Open Archive TOULOUSE Archive Ouverte (OATAO)
OATAO is an open access repository that collects the work of Toulouse researchers andmakes it freely available over the web where possible.
This is an author-deposited version published in : http://oatao.univ-toulouse.fr/
Eprints ID : 5919
To link to this article :
URL : http://www.electrochemsci.org/papers/vol7/7065429.pdf
To cite this version : Barus, Carole and Wetz-Torond, Fabienne and Brunel, Yves and Gros, Pierre Electrochemical Study of Antioxidant Regeneration Mechanisms – Application in Dermocosmetics. (2012) International Journal of Electrochemical Science, vol. 7 . pp. 5429-5441. ISSN 1452-3981
Any correspondence concerning this service should be sent to the repository administrator: [email protected]
Electrochemical Study of Antioxidant Regeneration
Mechanisms – Application in Dermocosmetics
Carole Barus1, Fabienne Wetz-Torond2, Yves Brunel2 and Pierre Gros1,*
1 Université de Toulouse, Laboratoire de Génie Chimique UMR CNRS/INP/UPS 5503, Université Paul Sabatier, 31062 Toulouse Cedex 9, France.
2 Laboratoire de Pharmacochimie Pierre Fabre Dermo-Cosmétique, Centre de Recherche Pierre Fabre, 3 avenue Hubert Curien, 31035 Toulouse Cedex 1, France.
*E-mail: [email protected]
Oxidative stress is associated to the massive generation of reactive oxygen species inducing fast oxidative reactions in chain. To overcome this problem, reasonable supplementations of antioxidants are widely practiced, mostly based on empirical protocols. An electrochemical process is proposed to choose the best molecules association inducing the greatest antioxidant capacity, by coupling homogeneous or heterogeneous catalytic reactions involving antioxidants with an electrochemical step. Cyclic voltammetry and constant potential electrolysis experiments were used to highlight regeneration reactions induced by specific associations of hydrophilic and lipophilic antioxidants in homogeneous medium or at liquid/liquid interface. Results showed that N-Acetyl-L-Cysteine (NAC) was effectively regenerated by both ascorbic acid (AA) and ascorbyl glucoside (AA-2G) but not by ascorbyl phosphate magnesium (AA-2P). The antioxidant properties of -tocopherol also increased to 40% when associated with AA. The process was successfully applied for the first time to the study of simple emulsions. Formulations involving NAC and AA-2G presented the highest synergic effect, the antioxidant capacity being amplified by more than 35%. On the other hand no catalytic mechanism was observed when introducing three antioxidants in the cream because it induced too low reaction kinetics. This electrochemical process can thus be exploited as a tool for the optimisation of “anti-aging” dermocosmetic formulations.
Keywords: electrochemical process – antioxidant capacity – catalytic mechanism – liquid/liquid
interface – dermocosmetics.
1. INTRODUCTION
Oxidative stress is the subject of tremendous attention, particularly since the end of the 20th century. It results from an imbalance between the production of oxidizing chemical species and their
effective removal by protective antioxidant molecules and scavenger enzymes. It is associated to the massive generation of reactive oxygen (ROS) and nitrogen (RNS) species inducing fast oxidative reactions in chain [1]. It is now clearly accepted that numerous pathologies and clinical disorders, i.e. aging, cancers, atherosclerosis, degenerative diseases... are directly or indirectly consecutive to continuous and/or repetitive exposure to oxidative stress [2]. Skin is one of the major targets since it provides the first line of defense against external oxidant aggressions such as UV radiations, ozone or chemicals, which contribute to the development of irreversible damages: psoriasis, premature aging, cancers... [3]. To prevent or reduce oxidative stress, reasonable supplementations of antioxidants are widely practiced [4,5]. In dermocosmetic field a large variety of creams or lotions are available on the market. However, the choice of antiradical constituent association to optimise the formulation performances is mostly based on empirical protocols, even if analytical strategies have been developed to detect and assay antioxidant properties in the final product [6,7].
Numerous chemical or biochemical protocols are been developed to evaluate the oxidative damages and antioxidant ability [8]. Several studies have made evidence antioxidant synergy and regeneration effect in antioxidant mixtures containing vitamin E [9-12], -carotene [13], ascorbic acid [14] or polyphenols [15,16] in various application fields such as clinical biology, pharmaceuticals, cosmetics and food. In most studies the analytical methods involve complex protocols and/or expensive materials such as UV spectrophotometry, IR spectroscopy, oximetry (electron spin resonance), high performance liquid chromatography or laser flash photolysis. Comparatively electrochemistry appears as a convenient way to determine the antioxidant status of a complex medium. Electrochemical techniques (four electrode system, three phase electrode, thin layer voltammetry, scanning electrochemical microscopy...) have been extensively used to study ion and/or charge transfer reactions at the interface between two immiscible electrolyte solutions [17-20]. Some of them have been applied to determine kinetic parameters associated with the oxidation of antioxidants (mainly ascorbic acid) at liquid/liquid interface [21-23]. More recently Bertolino et al. [24] highlighted the synergic effect of selenium with natural and synthetic antioxidants. However all these studies have been developed in model systems and no further experiments have been conducted in real complex media. On the other hand previous works performed in our laboratory have shown the possibility to define an indicator of the antioxidant global properties of real samples like wine, blood serum, skin or cosmetics by direct electrochemical measurements, without any pre-treatment of the samples [25-28]. Another electrochemical study has highlighted an increase in the global antioxidant capacity of a human skin treated by daily application of a dermocosmetic cream containing antioxidants [29]. Recently a catalytic electrochemical / chemical (EC’) mechanism of uric acid regeneration by ascorbic acid was demonstrated during the simultaneous assay of both antioxidant markers at the vicinity of a poly(3,4-ethylenedioxythiophene) modified gold electrode surface [30].
In the present work an electrochemical process is proposed to predict reactions induced by the association of antioxidants. Experiments were performed by coupling the electrochemical step with homogeneous or heterogeneous catalytic reactions involving antioxidants [31]. Catalytic currents resulting from cyclic voltammetry and chronoamperometry experiments were exploited to determine redox mechanisms induced by specific associations of hydrophilic and/or lipophilic antioxidants in homogeneous and biphasic (organic/water) media. In this latter case, a thin organic film deposited on
the electrode surface appeared to be a simple and powerful tool to study the electron transfer phenomena at liquid/liquid interface [32-34]. Finally, experiments in dermocosmetic creams were carried out to confirm the results obtained in previous model systems.
2. MATERIAL AND METHODS
2.1. Chemicals
Sulphuric acid (H2SO4)95 % w/v, potassium di-hydrogenophosphate (KH2PO4), di-potassium hydrogenophosphate (K2HPO4), tetrabutylammonium perchlorate ((Bu)4NClO4) (TBAP), ascorbic acid (AA) and N-Acetyl-L-Cysteine (NAC) were purchased from Acros Organics. Sodium perchlorate monohydrate (NaClO4, H2O) was obtained from Fluka, dichloromethane (CH2Cl2) from VWR and -tocopherol (Vit. E) from Calbiochem. Finally, dermocosmetic creams, Butylated hydroxyanisole (BHA), 6-Di-tert-butyl-4-methylphenol (BHT), ascorbyl glucoside (AA-2G) and ascorbyl phosphate magnesium (AA-2P) were made in Pierre Fabre's laboratory.
2.2. Materials
Electrochemical manipulations were performed at constant room temperature (298 K) with an Autolab Metrohm potentiostat interfaced to an HP omni-book XE 4500 microcomputer and using the GPES 4.9 software. A conventional three-electrode system was used for all the experiments. Several gold disks with different sizes were used as working electrodes. Platinum grid acted as counter electrode and the reference electrode was a saturated calomel electrode (SCE) (Hg/Hg2Cl2, KClsat) connected to the electrochemical cell by a Luggin capillary. All potentials were expressed versus this electrode. All the solutions were deaerated by bubbling nitrogen for 10 min.
2.2.1. Experiment in homogeneous media (aqueous or organic solutions)
The surface area of the electrode was 0.071 cm². Aqueous solution was 0.1 mol.L-1 phosphate buffer (PBS) pH 7. Organic solvent was dichloromethane containing 0.1 mol.L-1 TBAP as electrolyte.
2.2.2. Experiment in biphasic media
The surface area of the working electrode was higher than previously (1.96 cm²) in order to deposit easily the organic film on the electrochemical transducer. Biphasic solutions were basically constituted with 400 µL of water-saturated dichloromethane solvent containing 0.1 mol.L-1 TBAP as electrolyte and 25 mL of dichloromethane-saturated distilled water containing 1 mol.L-1 NaClO
4 as electrolyte. Water was saturated with dichloromethane to prevent dissolution of the organic film into the aqueous phase and vice versa. Lipophilic and hydrophilic antioxidants were added in organic and aqueous phases, respectively. 400 µL of the organic phase was necessary to cover the whole electrode surface. Counter and reference electrodes were placed into the aqueous phase without being in contact
with dichloromethane. Bubbling nitrogen directly into bulk solution was not possible because it caused too strong agitation of biphasic medium and destroyed the organic film. Therefore a nitrogen flow was maintained over the solution during all the experiments.
2.2.3. Experiment in creams
The creams studied were specially made by Pierre Fabre. The basic emulsion (background) was an oil phase dispersed in an aqueous phase (Dexeryl ®). For other formulations, one or more antioxidants were successively added in the emulsion. The working electrode was the same as that previously used for biphasic media study (1.96 cm²). The electrode surface was covered with a 4 mm thick cream film and the whole was immersed into 25 mL of 1 mol.L-1 NaClO
4 aqueous solution. The counter and references electrodes were placed into the aqueous phase. This configuration was preferred to that used previously by Guitton et al. [26] where the 3 electrode system was introduced into bulk creams, which induced in our case a strong ohmic resistance and resulted in irreproducible data. As previously a nitrogen flow was maintained over the solution during all the experiments.
2.3. Electrode activation and signal treatment
The gold electrode surfaces were polished with abrasive paper (262X imperial lapping film sheets) and rinsed with distilled water. Cathodic and anodic polarizations were then performed at -2 V and +2 V successively during 30 sec in 0.5 mol.L-1 H
2SO4. Finally, cyclic voltammograms were carried out in 0.5 mol.L-1 H
2SO4 at 200 mV.s-1 between -0.1 V and 1.4 V, until reproducible current-potential curves were obtained.
For all electrochemical measurements the potential range was chosen in order to avoid oxidation and reduction of the solvent. The background current was systematically recorded by plotting the current-potential curve with the supporting electrolyte only. To optimize the exploitation of the numerical data, the background was subtracted from the overall experimental curves by using GPES software. When some close anodic signals were present, peaks were deconvoluted using the Microcal Origin 7.0 software including the peak fitting module [35].
3. RESULTS AND DISCUSSION
3.1. Catalytic reactions in homogeneous media
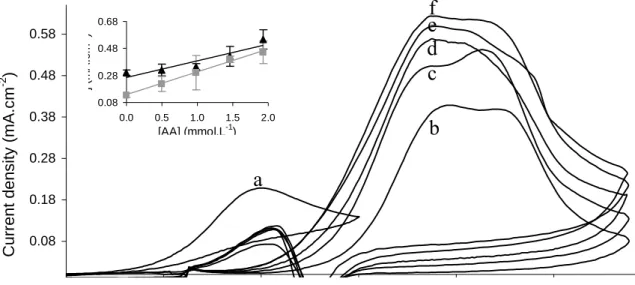
Fig. 1 presents cyclic voltammograms recorded in 0.1 mol.L-1 PBS pH 7 containing 1 mmol.L-1 ascorbic acid (AA) (curve a) and 1 mmol.L-1 N-acetyl-L-cysteine (NAC) (curve b). For both curves the background current was subtracted as described in section 2.3. On curve (a), an anodic peak corresponding to AA oxidation was observed at potential around 0.35 V. Two peaks corresponding to NAC oxidation appeared on curve (b) at 0.75 and 0.9 V, respectively. Previous works performed in our laboratory have shown that the first peak (0.75 V) was limited by diffusion of NAC in solution and the
second one (0.9 V) was controlled by adsorption phenomena [36]. For both molecules, no cathodic peak appeared in the reverse scan, indicating the irreversibility of the redox systems. On curve (b), the anodic peak observed at 0.42 V during the backward scan corresponded to the re-oxidation of NAC after the reduction of gold oxides occurring at 0.5 V [36].
Curves (c) to (f) correspond to cyclic voltammograms obtained with solutions containing both NAC and AA. The NAC concentration was kept constant whereas AA concentration increased. Although AA oxidised at lower potential than NAC, no peak was recorded for AA and only both anodic NAC peaks were observed. The strong affinity of NAC for gold and its spontaneous adsorption on electrode surface [36] could explain this phenomenon. To confirm this hypothesis, film transfer experiments were carried out. Briefly, the gold electrode was firstly immersed in 1 mmol.L-1 NAC during 1 hour, rinsed with water and immersed in PBS containing 1 mmol.L-1 AA. Successive cyclic voltammograms were then recorded (results not shown). The first cycle showed the anodic peak at 0.9 V corresponding to adsorbed NAC oxidation, whereas no AA signal was observed, neither on the following cycles. Obviously, the NAC adsorption on the electrode surface prevented AA oxidation. However, current density of both NAC anodic peaks increased linearly with AA concentration (Fig. 1, inset) while NAC concentration was kept constant. This result clearly highlights a catalytic EC’ mechanism: NAC is oxidized at the electrode surface and is further regenerated by AA by means of a coupled chemical reaction in solution close to the electrode. Both surface concentration and concentration gradient of NAC are then amplified, thus increasing the anodic current.
-0.12 -0.02 0.08 0.18 0.28 0.38 0.48 0.58 0.0 0.2 0.4 0.6 0.8 1.0 Potential (V/SCE) Current d ensity (mA .cm -2 )
b
c
e
d
f
0.08 0.28 0.48 0.68 0.0 0.5 1.0 1.5 2.0 [AA] (mmol.L-1) j (m A .cm -2 )a
Figure 1. Cyclic voltammograms recorded on gold electrode in 0.1 mol.L-1 deaerated PBS pH 7
containing: 1 mmol.L-1 AA (a); 1 mmol.L-1 NAC (b); 1 mmol.L-1 NAC and AA: 0.2 mmol.L-1 (c); 0.4 mmol.L-1 (d); 0.7 mmol.L-1 (e); 1 mmol.L-1 (f). Scan rate: 50 mV.s-1 (background current subtracted). Inset: variation of NAC oxidation current density with AA concentration. (▲) peak 1 at E = 0.75 V; (■) peak 2 at E = 0.9V
Similar experiments were performed by replacing AA by Ascorbyl Glucoside (AA-2G) and Ascorbyl Phosphate Magnesium (AA-2P) which are two AA derivatives less sensitive to light and oxygen than AA and consequently more commonly used in dermocosmetic formulations. Both species were found to be electrochemically inactive in the experimental conditions adopted (not shown). According to what observed with AA, results obtained in the first case made clearly evidence of a EC’ catalytic mechanism between NAC and AA-2G (not shown). However only the NAC oxidation peak at 0.9 V increased with AA-2G concentration, the diffusion controlled current at 0.75 V remaining roughly constant. This could be explained by a lower NAC regeneration reaction rate with AA-2G than with AA, the former being known to present a lower radical scavenging activity [37]. On the contrary, no evolution in the successive voltammograms was recorded when adding AA-2P in NAC solution. Obviously electrostatic hindrance and repulsion phenomena could strongly modify the reactivity as both AA-2P and NAC are negatively charged at pH 7.0 [36]. Consequently AA-2P can be used as antioxygen in dermocosmetics but is not efficient in antioxidant synergy when associated with NAC.
0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 0.3 0.5 0.7 0.9 1.1 1.3 1.5 Potential (V/SCE) Cu rr e n t d e n sity (m A .cm -2 ) b d c e f 0.0 0.4 0.8 1.2 0.0 1.0 2.0 3.0 4.0 [Vit E] (mmol.L-1) j (m A .cm -2) a
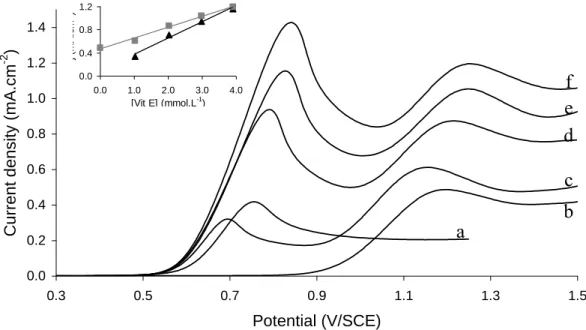
Figure 2. Linear voltammogram recorded on gold electrode in 0.1 mol.L-1 TBAP-dichloromethane
containing: 1 mmol.L-1 Vit. E (a); 1 mmol.L-1 BHT (b); 1 mmol.L-1 BHT and Vit. E: 1 mmol.L -1 (c); 2 mmol.L-1 (d); 3 mmol.L-1 (e); 4 mmol.L-1 (f). Scan rate: 50 mV.s-1 (background current subtracted). Inset: Variation of current density with Vit. E concentration. (▲) Vit. E oxidation peak at E = 0.75 V; (■) BHT oxidation peak at E = 1.2 V
The same procedure was exploited in organic solution in order to highlight possible regeneration mechanism between two lipophilic antioxidants. An example is given in Fig. 2 where voltammograms were plotted with Vit. E and 6-Di-tert-butyl-4-methylphenol (BHT) in 0.1 mol L-1 TBAP-containing dichloromethane. Vit. E and BHT exhibited a single anodic peak around 0.75 V (curve a) and 1.2 V (curve b), respectively. Successive additions of Vit. E into a 1 mmol.L-1 BHT solution resulted in the amplification in both amperometric signals (curves c to f). Current densities measured after signals deconvolution were reported as a function of Vit. E concentration (Fig. 2, inset).
The linear variation of BHT current density while its concentration was kept constant proves its regeneration by chemical reaction with Vit. E in organic solution.
3.2. Catalytic reactions in heterogeneous media
Synergic interactions between lipophilic and hydrophilic antioxidant compounds at liquid/liquid interface have been identified by depositing an organic film on the working electrode surface immersed in aqueous electrolyte. The organic phase was as thin as possible in order to minimize the diffusion barrier while covering entirely the electrode surface (see section 2.2.2) [34]. To ensure efficient regeneration reactions, the hydrophilic antioxidant concentration in the aqueous phase was 10 times higher than the lipophilic species one into organic phase.
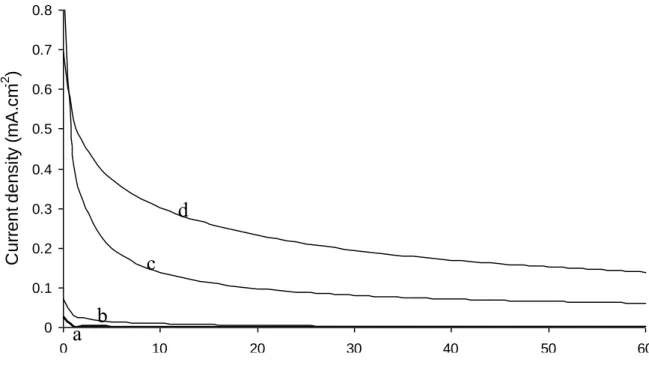
The electrochemical device was firstly validated by introducing 1 mmol.L-1 Vit. E (lipophilic) in dichloromethane and 10 mmol.L-1 AA (hydrophilic) in water. Vit. E is known to be regenerated by AA via a spontaneous chemical reaction [38-41]. However the cyclic voltammogram highlighted the same typical shape corresponding to the electrochemical activity of the Vit. E redox system whatever the experiment was realized with or without AA in the aqueous phase (not shown). Obviously the kinetics of the complete diffusion/reaction mechanism was too low to observe the antioxidant regeneration process in the time window used to plot the voltammogram. Consequently constant potential electrolyses were performed afterwards which offers a larger time window enabling slow chemical reactions to be studied. The electrode potential was deduced from previous voltammograms (Fig. 2) and maintained at 0.75 V during 2000 s in order the electrochemical oxidation of Vit. E to be diffusion controlled. Fig. 3 shows the typical chronoamperograms recorded during the first minute for a 1 mmol L-1 Vit. E solution without (curves c) and with (curves d) 10 mmol L-1 AA in aqueous phase, respectively. Similar experiment was performed with only both electrolytes containing no antioxidant compound (Fig. 3 curve a) for background current determination. A fourth electrolysis was realized with AA in the aqueous phase and only dichloromethane without any antioxidant as the organic phase (Fig. 3 curve b). This latter allowed to evaluate a possible contribution of the hydrophilic antioxidant compound which partitioned in organic phase and which could oxidize directly at the electrode surface. Curves obtained in both last cases were close, thus proving that the organic layer effectively prevented the direct AA oxidation at the electrode surface.
For all experiments, the electric charge (Q) was determined by integrating the chronoamperograms during 2000 s according to the Faraday’s law. The experimental charge obtained with the 1 mmol.L-1 Vit. E solution (Fig. 3 curve c) was Q = 0.083 ± 0.009 C after subtraction of the background charge. Considering that 2 electrons are exchanged for Vit. E oxidation [42] and assuming a reproducible organic layer deposit without any evaporation of dichloromethane, this value is very close to the theoretical charge (Qtheo = 0.077 C), indicating that the whole Vit. E has been consumed. Addition of 10 mmol L-1 AA into aqueous electrolyte significantly increased the electrolysis current (Fig. 3 curve d). After backgrounds subtraction, the electrical charge consumed during 2000 s was 39.8 % higher. The amount of lipophilic antioxidant being the same for both experiments, the enhanced
anodic charge resulted from a heterogeneous chemical reaction between oxidized Vit. E into organic solvent and AA into aqueous phase, leading to the regeneration of Vit. E as expected.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0 10 20 30 40 50 60 Time (s) C u rr e n t d e n si ty (m A .cm -2 )
a
b
c
d
Figure 3. Chronoamperograms recorded on gold electrode (1.96 cm2) covered with 400 µl of 0.1
mol.L-1 TBAP-containing dichloromethane and immersed in 25 mL of 1 mol.L-1 NaClO4 deaerated aqueous solution (a); with 10 mmol.L-1 AA (b); with 1 mmol.L-1 Vit. E (c); with 10 mmol.L-1 AA and 1 mmol.L-1 Vit. E (d). Electrode potential: 0.75 V
The electrochemical device was further exploited to study the synergic effect induced by others lipophilic / hydrophilic antioxidant associations. Three lipophilic antioxidants were used, i.e. Vit. E, Butylated hydroxyanisole (BHA) and BHT. In each case, the working electrode potential applied during the electrolysis (0.75 V, 1.27 V and 1.40 V, respectively) was chosen to ensure their maximal electrochemical oxidation rate and was determined experimentally by recording the cyclic voltammogram in the corresponding solution. It was verified that all three compounds were completely consumed when the electrolysis was performed during 2000 s without hydrophilic antioxidant, as previously explained. Table 1 gives the relative enhancement of the amount of charge Q recorded during the constant potential electrolysis with hydrophilic antioxidant in the aqueous phase. The highest is the charge enhancement, the most efficient is the regeneration mechanism and the best antioxidant capacity. Among hydrosoluble antioxidants tested, AA induced the best synergic effect in association with Vit. E or BHA. Similar spontaneous reactions were also observed, although at a lesser degree, by using AA-2G. This latter result confirms once again that AA-2G presents a lower radical scavenging and antioxidant activity than AA [37]. On the contrary, NAC did not regenerate Vit. E nor BHT while its presence amplified BHA oxidation current. From all these data it is possible to evaluate
the relative reactivity between lipophilic and hydrophilic antioxidants and thus select relevant associations promoting the best antioxidant capacity.
Table 1. Relative enhancement of the electric charge corresponding to the electrochemical oxidation
of lipophilic species in the presence of hydrophilic antioxidants. Electrode potential: Vit. E: 0.75 V; BHA: 1.27 V; BHT: 1.40V. Electrolysis time: 2000 s. Others experimental conditions are specified in section 2.2.2.
Lipophilic antioxidant (1 mM) Hydrophilic antioxidant (10 mM) Q (%) Vit E AA 39.8 AA-2G 4.9 NAC No increase BHA AA 8.7 AA-2G 3.4 NAC 7.4 BHT NAC No increase
3.3. Application in dermocosmetic creams
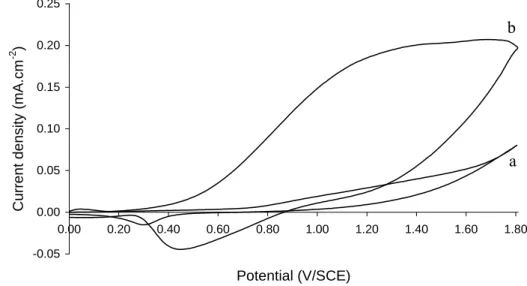
Several dermocosmetic creams were specially formulated based on a Dexeryl ® oil-in-water emulsion (see section 2.2.3). Lipophilic and/or hydrophilic antioxidants were added in the oil and/or the aqueous phase, respectively. Fig. 4 (curve b) shows the cyclic voltammogram obtained with a cream containing 0.1% NAC (w/w). Compared to the basic emulsion (Fig. 4, curve a) an anodic wave corresponding to NAC oxidation was clearly visible despite the very high viscosity of the cream which does not favour high diffusion transfer rate. The current rather resulted from the oxidation of spontaneously adsorbed NAC on gold surface. In the same experimental conditions, no noticeable amperometric signal was recorded by using AA-2G, Vit. E or BHA, although the last two species were electroactive on gold (see section 3.2).
Similarly to those performed for liquid/liquid interface study, electrolyses were then carried out during 2000 s and the electric charge Qcream was recorded. Background charge Q0 consumed with the basic emulsion was systematically removed from charges obtained with others creams. The working potential electrode was 1.4 V for all experiments in order to ensure the oxidation of the antioxidant species while avoiding oxygen evolution in the aqueous phase. Table 2 shows the charge enhancement resulting from the addition of one (entry 1 to 4) or several (entry 5 to 8) antioxidants in the cream. The electric charges were firstly compared to those obtained with the basic emulsion according to equation (1): (%) 0 0 1 Q Q Q Q cream (1)
-0.05 0.00 0.05 0.10 0.15 0.20 0.25 0.00 0.20 0.40 0.60 0.80 1.00 1.20 1.40 1.60 1.80 Potential (V/SCE) Curr e n t d e n sity (m A .cm -2 ) b a
Figure 4. Cyclic voltammograms recorded on gold electrode in Dexeryl cream without (a) and with
(b) 0.1 % NAC (w/w). Scan rate: 100 mV.s-1
Table 2. Relative enhancement of the anodic charge consumed during constant potential electrolysis
performed in dermocosmetic creams. Electrode potential: 1.4 V; Electrolysis time: 2000 s. Others experimental conditions are specified in section 2.2.3.
Cream composition
Water phase Oil phase Q1a (%) Q2b (%)
NAC (0.1 %) 39.3 AA-2G (0.1 %) 1.9 Vit. E (0.1 %) 19.3 BHA (0.02 %) 31.2 NAC (0.1%) + AA-2G (0.1%) 55.8 35.4 NAC (0.1%) Vit. E (0.1%) 74.9 27.8 NAC (0.1%) BHA (0.02 %) 86.3 22.5
NAC (0.1 %) + AA-2G (0.1 %) BHA (0.02 %) 67.6 No increase a Q
1: charge enhancement compared to that obtained with basic emulsion. Calculated from equation (1)
bQ
2: charge enhancement compared to the sum of the electric charges recorded with creams, each containing one of the corresponding antioxidants. Calculated from equation (2)
The chronoamperogram obtained with the cream containing AA-2G alone was very similar to that recorded with the basic emulsion. Consequently no change in the electric charge was observed (entry 2). It is not surprising since AA-2G was found to be electroinactive as previously mentioned. For all other formulations an enhanced electric charge was recorded. The lowest difference was obtained with samples containing only lipophilic antioxidants, i.e. Vit. E (entry 3) and BHA (entry 4). The basic emulsion being composed of an oil phase dispersed in an aqueous phase, the organic
compounds are encapsulated inside miscellaneous. Consequently lipophilic redox species could be hardly oxidized directly at the electrode surface. The highest electric charge enhancement has been obtained when more than one antioxidant were introduced into the cream (entry 5 to 8). In order to highlight catalytic phenomena promoted by the antioxidant association, the electric charge recorded Qcream2 was in these cases compared to the sum of the electric charge Qcream1 obtained with creams, each containing one of the corresponding antioxidants, according to equation (2):
1 0
(%) 0 1 0 2 2 Q Q Q Q Q Q Q cream cream cream (2)For example, the charge obtained for the cream containing NAC and AA-2G was 55.8 % higher than that recorded with the basic emulsion (entry 5). The charge enhancement induced by NAC and AA-2G being 39.3 % (entry 1) and 1.9 % (entry 2), respectively, the increase in the electric charge due to catalytic mechanism is (55.8-41.2)/41.2 = 35.4 %.
Among the formulations containing more than one antioxidant, that involving NAC+AA-2G presented the highest synergic effect. This is not surprising since two hydrophilic antioxidants were introduced in the aqueous phase which is the major part of the oil-into-water emulsion. Antioxidant regeneration process was also evidenced by introducing a hydrophilic and a lipophilic antioxidant species in the aqueous and the organic phases, respectively. From the experimental results it can be deduced that NAC was regenerated by Vit. E (entry 6) and that the former reacted with BHA (entry 7) as previously mentioned in model liquid/liquid interface (section 3.2.). It is not surprising that the catalytic mechanism was less efficient in both last cases since the electron transfer took place at the oil/water interface. Finally, no synergic effect was observed when three antioxidants (2 hydrophilic, i.e. NAC and AA-2G and 1 lipophilic species, i.e. BHA) were introduced into the cream (entry 8). In this last case the sum of the contribution of each antioxidant (entry 1, 2 and 4, respectively) was higher than the total electric charge Qcream2. The main reason could be that the regeneration process involved two spontaneous redox reactions occurring simultaneously and coupled with the diffusion of BHA inside the organic phase, this latter being oxidized at the electrode surface and regenerated at oil/water interface. This complex EC’ mechanism certainly induced too low reaction kinetics to have a real influence on BHA regeneration.
4. CONCLUSION
An electrochemical process has been developed to study antioxidant properties in homogeneous or heterogeneous model systems and in real media. On the one hand, cyclic voltammetry proved to be well adapted to highlight antioxidant regeneration reaction in aqueous and organic homogeneous solutions. AA and AA-2G was thus found to regenerate NAC in the former case whereas and Vit. E regenerated BHT in the latter case. On the other hand, constant potential electrolysis was necessary for experiments in biphasic systems and cosmetic emulsions since it offers a larger time window to study lower reaction kinetics. Chronoamperograms thus allowed heterogeneous
catalytic mechanism and antioxidant synergic effect to be clearly highlighted in such complex media. The antioxidant properties of NAC were thus amplified by 35.4%, 27.8% and 22.5% when associated with AA-2G, Vit. E and BHA, respectively. In conclusion electrochemistry appears to be a convenient tool for the optimisation of new “anti-aging” dermocosmetic formulations by helping in the choice of antioxidants molecules and relevant associations to induce the greatest antioxidant capacity. This electrochemical process can now be transposable to determine antioxidant potentialities of other complex media.
ACKNOWLEDGEMENTS
This work was financially supported by Pierre Fabre Dermo-cosmetic laboratories. The authors thank Laurent Massot from Laboratoire de Génie Chimique for the curves deconvolution, Laure Latapie for technical assistance and Maurice Comtat for scientific advices.
References
1. B. Halliwell and J.M.C. Gutteridge, Free Radicals in biology & medicine, Oxford University Press, Oxford (1989)
2. T. Finkel and N.J. Holbrook, Nature, 408 (2000) 239
3. J.J. Thiele, F. Dreher and L. Packer, J. Toxicol. Cutan. Ocul. Toxicol., 21 (2002) 119 4. A.Kumar, G. Singh, B.V.S. Kumar and S.K. Meur, Livest. Sci., 138 (2011) 299
5. K. Ezzedine, J. Latreille, E. Kesse-Guyot, P. Galan, S. Hercberg, C. Guinot and D. Malvy, Eur. J. Cancer, 46 (2010) 3316
6. J.F. Garcia-Jimenez, M.C. Valencia and L.F. Capitan-Vallvey, J. Chromatogr. Sci., 47 (2009) 485 7. T-F. Tsai and M-R Lee, Chromatogr., 67 (2008) 425
8. Z-Q. Liu, Chem. Rev., 110 (2010) 5675
9. E.U. Nwose, H.F. Jelinek, R.S. Richards and P.G. Kerr, Med. Hypotheses, 70 (2008) 1002 10. K. Mukai, T. Isozaki and S-I. Nagaoka, Bull. Chem. Soc. Jpn., 80 (2007) 1331
11. P. Pedrielli and L.H. Skibsted, J. Agric. Food Chem., 50 (2002) 7138
12. S-I. Nagaoka, M. Inoue, C. Nishioka, Y. Nishioku, S. Tsunoda, C. Ohguchi, K. Ohara, K. Mukai and U. Nagashima, J. Phys. Chem. B, 104 (2000) 856
13. R. Liang, C-H. Chen, R-M. Han, J-P. Zhang and L.H. Skibsted, J. Agric. Food Chem., 58 (2010) 9221
14. J.M. Irache, I. Ezpeleta and F.A.Vega, Chromatogr., 35 (1993) 232 15. F. Dai, W-F. Chen and B. Zhou, Biochimie, 90 (2008) 1499
16. K. Omura, J. Am. Oil Chem. Soc., 72 (1995) 1565
17. Z. Samec, V. Marecek, J. Koryta and M. W. Khalil, J. Electroanal. Chem., 83 (1977) 393 18. C. Wei, A. J. Bard and M. V. Mirkin, J. Phys. Chem. B, 99 (1995) 16033
19. M. Donten, Z. Stojek and F. Scholz, Electrochem. Commun., 4 (2002) 324
20. F. Quentel, V. Mirceski, M. L'Her, M. Mladenov, F. Scholz and C. Elleouet, J. Phys. Chem. B, 109 (2005) 13228
21. T. Sugihara, T. Kinoshita, S. Aoyagi, Y. Tsujino, and T. Osakai, J. Electroanal. Chem., 612 (2008) 241
22. X. Lu, L. Hu and X. Wang, Electroanalysis, 17 (2005) 953
23. T. Osakai, H. Jensen, H. Nagatani, D.J. Fermin and H.H. Girault, J. Electroanal. Chem., 510 (2001) 43
24. F.A. Bertolino, P.W. Stege, E. Salinas, G.A. Messina and J. Raba, Anal. Lett., 43 (2010) 2078 25. V. Castaignede, H. Durliat and M. Comtat, Anal. Lett., 36 (2003) 1707
26. C. Guitton, P. Gros, M. Comtat, R. Tarroux and P. Bordat, J. Cosmet. Sci., 56 (2005) 79
27. A.Ruffien-Ciszak, P. Gros, M. Comtat, A.-M. Schmitt, E. Questel, C. Casas and D. Redoules, J. Pharm. Biomed. Anal., 40 (2006) 162
28. F. Sekli-Belaidi, P. Temple-Boyer and P. Gros, J. Electroanal. Chem., 647 (2010) 159 29. A.Ruffien-Ciszak, J. Baur, P. Gros, E. Questel and M. Comtat, IRBM, 29 (2008) 162 30. F. Sekli-Belaidi, D. Evrard and P. Gros, Electrochem. Commun., 13 (2011) 423
31. H. H. Girault and D. J. Schiffrin, in Electroanalytical Chemistry, ed. A. J. Bard. Marcel Dekker, New York (1989)
32. C. Shi and F. C. Anson, J. Phys. Chem. B, 102 (1998) 9850
33. Y. Shao, M. V. Mirkin and J. F. Rusling, J. Phys. Chem. B, 101 (1997) 3202
34. V. Mirceski, F. Quentel, M. L'Her and C. Elleouet, J. Phys. Chem. C, 111 (2007) 8283 35. S. V. Romanenko, A.G. Stromberg and T. N. Pushkareva, Anal. Chim. Acta, 580 (2006) 99 36. C. Barus, P. Gros, M. Comtat, S. Daunes-Marion and R. Tarroux, Electrochim. Acta, 52 (2007)
7978
37. J. Takebayashi, A. Tai, E. Gohda and I. Yamamoto, Biol. Pharm. Bull., 29 (2006) 766 38. S. Nagaoka, T. Kakiuchi, K. Ohara and K. Mukai, Chem. Phys. Lipids, 146 (2007) 26 39. J. E. Packer, T. F. Slater and R. L. Willson, Nature, 278 (1979) 737
40. K. Mukai, K. Fukuda, K. Ishizu and Y. Kitamura, Biochem. Biophys. Res. Commun., 146 (1987) 134
41. K. Mukai, M. Nishimura and S. Kikuchi, Biochim. Biophys. Acta, 993 (1991) 168 42. R. D. Webster, Acc. Chem. Res., 40 (2007) 251