Les chimiothèques ciblant les interactions protéine-protéine
Texte intégral
Figure
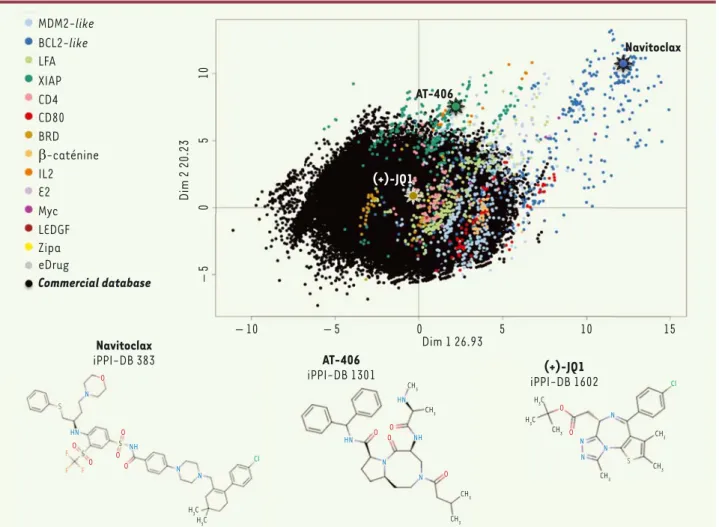


Documents relatifs
Il y aura donc lieu d'examiner dans quelle mesure et à quel titre (véritable droit privé ou droit public supplétif) le droit privé peut intervenir dans l'activité administrative.
Le droit non édicté ne peut être défini que négativement comme regroupant l'ensemble des normes qui sont bien des règles de droit, mais qui n'ont pas fait l'objet d'une
Selon le droit en vigueur jusqu’à fin 2006, qui est celui sur lequel la jurisprudence connue aujourd’hui s’est fondée, les décisions incidentes ne peuvent en effet être
101. Les juridictions administratives peuvent annuler une décision au motif que celle-ci est arbitraire. Cependant, comme l'arbitraire résulte d'une illégalité ou d'un abus du
Ce sera essentiellement le cas lorsqu'une collectivité publique est atteinte de la même manière qu’un administré, notamment si elle est touchée dans ses intérêts
71 PA, la plainte à l'autorité de surveillance (Aufsichtsbeschwerde) consiste dans le fait de dénoncer à l'autorité de surveillance les agissements (sous forme de décision ou
Pour des motifs d'économie de procédure, le droit suisse admet, sauf disposition légale contraire (p. 1 LEx), que l'autorité administrative ou judiciaire saisie tranche elle-même
Déterminer la distance de D aux trois points A, B et C, ainsi que le projeté orthogonal de D sur (ABC), et la distance de D à ce dernier. 7)En déduire le volume du tétraèdre
