HAL Id: tel-01468434
https://hal.archives-ouvertes.fr/tel-01468434
Submitted on 15 Feb 2017
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Catalyse électrochimique de l’activation des petites
molécules : analyse mécanistique et discrimination entre
voies réactionnelles compétitives
Hachem Dridi
To cite this version:
Hachem Dridi. Catalyse électrochimique de l’activation des petites molécules : analyse mécanistique et discrimination entre voies réactionnelles compétitives. Chimie théorique et/ou physique. Université Paris Diderot (Paris 7), 2016. Français. �tel-01468434�
U
NIVERSITE
P
ARIS
D
IDEROT
(P
ARIS
7)
Sorbonne Paris cité
Ecole Doctorale de Chimie Physique et Chimie Analytique de Paris Centre
(ED388)
Laboratoire d’Electrochimie Moléculaire
D
OCTORAT
E
LECTROCHIMIE
Hachem DRIDI
C
ATALYSE ELECTROCHIMIQUE DE L
’
ACTIVATION DES PETITES
MOLECULES
:
ANALYSE MECANISTIQUE ET DISCRIMINATION
ENTRE VOIES REACTIONNELLES COMPETITIVES
Thèse dirigée par Cyrille COSTENTIN
Soutenue le 08 juillet 2016 devant la commission d’examen composée de :
Rapporteurs
Vincent ARTERO
DR, CEA, Grenoble
Boniface KOKOH
PR, Université de Poitiers
Examinatrice
Florence GENESTE
CR, Université Rennes 1
Président
Jean-Michel SAVEANT
DRCE, Université Paris Diderot
à Youssef
“ايهلع ًإدئإز تن أ نكت ًائيش ةايلحإ لىع دزت لم إذ إ” “ليايِّللإ َرـِهَس لاُعلإ َبل َط ْنَمو لياــَعَلمإ ُب َسَتكُت ِِّدكلإ ِر ْدَقب”
Remerciements
Je tiens en tout premier lieu à remercier M. Benoît Limoges, directeur du laboratoire, et M. Marc Robert, responsable de l’équipe « Transferts d'électron et bouleversements moléculaires. Réactivité chimique fondamentale et Catalyse », pour m’avoir accueilli au sein du Laboratoire d’Électrochimie Moléculaire de l’Université Paris Diderot.
J’adresse mes sincères remerciements ensuite à mon directeur de thèse M. Cyrille Costentin pour son encadrement de grande qualité. Qu'il trouve ici le témoignage de ma profonde reconnaissance pour la confiance qu’il m’a portée ainsi que pour sa gentillesse, pour avoir répondu à toutes mes questions et pour m’avoir guidé efficacement dans mon doctorat avec autant de compétence et de disponibilité.
Je tiens à associer à ces remerciements M. Jean-Michel Savéant. Durant ces trois années passées au LEM, j’ai eu l’honneur et la chance de travailler et de partager avec vous des discussions de natures diverses mais toutes constructives et très enrichissantes tant sur le plan scientifique que sur le plan humain.
M. Vincent Artero et M. Boniface KoKoh m’ont fait l’honneur d’être les rapporteurs de cette thèse, je les en remercie vivement. J’exprime également ma gratitude à Mme Florence Geneste qui a pris le soin de lire et d'examiner ce manuscrit.
Qu'il me soit maintenant permis de remercier tous les gens que j’ai pu côtoyer au sein du laboratoire, notamment Cédric , François, Julien, Elodie, Sihem, Rabia…pour leur accueil et leur sympathie.
J’ai une pensée toute particulière pour ceux avec qui j’ai partagé mon bureau (Alexandra, Dany, Antoine, Arnaud et Hélène) et le labo (notamment Carlo, Irene, Iban, Tom et Jean-Claude). Que tous ceux qui m'ont aidé et accompagné dans ces années trouvent ici l'expression de ma reconnaissance et de mon amitié.
Je souhaite aussi remercier l’ensemble des thésards et post-doctorants qui sont passés au laboratoire durant ces années et que je n’ai pas cités.
Je tiens à exprimer ma gratitude à M. Philippe Schollhammer et Mme Catherine Elleouet qui m’ont initié à l’électrochimie moléculaire lors de mon stage de M2 et qui ont soutenu ma candidature pour effectuer cette thèse au sein du LEM.
Je ne saurais terminer sans remercier du fond du cœur mes chers parents qui m’ont toujours soutenu et qui durant toute ma vie ont été présent à mes côtés pour me réconforter, sans eux rien n’aurait été possible. Je dédie également ce travail à mes frères, les membres de ma grande famille, et à tous mes amis que je remercie infiniment pour leur soutien. Je pense à ma sœur Kalthoum qui n’a jamais cessé de me prodiguer conseils et encouragements tout au long de la période de mes études supérieures et à qui je ne saurais exprimer toute ma reconnaissance et ma gratitude.
Enfin, je remercie ma chère femme, Rania, pour son soutien, sa patience et sa présence à mes côtés.
Sommaire
INTRODUCTION GENERALE ... 5
1. Le défi énergétique mondial ... 6
2. Stockage réversible de l’énergie dans des liaisons chimiques ... 8
3. Catalyse de l’activation des petites molécules ... 10
3.1. Terminologie ... 10
3.2. Evaluation et comparaison des performances des catalyseurs moléculaires ... 11
3.3. La multiplicité des mécanismes ; une caractéristique inhérente aux réactions de catalyse électrochimique ... 14
3.4. Exemples de mécanismes compétitifs rencontrés dans des réactions d’intérêt technologique ... 15
4. But et cadre du présent mémoire ... 19
CHAPITRE I. Catalyse électrochimique moléculaire de la production de H2 dans le DMF par la Fe(0)TPP: compétition entre voies hétérolytique et homolytique ... 21
1. Introduction ... 22
2. Rappel des caractéristiques d’un mécanisme catalytique à l’aide d’un diagramme de zone ... 26
3. Différents schémas réactionnels envisageables pour la catalyse moléculaire de la HER 29 4. Dichotomie et compétition entre voies hétérolytique et homolytique dans le cadre d’un mécanisme de type EEPDimP (schéma I-4) en présence de la FeTPP comme catalyseur moléculaire ... 32
4.1. Caractéristiques générales et présentation des paramètres qui contrôlent le système ... 35
4.2. Situation SET ... 36
4.3. Situation ECE ... 38
4.4. Exploitation des diagrammes de zone ... 39
5. Application : catalyse de réduction de la triéthylamine protonée, du phénol et de l’acide acétique dans le DMF par la tétraphénylporphyrine de fer(0) électrogénérée ... 41
5.1. Cas de la triéthylamine protonée ... 41
5.2. Cas du phénol ... 44
5.3. Cas de l’acide acétique ... 46
5.4. Comparaison des trois acides ... 48
6. Conclusion ... 50
CHAPITRE II. Catalyse électrochimique moléculaire de la réduction de O2 dans l’eau par la FeTMPyP: compétition entre voies hétérogène et homogène ... 53
2. Comportement électrochimique du catalyseur en l’absence d’oxygène : choix des
conditions opératoires ... 56
2.1. Réponse courant-potentiel en voltammétrie cyclique : comment éviter l'interférence des dimères ... 59
2.2. Réponse courant-potentiel en voltammétrie cyclique : vérification de l’adsorption du catalyseur ... 63
2.3. Réponse courant-potentiel en voltammétrie sur électrode tournante à disque : vérification des conditions stationnaires (pour le catalyseur). ... 65
2.4. Réduction électrochimique du substrat (O2) seul dans les mêmes conditions opératoires ... 67
2.5. Vérification des conditions stationnaires en présence du substrat ... 69
3. Catalyse de la réduction du dioxygène par la FeIITMPyP ... 71
3.1. Remarques générales ... 71
3.2. Catalyse de la réduction de (O2) par la FeTMPyP adsorbée (catalyse hétérogène) . 74 3.3. Catalyse de la réduction de (O2) par la FeTMPyP en solution (catalyse homogène) 77 3.4. Effet de la concentration en O2 ... 80
3.5. Effet du pH sur les catalyses hétérogène et homogène ... 82
4. Discussion du mécanisme proposé ... 83
4.1. Catalyse hétérogène ... 83
4.2. Catalyse homogène ... 87
5. Conclusion ... 92
CHAPITRE III. Réduction électrocatalytique du dioxyde de carbone sur électrodes métalliques (Hg et Pt) en milieu aqueux : compétition avec la réduction des acides de Brønsted et phénomènes d’inhibition de l’électrode ... 93
1. Introduction ... 94
2. Réduction « électrocatalytique » du dioxyde de carbone sur Hg en milieu aqueux : compétition avec la réduction de H2CO3 ... 104
2.1. Voltammétrie cyclique : vague de réduction de H2CO3 ... 104
2.2. Electrolyses préparatives ... 106
3. Réduction « électrocatalytique » du proton sur Hg en milieu aqueux ... 109
3.1. Données obtenues à partir des expériences de voltammétrie cyclique ... 109
3.2. Comparaison avec les données de la littérature ... 110
3.3. Discussion ... 112
4. Réduction « électrocatalytique » de CO2 sur Pt dans l’eau en présence du pyridinium : compétition avec la réduction des acides de Brønsted et phénomènes d’inhibition de l’électrode ... 117
1.1. Electrolyse préparative et analyse de(s) produit(s) de la réaction ... 117
1.2. Phénomènes d’inhibition accompagnant les électrolyses préparatives sous CO2 ... 118
PARTIES EXPERIMENTALES ... 131
Partie expérimentale I ... 132
Partie expérimentale II ... 133
Partie expérimentale III ... 136
ANNEXES ... 141
Annexe I ... 142
Annexe II ... 166
Annexe III ... 183
1. Le défi énergétique mondial
L’évolution de la demande énergétique mondiale va de pair avec la croissance
démographique et économique.1 Pour réussir à satisfaire l’augmentation annuelle de la demande
globale en énergie, estimée à 2.6 % entre 2002 et 2012, 10 fois plus d’énergie devrait être
consommée en 2100 qu’en 2013 (9301 Mtoe i en 2013).2 Cela ne peut être possible dans des
sociétés modernes qui utilisent à plus de 86 % les combustibles fossiles (charbon, pétrole, gaz naturel et gaz de schiste) comme c’est le cas aujourd’hui. En effet, ces ressources énergétiques, d’origines non renouvelables, sont en quantités finies et seront un jour pratiquement épuisées. La rareté des combustibles fossiles incite nos sociétés à chercher des matières premières alternatives pour la production de l’énergie et de produits chimiques. A côté de cela, nous sommes confrontés à une deuxième contrainte qui résulte du changement climatique important dû, en particulier, à l’émission de gaz à effet de serre accompagnant l’utilisation des combustibles fossiles. Compte tenu de ces deux contraintes (épuisement des ressources fossiles,
et pollution atmosphérique), le défi énergétique du XXIe siècle a deux volets :
Réduire les émissions de CO2.
Remplacer progressivement les énergies fossiles par des sources d’énergies renouvelables et propres.
Figure 1. Les combustibles fossiles représentent 86% de l'énergie golobale consommée en 2014, en baisse de
1% par rapport à l'année précédente.3
La conversion (par le biais de cellules photovoltaïques, éoliennes…) des flux d’énergies propres, provenant des sources renouvelables et infinies (comme le soleil, le vent, les marées,
les vagues et la chaleur géothermique),4 en un flux d’électrons (énergie électrique) représente
une solution attractive de plus en plus utilisée dans les pays industrialisés pour remédier aux deux problèmes soulevés ci-dessus. Cependant, un inconvénient majeur de ce vecteur énergétique (l’électricité), c’est qu’il n’est pas facile de le stocker en grandes quantités et, quelle
que soit son origine, cela a un coût élevé.5 Le stockage de l’énergie demeure le point faible de
2. Stockage réversible de l’énergie dans des liaisons chimiques
Une des approches les plus prometteuses dans ce contexte consiste à s’inspirer de la nature (la photosynthèse) et du mode de fonctionnement d’un certain nombre d’enzymes (par
ex. l’hydrogénase,7-9 le monoxyde de carbone déshydrogénase) pour convertir et stocker les
flux d’énergies produits par des sources intermittentes dans des liaisons covalentes via la
transformation électrochimique de petites molécules abondantes telles que H2O, CO2, N2…en
combustibles chimiques (H2, CH3OH, CH4, NH3…).10
En particulier, du fait de sa combustion non polluante, le dihydrogène produit par électrolyse au moyen de l'énergie électrique provenant des sources renouvelables est considéré comme un des vecteurs énergétiques du futur. Par ailleurs, il permet un bon stockage des énergies propres de manière permanente et durable sous forme de dihydrogène comprimé ou
d’hydrures métalliques.11-12 Il pourra ensuite être utilisé, directement, comme combustible dans
un moteur thermique, au même titre que CH3OH, CH4, NH3…ou alimenter une pile à
combustible pour produire de l’électricité et de la chaleur.
Figure 2. un schéma général pour la conversion et le stockage des énergies produites par des sources
renouvelables (ici le soleil à titre d’exemple) sous forme de H2.
De plus, le dihydrogène forme avec le monoxyde de carbone un mélange gazeux combustible couramment appelé « gaz de synthèse » ou « syngas ». Ce mélange gazeux
(H2+CO) est souvent utilisé comme précurseur lors de la fabrication du pétrole brut synthétique
par le procédé Fischer-Tropsch, lui-même précurseur de l'essence synthétique « synfuel ». Il s’agit d’une méthode très performante en termes de rendement. Cependant, la grande quantité
de CO2 générée au cours de ce procédé représente un principal inconvénient d'un point de vue
environnemental.
L’hydrogénation de CO2 est un autre procédé similaire à la synthèse de Fischer-Tropsch
(hydrogénation du CO). Ce procédé offre la possibilité d’élaborer des molécules
énergétiquement valorisables (telles que CH3OH, HCOOH, CH3OCH3, CH4…) moyennant la
réaction du gaz à l’eau inversée (RWGS :CO2H2 CO + H O2 ) tout en contribuant à la
réduction du taux de CO2 émis dans l’atmosphère.
Schéma 1. Hydrogénation de CO2 en des produits chimiques qui peuvent être utilisés comme vecteurs
énergétiques.13
L’origine et le coût des précurseurs (H2 et CO) qui alimentent de tels procédés constituent
deux problèmes centraux. Le dihydrogène et le monoxyde de carbone doivent être absolument produits à partir de sources d’énergies propres, faute de quoi, il n’y aura aucun intérêt réel à
convertir le CO2 et le CO par ces deux techniques.
Malheureusement, à l’heure actuelle, on ne dispose pas d’une telle industrie de production de précurseurs et de combustibles chimiques « propres » à grande échelle pour servir à des applications énergivores. A titre d’exemple, la majorité du dihydrogène produit actuellement (96 %) provient du reformage du méthane (ou d’autres combustibles fossiles, selon la réaction
4 2 2 2
CH +2H O CO + 4H dans le cas de CH4), la petite partie (4 %) produite par électrolyse
de l’eau est destinée aux applications de haute technologie qui demandent un dihydrogène très pur.14
3. Catalyse de l’activation des petites molécules
L’hydrogène et le carbone sont deux éléments chimiques abondants. On trouve par exemple l’élément hydrogène dans l’eau, les hydrocarbures et la biomasse, mais il est toujours présent sous une forme oxydée, combiné avec d’autres éléments. La formation de combustibles
non fossiles (H2, CH3OH, CH4…) par électrolyse à partir de petites molécules particulièrement
stables, telles que H2O et CO2, demande d’appliquer de grandes surtensions (dépenser beaucoup
d’énergie) pour avoir lieu à des vitesses appréciables. Ceci est le cas pour un grand nombre de réactions électrochimiques d’intérêt technologique, notamment lorsqu’il ne s’agit pas seulement d'un simple transfert d'électrons (de type sphère externe) entre l'électrode et le réactif, mais plutôt d’un transfert entraînant la rupture et/ou la création de liaisons covalentes. Ces situations demandent l’utilisation d’un catalyseur efficace dont le but sera de fournir des chemins réactionnels alternatifs qui permettent d’éviter les étapes lentes et donc d’accélérer la cinétique de la réaction.
Figure 3. Profils d’énergie potentielle montrant l'effet d'un catalyseur dans une réaction chimique
exothermique (hypothétique) X + Y Z.15
3.1. Terminologie
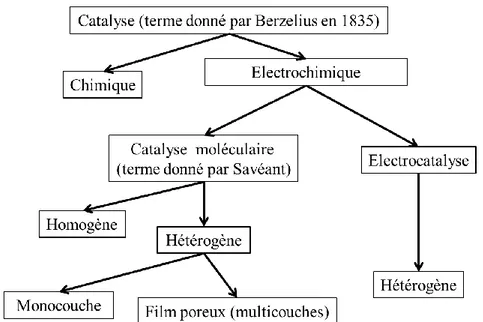
Différentes stratégies peuvent être envisagées pour atteindre ce but. Le terme « électrocatalyse » est traditionnellement utilisé pour décrire les réactions dans lesquelles le
matériau de l’électrode (souvent, mais pas toujours,16 un métal) est chimiquement impliqué
dans le processus catalytique. La production : de H2 sur Pt,17 de O2 sur Ni18-19 et de Cl2 sur
RuO2−TiO2,20-22 à titre d’exemples, sont trois réactions primordiales dans l’industrie
Une autre approche pour catalyser les réactions électrochimiques consiste à utiliser une
molécule, caractérisée par un potentiel standard ox red
0
Cat
E , comme catalyseur.23 La « catalyse
moléculaire » ainsi définie peut impliquer des molécules de catalyseur dispersées de manière homogène dans la solution contenant les réactifs et les produits ou bien immobilisées (adsorbées) sur la surface de l'électrode sous forme de monocouche ou de film poreux
(multicouches).24-26 A ce stade, il convient de préciser que le terme « électrocatalyseur» est
couramment utilisé dans la littérature pour décrire, à la fois les catalyseurs employés dans le cadre de la catalyse électrochimique moléculaire (sous ces deux sous-catégories ; homogène et hétérogène), ainsi que ceux utilisés en électrocatalyse. Sachant que, selon la définition donnée précédemment, une réaction électrocatalytique ne peut être qu’« hétérogène ».
Figure 4. Récapitulatif des principaux termes utilisés pour décrire les réactions de catalyse électrochimique.
3.2. Evaluation et comparaison des performances des catalyseurs moléculaires
3.2.1. En catalyse moléculaire
D’une manière générale, un « bon » catalyseur moléculaire doit opérer son transfert
d'électrons à un potentiel ox red
0 Cat
potentiel thermodynamique de la réaction Eproduit(s) substrat(s)0 . De plus, il doit permettre d'avoir, à
la fois une cinétique de transfert d'électrons hétérogène
kh et de réaction catalytique
kCatrapides.27 Cela se traduit par une amélioration de l’efficacité faradique et une diminution de la
surtension nécessaire pour réaliser une réaction électrochimique donnée (par ex. la réduction de CO2).28
Schéma 2. Mécanisme d’une réaction simple de catalyse homogène moléculaire.
L’efficacité de plusieurs électrocatalyseurs vis-à-vis d’une réaction de catalyse moléculaire donnée est comparée sur la base de plusieurs facteurs de mérite qui tiennent compte du potentiel standard de la réaction Eproduit(s) substrat(s)0 , des propriétés intrinsèques du
catalyseur (le potentiel standard ox red
0 Cat
E et la constante de vitesse kCat, ainsi que de la stabilité
et la sélectivité du catalyseur en question.10
De nouveaux outils d’électrochimie permettant une évaluation et une comparaison rationnelles des performances des catalyseurs moléculaires, au moyen de la voltammétrie cyclique (via la mesure du courant de plateau ou du pied de la vague catalytique), ont été
développés au sein du laboratoire d’électrochimie moléculaire,10, 24-25, 29-32 notamment, en ce
qui concerne la réaction de production de dihydrogène et la conversion électrochimique de CO2
en CO.33-34 L’application de ces outils requiert bien évidemment une connaissance précise des
mécanismes des réactions catalytiques en question. Electrode red Cat ox Cat Substrat Produit ox red 0 Cat E 0 produit substrat E appliqué V
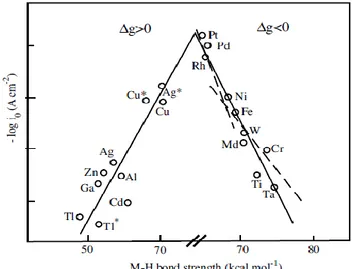
3.2.2. En électrocatalyse : énergie d’adsorption et activité électrocatalytique (les « courbes volcans »).
La corrélation entre l’activité catalytique (ou la vitesse de la réaction électrocatalytique i)
et la force de la liaison « métal‒intermédiaire clé » traduisant quantitativement le principe de Sabatier conduit dans le cas de la HER à une relation de type « courbe volcan ». Les réactions faisant intervenir un transfert de deux électrons, à l’exemple de la HER, donnent lieu
généralement à un seul intermédiaire clé.35 De ce fait, l’identification du meilleur catalyseur est
relativement simple. Pour la réduction de H+, il s’agit du platine occupant le sommet du
« volcan » et présentant une valeur intermédiaire d’énergie d’adsorption.36-37
Figure 5. Courant d'échange pour la réaction électrocatalytique de production de dihydrogène en
fonction de la force de la liaison hydrogène‒métal calculée à partir de la chaleur de formation de l'hydrure.36
En théorie des relations de type « courbes volcans », comme celle illustrée sur la Figure
5 pour la HER, peuvent être obtenues quel que soit le mécanisme de la réaction
électrocatalytique étudiée.37 Toutefois, les réactions mettant en jeu plus de deux électrons (et
protons) impliqueront, systématiquement,35, 38 plus d’un seul intermédiaire catalytique adsorbé.
Cela engendre des situations beaucoup plus difficiles à traiter, comme par exemple dans le cas
de la réduction électrocatalytique de CO2 dans l’eau qui fera l’objet principal des travaux
présentés dans le troisième chapitre.39
i Incluse dans l’expression du courant d’échange i 0.
3.3. La multiplicité des mécanismes ; une caractéristique inhérente aux réactions de catalyse électrochimique
Le cas idéal pour une réaction catalytique consiste à avoir une voie réactionnelle unique conduisant à un seul produit. Toutefois, comme nous le verrons plus en détail tout au long des prochains chapitres de ce mémoire, en présence d’un catalyseur, il est courant d’avoir plusieurs chemins réactionnels compétitifs qui ouvrent sur plusieurs possibilités de sélectivité. Tout cela conduit à une multitude de schémas mécanistiques à considérer. Ces schémas dans lesquels les transferts multi-électroniques et les étapes chimiques se lient étroitement, sont d’autant plus complexes avec l’augmentation du nombre d’électrons mis en jeu dans le processus
réactionnel.35 L’élucidation des différentes voies réactionnelles en compétition s’avère alors un
préalable nécessaire à la compréhension des principaux facteurs qui gouvernent ce type de réactions et à la détermination de la voie réactionnelle la plus efficace. De toute évidence, cela aidera à perfectionner les catalyseurs utilisés et à concevoir des réactions catalytiques plus efficaces.
De manière générale, les questions centrales qui se présentent sont de savoir :
Si les étapes clés du mécanisme ont lieu dans la solution (processus homogènes) ou bien si des espèces intermédiaires adsorbées à la surface interviennent (processus hétérogènes) ?
S’il existe plusieurs mécanismes compétitifs impliqués, ouvrant sur des possibilités de sélectivité, ou bien il s’agit d’une réaction dominée par un seul mécanisme ?
Quels sont les intermédiaires catalytiques clés impliqués et quelle est l’étape cinétiquement déterminante de la réaction ?
3.4. Exemples de mécanismes compétitifs rencontrés dans des réactions d’intérêt technologique
3.4.1. L’électrolyse de l’eau
Comme exemples de mécanismes compétitifs rencontrés dans des réactions d’intérêt technologique, nous pouvons considérer les réactions électrochimiques impliquées dans un processus d’électrolyse d’eau ou de dioxyde de carbone, induit par une source d’énergie renouvelable. Ce processus de conversion de l’énergie électrique en énergie chimique implique, souvent, la production du dioxygène dans le compartiment anodique de l’électrolyseur. Dans le deuxième compartiment, le carburant produit sur la cathode pourrait être soit du dihydrogène
(si nous réduisons H2O), soit une espèce organique comme le méthanol ou le méthane (si nous
réduisons CO2). Enfin, la recombinaison du carburant produit avec le comburant libéré (O2)
dans une pile à combustible permettra de reproduire l’énergie électrique à partir de l’énergie
chimique emmagasinée dans les liaisons moléculaires et de régénérer l'espèce originale (H2O
ou CO2). Ainsi, le cycle sera bouclé de façon neutre en carbone.
Regardons plus en détails, à titre d’exemple, la production alimentée par l’énergie
photovoltaïque du dihydrogène et de l’oxygène par électrolyse de l’eau.40-41 Il s’agit d’un
processus qui met en jeu des transferts d’électrons et de protons, en présence de catalyseurs,i
dans le but de casser les liaisons de H2O et former H2 et O2 suite à des réarrangements
moléculaires.42-44
Figure 6. Schéma d'une cellule photo-électrochimique : production du dihydrogène et de l’oxygène par
électrolyse de l’eau.45
Les bases scientifiques qui sous-tendent ce processus multi-électroniques d’apparence triviale soulèvent de nombreuses questions fondamentales de la chimie. Il suffit de savoir que trois prix Nobel ont été attribués pour des travaux de recherche qui s’articulent autour de l’étude
et l'élucidation des transferts électroniques.46-49 De même, les transferts de protons qui
pourraient s’associer aux transferts d’électrons dans le cadre d’un PCET (acronyme de Proton
Coupled Electron Transfer) ont suscité au cours des dernières années un vif intérêt dans la
mesure où des études ont montré qu’ils sont rencontrés, non seulement dans les processus de conversion et de stockage de l’énergie, mais également dans un grand nombre de réactions radicalaires et de processus biologiques incluant les réactions enzymatiques, la photosynthèse,
la respiration…50-52
Par ailleurs, outre les protons (issus de l’oxydation de H2O sur l’anode), pour former du
dihydrogène, l’électrolyse de l’eau fait intervenir au moins un catalyseur, le plus souvent
fabriqué à partir des métaux du groupe du platine (PGM ; Pt, Ir, Ru…).53 Cependant, les PGM
sont des métaux rares et onéreux. Pour pouvoir développer et rendre compétitifs ce type de procédés, il va falloir lever ce verrou majeur. Un vaste effort de recherche est entrepris depuis quelques années en vue de proposer, à terme, des catalyseurs moléculaires biomimétiques et synthétiques à base de métaux abondants dans l’écorce terrestre (comme Fe, Co, Ni…),
alternatifs à l’utilisation des PGM.54-57
De nos jours, un certain nombre de catalyseurs développés selon ladite approche est
connu et permet de catalyser efficacement la production électrochimique de H2 à des potentiels
raisonnables. Néanmoins, les détails mécanistiques des réactions catalytiques impliquées ne sont connus d’une façon précise que dans de rares cas. Par exemple, dans le cas d’un catalyseur
en solution agissant directement sur la molécule d’eau (voir Schéma 3), les différents chemins
réactionnels possibles pour former le dihydrogène, comme la protonation de l’espèce hydrure
(Mn+1−H) versus l’élimination réductrice biomoléculaire de H2 (par combinaison de deux
Schéma 3. Mécanisme proposé pour la production de H2 via la formation d'un intermédiaire
électrocatalyseur-hydrure commun.59
3.4.2. La pile à combustible
Au sein du compartiment cathodique des piles à combustible à membrane échangeuse de protons (PEMFC), la réaction de réduction de l'oxygène (ORR) en eau peut suivre deux voies principales : une voie à 4 électrons ou une voie à 2 électrons. Ces deux voies produisent
respectivement de l’eau ou du peroxyde d’hydrogène (H2O2). H2O2 est considéré comme
responsable de la dégradation des performances des PEMFC (en attaquant le support carboné,
le catalyseur et la membrane).64 Ainsi, mise à part son importance fondamentale, l’étude du
mécanisme de cette réaction est assez importante d'un point de vue technologique.
Figure 7. Schéma simplifié d’une pile à combustible à membrane échangeuse de protons (PEMFC).65
L’introduction de l’électrode tournante à disque et à anneau (RRDE) par Frumkin et Nekrasov à la fin des années 50 et l’élaboration de sa théorie dans les années qui ont suivi,
notamment avec Ivanov et Levich ,66-68 ont rendu possibles l’identification et la quantification
de l’intermédiaire H2O2 généré sur l’électrode-disque (rotative) et oxydé sur l’électrode-anneau
(électriquement isolée) qui l’entoure. De ce fait, la voltammétrie à RRDE est devenue une méthode de choix pour l’étude de la ORR et la distinction entre ces deux principales voies
réduction catalytique électrochimique du dioxygène sur une grande variété de catalyseurs (métaux, alliages et catalyseurs moléculaires), sont rarement connus d’une façon précise et constituent souvent un sujet de discussion. En effet, il s’agit d’un mécanisme complexe faisant intervenir quatre étapes de transfert d'électrons (couplées ou découplées à des transferts de protons) et une étape de rupture de la liaison O-O. Tenant compte, en plus de cela, des intermédiaires et des phénomènes d’adsorption éventuellement impliqués, il est possible
d'écrire un très grand nombre de schémas mécanistiques pour cette réaction ;69 Pletcher et
Walsh70 indiquent qu’« au moins 14 voies réactionnelles ont été considérées pour la réduction
de l’oxygène» et que « prenant en compte les différentes étapes cinétiquement déterminantes possibles, des courbes de Tafel anodiques et cathodiques ont été établies pour 53 mécanismes afférents au système d’électrode à oxygène ». Il n'empêche que, comme nous le verrons dans le
deuxième chapitre,i le mécanisme global peut être analysé en termes d’une voie réactionnelle
directe impliquant une réduction à 4 électrons du dioxygène, en compétition avec une deuxième
voie qui passe par la production du peroxyde d’hydrogène. La formation de H2O2 est suivie par
sa réduction en H2O, ou bien par sa décomposition en H2O et O2.
Schéma 4. Les deux voies principales correspondant à la réaction de réduction de l’oxygène (ORR).
2 O 2 2H O 2 2 H O + 4 H + 4 e + 2 H + 2 e 2H O2 + 2 H + 2 e
4. But et cadre du présent mémoire
Les travaux de recherches qui font l’objet de ce manuscrit s'inscrivent dans ce cadre général de l’étude des mécanismes compétitifs rencontrés au cours des réactions catalytiques de l’activation électrochimique des petites molécules. Nous nous sommes intéressés, particulièrement, à l’étude de trois réactions directement liées aux défis énergétiques contemporains, à savoir la réduction (à deux électrons) des protons en dihydrogène, la réduction (à quatre électrons) du dioxygène en eau et la réduction (multi-électroniques) du dioxyde de carbone en « acide formique, méthanol…». L'élucidation des mécanismes de ces réactions, impliquant plus d’un seul transfert de charge et plus d’une seule étape chimique, constitue souvent une tâche ardue, en raison de la multiplicité des chemins réactionnels compétitifs possibles. Le fil conducteur du présent exposé sera la complexité croissante des mécanismes étudiés corrélativement à l’augmentation du nombre d’électrons mis en jeu et à l’implication des phénomènes d’adsorptions dans les réactions étudiées.
Dans le premier chapitre, à travers l’étude de la réaction de production de H2 (HER) dans
le dimethylformamide catalysée par la tétraphénylporphyrine de fer (FeTPP) et de la compétition entre les voies hétérolytique et homolytique qui en résulte, nous traiterons le cas d’une réaction de catalyse moléculaire impliquant deux étapes de transfert d’électrons et un seul intermédiaire catalytique, en l'absence d'adsorption des espèces réactives. Le cas des réactions catalytiques faisant intervenir de tels phénomènes d’adsorption sera traité dans les chapitres 2 et 3.
De même, pour étendre l'analyse à une réaction impliquant un transfert de plus de deux électrons avec plus d’intermédiaires et d’étapes chimiques, nous nous sommes proposés, dans
le deuxième chapitre, de sonder la catalyse moléculaire de la réduction de O2 (ORR) par une
porphyrine soluble dans l’eau : la tétrakis(N-methyl-n-pyridyl)porphyrine de fer (FeTMPyP). L’examen détaillé de cette réaction a constitué un exemple particulièrement démonstratif pour l’étude de l’interférence entre processus homogène (volume) et processus hétérogène (surface) ayant lieu au cours d’une même réaction catalytique.
Enfin, le dernier chapitre sera principalement consacré à l’étude de la compétition entre
la réduction « électrocatalytique » de CO2 et celle des acides de Brønsted sur Pt et sur Hg en
CHAPITRE I
Catalyse électrochimique moléculaire de la production de H
2dans le
DMF par la Fe(0)TPP : compétition entre voies
hétérolytique et homolytique
1. Introduction
L'histoire de la cinétique électrochimique est intimement liée à l'étude du mécanisme de
la réduction du proton en hydrogène moléculaire (HER), 2H + 2e+ H2. Il s’agit
vraisemblablement de la réaction la plus largement étudiée en électrochimie71 et sa cinétique a
été examinée dans une grande variété de conditions expérimentales et en présence de nombreux électrocatalyseurs. Cela est dû à la fois à la grande importance pratique de la HER et à la croyance, autrefois, en sa simplicité apparente capable de faire d’elle un bon modèle pour
étudier d’autres réactions électrochimiques plus complexes.72 Ces dernières années, les efforts
investis dans la recherche de nouveaux matériaux peu coûteux pour la production catalytique des combustibles chimiques se sont intensifiés. Parmi les combustibles chimiques ciblés par ces travaux de recherche, le dihydrogène est un candidat prometteur de premier plan. La HER
a ainsi regagné l'attention de la communauté scientifique.73-82
Les études mécanistiques faites sur la HER depuis le début du XXe siècle (Tafel à partir
de 190583) ont montré que le mécanisme de cette réaction mettant en jeu deux protons et deux
électrons pour générer H2 n’est pas aussi simple que ce que l'on croyait. La preuve, nous voilà
aujourd’hui après plus d’un siècle de travaux, sans conclusions claires et finales sur les différentes étapes de la réaction dans le cadre de l’électrocatalyse. Néanmoins, sans connaître les détails mécanistiques de la réaction il est clair que les meilleurs électrocatalyseurs sont des métaux nobles rares et chers. Ainsi la catalyse électrochimique moléculaire apparaît comme une alternative possible pour la HER. Divers catalyseurs moléculaires homogènes ont donc été proposés pour la production du dihydrogène à partir de la réduction des acides de Brønsted. Parmi ceux-ci, on trouve principalement les complexes à base de métaux de transition comme
le nickel, le fer, le cobalt, le molybdène et plus récemment le manganèse.84-85 Cependant, une
conception rationnelle et un perfectionnement des électrocatalyseurs moléculaires passent
inévitablement par une compréhension minutieuse du mécanisme catalytique.59, 86
Dans le cadre de la catalyse électrochimique moléculaire, la HER a fait l’objet de
plusieurs études mécanistiques expérimentales et théoriques.61, 87-92 Le mécanisme
communément proposé pour la production de H2 fait intervenir un intermédiaire clé qui se
forme suite à des transferts d’électron et de proton respectivement depuis l’électrode et le
d’électron qui précèdent la formation de l’espèce intermédiaire (flèche verte, Schéma I-1)
peuvent être associées de manière séquentielle ou concertée.61, 93 Une fois l’intermédiaire clé
formé, une bifurcation de mécanisme peut avoir lieu entre deux voies réactionnelles possibles pour la création de la liaison H−H. La première voie implique un mécanisme homolytique où
deux molécules d’hydrure métallique se combinent pour générer H2 via une élimination
réductrice. Alternativement, une voie hétérolytique peut se produire, au cours de laquelle l’espèce intermédiaire initialement formée par la réaction entre l’acide et la forme active du
catalyseur se protone davantage pour libérer H2 et régénérer le catalyseur. Lors du transfert de
proton et de l’électron vers le complexe MN-1 dessiné par une flèche verte dans le Schéma I-1,
deux possibilités quant à la localisation la charge supplémentaire sont envisageables ; s’installer dans une orbitale d du métal de transition central ou alors occuper l’orbitale s de l’atome
d’hydrogène. Dans le premier cas le complexe formé N-1
M (H ) est un radical anion alors que
dans le deuxième, il s’agit d’un complexe métal-hydrure N
M (H ) . Il s’ensuit que l’espèce intermédiaire commune située au cœur de cette dichotomie homolytique/hétérolytique pourrait
être représentée sous forme mésomère. Ainsi,
N
N-1
M (H )
M (H )
occupera un rôle crucial dans le
processus de formation de la liaison H─H dès lors qu’il est capable soit de dimériser et d’agir en tant que radical hydrogène (voie homolytique), soit de déprotoner l’acide et de se comporter comme un simple hydrure (voie hétérolytique). Il est à noter, au passage, que le mécanisme général simplifié de la réaction catalytique illustré dans le Schéma I-1 est en fait l'équivalent moléculaire homogène exact de la réduction électrocatalytique des acides de Brønsted sur une électrode métallique (Schéma I-2), avec les deux premières étapes de transfert d’électron et de proton qui sont analogues à l'étape de Volmer. L'étape homolytique et l’étape hétérolytique quant à elles, sont respectivement comparables à l'étape de Volmer-Tafel et à l’étape de Volmer-Heyrovsky. Une discussion plus détaillée mettant l’accent sur l’analogie entre les deux types de catalyse de la HER peut être retrouvée dans un papier de Mark Koper publié
Schéma I-1. Voies homolytique et hétérolytique pour la réduction du proton en dihydrogène par un
catalyseur homogène moléculaire (M).
Schéma I-2. Electrocatalyse (hétérogène) de la réduction du proton en dihydrogène ; étapes Volmer,
Volmer–Heyrovsky et Volmer–Tafel. M désigne ici l’électrocatalyseur (souvent un métal).
Savoir si la réaction suit une voie homolytique ou hétérolytique, comme expliquée
précédemment, est une question qui a été abordée depuis longtemps95-96 et qui continue d'être
un sujet de débat.97-99 La distinction entre les deux voies est souvent difficile, d’autant plus si
on considère le fait que ces deux types de mécanismes pourraient se produire simultanément
100-102 et que jusqu’à nos jours aucun des intermédiaires de la réaction n’a été détecté en raison de
leur forte réactivité.103 Des conclusions divergentes ont souvent été obtenues, que ce soit avec
le même catalyseur ou avec des catalyseurs de structures similaires. Tel est particulièrement le cas pour les complexes de cobaloximes connues de pouvoir catalyser efficacement la réduction
des protons en dihydrogène.104-105 Gray et al.97, 103 et Peters et al.106 par exemple ont rapporté
que la HER suit une voie homolytique, tandis que d’autres études comme celles faites par Artero
et al.104, 107-108 favorisent une voie hétérolytique pour la HER en présence des mêmes catalyseurs. Plusieurs calculs théoriques des grandeurs thermodynamiques qui peuvent aider à
connaitre la voie réactionnelle préférée ont été réalisés, favorisant également la voie hétérolytique.57, 92, 109-110
La voltammétrie cyclique (CV) a été largement utilisée pour étudier le mécanisme de la réaction. Cependant, pour réussir à tirer des conclusions mécanistiques fiables au moyen de cette technique, il faut disposer au préalable des relations théoriques et des critères de diagnostic qui conviennent à l’analyse des schémas réactionnels comportant une compétition entre voies mono- et bi-métallique. Certes, il est possible de faire appel à la simulation numérique de
voltammogrammes cycliques expérimentaux,111 néanmoins en raison du grand nombre de
paramètres qu’il faut considérer, plusieurs schémas réactionnels différents (avec des valeurs de paramètres appropriées) peuvent s’adapter aux données expérimentales. L’emploi des simulations numériques est dans la plupart du temps très utile pour finaliser la détermination du mécanisme, à condition d’être précédé par une analyse détaillée du problème de diffusion-réaction de manière à identifier un nombre minimal de paramètres adimensionnels qui gouvernent le système.
Le but de ce premier chapitre est de fournir les relations théoriques et les critères de diagnostic nécessaires pour faire une analyse mécanistique fiable de la HER au moyen de la voltammétrie cyclique et en présence d’un catalyseur moléculaire homogène. Pour illustrer les prédictions formelles, nous avons étudié expérimentalement un complexe bien connu au laboratoire et dont le comportement électrochimique vis-à-vis de la catalyse de réduction du
proton112 et de CO
2 avait déjà fait l’objet d’un certain nombre d’études ; il s’agit de la
tétraphénylporphyrine de fer, appelée en abrégé FeTPP, que nous avons étudiée dans le DMF en présence de trois acides de forces différentes.
2. Rappel des caractéristiques d’un mécanisme catalytique à l’aide d’un
diagramme de zone
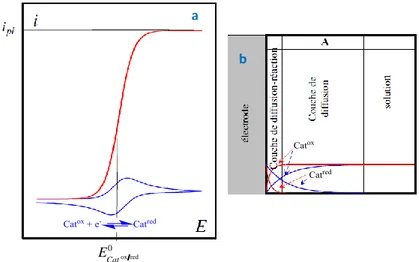
La catalyse moléculaire homogène de la HER est un processus multiélectroniques faisant intervenir plusieurs étapes électrochimiques (E) et chimiques (C). Il est néanmoins plus facile de rappeler les notions de base nécessaires à l’analyse de tels processus par voltammétrie cyclique à l’aide d’un schéma réactionnel simplifié comme celui représenté ci-dessous. Il s’agit d’un mécanisme catalytique impliquant un seul transfert d’électron suivi d’une réaction chimique de premier ordre.
Schéma I-3. Mécanisme catalytique simple. 0 ox red
cat
E et kcat sont respectivement le potentiel standard et la constante de vitesse intrinsèques caractérisant le catalyseur.
La réponse de voltammétrie cyclique attendue pour ce système peut présenter les différentes formes résumées dans la Figure I-1. La présence d’un catalyseur efficace, doté d’une
constante de vitesse catalytique kcat assez grande comparée à la vitesse de diffusion (que l’on
peut contrôler par le biais de v ; la vitesse de balayage), conduit à une réaction catalytique rapide et irréversible ayant lieu à l'intérieur d'une couche mince de diffusion-réaction située au
voisinage immédiat de l’électrode. L’épaisseur de cette couche est de l’ordre de Dcat kcat
(Dcat est le coefficient de diffusion du catalyseur).Au-delà de cette couche se développe une
couche de diffusion du substrat, puis se trouve le sein de la solution.10, 30 Dans de telles circonstances de catalyse, on peut considérer que les conditions de « cinétique pure » correspondant aux trois zones supérieures de la Figure I-1 sont réalisées ; c'est à dire que la
concentration de la forme active du catalyseur
CatRed
est pratiquement stationnaire dans lacouche de diffusion-réaction. Cette situation résulte d'une compensation mutuelle entre le flux
de production de CatRed à la surface de l’électrode par l’étape électrochimique et le flux de sa
disparition dans la couche de diffusion-réaction durant l’étape chimique.
cat k ox red 0 cat
E
Figure I-1. Diagramme de zone correspondant au schéma I.3.10
De plus, si le transfert d'électronsentre l'électrode et le couple Catox red est suffisamment
rapide pour satisfaire à la loi de Nernst et si le substrat est en grand excès par rapport au catalyseur de manière à rendre sa consommation négligeable au cours de l’expérience de voltammétrie cyclique, nous allons nous retrouvez dans des conditions de « cinétique pure avec excès de substrat ». Ces conditions de catalyse dite « canonique » conduisent, en l’absence de phénomènes secondaires sur lesquels nous reviendrons ultérieurement, à la réponse
courant-potentiel en forme de sigmoïde (Figure I-1, zone KS et Figure I-2a) donnée par
l'équation suivante : pl 1 2 1 exp ( ) i i F E E RT (I-1)
avec un potentiel de demi-vague :
0 1 2 cat
E E (I-2)
et un courant de plateau indépendant de la vitesse de balayage :
0 cat cat logRT k C F v 0 Subsrat 0 AH log C C
0 0
pl cat cat cat s
i FSC D k C (I-3)
où S est la surface de l’électrode et 0
s
C est la concentration du substrat au sein de la solution.
Figure I-2.a) Voltammogramme du catalyseur seul (bleu) et voltammogramme correspondant au Schéma I-3 (rouge) dans des conditions de catalyse canonique (zone KS). b) Profils de concentration pour un potentiel correspondant à ox red
0
Cat
E E pour le catalyseur seul (bleu) et pour une réaction catalytique correspondant au Schéma I-3 (rouge) montrant que les profils sont alors dans une couche de diffusion-réaction.10
La détermination expérimentale des valeurs du courant de plateau i et du potentiel de pl
demi-vague E1 2 spécifiques à cette réponse sigmoïdale permet d’accéder facilement à
l’ensemble des constantes cinétiques qui caractérisent le mécanisme.
ox red 0
Cat
3. Différents schémas réactionnels envisageables pour la catalyse
moléculaire de la HER
D’un point de vue mécanistique et compte tenu des deux chemins réactionnels compétitifs possibles (hétérolytique vs homolytique), le mécanisme de réduction du proton en dihydrogène par un catalyseur homogène moléculaire peut être simplifié en deux étapes de transfert d’électron (E), deux étapes de transfert de proton (P) et une étape de dimérisation (Dim). Sachant que ce processus catalytique est initié par un transfert d’électron (pour générer la forme active du catalyseur) et que, comme déjà expliqué dans l’introduction, l’étape de dimérisation doit être obligatoirement précédée d’au moins d’un transfert d’électron et d’un transfert de proton (pour produire l’intermédiaire clé de de la réaction), trois mécanismes différents sont envisageables pour la réduction du proton selon la succession des étapes E, P et Dim.
Schéma I-5. Séquence EPEDimP.
Nous savons à partir des travaux antérieurs réalisés dans des conditions identiques à nos
conditions expérimentales,112 qu’en présence de la FeTPP, l'hydrure de fer(II) formé suite à la
protonation du complexe de fer(0) est plus difficile à réduire que le complexe de fer(I). Cette information nous permet raisonnablement d’opter pour la séquence réactionnelle EEPDimP représentée dans le Schéma I-4 plutôt que les deux autres. Néanmoins pour parvenir à démêler la compétition hétérolytique/homolytique dans le cas des séquences EPEDimP et EPDimEP (Schéma I-5 et Schéma I-6) les données expérimentales correspondant à ces deux séquences pourraient être analysées d'une manière tout à fait similaire à celle que nous allons présenter dans les paragraphes suivantes pour la FeTPP.
4. Dichotomie et compétition entre voies hétérolytique et homolytique dans
le cadre d’un mécanisme de type EEPDimP (schéma I-4) en présence de la
FeTPP comme catalyseur moléculaire
La tétraphénylporphyrine de fer (Figure I-3) est un système complètement aromatique constitué d’un macro-hétérocycle tétrapyrrolique plan dont les quatre sous-unités de pyrrole
sont reliées sur les carbones α par des ponts méthines.113 Chacun de ces ponts est substitué par
un groupement phényle perpendiculaire au plan. Le fer(III) se trouve dans la cavité centrale de la porphyrine lié par complexation aux quatre atomes d’azote. La neutralité électrique est
assurée dans notre cas par un contre ionCl. On adoptera dans la suite de ce chapitre, l'écriture
J
Fe pour désigner le complexe de tétraphénylporphyrine avec l’ion de fer au degré d’oxydation J.
Figure I-3. La tétraphénylporphyrine de fer (FeTPP).
En l’absence d'acide, la réponse en voltammétrie cyclique dans le DMF met en évidence trois vagues réversibles d’oxydoréduction du complexe FeTPP qui résultent des étapes mono-électroniques représentées sur la Figure I-4 et dont l’intensité est proportionnelle à la vitesse de balayage (v), (on précise que dans tout ce chapitre, et sauf indication contraire, les courants des
différents voltammogrammes cycliques sont normalisés par rapport au courant de pic i de la p0
vague nernstienne à un électron Fe TPP obtenue à la même vitesse de balayage, en l’absence I 0
du substrat i ).
1 2
Figure I-4. Voltammétrie cyclique de FeIIITPP (1 mM) dans le DMF + 0.1 M de n-Bu
4NPF6 à 0.1 V/s,
T = 21 °C, sous Ar, sur électrode de carbone vitreux (diamètre 3 mm) en l’absence d’acide.
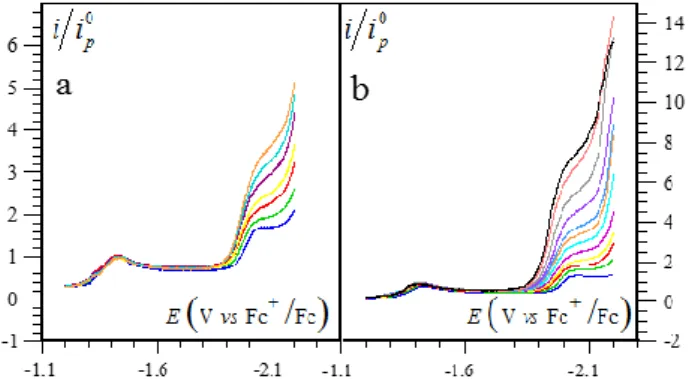
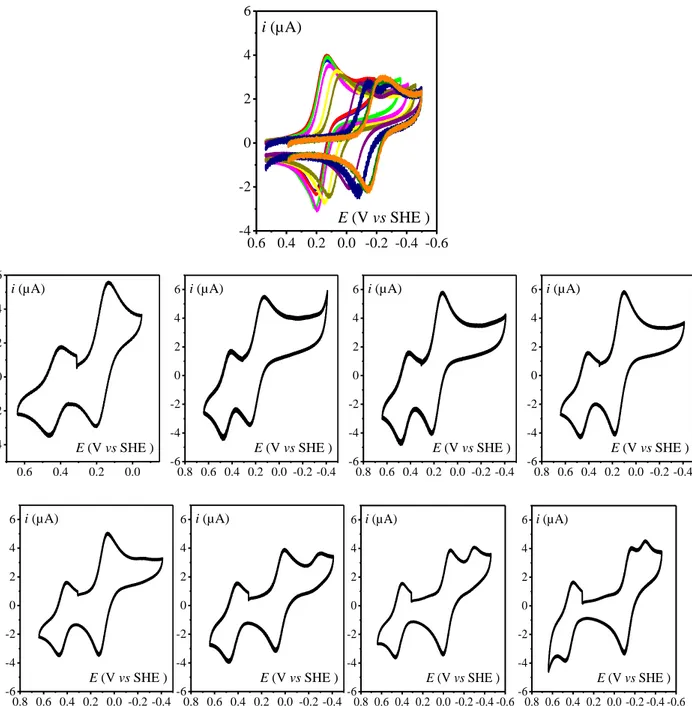
Comme le montre les voltammogrammes de la Figure I-5, l’addition de quelques
milli-moles d’un acide de force modérée, comme l'acide acétique (pKa = 13.2 dans le DMF114) par
exemple, entraîne une augmentation importante du courant de réduction au potentiel standard
0
E du couple I 0
Fe Fe accompagnée d’une perte du courant d’oxydation de la vague
correspondante. Cela indique que le complexe Fe (la forme active du catalyseur) est 0
consommé par une réaction chimique subséquente à la réduction électrochimique de Fe . I
L’augmentation observée du courant cathodique résulte de la régénération continuelle des deux
formes oxydées de la porphyrine (Fe et II Fe ) par la réaction catalytique ayant lieu à proximité I
de l’électrode dans la couche de diffusion-réaction et dont le mécanisme global est donné dans le Schéma I-7, ce mécanisme sera discuté en détail dans le paragraphe suivant.
Figure I-5. Voltammétrie cyclique de FeIITPP (1 mM) dans le DMF + 0.1 M de n-Bu
4NPF6 à 2 V/s, T = 21 °C,
sous Ar, sur électrode de carbone vitreux (diamètre 3 mm) en présence de AcOH (mM) : 0 (noir), 10 (rouge), 20 (vert), 50 (bleu). -1.2 -1.4 -1.6 -1.8 -2.0 -2.2 -2.4 -2 0 2 4 6 8 10 12 14 16 18 50 mM 20 mM 10 mM i pl / i 0 p 0 mM V Fc /Fc E vs
Schéma I-7. Séquence EEPDimP.
Comme attendu, en présence d'un excès de substrat (par exemple à 50 mM d’AcOH, Figure I-6), la vague cathodique ne présente plus de pic et prend la forme d’un sigmoïde « forme en S » dont l’intensité ne dépend pas de la vitesse de balayage. Ces conditions de catalyse canonique (cinétique pure + consommation négligeable de substrat) sont identiques à celles décrites plus haut pour un mécanisme catalytique simple. Sauf indication contraire, nous considèrerons que cette situation est toujours atteinte dans la discussion suivante qui concerne la compétition entre les voies hétérolytique et homolytique illustrées dans le Schéma I-7.
Figure I-6. Voltammétrie cyclique de FeIITPP (1 mM) dans le DMF + 0.1 M de n-Bu
4NPF6 à 2 V/s,
T = 21 °C, sous Ar, sur électrode de carbone vitreux (diamètre 3 mm) en présence de AcOH (50 mM).
4.1. Caractéristiques générales et présentation des paramètres qui contrôlent le système
Dans ce cadre, les réponses courant-potentiel « en forme de S » dépendent de trois
paramètres adimensionnels (voir annexe I) : 2
1 k k , 0 d cat 0 1 AH 2k C k C et 0 e cat 0 1 AH k C k C , où 0 cat C et CAH0 désignent
respectivement les concentrations du catalyseur et de l’acide, les constantes de vitesse k sont
telles que définies dans le Schéma I-7. Les deux premiers paramètres 2
1 k k et 0 d cat 0 1 AH 2k C k C mesurent
respectivement la rapidité des étapes de deuxième protonation (k2) et de dimérisation (kd) par
rapport à l’étape de la première protonation (k1) commune aux deux voies. Ils sont révélateurs
de la tendance de l’intermédiaire clé II I Fe (H ) Fe (H )
à être ou ne pas être en état stationnaire (ou, dit
autrement, à s’accumuler). Dans chacun de ces cas, le rapport des deux premiers paramètres donne une mesure de la compétition entre l’étape de la seconde protonation et celle de la dimérisation, autrement dit la compétition entre la voie hétérolytique et la voie homolytique. Le troisième paramètre 0 e cat 0 1 AH k C
k C relatif à cette séquence EEPDimP (que l’on peut écrire
autrement sous la forme : EPDimPE) est associé à la réduction du complexe Fe issu du II
processus hétérolytique. Cette étape de réduction, qui boucle le cycle catalytique, peut avoir lieu à la surface de l'électrode ou dans la solution (i.e. au sein de la couche de diffusion réaction)
-1.2 -1.4 -1.6 -1.8 -2.0 -2.2 -2 0 2 4 6 8 10 12 14 16 18 i pl 50 mM i pl / i 0 p
Une "cinétique pure" avec une consommation négligeable du substrat
E
1/2
V Fc /Fc
par réaction avec le complexe Fe . Si 0 k1 est grande, le complexe Fe , tout comme les autres II intermédiaires, est formé à proximité de la surface de l'électrode et est réduit avant d'avoir le
temps de rencontrer le complexe Fe et de réagir avec lui (situation de type ECE). En revanche, 0
une grande valeur de
0 e cat
0 1 AH
k C
k C implique que ce transfert d’électron a eu lieu dans la solution
(situation SET). En effet, comme le montre la Figure I-5, en l’absence d’acide les deux vagues
réversibles correspondant aux deux premières étapes de transfert d’électrons du Schéma I-7
sont observées à des potentiels standards
0E respectivement égaux à 1.34 et 2.04 V vs
Fc+/Fc). Ce séquençage de potentiel laisse augurer une thermodynamique très favorable
(enthalpie libre standard d’environ 0.7 eV ) pour la réaction de transfert d’électron entre les
complexes Fe et II Fe . Ainsi, il est raisonnable de considérer que l’étape SET apparaissant à 0
la fin du Schéma I-7 soit fortement favorisée et, par conséquent, dotée d'une constante de vitesse
de transfert électronique homogène
ke proche de la vitesse limite de diffusion en solution
10 -1 -1
dif 2 10 M s
k .
4.2. Situation SET
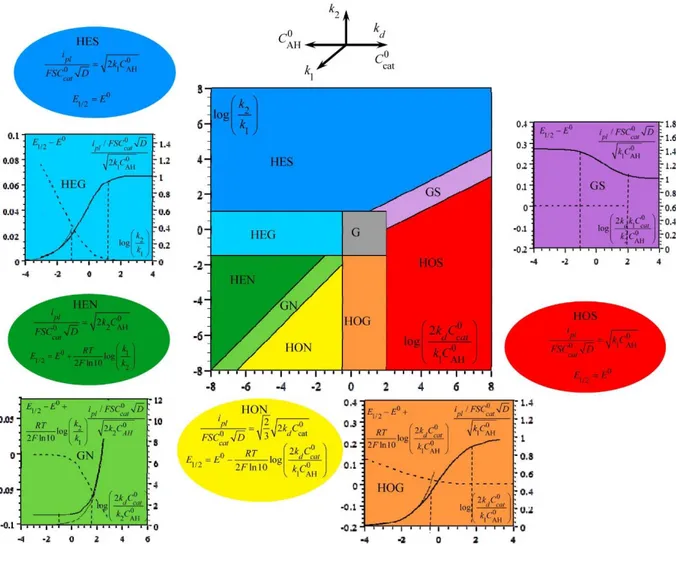
Nous examinerons tout d'abord la situation SET
0 e cat 0 1 AH k C k C . Le diagramme de zone
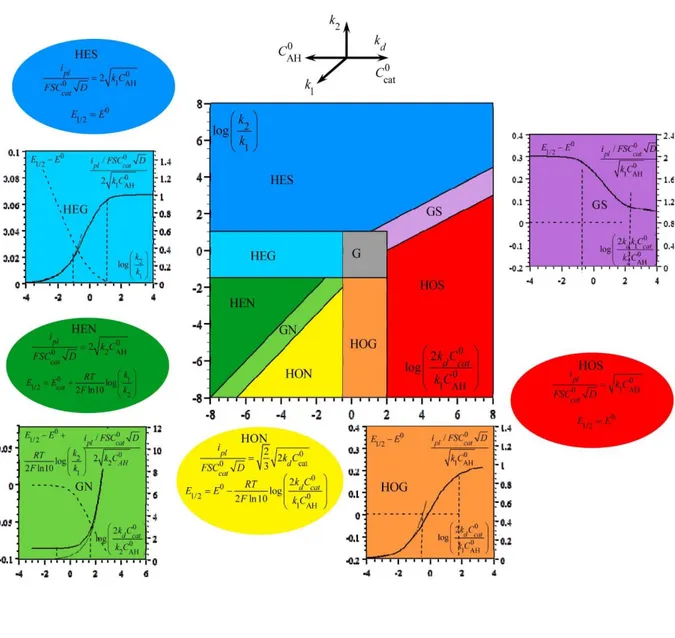
de la Figure I-7 résume les caractéristiques des réponses voltammétriques « en forme de S » en
fonction des deux paramètres 2
1 k k et 0 d cat 0 1 AH 2k C
k C . Les zones cinétiques représentées dans ce
diagramme sont obtenues en donnant des valeurs extrêmes à ces paramètres, respectivement 0 et ∞, de manière à atteindre des comportements simplifiés qui dépendent d'un nombre moins important de paramètres, respectivement zéro et un. En raison des incertitudes expérimentales dans la mesure des courants de plateau (~5%) et des potentiels de demi-vague (~5 mV), ces comportements simplifiés sont atteints expérimentalement respectivement pour des petites et des grandes valeurs finies des paramètres adimensionnels. Dans la détermination des frontières entre les zones, la zone la plus simple est privilégiée, c’est-à-dire la zone à zéro paramètre au détriment de la zone à un paramètre, la zone à un paramètre au détriment de la zone à deux paramètres et ainsi de suite. La superficie de la zone G à deux paramètres, où G signifie
« général », est ainsi réduite au minimum au profit des zones à zéro et à un paramètre. Les démonstrations de ces expressions sont détaillées dans l’annexe I.
Figure I-7. Compétition entre voies hétérolytique et homolytique dans le régime SET
0 e cat 0 1 AH k C k C .
Le diagramme de zone ci-dessus résume les différents comportements cinétiques en
fonction des deux paramètres adimensionnels 2
1 k k et 0 d cat 0 1 AH 2k C
k C . Les zones sont identifiées en
utilisant les abréviations suivantes : HE = hétérolytique ; HO = homolytique ; S = état stationnaire ; N = état non-stationnaire ; G = général. Les effets des paramètres expérimentaux contenus dans les paramètres adimensionnels sont résumés (sous une forme logarithmique) par l'ensemble de flèches en haut du diagramme, ce qui permet de voyager à travers les différentes zones. Les zones bleue (HES) et verte (HEN) correspondent aux voies hétérolytiques dans
lesquelles l’intermédiaire clé II I Fe (H ) Fe (H )
est ou n’est pas, respectivement, en état stationnaire. De
même, les zones rouge (HOS) et jaune (HON) correspondent aux voies homolytiques dans lesquelles le même intermédiaire est ou n’est pas, respectivement, en état stationnaire. Dans ces quatre cas, les expressions du courant de plateau et du potentiel de demi-vague à appliquer sont de formes assez similaires. Elles sont données dans l'encart ovale de la même couleur que la zone correspondante. Dans le cas hétérolytique, le passage du régime stationnaire au régime non-stationnaire se produit à travers la zone HEG. Les variations du courant de plateau (ligne noire épaisse) et du potentiel de demi-vague (ligne pointillée) avec le paramètre approprié sont représentées sous forme de courbes dans l'encart carré de la même couleur (cyan). La ligne fine représente la variation du courant de plateau dans la zone bornée correspondante. Les lignes verticales (en pointillé) indiquent les frontières entre les différentes zones. Il en va de même pour le passage du régime stationnaire au régime non-stationnaire à travers la zone HOG (orange) dans le cas homolytique. La zone en magenta (GS) traduit le passage de la voie hétérolytique vers la voie homolytique dans le régime stationnaire. Les variations du courant de plateau et du potentiel de demi-vague sont données dans ce cas en fonction d’un paramètre adimensionnel qui reflète la compétition directe entre les deux voies. De même pour la zone en vert clair (GN) qui décrit le passage de la voie hétérolytique vers la voie homolytique dans le régime non-stationnaire, néanmoins, il faut noter que le paramètre adimensionnel de compétition utilisé dans ce cas est diffèrent. Les limites entre les zones correspondent à une incertitude du courant de plateau de 10% et une incertitude du potentiel de demi-vague 5 mV. Les potentiels sont donnés en volts. S est la surface de l'électrode et D est le coefficient de diffusion du catalyseur. 4.3. Situation ECE La situation ECE 0 e cat 0 1 AH 0 k C k C
donne lieu à un autre diagramme de zone (Figure I-8)
analogue à celui obtenu dans la situation SET (Figure I-7) avec des expressions de courants de plateau et de potentiels de demi-vague qui sont similaires (quoique non identiques) dans les deux cas. Les mêmes définitions et caractéristiques utilisées dans le cas du premier diagramme de zone (Figure I-8) restent valables. Les expressions du courant de plateau dans les zones
(HES) et (HEN), ainsi que dans les zones de transition qui leurs correspondent sont modifiées en comparaison avec le régime SET.
Figure I-8. Compétition entre voies hétérolytique et homolytique dans le régime ECE
0 e cat 0 1 AH 0 k C k C .
4.4. Exploitation des diagrammes de zone
Le diagnostic du mécanisme est fondé sur l’observation combinée du courant de plateau
ipl et du potentiel de demi-vague
E1 2 et de leur variation ou absence de variation enfonction des concentrations de l’acide et du catalyseur, selon les équations données dans les encarts ellipsoïdes correspondant aux zones à zéro paramètre. Les courbes dessinées dans les encarts rectangulaires renseignent sur les variations attendues lors du passage d'une zone à zéro
paramètre à une autre en changeant les concentrations de l’acide et du catalyseur. Une
estimation des trois constantes de vitesse (k1, k2et kd) s’ensuit. La fiabilité du diagnostic est
ensuite vérifiée grâce à la simulation numérique et les valeurs des constantes de vitesse sont ajustées simultanément pour les adapter au mieux aux données expérimentales.
La situation la plus simple est rencontrée quand les expressions du courant de plateau et du potentiel de demi-vague changent lors du passage d'une zone à zéro paramètre à une autre zone de même type. Dans ce cas, il est possible de distinguer entre les deux zones en examinant
la variation ou l'absence de variation de ipl et de E1 2 lorsque les concentrations de l’acide et du
catalyseur changent. Dans le cas contraire, lorsque les expressions formelles de ipl et de E1 2
sont identiques comme c’est le cas dans les zones HES et HOS, la distinction entre les deux semble être irréalisable à première vue. Néanmoins, la stratégie suivante peut être appliquée :
les expressions du courant de plateau varient d'un facteur 2 (dans la situation SET) ou 2 (dans
la situation ECE) en traversant la zone de transition GS. Par conséquent, si la zone de transition est atteinte suite à un changement de concentration d'acide et/ou de catalyseur, la distinction entre les zones HES et HOS devient possible. Dans le cas contraire, l'absence d’une zone de transition implique que
0 d cat
0 1 AH
2k C
k C est soit <100 (zone HES), soit >100 (zone HOS). Ainsi, on
peut faire l’hypothèse que le système se trouve dans l’une des deux zones (HES ou HOS) et
estimer ensuite la valeur de k1 dans chaque cas. La comparaison des valeurs de kd qui en
découlent avec la constante de vitesse limite de diffusion permettra finalement de faire une distinction effective entre les deux zones.
5. Application : catalyse de réduction de la triéthylamine protonée, du
phénol et de l’acide acétique dans le DMF par la tétraphénylporphyrine de
fer(0) électrogénérée
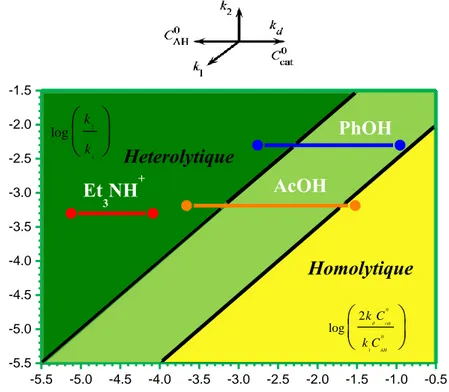
Pour illustrer expérimentalement la stratégie de diagnostic décrite dans le paragraphe précédent, nous avons réalisé une série d’expérience avec la FeTPP dans le DMF en présence
de trois acides de forces différentes, à savoir, la triéthylamine protonée (Et3NH+), l’acide
acétique (AcOH) et le phénol (PhOH).
5.1. Cas de la triéthylamine protonée
La très forte catalyse observée avec triéthylamine protonée entraîne l’apparition d’un pic arrondi dont la hauteur varie avec la vitesse de balayage et non pas d’une réponse « en forme de S » avec un palier indépendant de v comme attendu (Figure I-9). On peut noter également la présence d’une seconde vague catalytique située au niveau de la vague de réduction de l'hydrure de fer(II) ayant lieu à un potentiel plus négatif que celui de la réduction du complexe de fer(I).
112 La forme arrondie de la première vague catalytique, qui fait l’objet de cette étude, est en fait
due aux phénomènes secondaires qui accompagnent le processus catalytique et tout
particulièrement à la forte consommation du substrat (ici Et3NH+) dans la couche de
diffusion-réaction.115 Outre la consommation du substrat, les principaux phénomènes secondaires que
l’on peut rencontrer sont l'inhibition de la surface de l’électrode par les produits de la réaction et la désactivation du catalyseur. Ces effets augmentent avec la charge passée au cours de l'expérience de voltammétrie cyclique et sont donc réduits au minimum en augmentant la