which is enriched in DA neurons and reduced in SNpc of PD brains. We aim to evaluate if Rin can modulate LRRK2 activity to rescue alterations in autophagy and promote aSyn clearance. We used neuroblastoma cells expressing LRRK2-G2019S or LRRK2-WT and evaluated ALP deficits and aSyn pathology with or without Rin overexpression. We then tested our approach in vivo using viral vectors encoding aSyn and/or Rin in mice SNpc. We evaluated the locomotor impairment and performed histopathological analysis on brain sections. Rin overexpression in LRRK2-G2019S cells rescued the alterations in ALP and diminished aSyn inclusions. In vivo, viral mediated overexpression of Rin prevented motor deficits induced by AAV-aSyn injection. Overexpression of Rin also protected against the loss of dopaminergic axons in the striatum and neural degeneration. Our data indicate that Rin inhibits overactive LRRK2 to remove ALP impairment and counteract aSyn aggregation and related deficits. This suggests that targeting Rin signaling could represent a novel strategy to combat neuropathology in familial and idiopathic PD.
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des figures, tableaux, illustrations ... vi
Liste des abréviations, sigles, acronymes ... vii
Remerciements ... x
Introduction ... 1
1. Le système dopaminergique ... 1
1.1 La dopamine ... 3
1.2 La signalisation dopaminergique ... 3
2. Les ganglions de la base ... 6
2.1 Le contrôle moteur ... 7
3. La maladie de Parkinson ... 9
3.1 Origine des symptômes ... 9
3.2 Étiologie ... 10
3.3 Caractéristiques histopathologiques ... 11
3.4 L’alpha-synucléine ... 12
3.5 Mécanismes compensatoires ... 15
3.6 Les traitements actuels ... 16
3. Leucine-rich repeat kinase 2 (LRRK2) ... 17
4. L’autophagie ... 18
5. Rin ... 21
5.1 Les GTPases ... 21
5.2 La petite GTPase Rin ... 21
Objectif et hypothèse ... 24
Chapitre 1 – Méthodologie ... 25
1.1 Culture cellulaire et transfection ... 25
1.2 Immunobuvardage ... 25
1.3 Lysotracker Deep Red et coloration DQ-Red-BSA ... 25
1.6.1 Test de l’open field ... 27
1.6.2 Cylindre, rotations et test aux amphétamines ... 27
1.7 Hybridation in situ par fluorescence ... 27
1.8 Immunohistochimie et immunofluorescence ... 28
1.9 Proximity ligation assay (PLA) ... 29
Chapitre 2 – Résultats ... 30
2.1 RIT2 est exprimé dans les neurones dopaminergiques murins et son expression est
diminuée dans des échantillons humains de Parkinson idiopathique ... 30
2.2 Les inclusions de pS129-aSyn dans les cellules LRRK2-G2019S sont réduites par la
surexpression de Rin ... 31
2.3 La surexpression de Rin permet de renverser la dysfonction autophagique-lysosomale
observée dans les cellules LRRK2-G2019S ... 32
2.4 La surexpression de Rin induit la déphosphorylation de la sérine 1292, mais pas de la
sérine 935 de LRRK2 ... 34
2.5 La surexpression de Rin dans le mésencéphale de la souris préserve les neurones
dopaminergiques et stimule l’activité motrice ... 35
2.6 La surexpression de Rin réduit les niveaux de pS129-aSyn et d'aSyn totaux in vivo ... 38
2.7 In vivo, la surexpression de Rin permet de maintenir l’activité kinase de LRRK2 à un
niveau basal ... 39
Chapitre 3 : Discussion ... 41
Modulation de l’autophagie par l’action de Rin sur LRRK2 in vitro ... 41
Surexpression de Rin dans un modèle murin de la MP ... 43
Perspectives ... 46
Conclusion ... 47
Bibliographie ... 48
Annexe A – Le niveau protéique total de LRRK2 n’est pas affecté par la surexpression de
l’aSyn et de Rin ... 60
Annexe B – Mesure du taux de dopamine, de 3MT et de DOPAC dans le striatum des souris
AAV-RIT2 ... 61
Liste des figures, tableaux, illustrations
Figure 1: Le système dopaminergique et les structures impliquées. ... 2
Figure 2: La biosynthèse de la dopamine ... 3
Figure 3: Voies de signalisation activées par la dopamine. ... 5
Figure 4: Schématisation des voies directe et indirecte des ganglions de la base dans le
contrôle moteur. ... 7
Figure 5 : Les stages de Braak ... 12
Figure 6: Les principaux mécanismes compensatoires prenant place avant l'apparition des
symptômes moteurs. ... 15
Figure 7: Les trois types d'autophagie. ... 19
Figure 8: Gènes associés à la maladie de Parkinson ... 20
Figure 9 : RIT2 est exprimé dans les neurones dopaminergiques murins et son expression est
réduite dans la maladie de Parkinson idiopathique. ... 30
Figure 10: La surexpression de Rin diminue le nombre d'inclusions de pS129-aSyn dans les
cellules G2019S ... 31
Figure 11: La surexpression de Rin n'affecte pas le flux autophagique total ... 32
Figure 12 : La surexpression de Rin restaure le nombre de lysosomes et leur morphologie, et
augmente leur activité protéolytique. ... 33
Figure 13 : La surexpression de Rin diminue la phosphorylation du résidu S1292, mais pas du
résidu S935 et n'affecte pas le niveau total de la protéine LRRK2. ... 35
Figure 14: La surexpression de Rin stimule l'activité motrice et préserve les neurones
dopaminergiques et leurs projections dans des souris surexprimant A53T-aSyn. ... 37
Figure 15: La surexpression de Rin permet de réduire la quantité de pS129-
aSyn et d’
aSyn
totale dans les neurones dopaminergiques. ... 38
Figure 16: La surexpression de Rin maintient le niveau basal d'activation de la kinase LRRK2
endogène in vivo. ... 39
Figure 17: La surexpression de Rin dans la substance noire de souris ne réduit pas le niveau
total de la protéine LRRK2. ... 60
Figure 18: Les concentrations de dopamine et de ses métabolites 3MT et DOPAC ne sont pas
significativement augmentées suite à la surexpression de Rin dans les neurones
Liste des abréviations, sigles, acronymes
aSyn Alpha-synucléine
3MT
3-Methoxytyramine
6-OHDA 6-Hydroxydopamine
AAV Vecteur adéno-associé
ADN Acide désoxyribonucléique
ALP Voie autophagique-lysosomale
AMP Adénosine monophosphate
AMPc Adénosine monophosphate cyclique AMPK Adénosine monophosphate kinase ATG Gène relatif à l’autophagie
ATP Adénosine triphosphate
BH4 Tétrahydrobiopterine
BSA Sérum albumine bovin
Ca2+ Ion calcium
CAAX Cystéine-Acide aminé aliphatique x 2-Acide aminé CCPA Comité canadien de protection des animaux CMA Autophagie médiée par les chaperonnes
CO2 Dioxyde de carbone
CPAUL Comité de protection des animaux de l’Université Laval CpG Dinucléotide cytosine-guanine
CTCF Fluorescence cellulaire totale corrigée
CRISPR Clustered Regularly Interspaced Short Palindromic Repeats
DA Dopamine
DARPP-32 Phosphoprotéine régulée par l’AMPc et la DA de 32 kDa
DAT Transporteur de dopamine
DMEM Dulbecco’s modified Eagle’s medium
DOPAC
Acide-3,4-dihydroxyphenylacetic
DQ-Red-BSA “Dye Quenched-Bovine Serum Albumin”
DTT Dithiothréitol
FDA Food and Drug Administration
G2019S Substitution Glycine-2019-Sérine
GAP Protéine activatrice/accélératrice de l’activité GTPase
GBA β-glucocerebrosidase
GDP Guanosine diphosphate
GEF Facteur d’échange de nucléotide guanine GFP Protéine fluorescente verte
GPCR Récepteur couplé aux protéines G
GPe Globus pallidus externe
GPi Globus pallidus interne
GRK Kinase des récepteurs couplés aux protéines G
GTP Guanosine triphosphate
GTPCH GTP cyclohydrolase
GWAS “Genome Wide Association Study” Hsp70 “Heat shock protein 70”
IHC Immunohistochimie
K+ Ion potassium
LAMP1 Protéine membranaire associée aux lysosomes 1 LAMP2A Protéine membranaire associée aux lysosomes 2A LC3 Marqueur autophagique chaîne légère 3
LRRK2 “Leucine-rich repeat kinase 2” MATs Transporteurs des monoamines
MP Maladie de Parkinson
MPi Maladie de Parkinson idiopathique
MPTP 1-méthyl-4-phényl-1,2,3,6-tétrahydropyridine
MSA Atrophie multi-systémique
MSN Neurone épineux moyen
mTORC1 Mammalian target of rapamycin 1
NDS Sérum normal d’âne
NGF Sérum normal de chèvre
OCT « Optimal cutting temperature »
PBS Tampon phosphate salin
PFA Paraformaldéhyde
PI3K-III Phosphoinositide 3-kinase-III PI3P Phosphatidylinositol-3-phosphate
PKA Protéine kinase A
PKC Protéine kinase C
PLA “Proximity ligation assay”
PLC Phospholipase C
pS129-aSyn Alpha-synucléine phosphorylée à la sérine 129 pS1292 Sérine 1292 phosphorylée
pS935 Sérine 935 phosphorylé
RGS Régulateurs de la signalisation des protéines G RIPA Tampon de radioimmunoprecipitation ROS Espèces réactives d’oxygène
S1292 Sérine 1292
S935 Sérine 935
SDS Sodium dodecyl sulfate
SNARE “Soluble N-éthylmaleimide-sensitive-factor Attachment protein Receptor” SNCA Alpha-synucléine (gene)
SNpc Substantia nigra pars compacta
T73 Thréonine 73
TDAH Trouble du déficit de l’attention avec hyperactivité
TH Tyrosine hydroxylase
TOM20 Protéine membranaire mitochondriale 20 kDa ULK1 “Unc-51 like autophagy activating kinase” VMAT2 Transporteur vésiculaire de monoamine 2 VTA Aire tegmentale ventrale
Remerciements
Je vais évidemment remercier (mais avec raison) mes parents pour leur soutien inconditionnel pendant cette maîtrise et les (trop?) nombreuses années d’études la précédant. Votre confiance inébranlable m’a toujours encouragée et touchée. Évidemment, je dois remercier Martin de m’avoir donné la chance de mener à bien ce projet. Merci d’être une source inépuisable d’idées et de croire aux licornes avec autant de fougue. Un gros merci à Véro pour avoir répondu aux 132 857 384 questions que je lui ai posées pendant les deux dernières années et pour toute l’aide qu’elle m’a apporté. Et, finalement, merci aux étudiants présents et passés du laboratoire d’avoir contribué à créer une petite famille agréable à côtoyer au quotidien.
Introduction
La maladie de Parkinson (MP) fût identifiée pour la première fois en 1817 par le médecin britannique Sir James Parkinson. Dans une publication de 66 pages, il décrit ce qu’il appelle la « shaking palsy », que le neurologue français Jean Martin Charcot appellera plus tard la maladie de Parkinson. Il s’agit aujourd’hui de la deuxième maladie neurodégénérative en importance dans le monde après la maladie d’Alzheimer (1). Elle touche un peu plus de 67 000 personnes au Canada, dont la majorité sont âgés de plus de 65 ans (2). Le risque de développer la maladie augmentant avec l’âge, le vieillissement de la population au Canada et dans d’autres pays industrialisés en augmente progressivement la prévalence (3,4). Malheureusement, plus de 200 ans après sa découverte, nous ne détenons toujours aucun moyen avéré de traiter efficacement cette pathologie qui affecte significativement la qualité de vie des gens atteints. Heureusement, la recherche sur la maladie de Parkinson est abondante et des progrès colossaux ont été fait au cours des 50 dernières années dans la compréhension des processus pathologiques impliqués dans son développement.
L’une des stratégies thérapeutiques explorée par certaines équipes est la neuroprotection, dont il sera question dans le présent ouvrage. Pour bien comprendre ce concept dans le contexte de la MP, une description du système dopaminergique et de ses fonctions sera préalablement effectuée. Il sera ensuite question des fondements histopathologiques de la maladie ainsi que de ses causes. La protéine d’intérêt dans ce projet ainsi que le processus cellulaire dans lequel elle est impliquée seront ensuite abordés.
1. Le système dopaminergique
Notre cerveau est composé de plus de 80 milliards de neurones organisés en réseaux structurés qui nous permettent d’effectuer un nombre important de tâches différentes. Le système dopaminergique est formé de l’ensemble des connexions établies par les neurones libérant le neurotransmetteur dopamine (DA). Ces neurones sont peu abondants dans le cerveau humain, représentant environ 1% de la population neuronale totale (5). Malgré leur nombre réduit, leurs projections très étendues leur permettent de former des réseaux importants impliqués dans plusieurs fonctions cérébrales. En effet, les neurones dopaminergiques ont une longueur axonale moyenne impressionnante de 74 cm (6). La grande majorité des neurones dopaminergiques sont situés dans le mésencéphale, au niveau de la Substantia nigra pars compacta/Substance noire (SNpc) et de l’aire tegmentale ventrale (VTA). Chez l’humain et chez certaines autres espèces, ces neurones ont la particularité de contenir un pigment noir appelé neuromélanine. Ce pigment donne à la substance noire sa couleur caractéristique et est à l’origine de son nom (7).
Quatre différentes voies de communication composent le système dopaminergique: la voie mésocorticale, la voie mésolimbique, la voie tubéro-infudibulaire et la voie nigrostriée (Fig. 1). La voie mésocorticale prend son origine dans la VTA, située dans le mésencéphale, et projette principalement vers le cortex préfrontal et cingulaire. Elle serait impliquée dans la motivation et le comportement associé aux émotions. La voie mésolimbique prend également son origine dans la VTA, mais projette principalement au noyau accumbens. Cette voie joue un rôle très important dans le système de la récompense (8). Les voies mésocorticale et mésolimbique étant fortement interconnectées, elles sont souvent regroupées ensemble sous le nom de système mésocorticolimbique. Elles sont fortement impliquées dans les processus de récompense et d’addiction, et sont affectées dans plusieurs maladies psychiatriques incluant la schizophrénie et le trouble de déficit de l’attention et d’hyperactivité (TDA/TDAH) (9). La voie tubéro-infundibulaire débute dans l’hypothalamus et projette vers l’hypophyse. Dans cette voie, la dopamine agit comme une neurohormone qui inhibe la libération de prolactine (10). Finalement, la voie nigrostriée prend son origine dans la SNpc et projette vers lestriatum. Cette voie est impliquée dans le contrôle des mouvements volontaires, qui sera abordé plus en détails dans la prochaine section. De leurs noyaux d’origine, les axones des neurones dopaminergiques empruntent ensemble le faisceau médian du télencéphale pour rejoindre la capsule interne. Ils se séparent ensuite pour aller innerver leurs cibles respectives (6).
Figure 1: Le système dopaminergique et les structures impliquées. La voie mésocorticale débute dans l’aire
tegmentale ventrale et projette vers le cortex préfrontal et cingulaire. La voie nigrostriée origine dans la substance noire et projette vers le striatum. La voie tubéro-infudibulaire débute dans l’hypothalamus et projette vers
Hypothalamus Cervelet Striatum dorsal Thalamus Noyau accumbens Hypothalamus Cervelet Substance noire Aire tegmentale ventrale Hypophyse Voie mésocorticale Voie nigrostriée Voie tubéro-infundibulaire Voie mésolimbique
1.1 La dopamine
La dopamine est un neurotransmetteur de la famille des catécholamines, dont font également partie l’adrénaline et la noradrénaline. Ces trois neurotransmetteurs sont synthétisés à partir de l’acide aminé tyrosine. Dans le système nerveux central, leur principale voie de biosynthèse débute dans le cytoplasme d’un neurone catécholaminergique par l’hydroxylation de la L-tyrosine en L-DOPA (Figure 2). Cette étape hautement régulée est effectuée par l’enzyme tyrosine hydroxylase (TH) et dépend de la présence du cofacteur tétrahydrobiopterine (BH4). Ce dernier est synthétisé à partir de la guanosine triphosphate (GTP) par la GTP cyclohydrolase (GTPCH). La transformation de la tyrosine en L-DOPA est l’étape limitante dans la synthèse de la dopamine. Par la suite, la L-DOPA est décarboxylée par la DOPA-décarboxylase pour générer la dopamine (Fig. 2). Une autre étape enzymatique permet d’obtenir la noradrénaline, qui peut subséquemment être transformée en adrénaline (11)
.
La dopamine peutégalement être indirectement synthétisée à partir de l’acide aminé phénylalanine, puisque ce dernier peut être transformé en L-tyrosine par l’enzyme phénylalanine hydroxylase. Après sa synthèse, la dopamine est transportée à l’intérieur des vésicules synaptiques par le transporteur vésiculaire de monoamine 2 (VMAT2), où elle est entreposée jusqu’à son utilisation (12). L’environnement acide du lumen des vésicules synaptiques aide à maintenir la stabilité de la molécule et à prévenir son oxydation. Dans un environnement plus basique, la dopamine tend à s’oxyder ou à être métabolisée par d’autres enzymes, générant des métabolites détectables dans le sang (13,14).
1.2 La signalisation dopaminergique
Dans la majorité des cas, la dopamine est libérée dans la fente synaptique par le neurone dopaminergique par exocytose à la suite d’un potentiel d’action. Une fois dans l’espace extracellulaire, elle peut se lier aux récepteurs postsynaptiques sur le neurone cible ou à certains récepteurs présynaptiques. Lorsque la transmission du signal est terminée, la dopamine est recapturée par le neurone présynaptique grâce aux transporteurs de la dopamine (DAT) ou par les transporteurs des monoamines (MATs)(15).
Figure 2: La biosynthèse de la dopamine. L’acide aminé tyrosine est hydroxylé en L-DOPA par la tyrosine
hydroxylase. Le L-DOPA est ensuite décarboxylé par la DOPA-décarboxylase pour former la dopamine (modifié à partir de Hare & Loer, 2004).
La dopamine produit ses différents effets par l’activation de l’un des cinq récepteurs dopaminergiques. Il n’est pas possible de classifier ce neurotransmetteur dans la catégorie excitateur ou inhibiteur, puisque ses effets dépendent du récepteur activé. Les récepteurs à la dopamine sont des récepteurs couplés à une protéine G (GPCRs) contenant sept domaines transmembranaires. Les protéines G sont composées de trois sous-unités : a, b et g, qui forment un complexe trimérique. Dans l’état non-activé, la sous-unité a est liée à un guanosine diphosphate (GDP). Lors de la liaison du ligand sur le récepteur, il se produit un changement de conformation qui a pour effet de faciliter l’échange du GDP pour le guanosine triphosphate (GTP). La sous-unité a liée au GTP est en mesure de se dissocier du complexe trimérique. La sous-unité a-GTP et le complexe bg sont par la suite tous les deux en mesure d’activer des effecteurs cellulaires. L’hydrolyse du GTP permet ensuite la réassociation des trois sous-unités temporairement inactivées (16).
Les récepteurs à la dopamine sont séparés en deux classes principales, soit les récepteurs de type D1 et les récepteurs de type D2. Les récepteurs de type D1 (D1 et D5) sont exprimés exclusivement sur la membrane postsynaptique des neurones activés par la dopamine. Ils sont couplés à une protéine GaS qui
active l’adénylate cyclase et permet donc la synthèse d’adénosine monophosphate cyclique (AMPc). L’AMPc active la protéine kinase A (PKA), qui phosphoryle une variété de substrats, module l’activité de plusieurs canaux ioniques et stimule l’expression de certains gènes (17). Les récepteurs de type D2 (D2, D3 et D4) sont exprimés à la fois sur les membranes postsynaptiques des neurones cibles et sur les membranes présynaptiques des neurones dopaminergiques (18). Ils sont couplés à une protéine Gai qui
inhibe l’adénylate cyclase et diminue donc les taux d’AMPc. Il en résulte une baisse de l’activité de la PKA et une activation des canaux potassiques (K+) (Fig. 3). Les autorécepteurs de type D2 peuvent être situés
sur le soma ou les dendrites présynaptiques et diminuer l’excitabilité neuronale par l’augmentation de la conductance des canaux K+. Ils peuvent également être situés sur les terminaisons présynaptiques et
inhiber la libération de la dopamine. Ils ont donc une fonction d’inhibition et de contrôle de la transmission dopaminergique (19–21). De façon générale, les récepteurs de type D1 sont donc excitateurs alors que les récepteurs de type D2 sont inhibiteurs. Le substrat le mieux caractérisé de la PKA est la phosphoprotéine régulée par l’AMPc et la DA de 32 kDa (DARPP-32), principalement exprimée dans les neurones épineux moyens du striatum (MSNs). La DARPP-32 est un inhibiteur des phosphatases qui amplifie la signalisation par la PKA et agit de manière intégrative pour moduler la signalisation dopaminergique dans le striatum (17).
La signalisation par la dopamine est complexe et diversifiée. La voie impliquant la liaison à des protéines Gai/S qui module l’activité de l’adénylate cyclase est la plus importante et la mieux caractérisée.
Cependant, la dopamine peut également activer des récepteurs couplés à une protéine Gaq qui active la
phospholipase C (PLC). Cette voie de signalisation conduit ultimement à une augmentation du calcium intracellulaire, ce qui a des effets très variés dans la cellule par l’activation de plusieurs enzymes (Fig. 3) (22). Les récepteurs à la dopamine peuvent également interagir avec des canaux sodiques, les récepteurs au GABA ou l’ATPase Na+/K+, entre autres (15,23). Il existe également de voies de signalisation
indépendantes des récepteurs de type GPCR. La dopamine peut par exemple inhiber la voie PI3K-AkT-GSK3 par l’activation des récepteurs de type D2. Cette voie impliquant le recrutement de la protéine adaptatrice arrestine mène à l’inhibition de GSK3, une kinase ayant comme substrats connus la b-catenine, les récepteurs ionotropiques au glutamate (récepteurs NMDA) et des gènes horloges impliqués dans la régulation des cycles circadiens (24). Ainsi, le type de récepteur, la localisation dans le système nerveux central, l’état physiologique et l’interaction entre ces différentes voies de signalisation influencent l’effet physiologique observé lors de la signalisation dopaminergique (23).
Figure 3: Voies de signalisation activées par la dopamine. La liaison aux récepteurs de type D1 mène à
l’activation de l’adénylate cyclase et de la PKA, qui phosphoryle une variété de substrats, incluant DARPP-32. La liaison aux récepteurs de type D2 mène à l’inhibition de l’adénylate cyclase et de la PKA. La dopamine peut également activer des récepteurs liés à une protéine Gaq, qui active la PLC et conduit à une augmentation du calcium
intracellulaire. La liaison aux récepteurs de type D2 sur le neurone présynaptique peut également diminuer l’excitabilité du neurone (inhibition présynaptique).
Par ailleurs, l’activité des GPCRs est étroitement régulée. Ils peuvent tout d’abord être inactivés par les membres de la famille des régulateurs de la signalisation des protéines G (RGS). Les protéines RGS accélèrent la vitesse d’hydrolyse du GTP lié à la sous-unité a, limitant la durée de la signalisation (25). La sensibilité de ces récepteurs change également en fonction du degré de stimulation auquel ils sont exposés. Ils peuvent être désensibilisés lors d’une stimulation prolongée ou resensibilisés lors de l’absence de stimulation pendant une longue période. Un mécanisme important de désensibilisation se produit via une kinase des récepteurs couplés aux protéines G (GRKs). Après l’activation du récepteur, la GRK le phosphoryle à certains sites spécifiques et recrute des protéines adaptatrices de type arrestines. La liaison de ces protéines permet de bloquer l’activation du récepteur, même lorsqu’il y a encore présence de ligands (16). Les arrestines permettent également l’internalisation des GPCRs par la liaison à une protéine adaptatrice des clathrines et aux clathrines elles-mêmes. Ce procédé permet l’endocytose du récepteur, qui sera subséquemment retourné à la membrane ou dégradé par la fusion avec un lysosome (26,27). Le recrutement des arrestines joue donc un rôle primordial dans la modulation négative de la signalisation dopaminergique. Elles permettent également la liaison de protéines de signalisation intracellulaire telles que Akt, les MAP kinases ou c-Src (18).
2. Les ganglions de la base
La génération de mouvements est évidemment essentielle dans notre quotidien. De façon très simplifiée, le cortex moteur et la moelle épinière sont associés à la génération et la transmission des commandes motrices volontaires. Le tronc cérébral, qui reçoit une variété d’informations sensorielles, est généralement associé aux mouvements involontaires permettant le maintien de l’équilibre et l’ajustement postural de manière réflexe. La moelle épinière est également responsable de certains mouvements réflexes en réponse à des stimuli sensoriels (douleur, étirement musculaire ou autre) (28).
La génération des mouvements ne se résume cependant pas à la délivrance d’une commande motrice par le cortex moteur, puis sa transmission directe vers les muscles. Il existe des boucles de contrôle sous-corticales qui ont un impact indirect sur l’issue motrice. Un groupe de structures situées dans le télencéphale appelé ganglions de la base sont principalement importantes pour l’initiation des mouvements volontaires. Des lésions au niveau de ces structures affectent le tonus musculaire, la posture, les mouvements volontaires et génèrent même parfois des mouvements involontaires (29). Les ganglions de la base incluent le noyau caudé, le putamen, le globus pallidus interne et externe, le noyau sous-thalamique et la substance noire. Le noyau caudé et le putamen forment ensemble le striatum. Ce dernier est la cible principale des afférences corticales aux ganglions de la base. La substance noire est
qui sécrètent le neurotransmetteur dopamine. Ces neurones, appelés dopaminergiques, projettent vers le striatum dorsal et constituent la voie nigrostriée du système dopaminergique (30).
2.1 Le contrôle moteur
Toutes les structures décrite précédemment sont reliées ensemble par des connexions excitatrices et inhibitrices qui forment deux voies distinctes : la voie directe et la voie indirecte. Les deux voies ont un effet net opposé sur le cortex moteur et il s’agit apparemment de la balance contrôlée d’activité entre les deux qui permet un fonctionnement normal des ganglions de la base et du contrôle moteur (28).
Voyons plus en détail comment sont organisées ces différentes structures (Figure 4). Le cortex cérébral émet des efférences excitatrices glutamatergiques vers le striatum. Ce dernier projette vers le globus pallidus interne (GPi) et externe (GPe). Les connexions avec le GPi et le GPe sont de type inhibitrices GABAergiques. Le GPe émet des efférences GABAergiques vers le noyau sous-thalamique qui projette ensuite vers le GPi de manière excitatrice. Le GPi émet quant à lui des efférences inhibitrices vers le thalamus, qui retourne des efférences excitatrices vers le cortex. Finalement, la substance noire du
Figure 4: Schématisation des voies directe et indirecte des ganglions de la base dans le contrôle moteur.
La voie directe mène à l’activation du cortex par le thalamus et passe directement du striatum au globus pallidus interne. La voie indirecte mène à l’inhibition du cortex par le thalamus et passe du striatum au globus pallidus externe, puis au noyau sous-thalamique avant d’atteindre le globus pallidus interne. La substance noire augmente l’excitabilité du cortex en activant la voie directe et en inhibant la voie indirecte par l’activation des récepteurs D1 et D2 au striatum.
Cortex cérébral
Striatum (noyau caudé et putamen)
Globus pallidus interne (Gpi)
Globus pallidus externe (Gpe)
Thalamus Noyau sous-thalamique Substance noire (SNpc) + Récepteurs D1 - Récepteurs D2 + Glutamate + G lu tam at e + Glutamate - GABA - GABA - GABA - GABA
Voie indirecte inhibitrice Voie directe excitatrice
mésencéphale établit des connexions dopaminergiques avec le striatum. Le type de récepteur impliqué détermine ensuite l’effet obtenu (excitateur ou inhibiteur) (28,30).
La voie directe a comme effet final de faciliter l’initiation des mouvements volontaires par le cortex. Elle débute par l’excitation du striatum par le cortex, puis implique l’inhibition directe du GPi par le striatum. Puisque le GPi a un effet inhibiteur sur le thalamus, son inhibition par le striatum se traduit par une désinhibition du thalamus. Le thalamus étant désinhibé, il est libre de stimuler le cortex cérébral, ce qui permettra la génération du mouvement (Fig. 4). Dans la voie directe, les afférences dopaminergiques de la substance noire excitent les récepteurs dopaminergiques de type D1 au striatum, qui sont excitateurs. La voie nigrostriée, lorsqu’elle est active, a donc pour effet de stimuler davantage la voie directe (30,31).
La voie indirecte a comme effet final d’inhiber l’initiation des mouvements volontaires par le cortex. Dans cette voie, le striatum excité par le cortex inhibe le GPe plutôt que le GPi comme dans la voie directe. Le GPe a des projections GABAergiques vers le noyau subthalamique. Puisque les cellules du GPe sont inhibées par le striatum, le noyau subthalamique est désinhibé et est en mesure d’exciter le GPi. On se rappelle que le GPi a des projections inhibitrices vers le thalamus. Ce dernier est donc activement inhibé et son effet excitateur sur le cortex est réduit, inhibant la génération du mouvement (Fig. 4). Dans la voie indirecte, les afférences dopaminergiques de la substance noire excitent les récepteurs dopaminergiques de type D2 au striatum, qui sont inhibiteurs. Ainsi, lorsque la voie nigrostriée est activée, il se produit une inhibition de la voie indirecte, ce qui a pour effet net de favoriser l’excitation du cortex. L’activation de la voie nigrostriée favorise donc l’excitabilité du cortex de deux manières opposées : par l’activation de la voie directe excitatrice et l’inhibition de la voie indirecte inhibitrice (30,31).
Aujourd’hui, ce modèle d’organisation des ganglions de la base peut cependant être considéré comme simpliste, bien qu’il soit toujours valable. De nombreuses études neuroanatomiques dans les 30 dernières années suggèrent que l’architecture de ce système est en réalité sensiblement plus complexe. Les différentes régions anatomiques sont beaucoup plus interconnectées que ce qui avait été proposé initialement et sont également subdivisées en territoires fonctionnels (32,33). Par ailleurs, les neurones épineux moyens du striatum ne seraient pas ségrégés en deux populations distinctes exprimant uniquement des récepteurs de type D1 ou D2 et innervant chacune des cibles différentes. Des études ont montré que la majorité des neurones projettent à la fois vers le GPe, le GPi et la SNpr (substantia nigra pars reticula), et que certains expriment les deux types de récepteurs à la dopamine. Cela remet en question
le modèle traditionnel des voies directe et indirecte anatomiquement et fonctionnellement séparées (34,35).
3. La maladie de Parkinson
3.1 Origine des symptômes
La maladie de Parkinson est caractérisée par une dégénérescence des neurones dopaminergiques de la substance noire du mésencéphale (36). Les principaux neurones affectés sont impliqués dans la voie nigrostriée du système dopaminergique qui, comme mentionné précédemment, est impliquée dans le contrôle moteur (37). Le corps cellulaire des neurones de cette voie sont situés dans la SNpc, et leurs axones projettent vers le striatum dorsal. La perte des neurones au niveau de la SNpc provoque donc une diminution de la quantité de dopamine libérée dans le striatum dorsal (38). Selon le modèle traditionnel, cela provoque un débalancement de l’activité des voies directe et indirecte en faveur de la voie indirecte inhibitrice. En effet, la perte de dopamine au niveau du striatum empêche l’activation à la fois des récepteurs D1 et D2. Par conséquent, il se produirait une diminution de l’excitation de la voie directe et une diminution de l’inhibition de la voie indirecte résultant en une inhibition pathologique du cortex moteur (39). En considérant un modèle architectural plus récent des ganglions de la base, la perte d’innervation dopaminergique provoquerait cependant un débalancement fonctionnel beaucoup plus complexe impliquant des territoires spécifiques distincts du striatum (34). Ce sont ces changements dans l’innervation qui sont responsables de la majorité des symptômes moteurs caractéristiques de la maladie de Parkinson : rigidité posturale, bradykinésies, tremblements au repos et dyskinésies. On observe également l’apparition d’une posture inclinée vers l’avant et la disparition de mouvements posturaux réflexes, menant souvent à des chutes (3).
Il est important de mentionner que des symptômes non moteurs sont également très présents chez les patients parkinsoniens et que certains d’entre eux devancent l’apparition des symptômes moteurs de plusieurs années. C’est ce qu’on appelle la phase prodromale de la maladie. Les symptômes non moteurs comprennent des problèmes d’odorat, des problèmes de sommeil, des dysfonctions du système nerveux autonome et des problèmes cognitifs et/ou psychiatriques (40). Ils seraient associés à la perte de types cellulaires autres que les neurones dopaminergiques tels que les neurones cholinergiques ou sérotoninergiques (41). L’ensemble de ces symptômes affectent considérablement la qualité de vie des patients atteints en limitant progressivement leur autonomie et en les confinant ultimement à l’immobilité.
3.2 Étiologie
L’étiologie de la maladie de Parkinson est très hétérogène. On considère que des facteurs environnementaux et génétiques peuvent interagir et prédisposer au développement de la maladie, bien que la grande majorité des cas soient idiopathiques (MPi).
En 1983, de jeunes adultes ont développé des symptômes parkinsoniens permanents suite à la contamination de leur drogue injectable avec du 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). On découvrit subséquemment que le MPTP induit la mort des neurones dopaminergiques dans la substance noire, supportant l’hypothèse que des toxines environnementales pourraient être impliquées dans le développement de la maladie. Plusieurs études épidémiologiques ont été faites et ont menées à l’identification de certains facteurs de risque environnementaux. Ces derniers incluent l’exposition aux pesticides, une blessure antérieure à la tête, l’utilisation de bêtabloquants et la consommation d’eau provenant de puits (42). Par ailleurs, plusieurs études ont montré une diminution du risque de MP chez les personnes fumant la cigarette et chez les personnes consommant du café. Il est également important de mentionner que la prévalence de la MP est plus élevée chez les hommes que chez les femmes. Il est possible que les œstrogènes jouent un rôle protecteur, bien que des études additionnelles sont nécessaires afin de prouver cette association et d’élucider les mécanismes impliqués (43).
Dans un autre ordre d’idées, de nombreuses mutations génétiques ont été associées à différents niveaux au développement de la MP. Un certain nombre de mutations sont dites causatives (ou monogéniques), c’est-à-dire qu’elles sont associées à un risque très élevé de développer la maladie. C’est le cas des mutations dans les gènes SNCA (encodant l’alpha-synucléine), LRRK2 (encodant la Leucine-rich repeat kinase II), PINK1, GBA (encodant β-glucocerebrosidase) et Parkin, entre autres (42,44). Souvent, ces formes familiales ont une présentation clinique et histopathologique différente des cas idiopathiques, avec une apparition précoce et un risque plus élevé de démence, par exemple (43). Il existe également une multitude de polymorphismes dans d’autres gènes qui augmentent le risque de MP dans la population générale. Ce sont des gènes de susceptibilité. Il est possible que la mutation d’un de ces gènes ne soit pas en mesure à elle seule de causer la MP, mais que sa combinaison avec d’autres mutations ou avec des facteurs de risque environnementaux puisse prédisposer à la maladie. L’identification de ces mutations a permis le développement de modèles animaux récapitulant les caractéristiques histopathologiques et parfois les symptômes moteurs de la MP. Elles ont également permis la découverte de voies moléculaires qui sont altérées dans la maladie (autant sporadique que familiale) par l’étude du rôle de ces gènes dans la cellule, et par leur manipulation (42).
3.3 Caractéristiques histopathologiques
De façon générale, la MP affecte donc principalement les neurones dopaminergiques. Ces derniers présentent une variété de dysfonctionnements intracellulaires qui, ensemble, contribueraient à leur dégénérescence. Les deux principaux mécanismes qu’on suppose être impliqués dans la mort cellulaire sont 1) une dysfonction mitochondriale menant à un stress oxydatif, et 2) un défaut dans les voies de dégradation intracellulaire menant à l’agrégation anormale de protéines. La pathogénèse de la MP est complexe et il est fort probable qu’elle regroupe ces deux éléments, bien que l’ordre dans lequel ils entrent en jeu soit incertain.
L’agrégation protéique a historiquement retenu beaucoup l’attention. En effet, en 1912, Fritz Jacob Heinrich Lewy décrit pour la première fois la présence d’inclusions intracytoplasmiques dans certains neurones de patients atteints de la maladie de Parkinson (45). Ces inclusions toxiques, présentes dans les neurones dopaminergiques en dégénérescence, seront subséquemment nommées « corps de Lewy ». C’est suite à la découverte, en 1997, d’une mutation dans le gène codant pour la protéine alpha-synucléine (aSyn) que l’on découvrit que cette dernière est le principal constituant des corps de Lewy. D’autres protéines sont également présentes dans ces inclusions toxiques, incluant l’ubiquitine et la parkin (46). Ces agrégats ne sont par ailleurs pas restreints aux corps cellulaires. On les retrouve également dans les neurites, où ils sont appelés « neurites de Lewy » (47). Ces derniers sont plus abondants que les agrégats cytoplasmiques, et ils apparaissent plus précocement (48). Cette pathologie impliquant des corps et des neurites de Lewy ne se développe pas simultanément dans toutes les régions du cerveau. Certaines études ont suggéré que les premières régions à être touchées sont le bulbe olfactif et le noyau moteur dorsal du nerf vague (49,50). Selon l’hypothèse de Braak, la pathologie se propagerait à travers le cerveau en six stages, débutant dans les régions périphériques et progressant de manière postéro-antérieure (Fig. 5) (51). L’agrégation d’aSyn dans les régions autres que la substance noire pourrait être responsable des symptômes de la phase prodromale de la maladie (troubles du sommeil, constipation, dépression, etc.) (47,52). Finalement, les corps de Lewy ne sont pas spécifiques à la maladie de Parkinson et leur quantité
ne corrèle pas avec le niveau de perte neuronale. Leur contribution directe à la mortalité cellulaire est donc discutée (40,53).
3.4 L’alpha-synucléine
Des mutations ponctuelles (A53T, A30P, E46K, etc.) ou une multiplication du locus complet du gène SNCA encodant l’aSyn sont fortement associées au développement de la MP (54). Puisqu’il s’agit également de la principale protéine présente dans les corps de Lewy, son implication dans la dégénérescence neuronale a été largement étudiée. Les agrégats d’aSyn ne sont cependant pas unique à la MP. On les retrouve également dans d’autres maladies, incluant la démence avec corps de Lewy, l’atrophie multisystémique (MSA) et certains cas de maladie d’Alzheimer. On regroupe ces affections sous le terme synucléinopathies. Bien qu’un niveau élevé d’aSyn a très certainement une implication dans la pathogénèse de la MP, son rôle exact dans la dégénérescence cellulaire n’est pas connu. Notons
Stages 5 et 6
Plusieurs régions corticales (cortex insulaire, aires corticales associatives, etc.)
Stage 4
Système limbique (noyaux basolatéral et accessoires central de l'amygdale, claustrum ventral), thalamus (noyau intralaminaire), cortex temporal
Stage 3
Pont (noyau pédonculopontin), mésencéphale (substantia nigra pars compacta), cerveau antérieur basal, système limbique (noyau central de l'amygdale)
Stage 2
Pont (locus ceruleus, portion magnocellulaire de la formation réticulée, noyau raphé postérieur), substance grise de la moelle épinière
Stage 1
Système nerveux autonome, système olfactif (bulbe olfactif et noyau olfactif antérieur), bulbe rachidien (noyau moteur dorsal et nerfs glossopharyngiens)
Figure 5 : Les stages de Braak. Selon l’hypothèse de Braak, la pathologie se propage du système
d’ailleurs que les niveaux d’aSyn augmentent avec l’âge, même en l’absence de pathologie clinique (55). Ainsi, il n’est pas clair si l’accumulation d’aSyn est un « symptôme » de défaillances sous-jacentes, ou si elle en est la cause.
Tout d’abord, voyons un résumé de ce qui est connu de cette mystérieuse protéine. L’alpha-synucléine est composée de 140 résidus et est exprimée de manière ubiquitaire dans le cerveau. Son expression a également été rapportée dans des tissus non neuronaux incluant les jonctions neuromusculaires et les globules rouges (56,57). Dans le cerveau, elle est presque exclusivement localisée dans les terminaisons présynaptiques. L’aSyn fait partie de la famille des synucléines qui contient deux autres membres, soit la b-Syn et la g-Syn. Ces dernières sont également situées dans les terminaisons présynaptiques, mais leur patron d’expression n’est pas identique (58). Les trois synucléines semblent avoir des fonctions qui se chevauchent, puisque la délétion de l’une des synucléines ou d’une combinaison de deux ne cause aucun phénotype particulier chez la souris (59). L’aSyn n’a donc pas une fonction essentielle pour le fonctionnement et la survie cellulaire. La délétion des trois synucléines cause cependant des problèmes au niveau de la transmission et de la structure synaptique (60).
L’aSyn existe sous une forme soluble cytosolique et une forme associée aux membranes. Il existe un équilibre dynamique entre ces deux états, qui influencent la structure secondaire de la protéine. Dans sa forme soluble, l’aSyn est naturellement non repliée et monomérique (61). Certaines études ont suggéré qu’elle pouvait également exister sous forme de tétramère stable dans le cytoplasme (62,63). En présence d’une membrane lipidique, sa région N-terminale adopte une structure alpha-hélicoïdale qui lui permet de s’associer à la membrane. L’aSyn a tendance à s’associer préférablement aux membranes à courbure prononcée et est donc souvent liée aux vésicules synaptiques (64,65). Le repliement de l’aSyn et son association aux membranes est nécessaire à ses fonctions et prévient son agrégation (66). Dans un contexte pathologique, elle a tendance à former des feuillets beta qui s’agrègent, forment des fibrilles et se regroupent en corps de Lewy (67,68). Ces conformations toxiques dérivent plausiblement de la forme soluble non-repliée présente dans le cytoplasme (69). Plus de 90% de l’aSyn présente dans les corps de Lewy est phosphorylée au niveau de la sérine 129 (pSer129). Cela suggère un rôle important de cette modification post-traductionnelle dans les mécanismes qui contrôlent son agrégation. Cependant, le rôle pathologique de la pSer129-aSyn est débattu puisque plusieurs études ont obtenu des résultats contradictoire quant à son implication dans la mortalité cellulaire (70).
Les fonctions de l’aSyn sont largement inconnues. Ses deux implications les mieux décrites ont rapport au métabolisme de la dopamine et à la transmission synaptique. Tout d’abord, l’aSyn se lie au transporteur de la dopamine DAT et module son activité (71). Elle inhibe également l’expression et l’activité de la tyrosine hydroxylase, ce qui diminue la synthèse de la dopamine (72,73). La surexpression de l’aSyn inhibe l’activité des transporteurs VMAT2, ce qui dérègle l’homéostasie de la dopamine en augmentant ses taux cytosoliques (74). Ces éléments corrèlent avec la susceptibilité singulière des neurones dopaminergiques à la toxicité par l’aSyn. Par ailleurs, la localisation présynaptique de l’aSyn, son association aux membranes à courbure prononcée et son interaction avec d’autres protéines synaptiques telle la synaptobrevin-2 supportent un rôle dans la transmission synaptique (58,75). Elle ne semble pas avoir un rôle direct et essentiel dans la libération des neurotransmetteurs. Cependant, l’aSyn promeut le regroupement des vésicules synaptiques par sa capacité à former des multimères à la surface des vésicules (76). Ce procédé contribue également à stimuler la formation des complexes de protéines SNAREs, qui sont nécessaires à la fusion des membranes permettant la libération des neurotransmetteurs. L’aSyn jouerait donc un rôle important dans la régulation du nombre de vésicules synaptiques présentes à la membrane lors de la libération des neurotransmetteurs (75,77).
Finalement, plusieurs études ont récemment montré que certaines formes d’aSyn peuvent être libérées dans l’espace extracellulaire et stimuler l’agrégation de la protéine endogène dans les neurones avoisinants. Cette hypothèse a émergé après la découverte de corps de Lewy dans des transplants de tissus mésencéphalique fœtaux, plus de 10 ans après l’intervention (78,79). L’aSyn agirait donc comme une molécule de type prion. La majorité de l’aSyn serait libérée par exocytose, alors qu’une petite portion serait liée aux exosomes (80). Elle serait ensuite principalement intégrée par les autres neurones par endocytose, bien que le mécanisme de sa liaison aux membranes ne soit pas encore connu (81,82). Les agrégats toxiques peuvent être transportés dans les axones, ce qui expliquerait la propagation de la pathologie à des régions du cerveau très éloignées (83).
3.5 Mécanismes compensatoires
Il est bien connu que les symptômes moteurs de la MP apparaissent uniquement lorsque près de 60% des neurones dopaminergiques ont disparu et que la concentration de dopamine a diminué d’environ 70% (84). Il existe donc des mécanismes compensatoires qui camouflent la dégénérescence prenant place dans le mésencéphale et qui retardent le développement des symptômes moteurs (Fig. 6). Ces mécanismes ne sont toujours pas bien expliqués en raison des résultats contradictoires obtenus dans plusieurs études et de la difficulté à ségréguer les effets résultant d’une compensation de ceux découlant de la pathologie (85). D’abord, l’augmentation du métabolisme de la dopamine est l’un des premiers mécanismes suggérés
Figure 6: Les principaux mécanismes compensatoires prenant place avant l'apparition des symptômes moteurs. A) Conditions physiologiques B) Augmentation
de la synthèse et du métabolisme de la dopamine C) Diminution de l’expression de DAT
D) Augmentation du nombre de récepteurs de type D2 E) Diffusion extrasynaptique de
et celui-ci est maintenant largement accepté. L’augmentation du contenu dopaminergique et le ralentissement de son inactivation permettent d’augmenter la libération d’un facteur cinq par neurone (86,87). Ce mécanisme est possiblement couplé à une diminution de la recapture de la dopamine afin d’augmenter la quantité de dopamine disponible dans les fentes synaptiques. Une diminution de l’expression du transporteur DAT a d’ailleurs été observé dans le cerveau de patients parkinsoniens (88,89). Les récepteurs striataux à la dopamine semblent également affectés lors de la phase asymptomatique. L’un des mécanismes les mieux étudiés est l’augmentation du nombre de récepteurs de type D2 sur les neurones postsynaptiques (90,91). Finalement, la diffusion de la dopamine à l’extérieur de la fente synaptique est également un mécanisme probable de compensation. La réduction de la recapture et du catabolisme augmente la capacité de diffusion de la dopamine et permet une transmission extrasynaptique (92–94). Ces mécanismes ne sont pas exclusifs et il n’est pas clair à quel moment ils prennent place au cours du développement de la maladie. L’étendue de leur rôle dans la compensation n'est également pas connue.
3.6 Les traitements actuels
Actuellement, il n’existe aucun traitement permettant de stopper ou de retarder la progression de la maladie de Parkinson. La MP n’est pas causée par le défaut d’un seul processus cellulaire, mais bien d’une combinaison de facteurs. Sa pathogenèse est complexe et hétérogène. Il serait donc surprenant qu’une seule molécule thérapeutique soit en mesure de traiter tous les patients atteints de la MP (42). Les thérapies existantes ne visent qu’au contrôle des symptômes moteurs et non moteurs afin d’améliorer la qualité de vie des patients. Les médicaments les plus fréquemment utilisés sont le levodopa, les agonistes de la dopamine et les inhibiteurs de la monoamine oxydase de type B (95). Le levodopa est en réalité du L-DOPA, un précurseur de la dopamine qui, contrairement à cette dernière, est en mesure de traverser la barrière hémato-encéphalique. Le levodopa est le traitement qui a montré le meilleur effet sur les symptômes moteurs. Cependant, à long terme, il provoque des effets secondaires incapacitants tels que des dyskinésies et des fluctuations motrices. Les agonistes de la dopamine permettent de « remplacer » la dopamine par l’activation des récepteurs dopaminergiques. Cependant, ils sont associés à des comportements impulsifs tels que le jeux pathologique, l’hypersexualité ou la frénésie alimentaire. Ils sont également davantage associés au développement d’hallucinations et sont donc contre-indiqués chez les patients âgés et ayant déjà souffert d’addictions. Les inhibiteurs de la monoamine oxydase de type B permettent de ralentir la dégradation de la dopamine afin de prolonger son effet. Ils n’ont cependant qu’un effet bénéfique modéré (42,95,96). Finalement, la stimulation crânienne profonde est un traitement nécessitant une intervention chirurgicale qui s’est montré efficace pour contrôler les symptômes dans les cas avancés de la maladie. Elle consiste en la stimulation avec un électrode du noyau subthalamique ou
symptômes non-moteurs tels que les troubles du sommeil et les troubles comportementaux. Il est cependant possible qu’une partie de ces effets soient modulés par l’amélioration des symptômes moteurs et la diminution de la dose des médicaments administrés (97).
3. Leucine-rich repeat kinase 2 (LRRK2)
Comme mentionné précédemment, des mutations dans le gène LRRK2 ont été associées à la forme familiale de la MP. LRRK2 est une protéine volumineuse et multifonctionnelle qui contient un domaine GTPase et un domaine kinase, entourés de domaines d’interaction protéique. Sept mutations dans des régions codantes de ce gène ont été identifiées (N1437H, R1441G, R1441C, R1441H, Y1699C, G2019S and I2020T) et causent une MP à développement tardif. Elles ont globalement une présentation clinique et histopathologique semblable à celle du Parkinson idiopathique (98). La MP familiale causée par LRRK2 pourrait donc représenter une fenêtre intéressante pour l’étude du Parkinson idiopathique. D’autres polymorphismes dans des régions codantes et non-codantes ont également été associées à un risque plus élevé de MP (99). La mutation G2019S, une substitution d’une glycine par une sérine dans le domaine kinase de la protéine, représente 4% des formes familiales de MP dans le monde (100). Sa pénétrance est variable, mais atteint plus de 30% des formes familiales dans certaines populations telles que les Juifs Ashkenazi (101). Cette mutation entraîne un gain de fonction pathologique de la kinase, de deux à trois fois le niveau normal d’activité (102).
L’ensemble des rôles physiologiques de LRRK2 ne sont pas connus. La recherche extensive sur cette protéine a mené à l’identification de plus de 250 interacteurs potentiels (103). Ces données suggèrent que LRRK2 pourrait être en mesure d’interagir avec une multitude de protéines et de réguler des fonctions cellulaires et voies de signalisation diverses en fonction du type cellulaire, du stage développemental, du stimulus spécifique et du substrat impliqué (104). Ainsi, elle serait entre autres impliquée dans le trafic de vésicules, l’autophagie, la dynamique du cytosquelette, la transmission synaptique, les fonctions mitochondriales et la réponse immunitaire (42,105–107). Les mutations dans LRRK2 prédisposent donc à l’apparition de la MP en raison de ses fonctions variées, dont plusieurs sont affectées dans la maladie.
Parmi tous ces rôles présumés, l’interaction de LRRK2 avec les GTPases Rab et son implication dans l’autophagie sont les plus pertinents pour ce projet. D’abord, LRRK2 phosphoryle directement Rab10 au niveau de la thréonine 73 (T73). Des analogues de Rab10 qui possèdent une T73 sont également efficacement phosphorylées par LRRK2. La phosphorylation de ce résidu module l’interaction des GTPases Rab avec certaines molécules régulatrices, ce qui affecte leur localisation cellulaire et leur activité. Par exemple, la mutation G2019S augmente le niveau de phosphorylation des Rabs, ce qui favorise leur insertion membranaire sous une forme inactive. Puisque les GTPases Rab sont impliquées dans le
transport de vésicules intracellulaires, un débalancement dans leur homéostasie perturbe le transport vésiculaire (108) et pourrait conséquemment avoir un effet sur l’autophagie et les fonctions lysosomales (109). Il est important de mentionner que les GTPases Rab font partie de la superfamille des GTPases Ras, dont il sera question dans une prochaine section. Par ailleurs, de nombreuses études ont montré que LRRK2 joue un rôle dans la régulation de l’autophagie, bien qu’il n’y ait pas de consensus sur la direction de son effet. Des études in vitro ont donné des résultats contradictoires quant à sa capacité à activer ou inhiber l’autophagie (107). La déplétion complète de LRRK2 ne semble pas avoir d’effet sur l’autophagie dans le cerveau, mais plusieurs mutations dans son gène, incluant la mutation G2019S, semblent altérer le processus autophagique normal et provoquent l’accumulation de pS129-aSyn (110,111). De plus, il apparait que LRRK2 soit importante pour le fonctionnement des lysosomes. Dans des souris et dans des neurones primaires, la surexpression de la mutation G2019S provoque l’apparition de lysosomes anormalement volumineux et avec un pH plus bas (112). L’utilisation d’inhibiteurs pharmacologiques de LRRK2 ont été en mesure de rétablir le pH et la morphologie normale des lysosomes, et ont montré une capacité générale à activer l’autophagie. Cela suggère qu’une hyperactivation de la kinase altère l’efficacité de la mécanique autophagique (113,114). La complexité du réseau de signalisation par lequel LRRK2 module l’autophagie rend difficile l’étude de ses effets exacts (107).
Il est intéressant de constater qu’il semble exister un lien reliant l’alpha-synucléine et LRRK2 dans la MP. En effet, on a montré que la surexpression de LRRK2-G2019S décuplait la pathologie observée à la suite de l’injection de fibrilles d’aSyn préformés, et que la délétion ou l’inhibition de LRRK2 empêchait la neurodégénérescence causée par la surexpression virale de l’aSyn. Ces éléments suggèrent que LRRK2 module de manière inconnue l’agrégation pathologique de l’aSyn, possiblement par une action au niveau du système autophagique (115,116).
4. L’autophagie
L’accumulation anormale de protéines dans les cellules pourrait résulter d’un défaut au niveau des procédés de dégradation intracellulaires. Les études réalisées sur LRRK2 et ses cibles identifiées jusqu’à maintenant supportent son implication dans la régulation de l’autophagie et donc son rôle possible dans la pathogénèse de la MP lorsque son fonctionnement est altéré. Pour bien comprendre les résultats du présent projet, un bref survol du processus autophagique est nécessaire.
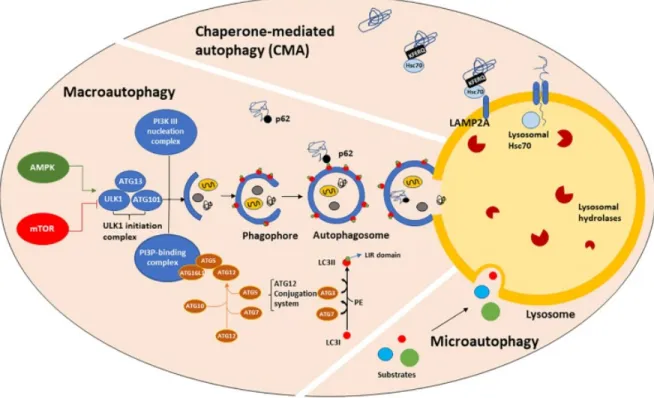
L’autophagie est un processus conservé dans toutes les cellules et est essentiel à leur survie. Ce processus dégrade et recycle les organelles et protéines endommagées afin d’éviter qu’elles n’interfèrent avec le fonctionnement cellulaire normal. Il existe trois types d’autophagie : la microautophagie, l’autophagie médiée par des chaperonnes (CMA) et la macroautophagie (Fig. 7). La microautophagie consiste en une petite invagination de la membrane lysosomale afin de dégrader du contenu cytoplasmique. La CMA permet la dégradation ciblée d’éléments intracellulaires par leur marquage avec un peptide spécifique, leur reconnaissance par une protéine chaperonne et leur translocation dans le lysosome. La macroautophagie, dont il sera question dans ce travail et qui sera donc appelée simplement autophagie, est activée en fonction de l’état nutritionnel de la cellule (niveaux de nutriments, d’oxygène, de facteurs de croissance et d’AMP/ATP). Elle se caractérise par la formation d’une double-membrane, le phagophore, qui se referme autour du cargo à éliminer. La vésicule ainsi formée est appelée l’autophagosome. Ce dernier fusionne ensuite avec un lysosome contenant des hydrolases qui permettent la dégradation du cargo (112). Une famille de gènes relatifs à l’autophagie (gènes ATG) code pour des protéines qui régulent l’initiation du processus.
Figure 7: Les trois types d'autophagie. L’autophagie médiée par les chaperonnes permet l’élimination sélective
d’un cargo par sa reconnaissance par une protéine chaperonne, qui l’achemine à la membrane lysosomale. La macroautophagie implique la formation d’une membrane se refermant autour des éléments à dégrader dans le cytoplasme : l’autophagosome. La fusion de l’autophagosome avec un lysosome permet la dégradation du cargo. Il s’agit d’un processus étroitement régulé par les gènes ATG et dépendant du contexte nutritionnel de la cellule. La microautophagie consiste en de petites invaginations dans la membrane du lysosome pour digérer des substrats cytoplasmiques.
De manière plus détaillée, ce processus comprend quatre étapes principales : l’initiation, la nucléation, l’élongation et la fusion. L’initiation prend place en cas de carence nutritionnelle. Elle débute par l’inhibition du complexe mTORC1 (mammalian target of rapamycin complex 1) qui provoque la dissociation du complexe ULK1 (Unc-51 like autophagy activating kinase). Ce dernier est subséquemment activé par l’AMPK (adénosine monophosphate kinase) et provoque l’apparition d’une membrane recourbée appelée phagophore. ULK1 active ensuite le complexe de nucléation PI3K-III (phosphoinositide 3 kinase), qui comprend ATG14, Beclin-1 et VSP34 (vesicular sorting protein 34), entre autres. VSP34 permet la synthèse du phosphatidylinositol triphosphate (PI3P), un phospholipide important pour l’élongation du phagophore et le recrutement d’autres protéines ATG à la membrane (107,112,117) (Fig. 7). Par la suite, l’élongation dépend de deux complexes de conjugaison : Atg5-Atg12 et LC3B-PE (phosphatidyléthanolamine). Le complexe Atg5-Atg12 (qui comprend également Atg16L) permet la courbure de la membrane et est également nécessaire pour le recrutement de LC3B à la membrane. Ce dernier est présent dans le cytoplasme sous forme de LC3B-I. Sa conjugaison au PE le transforme en LC3B-II, qui est ensuite intégré à la membrane de l’autophagosome où il joue un rôle dans la fusion des membranes et dans la sélection des cargos (117). En effet, en plus de l’engloutissement d’organelles et de protéines directement dans le cytoplasme par le phagophore, certains cargos peuvent être spécifiquement reconnu et
transportés dans l’autophagosome. La forme lipidée LC3B-II agit comme récepteur de protéines adaptatrices telles p62, qui reconnaissent des substrats spécifiques et les acheminent vers la membrane de l’autophagosome (118). La synthèse de LC3 augmente lors de l’induction de l’autophagie, ce qui en fait un bon marqueur du niveau d’autophagie dans les cellules (117).
Un processus autophagique normal est primordial afin de maintenir l’homéostasie cellulaire. En raison de leur état post-mitotique, les neurones
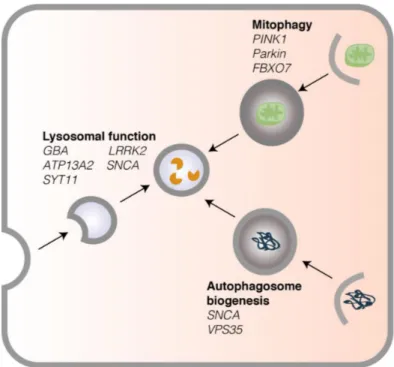
Figure 8: Gènes associés à la maladie de Parkinson impliqués
dans la mitophagie, la biogénèse des autophagosomes, les fonctions lysosomales ou la formation des lysosomes. Source : Essays
dégradation intracellulaire (112). Le vieillissement normal entraîne une baisse de l’efficacité de l’autophagie (119). Cela se traduit par l’accumulation de radicaux libres, de protéines mal repliées, inactives ou oxydées et de mitochondries endommagées. Il peut en résulter un stress oxydatif et la formation d’agrégats cytotoxiques qui altèrent les fonctions cellulaires (107,120). Dans la MP idiopathique, on a observé une baisse des marqueurs lysosomaux (LAMP1, LAMP2A) et une augmentation des marqueurs des autophagosomes (LC3II, p62)(121,122). Des altérations dans les marqueurs autophagiques indiquent une dérégulation du processus autophagique normal. D’ailleurs, la majorité des mutations associées à la MP sont des gènes impliqués dans les voies de dégradation intracellulaires, que ce soit l’autophagie ou la dégradation par le protéasome (Fig. 8)(112).
5. Rin
5.1 Les GTPases
Une GTPase est une protéine qui agit comme un interrupteur moléculaire à l’intérieur de la cellule pour activer d’autres protéines ou des voies de signalisation. Elle remplit sa fonction en alternant entre sa forme active, liée au GTP, et sa forme inactive, liée au GDP. La grande famille des GTPase Ras est composée de plusieurs protéines structurellement semblables et de faible poids moléculaire impliquées dans la transduction de signal et dans la régulation de plusieurs processus cellulaires incluant la croissance, la différenciation, le transport vésiculaire et l’apoptose (123). Pour alterner entre leur forme active et inactive, les GTPases Ras nécessitent l’intervention des protéines GEF (facteur d’échange de nucléotide guanine) et GAP (protéine activatrice/accélératrice de l’activité GTPase). Ce sont également ces protéines qui permettent la régulation étroite de leur activité en fonction de différents signaux activateurs (124). De façon générale, les GTPases Ras sont liées à la membrane dans leur forme inactive liée au GDP. Le recrutement d’une GEF induit un changement de conformation qui réduit l’affinité du GDP et permet sa libération. Puisque la quantité de GTP dans les cellules est plus importante que la quantité de GDP, le GTP remplace le GDP sur la GTPase. Cet échange provoque un changement de conformation dans deux régions spécifiques de la protéine, Switch 1 et Switch 2, qui sont ensuite en mesure d’interagir avec des protéines effectrices. Le retour à l’état inactif se produit en partie par l’activité GTPase de Ras. Cependant, puisque son efficacité enzymatique est faible, la présence d’une protéine GAP permet d’accélérer considérablement la vitesse d’hydrolyse du GTP et le retour à l’état inactif lié au GDP (124,125). Dans la majorité des cas, l’association membranaire des GTPases Ras est nécessaire à leur activité. Elle doivent donc subir une modification post-traductionnelle permettant l’ajout d’un groupement isoprényl dans un domaine conservé de leur région C-terminale qui permet leur liaison à la membrane (126).
5.2 La petite GTPase Rin