HAL Id: pastel-00601961
https://pastel.archives-ouvertes.fr/pastel-00601961
Submitted on 21 Jun 2011HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Effets de régimes hyperlipidique et cafeteria sur le
développement de l’obésité et ses désordres associés chez
la souris
Charles Desmarchelier
To cite this version:
Charles Desmarchelier. Effets de régimes hyperlipidique et cafeteria sur le développement de l’obésité et ses désordres associés chez la souris. Alimentation et Nutrition. AgroParisTech, 2010. Français. �NNT : 2010AGPT0022�. �pastel-00601961�
N°: 2009 ENAM XXXX
AgroParisTech
Molecular Nutrition Unit, Technische Universität München
présentée et soutenue publiquement par
Charles DESMARCHELIER
Le 8 mars 2010Effects of high fat and cafeteria diets on obesity development and
associated metabolic disturbances in mice
Doctorat ParisTech
T H È S E
pour obtenir le grade de docteur délivré par
L’Institut des Sciences et Industries
du Vivant et de l’Environnement
(AgroParisTech)
Directeur de thèse : Hannelore Daniel
Jury
M. Daniel TOME, Professeur, Agroparistech Examinateur
M. Dominique BAUCHART, Directeur de Recherche, INRA Rapporteur
M. Xavier BIGARD, Professeur, CRSSA Rapporteur
M. Mihai COVASA, Professeur, Pennsylvania State University Examinateur
Acknowledgments
I would like to thank my supervisor Prof. Hannelore Daniel for giving me the chance
to do this project and for her support and guidance over the years.
I would like to thank the members from the Nusisco program and committee for
funding this project and for all the meetings and events.
I would like to thank Daniel Tome and Gilles Fromentin for their help for my
inscription in this PhD program and for the organization of my PhD defense.
I would like to thank my examiners and my reporters for accepting to examine my
thesis.
I would like to thank everyone at ZIEL, especially Dr. Thomas Clavel, Adelmar
Stamfort, Tobias Ludwig, Christoph Dahlhoff and Tanja Heidler for the work done in
common and all the discussions.
I would like to thank Prof. Jimmy Bell, Jelena Anastasovska, Mohammed Hankir,
Michael Russ and everyone at the BIC for having hosted me 6 months in their lab in
London.
I would like to thank my mother and my sister for their constant support over these
Finally, I would like not to thank all the people who kept enticing me into climbing and
were often successful at it during these last 3 years, especially Tobias, Korbi,
Thomas, Hase, Tilo, Alex, Frosch, Claus, Dave, Simon, Michael, Trev, Andy and all
Abstract
Introduction: Obesity results from a prolonged imbalance between energy intake and energy expenditure, as depending on basal metabolic rate, heat production,
thermogenic effects of the diet and physical activity. Diet-induced obesity (DIO) in
rodents can be achieved by different regimens and approaches. Diets providing a high fat intake have been established as a “gold standard” to generate obese rodent
models and have proven to initiate pathologies similar to those encountered in
humans. However, this dietary treatment is far from being standardized and its
relevance has been criticised on the basis of findings in humans that total energy
intake rather than fat per se determines body fat accumulation in humans. Hence,
cafeteria diets have been introduced by providing a choice of several palatable food
items of variable composition, appearance and texture in addition to a non-purified
diet. Those approaches have been shown to induce obesity by a hyperphagia.
Objective: This thesis aimed at comparing the effects of a high fat vs. a cafeteria diet on food intake, weight gain and determinants of energy homeostasis and metabolism
in obese mice. Results: Our key findings demonstrate that both a high fat and a
cafeteria diet were almost equally efficient in driving an obese phenotype but did not
necessarily elicit the same metabolic changes. The cafeteria diet as characterised by
a higher carbohydrate (mainly sucrose) and lower fat content seemed to be more
deleterious for liver steatohepatosis and provoked more pronounced changes in the
gut microbiota. Despite a lower cholesterol content than in the high fat diet, mice fed
the cafeteria diet presented levels of circulating cholesterol as high as animals on a
high fat diet. Changes in gene expression in liver and intestine suggested an
pronounced effects of the two high calorie diets causing obesity when compared to
animals on control diet remaining lean vanished when diets with identical
composition were supplied in powder form and not as standard pellets. Here, even
the control diet with a high starch but very low fat content caused a substantial weight
gain with only minor differences to the two other high-calorie diets. Conclusion: The
results presented here raise the question of whether high fat diets used for induction
of obesity are the proper models to simulate human obesity and its pathologies.
Résumé
Introduction : L’obésité est causée par un déséquilibre prolongé entre les apports
énergétiques et l’activité physique, dépendant du métabolisme de base, de la
production de chaleur et des effets thermogéniques du régime et de l’activité
physique. Chez les rongeurs, l’obésité induite par le régime peut être obtenue par
différents régimes et approches. A cet égard, les régimes hyperlipidiques sont considérés comme les régimes de référence pour générer des modèles de l’obésité
chez le rongeur et engendrent des pathologies similaires à celles rencontrées chez l’homme. Cependant, ce régime alimentaire est loin d’être standardisé et a été
critiqué sur le fait que la prise énergétique totale et non uniquement les lipides
régissait l’accumulation de graisse corporelle chez l’homme. Ainsi, les régimes
cafétéria ont été introduits : en offrant en plus d’un régime non purifié un choix de
plusieurs aliments appétants, de composition, d’apparence et de texture différentes, ils permettent le développement de l’obésité en déclenchant l’hyperphagie. Objectif :
L’objet de ces travaux a été de comparer chez des souris obèses les effets d’un
régime hyperlipidique à ceux d’un régime cafétéria sur la prise de nourriture, la prise
de poids et les déterminants du métabolisme et de l’homéostase énergétique.
Résultats : Nos résultats démontrent qu’un régime hyperlipidique et un régime
cafeteria permettent tous deux d’obtenir un phénotype obèse mais sans causer
nécessairement les mêmes changements métaboliques. Le régime cafétéria,
caractérisé par un contenu en glucides (principalement le sucrose) plus élevé et un
contenu en lipides plus faible, semble avoir des conséquences plus néfastes pour le
foie et provoque des changements plus prononcés au niveau du microbiote
cholestérolémie similaire. Les niveaux d’expression des gènes impliqués dans le
métabolisme du cholestérol dans l’intestin grêle et le foie suggèrent une
augmentation de la synthèse de cholestérol de novo et une modification de son
transport, ces effets étant plus marqués chez les souris nourries au régime
hyperlipidique. Conclusion : Ces résultats remettent en question le statut des régimes hyperlipidiques pour déclencher l’obésité et pour générer ses pathologies
associées. Les régimes cafétéria sont aussi efficaces à cet égard et sont plus proches des régimes consommés chez l’homme.
Keywords
High fat diet, cafeteria diet, food texture, food intake, adipose tissue, small intestine,
intrahepatic triacylglyceride, cholesterol metabolism, gut microbiota, microarrays,
Fourier transform infrared spectroscopy, obesity
Mots clés
Régime hyperlipidique, régime cafeteria, texture alimentaire, prise alimentaire, tissu
adipeux, intestin grêle, triacylglyceride intrahépatique, métabolisme du cholestérol,
microbiote intestinal, puce à ADN, Spectroscopie infrarouge à transformée de
Declaration of contributors
Experiments of Study 1 of this thesis were conducted at:
Molecular Nutrition Unit
ZIEL - Research Center for Nutrition and Food Sciences
Technische Universität München (TUM)
Gregor-Mendel-Str. 2
85350 Freising - Weihenstephan
Germany
Clinical Nutritional Medicine
ZIEL - Research Center for Nutrition and Food Sciences
Technische Universität München (TUM)
Gregor-Mendel-Str. 2
85350 Freising - Weihenstephan
Germany
Molecular Nutritional Medicine
ZIEL - Research Center for Nutrition and Food Sciences
Technische Universität München (TUM)
Gregor-Mendel-Str. 2
85350 Freising - Weihenstephan
Experiments of Study 2 of this thesis were conducted at:
Molecular Nutrition Unit
ZIEL - Research Center for Nutrition and Food Sciences
Technische Universität München (TUM)
Gregor-Mendel-Str. 2
85350 Freising - Weihenstephan
Germany
Friedrich Schiller University
Institute of Nutrition
Dornburger Str. 24
07743 Jena
Germany
Experiments of Study 3 of this thesis were conducted at:
Molecular Nutrition Unit
ZIEL - Research Center for Nutrition and Food Sciences
Technische Universität München (TUM)
Gregor-Mendel-Str. 2
85350 Freising - Weihenstephan
Biofunctionality
ZIEL - Research Center for Nutrition and Food Sciences
Technische Universität München (TUM)
Gregor-Mendel-Str. 2
85350 Freising - Weihenstephan
Germany
Microbiology
ZIEL - Research Center for Nutrition and Food Sciences
Technische Universität München (TUM)
Weihenstephaner Berg 1
85350 Freising – Weihenstephan
Germany
Lehrstuhl für Biologische Chemie
Technische Universität München (TUM)
Emil-Erlenmeyer-Forum 5
85350 Freising-Weihenstephan
Table of contents
Acknowledgements 2 Abstract 4 Résumé 6 Keywords 8 Mots clés 8 Declaration of contributors 9 Table of contents 12 List of figures 15 List of tables 17 Abbreviations 19Publications and scientific communications 23
General Introduction 25
I. Literature review 27
I.I. Diet-induced obesity in animal models 28
I.I.1. Animal models of obesity 28
I.I.2. High fat diets in diet-induced obesity 29
I.I.2.1. The effect of high fat diets on the development of obesity 29
I.I.2.2. High fat diets and hyperphagia 30
I.I.2.3. Diet-induced thermogenesis 31
I.I.2.4. The effect of the dietary fat intake on the development of obesity 32
I.I.2.5. Ketogenic diets 33
I.I.2.6. The effect of dietary fat composition on the development of obesity 34
I.I.3. Cafeteria diet in diet-induced obesity 35
I.I.3.1. Is a high fat diet the most appropriate model to simulate a Western
diet in human obesity?
35
I.I.3.2. The cafeteria diet: a high fat / high sugar diet inducing hyperphagia 36
I.I.3.3. The effect of flavour on hyperphagia 37
I.I.3.4. The limits of the cafeteria diet 37
I.II. Physiological effects of high fat diets 38
I.II.1. The adipose tissue 38
I.II.1.1. Remodelling of the adipose tissue 38
I.II.1.2. Insulin resistance 38
I.II.1.3. Lipoprotein lipase activity 39
I.II.1.4. De novo lipogenesis 39
I.II.1.5. Adipokines 40
I.II.1.6. Inflammation 41
I.II.2. Liver 41
I.II.2.1. Hepatic steatosis 41
I.II.2.2. Hepatic insulin resistance 42
I.II.2.3. Hepatic de novo lipogenesis 42
I.II.3. Intestine 43
I.II.3.1. Fat absorption 43
I.II 3.2. Lipoprotein secretion 45
I.II.3.3. Lipid oxidation 45
I.II.3.4. Hormone secretion 46
I.III. The gut microbiota and obesity 47
I.III.1. Role of the gut microbiota in fat storage 48
I.III.3. Gut microbiota and lipoprotein lipase (LPL) activity 50
I.III.4. Gut microbiota and hepatic lipogenesis 51
I.III.5. Gut microbiota and AMP-activated protein kinase 51
I.III.6. Gut microbiota and metabolic endotoxemia 52
I.III.7. Composition of the gut microbiota in obese states 53
I.III.7.1. In genetically obese mice 53
I.III.7.2. In diet-induced obese mice 54
I.III.7.3. In humans 56
II. Experimental work conducted 57
Study 1: Influence of high fat diets on pathophysiological changes in obese mice
60
Study 2: A cholesterol-paradox: high fat diets induce major changes in intestinal cholesterol metabolism with reduced tissue levels despite a plasma hypercholesterolemia
90
Study 3: Chemical and phylogenetic alterations of mouse cecal microbiota induced by diet and obesity
131
III. Final discussion and conclusion 166
IV. References 171
List of figures
Part 1: Literature review
Figure 1.1. Body fat mass as a function of body weight in 43 strains of inbred mouse
fed a high fat diet for 8 weeks.
Figure 1.2. Summary of the effects of high fat diets on the adipose tissue.
Figure 1.3. Summary of the effects of high fat diets on the liver.
Figure 1.4. Summary of the effects of high fat diets on the small intestine.
Figure 1.5. Summary of the effects of gut microbiota on host metabolic and
inflammatory processes.
Part 2: Experimental work conducted Study 1
Figure 2.1. Body weight developments in animals receiving the different diets during
12 or 18 weeks of feeding.
Figure 2.2. Liver weight and IHTG in animals receiving the different diets.
Study 2
Figure 3.1. Body weight developments in animals fed the different diets.
Figure 3.2. Glucose tolerance in animals fed for 9 weeks the different diets.
Figure 3.3. Selected blood parameters.
Figure 3.4. Cholesterol balance data obtained from animals fed the different diets for
12 weeks.
Figure 3.5. Cholesterol and TG contents in intestine and liver of animals fed the
different diets for 12 weeks.
Figure 3.7. The effect of dietary fat on the expression of genes related to cholesterol
and lipid metabolism in the small intestine.
Study 3
Figure 4.1 Cumulative body weight gain over the 12 week feeding trial.
Figure 4.2 Spectral analysis revealed distinct chemical patterns.
Supplemental figure 4.1 Calculation of bacterial proportions using FISH coupled with
flow cytometry.
Supplemental figure 4.2 Dot plots showing fluorescence signals obtained with the
16S rRNA probe Ecyl-0387 in both cohorts of mice.
Supplemental figure 4.3 Dot plots showing the purity of sorted bacteria hybridized
List of tables
Part 1: Literature review
Table 1.1. Single gene mutations associated with an obesity phenotype
Study 1
Table 2.1. Composition of the different diets employed.
Table 2.2. Final body weight, cumulative food, energy, water and macronutrient
intake in animals receiving the different diets either provided in pellet or powder form.
Table 2.3. Serum clinical chemistry and adipokine levels.
Table 2.4. Organ weight.
Table 2.5. Relative expression of selected target genes in visceral adipose tissues.
Study 2
Table 3.1. Diet composition.
Table 3.2. Primer sequences.
Table 3.3. Effect of a chronic high fat and cafeteria diet on final body weight,
cumulative food, energy, water, macronutrient and cholesterol intake and energy
assimilation according to the dietary treatment.
Table 3.4. Effect of a chronic high fat and cafeteria diet on the expression of genes
related to cholesterol and lipid metabolism in the small intestine.
Table 3.5. Effect of a chronic high fat and cafeteria diet on the expression of genes
related to cholesterol in the liver.
Study 3
Table 4.2 Group-specific 16 rRNA oligonucleotide probes used for in situ
hybridization.
Table 4.3 Final body weight, cumulative energy, water and macronutrient intake
according to the dietary treatment.
Abbreviations
11-β-HSD-1: 11β-hydroxysteroid dehydrogenase type 1
Abca1: ATP-binding cassette, sub-family A, 1
Abcg5 : ATP-binding cassette, sub-family G, 5
Abcg8 : ATP-binding cassette, sub-family G, 8
ACC: acetyl CoA carboxylase
Acc1: acetyl-CoA carboxylase 1 Actb: β-actin
ALT: alanine aminotransferase
AMP: adenosine monophosphate
AMPK: AMP-activated protein kinase
Apo: apoplipoprotein
ARH(1): heterogeneous autoregressive order one
AST: aspartate aminotransferase
ATGL: adipose triacylglycerols lipase
ATP: adenosine triphosphate
BAT: brown adipose tissue
BMI: body mass index
Bp: base pairs
Cal: calorie
CD14: cluster of differentiation 14
CD36: cluster of differentiation 36
ChREBP: carbohydrate response element binding protein
CLA: conjugated linoleic acid
CoA: coenzyme A
CPT1: carnitine palmitoyl transferase-1
Cyp27a1: cytochrome P450, family 27, subfamily a, polypeptide 1
Cyp51: cytochrome P450, family 51
DHA: docosahexaenoic acid
Dhcr7: 7-dehydrocholesterol reductase
DIO: diet-induced obesity
EAT: epididymal adipose tissue
EDTA: ethylenediaminetetraacetic acid
EPA: eicosapentaenoic acid
Fas: fatty acid synthase
FATP-4: fatty acid transport protein 4
Fiaf: fasting-induced adipose factor
FISH: fluorescent in situ hybridization
FT-IR: Fourier transform infrared
GAPDH: glyceraldehyde 3-phosphate dehydrogenase
GE: gross energy
GF: germ-free
GIP: gastric inhibitory polypeptide
GLP-1: glucagon-like peptide-1 Gpr41: G protein-coupled receptor 41 Gpr43: G protein-coupled receptor 43 HDL: high-density lipoprotein Hmgcr: 3-hydroxy-3-methylglutaryl-Coenzyme A reductase Hmgcs2: 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 Hprt: hypoxanthine phophoribosyltransferase
Idh1: isocitrate dehydrogenase 1 (NADP+), soluble
I-FABP: intestinal fatty acid binding protein
IHTG: intrahepatic triacylglyceride
IRS-1: insulin receptor substrate 1
IRS-2: insulin receptor substrate 2
LDL: low-density lipoprotein
LDLr: LDL receptor
L-FABP: liver fatty acid binding protein
LPL: lipoprotein lipase
LPS: lipopolysaccharide LXRα: liver X receptor α
MAT: mesenteric adipose tissue
MCP-1: monocyte chemotactic protein-1
ME: metabolizable energy
Me1: malic enzyme 1, NADP(+)-dependent, cytosolic
MTP: microsomal triacylglyceride transfer protein
mRNA: messenger ribonucleic acid
MUFA: mono-unsaturated fatty acid
Mvd: mevalonate decarboxylase
N: nitrogen
NADP: nicotinamide adenine dinucleotide phosphate
NAFLD: non-alcoholic fatty liver disease
Npc1l1: Niemann-Pick C1-like protein 1
Nsdhl: NAD(P) dependent steroid dehydrogenase-like
PBS: phosphate buffered saline
Pgc-1α: peroxisomal proliferator activated receptor coactivator 1α
Pmvk: phosphomevalonate kinase
PUFA: poly-unsaturated fatty acid
PYY: peptide YY
qPCR: real-time quantitative polymerase chain reaction
RT: room temperature
Scarb1: scavenger receptor class B, member 1
Scd1: stearoyl-Coenzyme A desaturase 1
SCFA: short-chain fatty acids
SDS: sodium dodecyl sulfate
SFA: saturated fatty acid
Slc25a1: solute carrier family 25 (mitochondrial carrier, citrate transporter), member 1
SR-B1: scavenger receptor class B1
SREBP-1: sterol response element binding protein 1
SREBP-2: sterol response element binding protein 2
TG: triacylglycerols
TLR-4: toll-like receptor 4
Tm7sf2: transmembrane 7 superfamily member 2
UCP: uncoupling protein
UN: unstructured
V/v: ratio volume to volume
VLDL: very low-density lipoprotein
W/v: ratio weight to volume
W/w: ratio weight to weight
WAT: white adipose tissue
Publications and scientific communications
Publications:
Clavel T., Desmarchelier C., Binder U., Wenning M., Skerra A. ,Haller D., Daniel H.
Chemical and phylogenetic alterations of mouse cecal microbiota induced by diet and
obesity. Submitted to Journal of Nutrition (in revision).
Desmarchelier C., Ludwig T., Bader B.L., Klingenspor M., Daniel H. Diet-induced
obesity in ad libitum fed mice: food texture overrides the effect
of macronutrient composition. Submitted to Journal of Nutrition (in revision).
Desmarchelier C., Dahlhoff C., Keller S., Sailer M., Daniel H. A cholesterol-paradox:
a high fat diet induces major changes in intestinal
cholesterol metabolism with reduced tissue levels despite a plasma
hypercholesterolemia. Submitted to Journal of Lipid Research.
Communications:
Desmarchelier C., Dahlhoff C., Keller S., Sailer M., Daniel H. A cholesterol-paradox: a high fat diet induces major changes in intestinal
cholesterol metabolism with reduced tissue levels despite a plasma
hypercholesterolemia. Poster communication at the XI International Conference on
Obesity, July 2010, Stockholm, Sweden.
Desmarchelier C., Ludwig T., Bader B.L., Klingenspor M., Daniel H. Diet-induced obesity in ad libitum fed mice: food texture overrides the effect
of macronutrient composition. Poster communication at the XI International
Conference on Obesity, July 2010, Stockholm, Sweden.
Desmarchelier C., Daniel H. Intestinal cholesterol metabolism in obese mice. Poster communication at the annual meeting of Société Française de Nutrition (SFN),
December 2009, Montpellier, France.
Desmarchelier C., Daniel H. Comparison of the effects of different obesigenic diets on mice. Poster presentation for scientist of Unilever R&D September 2009,
Vlaardingen, The Netherlands.
Desmarchelier C., Daniel H. The intestine as a target for obesity-associated disturbances in signalling and function. Poster presentation for scientist of Unilever
R&D September 2008, Vlaardingen, The Netherlands.
Desmarchelier C., Daniel H. The intestine as a target for obesity-associated disturbances in signalling and function. Poster presentation for scientist of Unilever
General introduction
Obesity incidence has increased rapidly over the last two decades, reaching an
epidemic state. An incidence of above 20% has been observed in most Western
countries (WHO 2007), particularly in the US and UK, with an alarmingly high
incidence among children (Rocchini 2002). The estimated number of overweight
children globally in 2005 was at least 20 million (WHO 2005). Obesity is defined as a
body mass index (BMI) of 30 kg/m2 or more and overweight is defined as a BMI of 25 kg/m2 or more by the World Health Organization (WHO) (WHO 2005). However, BMI is not necessarily the best parameter in defining obesity, and in particular not for
predicting obesity-associated metabolic problems.
Obesity is known to increase the risk of developing a variety of disorders including
type 2 diabetes, coronary heart disease, osteoarthritis, as well as certain types of
cancer and psychological problems (Pi-Sunyer 1993; Must et al. 1999; Visscher and
Seidell 2001; Calle and Kaaks 2004). It is also highly associated with the metabolic
syndrome, which includes conditions such as impaired glucose tolerance, insulin
resistance, dyslipidemia and hypertension (Grundy 2004; Grundy et al. 2004). This
condition is a worldwide problem, and is showing a worrying increase in developing
countries as well as developed countries (Prentice 2006).
The dramatic rise in obesity and the metabolic syndrome are a consequence of
several lifestyle factors in modern societies. Factors such as nutrition, physical
activity, smoking, alcohol and stress are well known lifestyle components associated
with the development of obesity-associated diseases (Ueno et al. 1997). Modern
nature of many jobs with technological developments and the increasing use of
computers for everyday tasks as well as readily available, high calorie, ready-made
diets.
Thus, there is currently a great need fot effective therapies. Numerous medical and
behavioural interventions have been tried to treat obese patients but only a few were
successful. Pharmacological compounds often had to be withdrawn, due to severe
undesired side effects (Farrigan and Pang 2002). Bariatric surgery is considered the
most successful treatment in highly obese patients but the significant risk of complications does not allow its wide‐range use (Sjostrom et al. 2004). Therefore, there is a huge challenge for the scientific community to search for more effective
I.I. Diet-induced obesity in animal models
I.I.1. Animal models of obesity
To understand the genetic and environmental basis of obesity, animal models have
proven useful by allowing manipulations technically or ethically not feasible in
humans (Speakman et al. 2008). In these models, obesity can be induced by genetic
mutations (See Table 1.1 for spontaneous single gene loss-of-function mutations),
pharmacologically, by injecting gold thioglucose for example (Brecher and Waxler
1949) or by a variety of dietary manoeuvres.
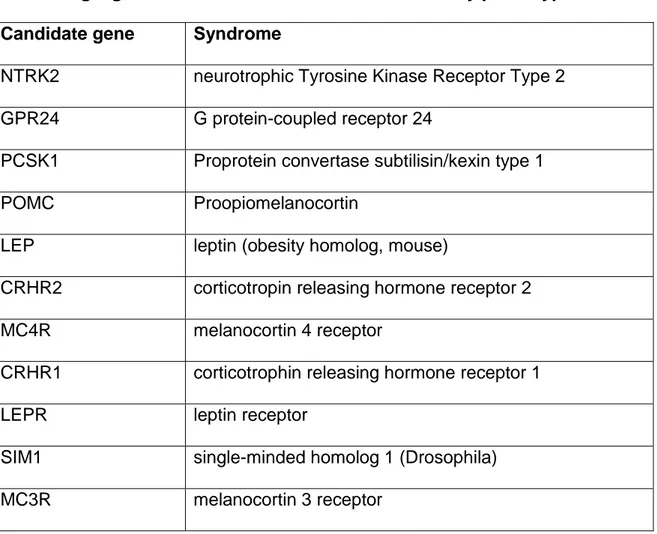
Table 1.1. Single gene mutations associated with an obesity phenotype
Candidate gene Syndrome
NTRK2 neurotrophic Tyrosine Kinase Receptor Type 2
GPR24 G protein-coupled receptor 24
PCSK1 Proprotein convertase subtilisin/kexin type 1
POMC Proopiomelanocortin
LEP leptin (obesity homolog, mouse)
CRHR2 corticotropin releasing hormone receptor 2
MC4R melanocortin 4 receptor
CRHR1 corticotrophin releasing hormone receptor 1
LEPR leptin receptor
SIM1 single-minded homolog 1 (Drosophila)
MC3R melanocortin 3 receptor
These models of obesity have allowed insights into some critical pathways but their
overall relevance is nonetheless questionable since common obesity cannot be
attributed to a single gene or single pathway. Thus, the polygenic nature of obesity
calls for a more realistic approach to generate animal models of obesity. In this
respect, diet-induced obesity allows to mimic situations more closely related to what
can be observed in humans.
I.I.2. High fat diets in diet-induced obesity
I.I.2.1. The effect of high fat diets on the development of obesity
As early as 1951, Fenton and Carr observed that, when providing diets with
increasing fat content to rodents, some strains showed marked weight gains, while
others had a much less pronounced response. They reported elevated food
utilization with diets high in fat and showed by carcass analysis that the strain
responding well to high fat feeding accumulated most of the excess weight as fat
(Fenton and Carr 1951). In 1955, Mickelsen et al. showed for the first time in rats that
obesity could be achieved by feeding a diet high in fat and implied that this could be
due to an excess consumption of calories (Mickelsen et al. 1955). High fat diets are
now fairly well accepted to model the disorders of human obesity in rodents (Buettner
et al. 2007) and have since then been extensively used to induce obesity in animal models. A PubMed search in December 2009 with the keywords “high fat diet” and
“obesity” retrieved more than 1500 results, mainly animal studies. To understand the
mechanisms behind the excess storage of energy usually associated with feeding
I.I.2.2. High fat diets and hyperphagia
The most obvious, and possibly easiest, parameter to look at is the energy intake,
which is simply calculated by measuring the food consumption and multiplying it by
the energy density of the diet. High fat feeding has usually been associated with
hyperphagia, meaning the group given the high fat diet tended to consume more
calories than the control group. This effect has been observed in mice (Mercer and
Trayhurn 1987; West et al. 1995; Gallou-Kabani et al. 2007), rats (Ramirez and
Friedman 1990; Shafat et al. 2009) and humans (Lissner et al. 1987). Therefore, it
seems that subjects fed a high fat diet are unable to regulate their food intake to
meet their needs and develop obesity as a consequence.
Interestingly, the hyperphagia associated with high fat diet does not seem to be due
to fat itself but rather to the energy density of the diets. Fat is characterised by a high
energy density (in kcal per g of macronutrient: fat, 9; carbohydrate, 4; protein, 4) and
thus, high fat diets are often high in energy density. Ramirez and Friedman fed rats
either a low fat or a high fat diet but presenting the same energy density. Rats fed the
high fat diet then presented decreased body weight and energy intake compared to
the mice fed the low fat diet (Ramirez and Friedman 1990). This result has been
confirmed by others in rats (Paulino et al. 2008). Therefore, as underlined by
Warwick and Schiffman, who reviewed 40 studies comparing the effects of high fat to
high carbohydrate diets, when the caloric density of the diets was similar (density of
the high fat diet less than 25 % greater than high carbohydrate diet), only 5 out of 10
studies observed greater weight gain in high fat fed animals whereas when the high
fat diet had an energy density at least 25 % greater than the high carbohydrate diet,
then 28 out of 30 studies observed a greater weight gain in the high fat fed animals
(Warwick and Schiffman 1992). These findings have been confirmed in humans as
associated with high fat diet feeding is abolished when the diets provided are
matched with respect to caloric density. To summarize, the hyperphagia associated
with high fat diet seems to be due to the high energy densities of high fat diets and
not because of the fat content of the diet per se. Of note, contradictory results have
been reported in rats when using liquid diets (Warwick 2003).
I.I.2.3. Diet-induced thermogenesis
Diet-induced thermogenesis (DIT) has been shown to have a significant effect on the
regulation of energy balance (Himms-Hagen 1985) and mainly takes place in the
brown adipose tissue in rodents (Rothwell and Stock 1979; Cannon and Nedergaard
2004). Mercer and Trayhurn showed that mice fed a high fat diet rich in corn oil,
meaning a high content of PUFA, exhibited increased energy expenditure, as
revealed by an enhanced total thermogenic activity of the BAT, compared to mice fed
a low fat diet (Mercer and Trayhurn 1987). Interestingly, the mice fed a high fat diet
rich in beef tallow, meaning a high content of SFA, did not show any evidence for an
increased DIT which could partly explain why they displayed greater body weight
compared to the 2 other groups. Differences in DIT could also partly account for the
differences in energy assimilation efficiencies since mice fed the corn oil diet retained
only 18 % of the excess energy intake in the carcass whereas mice fed the beef
tallow diet retained 77 %, despite similar total energy intakes. A decrease in DIT
induced by a diet rich in SFA as compared to diets rich in MUFA or PUFA has been
confirmed in rats as well (Takeuchi et al. 1995). Altogether, these results point at
differences in DIT as a function of the fat amount, or possibly the total energy intake
since DIT has been associated with overfeeding, and the fatty acid composition of
the diet which influences the obesity state as well (Corbett et al. 1986). BAT has long
activity, and its role in the energy balance has been neglected until recently where
positron emission tomography demonstrated that adult humans had significant
depots of metabolically active BAT (Cypess et al. 2009; Saito et al. 2009; van Marken
Lichtenbelt et al. 2009; Virtanen et al. 2009). Therefore, there is currently a renewal
of interest for the role of the BAT in obesity in humans.
The term high fat diet actually encompasses a fairly wide spectrum of diets. In a 1992
review, Warwick analysed 40 studies comparing the effects of high fat and high
carbohydrate diets (Warwick and Schiffman 1992) and reported differences in fat
content of the diets used ranging from 28 to 84 % of total energy. The diets were also
characterised by different fat sources (corn oil, lard, tallow and others, hence
providing quite different fatty acid profiles). Therefore, an analysis of feeding trials
with animals on high fat diets has to take into account not only the energy density
and the fat content but also the role of the fatty acids provided on the development of
obesity.
I.I.2.4. The effect of the dietary fat intake on the development of obesity
The fat content of the diet has been shown to be a major determinant of body weight
in mice fed ad libitum (West et al. 1995). In this study, the authors fed two strains of
mice, AKR/J and SWR/J, with increasing levels of fat (15, 30, 45 kcal %) and
observed that dietary fat content was strongly associated with body weight gain and
a very marked increase in the weight of adipose tissue depots. However, these
effects did not become obvious in the SWR/J mice, addressing the importance of
genetic predisposition on the development of obesity. Boozer et al. fed rats
increasing amounts of dietary fat (12, 24, 36, and 48 energy %) in quantities matched
body weight (although the diet by time interaction was significantly different) but the
absolute weights of the white adipose tissue correlated with the amount of dietary fat.
The authors concluded that dietary fat promoted adiposity, independently of the
energy intake. Pair-feeding studies are the gold-standard to determine the relative
contributions of hyperphagia versus metabolic effects of dietary fat in inducing
obesity. Although some studies reported that animals fed an isocaloric high fat diet
had greater body weight gain than animals fed a control or low fat diet (Wade 1982;
Oscai et al. 1987), other data led to the conclusion that there was no difference
(Woods et al. 2003).
I.I.2.5. Ketogenic diets
If there is a positive relationship between increasing levels of dietary fat and the
development of obesity, it actually only holds true within a defined range of fat
content as shown by experiments using ketogenic diets. Ketogenic diets typically
consist of at least 80 % of calories from fat with minimal requirements for protein and
marginal levels of carbohydrates. In mice fed ad libitum, a ketogenic diet (95 kcal %)
has been shown to promote weight loss compared to mice fed a control or a high fat
diet (45 kcal %) although they ingested similar levels of energy. This effect was due
to increased energy expenditure. Interestingly, the ketogenic diet not only prevented
mice to develop obesity but was also able to reverse obesity in mice previously fed a
high fat diet (Kennedy et al. 2007). Such ketogenic diets have proven to be more
efficient for weight loss compared to low fat diets in obese humans, even if only 68 %
I.I.2.6. The effect of dietary fat composition on the development of obesity Diets high in fat not only differ with respect to total fat content but also in their fat
sources and thereby their fatty acid profiles. For example, beef tallow, butterfat or
pork lard are rich in SFAs, coconut oil is rich in medium-chain SFAs, olive oil is rich in
MUFAs, corn oil is rich in omega-6 PUFAs and fish oil is rich in omega-3 PUFAs.
Therefore, it is difficult to determine the effects of fat quality (SFA, MUFA, PUFA) on
the development of obesity, as the fat sources used always consist of a mixture of
different fatty acids. Moussavi et al. reviewed the possible association between types
of fatty acids in diets and weight change (Moussavi et al. 2008). In animal studies,
diets rich in SFA (beef tallow and lard) seem to initiate a greater weight gain, as
reported for mice (Buettner et al. 2006) and for rats (Mercer and Trayhurn 1987;
Takeuchi et al. 1995). Fish oil, rich in PUFA, notably the omega-3 fatty acids
eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), have mainly been
shown to promote weight loss, except in one study (Awad et al. 1990). Only a few
epidemiological studies have been carried out in humans and the results are still
contradictory with regard to the quality of the dietary fatty acid patterns provided in
the diet and the effects on obesity development (Moussavi et al. 2008).
I.I.2.7. Strain differences
As already addressed, the association of dietary fat intake and obesity in mice is
strain-dependent (Fenton and Carr 1951). West et al. showed that the murine AKR/J
strain is obesity-prone whereas the murine SWR/J strain is obesity-resistant,
although when enough fat was provided in the diet, their body fat eventually
increased (West et al. 1995). Interestingly, in a following study, the AKR/J mice were
shown to have preference for fat while the SWR/J mice preferred other diets
of high fat diets on the development of obesity has been observed in rats as well
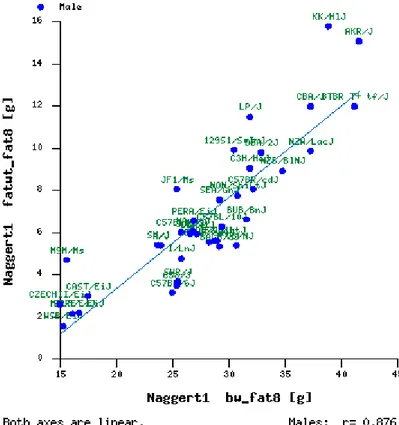
(Schemmel et al. 1970; Svenson et al. 2007). Svenson et al., from the Jackson
Laboratory, carried out a comprehensive study in 2007 where they fed 43 inbred
mouse strains for 8 weeks with a high fat diet, revealing large differences with
regards to body weight or fat mass gain as shown in Figure 1.1.
Figure 1.1. Body fat mass as a function of body weight in 43 strains of inbred mouse fed a high fat diet for 8 weeks (Svenson et al. 2007).
I.I.3. Cafeteria diet in diet-induced obesity
I.I.3.1. Is a high fat diet the most appropriate model to simulate a Western diet in human obesity?
High fat diet feeding in rodents induces changes in body weight and food intake
ongoing over whether dietary fat per se determines body fat accumulation or not.
Willett has pointed out, based on large epidemiological studies, that obesity has
increased in the last decades in the US although fat intake has decreased. Moreover,
some studies did not reveal any relation between dietary fat intake and body fatness ,
and furthermore, a reduction in fat intake had little effect, if any, on the reduction in
body weight (Willett 1998b, 1998a). To date, this debate is still open (Bray and
Popkin 1998; Willett 2002). However, a Western diet not only provides a higher
dietary fat intake but may be best characterized by a hyperphagia, providing also
high intakes of carbohydrates and proteins.
I.I.3.2. The cafeteria diet: a high fat / high sugar diet inducing hyperphagia It appears more sensible to induce obesity not only by increasing the amount of
dietary fat but also by inducing hyperphagia. Cafeteria diets have been introduced in
this respect: animals are offered a choice of several palatable food items of varied
composition, appearance and texture in addition to their nonpurified diet (Sclafani
and Springer 1976). These diets have been shown to induce obesity in a very
efficient manner, driven by hyperphagia, in rats and mice (Sclafani and Springer
1976; Rothwell and Stock 1988). For example, Rothwell and Stock reported an 80 %
increase in energy intake in rats fed a cafeteria diet compared to animals on control
diets, although the weight gain was only 27 % greater than that of the control
animals. (Rothwell and Stock 1979) This paradox was explained by an increase in
DIT in the cafeteria-fed animals, which could thus partly compensate for the excess
I.I.3.3. The effect of flavour on hyperphagia
Since cafeteria diet items are characterised by variations in flavour, texture or
macronutrient composition, it is difficult to determine what factor(s) induce
hyperphagia. The question of whether flavour variations induce hyperphagia remains
controversial. Treit et al. fed rats 2 hours per day with powdered chow flavoured with
one of 4 options, either changed every 30 min (variety day) or not (control day). The
rats given different flavours showed an approximate increase of 25 % in food intake
(Treit et al. 1983). Rolls et al. also showed that this effect was more pronounced
when rats were given the palatable items simultaneously than in succession (Rolls et
al. 1983). Nevertheless, Naim et al. did not report any effect of flavour variety on
body weight gain or energy intake when they fed rats for 23 days three control diets
with different added flavours as compared to a control diet (Naim et al. 1985). Thus, it
is not yet clear if flavour variety has an effect on energy intake or body weight gain.
I.I.3.4. The limits of the cafeteria diet
In a 1987 article in the Journal of Nutrition, Moore critically assessed the use of
cafeteria diets for studies on thermogenesis (Moore 1987). As cafeteria foods are low
in vitamins and minerals and the animals tended not to consume enough of the
nutritionally adequate nonpurified diet, the animals could face deficiencies. Moreover,
the animals usually did not eat the same items and therefore the composition of the
diet could greatly differ from one animal to another, which could affect the outcomes
of the study since the diet factor was not fully controlled. In a subsequent issue of the
Journal of Nutrition, Rothwell and Stock, the most prolific users of the cafeteria diet, convincingly dismissed Moore’s criticisms, reporting a 20 % energy intake from the
nonpurified diet and similar coefficients of variation for the different macronutrients in both control and cafeteria fed animals. Nonetheless, they acknowledged that “the
major drawbacks of the cafeteria diet are the variations in nutrient composition and the poor control over this factor” (Rothwell and Stock 1988). Controlling this factor is
actually possible, albeit painstaking and tedious (Shafat et al. 2009).
I.II. Physiological effects of high fat diets
I.II.1. The adipose tissue
Feeding a high fat diet induces a weight gain and most of this excess weight is based
on accumulated fat. However, this fat accumulation has a variety of physiological
effects, since not only does the adipose tissue expand but this organ also secretes a
large number of endocrine and paracrine factors.
I.II.1.1. Remodelling of the adipose tissue
High fat feeding elicits an increase in adipocyte size (hypertrophy) and number
(hyperplasia) (Faust et al. 1978; Berke and Kaplan 1983; Corbett et al. 1986). This
hyperplasia is not affected by food restriction, contrary to the adipocyte size, which
might indicate permanent deleterious effects of high fat diets on body weight (Rolls et
al. 1980). The consequences of this remodelling are also linked to the capacity of the
adipose tissue for the secretion of adipokines and cytokines (Huber et al. 2006).
I.II.1.2. Insulin resistance
Lavau et al. fed rats for one week either a low fat or a lard-based high fat diet. They
reported significantly decreased rates of glucose transport into the adipocytes of rats
fed the high fat diet compared to rats fed the low fat diet but the effect of insulin on
glucose uptake in high fat fed rats, but far less marked and only restricted to the
epididymal fat pad when stimulated by insulin, has been reported by Storlien et al.
(Storlien et al. 1986). Maegawa et al. also presented data on a decreased glucose
uptake but only upon insulin stimulation in high fat fed rats (Maegawa et al. 1986).
Wilkes et al. did not observe any effect of a high fat diet with a balanced fatty acid
profile but a decrease of the insulin-stimulated glucose uptake at high insulin
concentrations was seen with a high fat diet rich in PUFA (Wilkes et al. 1998). The
adipose tissue is generally considered to develop a mild insulin resistance upon high
fat feeding but very little is actually known on the impairments in the underlying
insulin signalling mechanism in the adipocyte (Anai et al. 1999; Park et al. 2005).
I.II.1.3. Lipoprotein lipase activity
Lipoprotein lipase (LPL) is the enzyme catalyzing the release of free fatty acids and
triacylglycerol from circulating triacylglyceride-rich lipoproteins to adipose tissue and
muscle. LPL activity has been shown to be enhanced in high fat fed mice in inguinal
and mesenteric fat pads but lipase activity did not show any association with insulin
levels (Surwit et al. 1995). Rossmeisl et al. observed an increase in LPL activity in
the epididymal fat depots of mice fed a high fat diet for 12 weeks. LPL activity
showed a 2-fold increase per unit of weight of tissue and a 4-fold increase if the
entire depot was considered (Rossmeisl et al. 2005). This increase in lipoprotein
lipase activity could promote the storage of excess lipids in adipose tissue.
I.II.1.4. De novo lipogenesis
Lavau et al. also determined the fate of glucose and observed that glucose
incorporation into CO² and fatty acids was decreased in rats fed a high fat diet.
dramatically increased, including for glucose incorporation into lipids, and the authors therefore concluded that “high fat feeding markedly decreases the adipocyte’s
responsiveness to insulin”. They subsequently measured lipogenic enzyme activities
which were all massively reduced. Hence, lipogenesis is already reduced in mice fed
a high fat diet for one week (Lavau et al. 1979). This effect on adipocyte lipogenesis
and lipogenic enzymes was confirmed in rats fed during 3 or 7 weeks from weaning
onwards with a high fat diet (Berke and Kaplan 1983) and was also partly confirmed
on transcription level in mice based on microarray analysis (Moraes et al. 2003).
I.II.1.5. Adipokines
The main function of adipose tissue has long been thought to be solely its energy
storage capacity. However, during the past years, considerable advances have been
made in defining its functions as an endocrine organ (Zhang et al. 1994; Kershaw
and Flier 2004) Leptin was the first adipokine discovered (Zhang et al. 1994) and was
shown to inhibit food intake and stimulate energy expenditure (Havel 2000). In mice
fed a high fat diet, leptin levels were elevated and positively correlated with body
weight (Ahrén 1999; Bullen et al. 2007). Adiponectin is the only adipokine currently
known to be negatively correlated with body mass and its decrease has been
associated with the progression of metabolic syndrome (Hauner 2005). The effects of
high fat diet feeding on adiponectin secretion are still controversial since adiponectin
plasma levels have been reported to be either unaffected upon high fat diet feeding
or elevated (Barnea et al. 2006; Bullen et al. 2007; Lee et al. 2009). Resistin, at least
in rodents, has been shown to counteract insulin activity (Steppan et al. 2001) and its
secretion has been shown to be increased in mice fed a high fat diet (Steppan et al.
I.II.1.6. Inflammation
Analysis of gene expression levels using microarray in adipose tissue of mice fed a
high fat diet identified numerous genes significantly up-regulated that belong to
inflammatory pathways (Moraes et al. 2003). Indeed, it was shown with
immunohistochemical methods that mice fed a high fat diet showed an increased
infiltration of macrophages into their adipose tissues, forming aggregates named
Crown-like structures (CLS), and their number was significantly correlated to the
adipocyte size (Weisberg et al. 2003; Xu et al. 2003). This infiltration has been
associated with increased levels of secretion of the monocyte chemotactic protein-1
(MCP-1), a chemoattractant specific for monocytes and macrophages (Takahashi et
al. 2003; Chen et al. 2005). Interestingly, this macrophage infiltration seems to be
prevented by a diet containing fish oil, rich in omega-3 PUFA (Todoric et al. 2006).
Figure 1.2. Summary of the effects of high fat diets on the adipose tissue.
I.II.2. Liver
I.II.2.1. Hepatic steatosis
It is well established that diet-induced obesity in animals is associated with the
disease), characterized by large vacuoles of triacylglycerides accumulating in
hepatocytes (Clarke et al. 1977; Yaqoob et al. 1995). In humans, an increase in
intrahepatic triacylglycerides (IHTG) has been associated with hepatic and peripheral
insulin resistance (Hwang et al. 2007; Korenblat et al. 2008) and is considered a
major determinant of the metabolic syndrome (Marchesini et al. 2003). Recently,
Fabbrini et al. demonstrated that IHTG content, but not visceral adipose tissue size,
was a marker of obesity-related metabolic alterations in humans (Fabbrini et al.
2009). To date, the mechanisms underlying ectopic fat distribution are not known.
I.II.2.2. Hepatic insulin resistance
The association of NAFLD and hepatic insulin resistance has been shown in
diet-induced obese animals. Using the hyperinsulinemic-euglycemic clamp technique, it
was demonstrated that the insulin-stimulated suppression of hepatic glucose
production was drastically impaired in rats fed a high fat diet (Storlien et al. 1986;
Anai et al. 1999; Li et al. 2006). The mechanisms for insulin resistance in the liver
upon high fat feeding seem to be different from those encountered in the muscle and
adipose tissue since neither the insulin receptor substrate 1 and 2 (IRS-1 and -2)
protein levels, nor their phosphorylation status, are altered. However,
phosphoinositide-3-kinase activity, acting downstream of IRS-1 and -2 to translate
insulin receptor activation into metabolic responses, is increased (Anai et al. 1999).
I.II.2.3. Hepatic de novo lipogenesis
As shown by Lavau et al. in adipocytes, glucose incorporation into fatty acids was
also decreased in the liver of rats given a high fat diet when compared to rats given a
control diet. This difference was even more marked when the incorporation was
hepatic lipogenesis (Storlien et al. 1986). Clarke et al. showed that this reduction of
de novo hepatic lipogenesis was more pronounced in rats receiving a PUFA-rich rather than a SFA-rich diet (Clarke et al. 1977).
Figure 1.3. Summary of the effects of high fat diets on the liver.
I.II.3. Intestine
The role of the intestine in the genesis of obesity has been quite underestimated,
although it is responsible for fat absorption into the circulation. Very little is known on
the effects of feeding high fat diets on the intestine and it only recently received
increased interest, notably through the use of microarrays, which allow access to the
transcriptome.
I.II.3.1. Fat absorption
In an early study, Singh et al., showed that when feeding rats for 4 weeks a
lard-based high fat diet, fecal excretion of radiolabelled lipids was significantly decreased.
Moreover, radioactive uptake of oleic acid in everted gut sacs from both jejunum and
ileum and its rate of reesterification were found to be greater in the rats fed the high
fat diet. This was confirmed by an enhanced activity for the jejunal monoglyceride
acyltransferase, a reesterifying enzyme which allows the synthesis of diglycerides in
the enterocytes from monoglycerides, a necessary step in triacylglyceride synthesis
for their export in chylomicrons (CM). Altogether, these results pointed to an
1972). This was confirmed in mice where it was shown that the fecal lipid content
was not affected by a 6-week-long high fat feeding. Thus, whether or not through an
adaptation in its absorptive capacity, the small intestine shows a very high efficiency
for fat absorption (Petit et al. 2007).
Fat absorption is a function of the total absorptive surface area times the absorptive
capacity of each enterocyte. In the work by Petit et al., microarray analysis revealed
an upregulation in genes involved in fatty acid uptake (such as the transporters
FATP-4 and CD36), intracellular fatty acid processing (the fatty acid binding proteins
I-FABP and L-FABP for example) and lipoprotein secretion (ApoA-IV and MTP for
example). This demonstrated an adaptive upregulation of genes / proteins in the
machinery to allow an enhanced lipid absorption and processing (Petit et al. 2007).
The intestinal absorptive surface area has also been found to be increased in high fat
fed mice. Petit et al. and de Wit et al. both observed an increase in the proliferation
rate of the enterocytes which could lead to an increase in villus size and ultimately in
the absorptive area (Petit et al. 2007; de Wit et al. 2008). Interestingly, de Wit et al.
also showed a downregulation of genes involved in apoptosis and an upregulation of
genes involved in cell cycle, especially in the mid and distal parts of the small
intestine. They also confirmed the increase in villus length and the total number of
cells per villus in the distal small intestine. These changes could constitute a
mechanism to support the small intestinal capacity in absorbing the bulk of dietary
I.II.3.2. Lipoprotein secretion
It has been shown that after long term feeding of a high fat diet containing long chain
fatty acids, postprandial intestinal lipoprotein secretion is increased (Cartwright and
Higgins 1999). Interestingly, this effect of diet-induced changes in postprandial
lipoprotein secretion was observed as early as after 7 days of feeding and was
characterised in the small intestine with a secretion of a smaller number of CM but of
larger size (Hernandez Vallejo et al. 2009). As the lipoprotein lipase shows a higher activity towards larger-sized particles, this could be interpreted as a way to “manage
the lipid overloading”. An adaptation of CM assembly in the intestine and secretion
was confirmed also on the transcriptome basis by increased levels of apolipoprotein
B (apoB) or the microsomal triacylglyceride transfer protein (MTP). Surprisingly, mice
fed a high fat diet for 6 weeks showed decreased blood triacylglyceride levels which
were shown to be due to an enhanced clearance from the blood, possibly through an
elevated apoCII / apoCIII ratio. It underlines the prominent role of the small intestine
in postprandial triglyceridemia (Petit et al. 2007).
I.II.3.3. Lipid oxidation
As addressed above, the small intestine reacts to a high fat diet by trying to export
more efficiently excess lipids, which can be toxic for the enterocyte (Unger and Orci
2002). De Wit et al., analysed the small intestinal transcriptome of mice fed either a
low fat or a high fat diet. They reported a significant number of biological processes
linked to lipid metabolism, cell cycle and inflammation / immune response being
significantly affected by the dietary treatment. Interestingly, they found several genes
associated with fatty acid oxidation to be upregulated in the mice fed the high fat diet,
speculating that this might act as a detoxifying process, to prevent free fatty acids
observed an upregulation of genes associated with fatty acid oxidation and
interestingly, this effect was more prominent in obesity-resistant A/J mice than in
obesity-prone C57Bl/6 mice, therefore suggesting a role of the small intestine in the
development of obesity (Kondo et al. 2006). Nonetheless, according to Gniuli et al.,
this increase in expression of genes linked to fatty acid oxidation and the increase in
enterocyte mitotic rate is not sufficient to counteract the lipotoxic effect of the high fat
diet since such a diet has been shown to induce apoptosis in rat enterocytes, the
longer the diet was being fed, the more prone the enterocytes were to lipid-induced
apoptosis (Gniuli et al. 2008). Interestingly, PUFA of marine origin (EPA and DHA)
have been found to increase lipid oxidation rates in the small intestine and might
therefore protect the small intestine from lipo-apoptosis and reduce the amount of
lipids for export via CM to the other organs (van Schothorst et al. 2009).
I.II.3.4. Hormone secretion
When de Wit et al. analysed the intestinal transcriptome of mice fed a high fat diet
compared to mice fed a low fat diet, they also found several transcripts of secreted
proteins being affected by the high fat feeding, suggesting alterations in the
communication of the gut with other organs, such as liver, muscle and adipose
tissue, via hormones and thereby underpinning the role of the small intestine in
metabolic perturbations (de Wit et al. 2008). The gut is not only an absorptive organ
but also secretes various hormones involved in energy homeostasis and satiety
(Chaudhri et al. 2008). For example, feeding a high fat diet for 30 days has been
shown to cause an elevated gastric inhibitory polypeptide (GIP) secretion (an incretin
that amplifies insulin secretion) but this response was blunted after 90 days of
feeding and was associated with a decreased insulin secretion (Gniuli et al. 2008).
glucagon-like peptide-1 (GLP-1) (another incretin) levels and showed a reduced GLP-1
response following an oral glucose load (Anini and Brubaker 2003). It seems
therefore that feeding a high fat diet blunts the response of the small intestine with
respect to secreted peptide hormones that are involved in satiety control and in
energy homeostasis.
Figure 1.4. Summary of the effects of high fat diets on the small intestine.
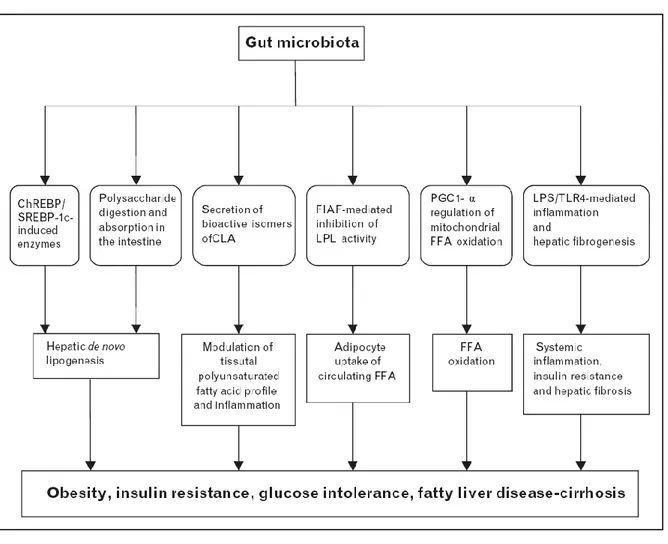
I.III. The gut microbiota and obesity
The human gut hosts as many as 100 trillion microbes – collectively called microbiota
- mainly located in the colon where densities approach 1011 – 1012 cells/ml. This makes the human gut one of the most densely populated microbial habitats on Earth.
There might be thousands of species, dominated by anaerobic microorganisms,
which contain an estimated 100 times more genes than the human genome (Ley et
al. 2006a). The microbiota is able to perform functions that humans cannot
accomplish by converting undigested food components and endogenous substrates,
such as plant polysaccharides, phenolic compounds, mucin, cholesterol, biliary acids
and releases large quantities of short chain fatty acids, considered to be beneficial for
gut health. However, the gut microbiota also produces metabolites that can be
harmful. For instance, diets characterized by high saturated fat or low fibre content
have been associated with changes in the bacterial metabolism of steroids and bile
acids. Resulting metabolites, such as secondary bile acids, have been linked to
pathologies, e.g. colon cancer and cholesterol gallstone disease (Blaut and Clavel
2007). The next section will briefly review recent studies linking the gut microbiota to
obesity. This field has received great interest in the very recent years, notably through the work of Jeffrey Gordon’s group.
I.III.1. Role of the gut microbiota in fat storage
The first study that established a link between the gut microbiota and obesity was
published in 2004 by the group of Jeffrey Gordon. In this comprehensive work,
Bäckhed et al. compared parameters of energy balance in germ-free (GF) (i.e. raised
in the absence of microorganisms), conventionally raised and conventionalized
C57Bl/6 mice fed a standard rodent chow. Conventionalized mice were obtained by
spreading resuspended cecal contents of conventionally raised mice on the fur of GF
animals. 2 weeks after the colonization of the GF mice, conventionalized and
conventionally raised animals showed a 42 % increase in total body fat compared to
the GF animals. Surprisingly, the GF mice presented a 40 % higher food intake and a
27 % lower metabolic rate (as measured by O² consumption) compared to the mice
bearing a gut microbiota. As muscle and liver high-energy phosphate stores were not
affected by the microbiota, the authors concluded that the presence of
microorganisms induced futile cycles, therefore allowing a dissipation of energy
(Bäckhed et al. 2004). Moreover, GF mice appeared to be protected against
gain in GF animals when compared to GF mice fed a low fat diet. Conventionalized
mice did not present any protection against diet-induced obesity (Bäckhed et al.
2007). Whether fed a low fat or a high fat diet, GF mice displayed an increased
locomotor activity compared to conventionalized mice, seemingly not due to the
difference in adiposity, which could contribute to the energy balance of these animals
(Bäckhed et al. 2007). Some of the proposed mechanisms which could be underlying
these effects on energy balance will be discussed next.
I.III.2. Gut microbiota and short-chain fatty acids absorption
Polysaccharide fermentation by the gut microbiota produces SCFA, e.g. acetate,
propionate or butyrate. These SCFA appear to serve as ligands for the G
protein-coupled receptor 41 and 43 (Gpr41 and Gpr43 respectively) which are located in
enteroendocrine cells of the gut. To study the contribution of Gpr41 to energy balance, Jeffrey Gordon’s group compared wild-type and Gpr41 knock-out mice
cocolonized or not with 2 organisms promoting SCFA production from dietary
polysaccharides. Knocking out Gpr41 abolished the effect of gut microbiota on fat
storage (as described in the previous section I.III.1.): GF mice, whether expressing
Gpr41 or not, had the same body weights, fat pad weights and adiposity as
cocolonized mice not expressing Gpr41. Cocolonized mice expressing Gpr41
presented significantly higher values for these phenotypic changes. Moreover,
deletion of Gpr41 reduced leptin secretion more than expected if only the associated
decrease in adiposity was considered, therefore suggesting a distinct role of Gpr41 in
microbiota-mediated leptin production. Moreover, an increased fecal energy output,
an increased uptake of monosaccharide from the gut and an increased intestinal
transit time were also observed. The latter was discussed as a consequence of a