Effet du diabète sur la pathologie de la protéine
Tau in vivo
Thèse
NOURA EL KHOURY
Doctorat en Neurobiologie
Philosophiæ Doctor (Ph.D.)
Québec, Canada
Résumé
La maladie d‟Alzheimer (MA) représente aujourd‟hui la forme de démence la plus commune pour laquelle il n‟existe toujours pas de traitement curatif. Dans le cerveau, elle se caractérise par la présence de deux agrégats protéiques majeurs : les plaques amyloïdes qui résultent de l‟accumulation extracellulaire d‟un peptide nommé peptide amyloïde, et les enchevêtrements neurofibrillaires correspondant à l‟agrégation intra-neuronale d‟une protéine nommée Tau qui se trouve dans un état anormalement hyperphosphorylé. La pathologie Tau est importante puisque son étendue corrèle avec le degré du déficit cognitif retrouvé dans la MA.
Seule une petite proportion des cas de la MA est causée par des mutations génétiques. En revanche, l‟étiologie de la majorité des cas (~99%), qui est d‟origine sporadique et à apparition tardive, semble être multifactorielle, avec des facteurs externes pouvant interagir avec des susceptibilités biologiques ou génétiques afin d‟accélérer la manifestation de la maladie.
Au cours de la dernière décennie, des données cliniques et précliniques émergentes suggèrent que le diabète sucré et la dysfonction de l‟insuline qui l‟accompagne pourrait représenter l‟un de ces facteurs. En plus de son rôle métabolique, l‟insuline a été rapportée pour avoir un rôle neurotrophique et régulateur dans le cerveau humain. Plus particulièrement, des études in vitro ont montré que l‟insuline est capable de moduler la phosphorylation de Tau dans les cellules neuronales. Cette hypothèse a été par la suite renforcée par les observations d‟hyperphosphorylation de Tau dans les cerveaux de souris montrant des anomalies au niveau de la signalisation de l‟insuline. Malgré toutes ces données, on connaît très peu concernant l‟impact du diabète sur la pathologie Tau in vivo. Le but global de ce projet de doctorat a été donc de clarifier l‟impact du diabète sur la pathogenèse de la protéine Tau, dans deux modèles génétiques de diabète de type 1 (DT1) et diabète de type 2 (DT2), qui sont les souris NOD (non-obese-diabetic) et les souris ob/ob, respectivement.
Nos résultats montrent que le DT1 entraine une hyperphosphorylation progressive de Tau qui commence à être détectée même en absence de toute dérégulation dans le métabolisme du glucose. De plus, cette hyperphosphorylation est plus prononcée en présence des caractéristiques du DT1 (hyperglycémie et glycosurie) et encore plus amplifiée en présence de l‟hypothermie. D‟une manière intéressante, nos résultats suggèrent que l‟hyperphosphorylation de Tau chez ces souris corrèle avec une dérégulation de PP2A (protein phosphatase 2A), l‟une des phosphatases les plus importantes de Tau in vivo. Quant au DT2, nos résultats montrent une hyperphosphorylation de Tau chez les souris ob/ob à 4 et 26 semaines, au niveau de plusieurs sites spécifiques. De plus, ces souris développent une hypothermie modérée, mais le rétablissement de la normothermie ne restaure pas les niveaux de phosphorylation de Tau, ce qui suggère que cette hyperphosphorylation serait plutôt la conséquence des composantes du DT2, et non pas de l‟hypothermie qui en résulte. D‟une manière intéressante, nos résultats ne montrent pas de dérégulation au niveau des protéines impliquées dans la voie de signalisation de l‟insuline, suggérant par conséquent que, d‟autres facteurs, probablement associés à l‟obésité, pourraient contribuer à l‟hyperphosphorylation de Tau dans le DT2.
La compréhension des mécanismes qui sous-tendent la corrélation entre la dysfonction de l‟insuline et la pathologie Tau aidera par la suite à trouver de nouvelles cibles thérapeutiques visant à contrôler la progression de la maladie.
Abstract
Alzheimer‟s disease (AD) is the leading form of dementia. There is actually no cure for AD, but even a treatment that would slow down the progression of the disease by 5 or 10 years will have a tremendous socio-economic impact for Canada.
The neuropathological hallmarks of Alzheimer's disease include senile plaques of -amyloid (A) peptides (a cleavage product of the -amyloid precursor protein, or APP), and neurofibrillary tangles (NFT) of hyperphosphorylated Tau protein assembled in paired helical filaments (PHF). NFT pathology is important since it correlates with the degree of cognitive impairment in AD.
Only a small proportion of AD is due to genetic variants, the large majority of cases (~99%) is late onset and sporadic in origin. The cause of sporadic AD is likely to be multifactorial, with external factors interacting with biological or genetic susceptibilities to accelerate the manifestation of the disease.
Diabetes mellitus (DM) might be such factor, as there is extensive data from epidemiological studies suggesting that DM is associated with an increased relative risk for AD. Type 1 diabetes (T1DM) and type 2 diabetes (T2DM) are known to affect multiple cognitive functions in patients. However, the consequences of both type of diabetes on AD pathology are not well understood. The challenge is therefore to better understand the mechanisms of AD pathology and how they are affected by factors such as diabetes.
The overall goal of this project is therefore to clarify the impacts that diabetes have on Tau protein pathogenesis, in two well-characterized mouse models of T1DM and T2DM: NOD (non-obese diabetic) and ob/ob mice, respectively.
Our data suggest that spontaneous T1DM provokes a progressive Tau hyperphosphorylation that begin to be detectable in adult mice even during the non-diabetic stage, where there is no apparent deregulation of glucose metabolism. We further show that
demonstrate that Tau hyperphosphorylation during T1DM is likely attributable to a deregulation in PP2A (protein phosphatase 2A), the major Tau phosphatase in vivo.
Furthermore, we show that ob/ob male mice aged 4 and 26 weeks present Tau hyperphosphorylation at specific sites, but also have mild hypothermia. However, restoring normothermia did not rescue Tau hyperphosphorylation to control levels. These data indicate that Tau hyperphosphorylation accompanies major features of T2DM. Interestingly, we did not observe any deregulation in the proteins implicated in the insulin-signaling pathway, suggesting that other obesity-associated factors, contribute to Tau phosphorylation in ob/ob mice.
In turn, this understanding will help the development of treatments or life-style strategies destined to check the advance of the disease.
Avant-propos
Au cours de mon doctorat, j‟ai eu la chance de participer à différents projets qui m‟ont permis d‟apprendre et d‟apprécier plusieurs aspects de la recherche sur la phosphorylation de Tau, une protéine majoritairement impliquée dans plusieurs maladies neurodégénératives, notamment la maladie d‟Alzheimer. Mon projet principal de doctorat m‟a amenée à publier un article dont je suis co-première auteure (en contribution égale) qui sera présenté dans le Chapitre II, et un autre qui sera soumis bientôt pour publication et qui sera présenté dans le chapitre III. Une revue de littérature dont je suis la première auteure et qui discute de l‟impact de la dysfonction de l‟insuline sur la maladie d‟Alzheimer, est aussi en cours de préparation et sera présentée dans la partie annexe. Tous les autres projets auxquels j‟ai participé seront également présentés dans cette partie. Ces projets ont fait l‟objet de plusieurs publications dont je suis co-auteure :
5. Robert A. Whittington, László Virág, Charles W. Emala, Thomas O. Maurin, François
Marcouiller, Carl Julien, Franck Petry, Noura B. El Khoury, Alexis Bretteville, Emmanuel Planel (2013). Anesthesia-induced hypothermia mediates decreased ARC gene and protein expression through ERK/MAPK inactivation. Sci. Rep, Accepted
4. Carl Julien, François Marcouiller, Alexis Bretteville, Noura B. El Khoury, Joanie
Baillargeon, Sébastien S. Hébert, Emmanuel Planel (2012). Dimethyl sulfoxide induces both direct and indirect Tau hyperphosphorylation. PLoS ONE 7(6): e40020
3. Alexis Bretteville, François Marcouiller, Carl Julien, Noura B. El Khoury, Franck R.
Petry, Isabelle Poitras, Didier Mouginot, Georges Lévesque, Sébastien S. Hébert, Emmanuel Planel (2012). Hypothermia-induced hyperphosphorylation: a new model to study Tau kinase inhibitors. Sci. Rep. 2, 480; DOI: 10.1038/srep00480
2. Robert A. Whittington, László Virág, François Marcouiller, Marie-Amélie Papon, Noura B. El Khoury, Carl Julien, Françoise Morin, Charles W. Emala, Emmanuel Planel (2011).
1. Marie-Amélie Papon, Robert A. Whittington, Noura B. El Khoury, Emmanuel Planel
(2011). Alzheimer's disease and anesthesia. Frontiers in Neuroscience (4): 1-7 (invited review article).
Remerciements
Je profite de ces quelques lignes pour remercier tous les gens qui m‟ont entourée, supportée et encouragée pour obtenir ce doctorat.
Merci d‟abord aux membres du jury d‟avoir accepté d‟évaluer mon travail.
J‟exprime aussi mes remerciements les plus sincères à mon directeur de thèse, le Dr. Emmanuel Planel, qui m‟a accordée sa confiance et son soutien tout le long de mon doctorat. J‟ai eu le grand honneur d‟être la première doctorante dans son laboratoire, et j‟ai pu compter sur sa grande expertise dans le domaine de la protéine Tau. J‟ai énormément apprécié sa patience, son dévouement, sa bonne humeur et sa disponibilité tout le long de ces trois années inoubliables. C‟est la vraie science que j‟ai apprise avec lui et je lui en suis très reconnaissante.
J‟adresse mes remerciements à tous les membres de mon laboratoire : Françoise Morin qui m‟a fournie tout le support technique et personnel dont j‟avais besoin ; François Marcouiller qui m‟a aidée à faire mes premiers pas ; Marie-Amélie Papon, Alexis Bretteville et Carl Julien qui m‟ont incitée à élargir mes connaissances et à développer mon esprit critique. Je remercie également Isabelle Poitras, Franck Petry, Maya Dickler, François Bezeau, Maude Gratuze et Anastasia Noel pour tous les commentaires constructifs et les bons moments qu‟on a passés ensemble. On faisait tous une belle « Tau Team » coopérative, responsable, honnête et agréable.
Mes remerciements s‟adressent également à la direction du programme de doctorat en neurobiologie à l‟Université Laval pour les excellentes conditions de travail qu‟elle m‟a offertes.
Je suis également reconnaissante aux organismes subventionnaires qui m‟ont accordé leur appui financier. Merci à la Société Alzheimer du Canada (SAC), aux Fonds de Recherche en Santé du Québec (FRSQ), aux Instituts de Recherche en Santé du Canada (IRSC) et à la faculté de médecine de l‟Université Laval.
La réalisation de cette thèse n‟aurait jamais été possible sans l‟amour et le dévouement inconditionnel de ma famille, spécialement ma mère. Ma famille a toujours accepté, encouragé et supporté mon choix de partir à l‟étranger et développer mon ambition. Seuls ceux qui ont vécu loin de ceux qui les aiment peuvent comprendre cette difficile épreuve. Enfin, je ne saurais terminer sans adresser un petit mot au Dr. Didier Mouginot, ancien directeur du programme de neurobiologie et membre de mon comité d‟encadrement, qui nous a quitté pour l‟autre monde dans des conditions tragiques. Merci Didier pour ton appui, merci pour tes recommandations, merci pour tout ce que tu m‟as apprise.
À mes parents Je vous dois énormément… À mes frères Nadim et Gebran Vous n’étiez jamais loin….
Table de matières
Résumé ...iii Abstract ...v Avant-propos ...vii Remerciements ...ix Table de matières...xiiiListe des figures ...xvii
Liste des tableaux ...xix
Liste des abréviations...xxi
Chapitre I ...1
Introduction...1
1 La maladie d‟Alzheimer ... 3
1.1 Prévalence ... 7
1.2 Signes cliniques et diagnostic ... 7
1.3 Génétique ... 10 1.3.1 APP ... 10 1.3.2 Présénilines ... 14 1.3.3 Apolipoprotéine E ... 18 1.4 Neuropathologie... 22 1.4.1 Atrophie cérébrale ... 22
1.4.2 Les plaques amyloïdes ... 23
1.4.2.1 Classification et extension des plaques amyloïdes ... 24
1.4.2.2 L‟hypothèse de la cascade amyloïde ... 26
1.4.2.3 Cytotoxicité des différentes formes d‟A ... 28
1.4.3 Les enchevêtrements neurofibrillaires ... 31
1.4.3.1 Évolution des NFT... 31
1.4.3.2 Tau and tangles hypothesis ... 36
1.4.4 L‟interrelation entre les deux lésions ... 37
1.4.5 Réduction du métabolisme du glucose... 39
1.4.6 Autres caractéristiques neuropathologiques ... 39
2 La protéine Tau ... 41 2.1 Du gène à la protéine ... 41 2.2 Structure et localisation... 43 2.2.1 Structure ... 43 2.2.2 Localisation ... 44 2.3 Fonctions de Tau ... 45
2.3.1 Tau : une protéine associée aux microtubules ... 45
2.3.2 Tau et partenaires protéiques ... 47
2.3.3 Tau et organites cellulaires ... 47
2.4 Tauopathies et mutations sur Tau ... 47
2.4.1 Différences entre les DFTP-17 et la MA... 48
3 Phosphorylation de Tau ... 51
3.3.1 Phosphorylation et liaison aux microtubules ... 54
3.3.2 Phosphorylation de Tau et autres fonctions potentielles ... 55
4 Régulation de la phosphorylation de Tau ... 57
4.1 Kinases ... 57
4.1.1 Glycogène synthase kinase-3β ... 61
4.1.2 Mitogen-activated protein kinases ... 64
4.1.2.1 c-Jun N-Terminal kinases ... 64
4.1.2.2 Les p38 ... 65
4.1.2.3 Extracellular signal-regulated kinases... 66
4.1.3 Les cyclines-dependent kinases... 67
4.1.4 Autres kinases ... 69
4.2 Phosphatases ... 70
4.2.1 La phosphatase PP2A... 71
5 Maladie d‟Alzheimer et dysfonction de l‟insuline ... 75
5.1 L‟insuline : fonctions et voie de signalisation ... 75
5.2 L‟insuline dans le SNC : une autre histoire ... 78
5.3 Le diabète sucré... 79
5.3.1 Diabète de type 1 ... 79
5.3.2 Diabète de type 2 ... 79
5.4 Lien entre la maladie d‟Alzheimer et le diabète : évidences cliniques ... 80
5.4.1 DT1 et dysfonctions cognitives ... 80
5.4.2 DT2 et maladie d‟Alzheimer... 81
5.5 Effet de l‟insuline sur la phosphorylation de Tau ... 82
5.5.1 Études in vitro ... 82
5.5.2 Études in vivo ... 83
5.5.2.1 Hyperphosphorylation de Tau dans des modèles de DT1 induit par la STZ ... 83
5.5.2.2 Hyperphosphorylation de Tau dans des modèles génétiques de DT1... 85
5.5.2.3 Hyperphosphorylation de Tau dans des modèles de DT2 ... 85
5.5.2.3.1 Modèles génétiques de DT2 ... 85
5.5.2.3.2 Modèles de DT2 induit par la diète riche en gras ... 86
5.5.2.4 Hyperphosphorylation de Tau dans des modèles de diabète de type 3 ... 87
5.5.3 Métabolisme de l‟APP dans les modèles de diabète ... 89
5.5.3.1 Métabolisme d‟A dans les modèles de DT1 induit par la STZ ... 89
5.5.3.2 Métabolisme d‟A dans les modèles de DT2 ... 89
5.5.3.3 Métabolisme de l‟A dans le modèle de DT3... 90
6 Hypothèse et objectif du projet... 95
Chapitre II ... 97
Dérégulation de PP2A et hyperphosphorylation de Tau après l’apparition du diabète dans les souris NOD ... 97
Chapitre III... 103
Phosphorylation de Tau dans les souris ob/ob ... 103
Annexe ... 109
Autres contributions ... 109
1 Effet du diabète sur les pathologies Aβ et Tau ... 111
2 Maladie d‟Alzheimer et anesthésie ... 115
4 Le diméthyl sulfoxide induit l‟hyperphosphorylation de Tau à la fois directement et
indirectement ... 123
5 L‟hyperphosphorylation induite par l‟hypothermie : un nouveau modèle pour tester les inhibiteurs des kinases de Tau ... 127
6 L‟hypothermie induite par l‟anesthésie entraine une diminution de l‟expression du gène et de la protéine ARC via l‟inactivation de ERK/MAPK. ... 131
Chapitre IV : Discussions et perspectives...135
1 Remise en contexte ... 137
2 Mécanismes liant la dysfonction de l‟insuline à la MA ... 139
2.1 Effet de l‟insuline sur le métabolisme de l‟APP ... 139
2.1.1 Effet de l‟insuline sur la sécrétion d‟A ... 139
2.1.2 Effet de l‟insuline sur la dégradation d‟A ... 139
2.1.2.1 IDE et dysfonction de l‟insuline... 140
2.1.3 Effet de l‟insuline sur la toxicité des oligomères A... 140
2.2 Mécanismes liant le DT1 à la phosphorylation de Tau... 141
2.2.1 Activation des kinases... 141
2.2.2 Inhibition de PP2A ... 141
2.3 Mécanismes liant le DT2 à la phosphorylation de Tau... 142
2.3.1 Résistance à l‟insuline... 143
2.3.2 Obésité ... 143
2.3.2.1 Dyslipidémie ... 144
2.3.2.2 Adipokines... 144
2.3.3 Autres mécanismes potentiels ... 145
2.3.3.1 La glycosylation ... 146
2.3.3.2 Inflammation... 146
3 Dysfonction de l‟insuline et MA : Implications thérapeutiques... 149
3.1 Traitement avec l‟insuline et phosphorylation de Tau... 149
3.2 Médicaments antidiabétiques et phosphorylation de Tau ... 149
3.3 Médicaments antidiabétiques et Aβ ... 150
3.4 L‟insuline intranasale : Une voie prometteuse ? ... 150
4 Conclusion générale ... 151
Liste des figures
Figure 1-1. Éléments du dossier médical original de la patiente Auguste D (Deter)... 5
Figure 1-2. Dr. Alois Alzheimer... 6
Figure 1-3. Comparaison de la rétention du PIB et du métabolisme de glucose dans un cerveau normal et un cerveau de patient atteint de la MA. ... 9
Figure 1-4. Structure et métabolisme de la protéine précurseur du peptide β-amyloïde (APP)... 12
Figure 1-5. Structures des présénilines et mutations liées aux FAD. ... 15
Figure 1-6. Rôle du complexe PS1/GSK-3β dans la MA. ... 17
Figure 1-7. Structure et isoforme de l‟Apolipoprotéine E. ... 18
Figure 1-8. Rôle de l‟apolipoprotéine-E4 dans la MA ... 21
Figure 1-9. Atrophie cérébrale observée au cours de la MA ... 22
Figure 1-10. Marqueurs neuropathologiques de la MA et techniques de marquage. ... 23
Figure 1-11. Distribution des plaques amyloïdes au cours de la MA ... 25
Figure 1-12. Illustration de la cascade d‟évènements reliant la pathologie amyloïde à la dégénérescence neurofibrillaire ... 26
Figure 1-13. Effets pathologiques des oligomères d'A ... 30
Figure 1-14. Les trois stades d‟évolution cytologique de la dégénérescence neurofibrillaire au cours de la MA... 32
Figure 1-15. Progression de la dégénérescence neurofibrillaire au cours de la MA ... 34
Figure 1-16. Progression des enchevêtrements neurofibrillaires et des plaques amyloïdes dans 2,661 cas d‟autopsie calculés d‟après Braak et Braak ... 35
Figure 1-17. « The Tau and tangle hypothesis ». ... 37
Figure 2-1. Représentation schématique du gène, des ARNm(s), et des différentes isoformes de Tau ... 42
Figure 2-2. Représentation schématique des domaines fonctionnels de l‟isoforme la plus longue de Tau (2+3+10+). ... 44
Figure 3-1. Dérégulation de la phosphorylation de Tau et sondes immunologiques. ... 53
Figure 3-2. L‟équilibre dynamique de la liaison de Tau aux microtubules. ... 55
Figure 4-1. Structure de PP2A ... 72
Figure 5-1. Voies de signalisation de l‟insuline. ... 77
Figure 2-1. Les adipocytes secrètent des protéines ayant des effets variés sur l‟homéostasie du glucose. ... 145
Liste des tableaux
Tableau 1-1. Gènes impliqués dans les FAD... 10 Tableau 1-2. Effet de l‟allèle Apoε4 sur la fréquence et l‟âge d‟apparition de la MA ... 20 Tableau 2-1. DFTP-17 : Effets des mutations de Tau... 50 Tableau 4-1. Sites putatifs de phosphorylation de Tau et les différentes kinases et phosphatases
impliquées. ... 59 Tableau 5-1. Résumé des études in vivo montrant l‟hyperphosphorylation de Tau et ses
Liste des abréviations
Unités de mesure: °C Degré Celsius g gramme h Heure kDa Kilodalton kg Kilogramme L Litre m mètre M Molaire mg Milligramme min Minute mM Millimolaire mmol Millimole μl Microlitre μM Micromolaire μU Microunit Modèles animaux:3xTg-AD Tripe transgenic Alzheimer's disease
BB/Wor Bio-Breeding/Worcester
BBZDR/Wor Bio-Breeding Zucker Diabetic Rats/Worcester ICR Institute of cancer research
NIRKO Neuron insulin receptor knockout
NOD Non-obese diabetic
NODG NOD glycosuriques
NODH NOD hypothermiques
OLETF Otsuka Long-Evans Tokushima Fatty Protéines et peptides:
Aβ Peptide β amyloide
AChE Acetylcholinesterase
ADAM α disintegrin and metalloprotease ADDL Aβ-derived diffusable ligands
AICD Amyloid precursor protein intracellular domain APLP Amyloid precursor-like proteins
ApoE Apolipoprotéine E
APP Amyloid Precursor Protein APP-CTFs APP-C-terminal fragments
Cdc2 Cell cycle division 2
Cdk Cyclin-dependent kinases
ChAT Choline acetyltransferase
CTFs C-Terminal fragment
CX3CL1 Chemokine (C-X3-C) motif ligand 1 CX3CR1 CX3C chemokine receptor 1
ERK Extracellular signal-regulated kinase
GLUT Glucose transporters
GSK-3 Glycogène synthase kinase-3β
IDE Insulin degrading enzyme
IL-6 Interleukin-6
IRS Insulin receptor substrate
JNK c-Jun N-terminal kinase
LDLR Low-density lipoprotein receptor
LEPR Leptin receptor
MAP Microtubule associated proteins MAPK Mitogen-activated protein kinases MARK Microtubule-affinity regulated kinases
MKK MAPK kinases
Nct Nicastrine
N-PDPK Non proline-directed protein kinases
PDK1 3-phospho-inositide-dependent protein kinase-1 PDPK Proline-directed protein kinases
PI3K Phosphatidyl-inositol 3 kinase
PKA Protein kinase A
PKB Protein kinase B
PKC Protein kinase C
PKN Protein kinase N
PLC- Phospholipase C-
PP Ser/Thr protein phosphatases
PS1 Préséniline 1
PS2 Préséniline 2
PTB Phospho tyrosine binding RBP4 Retinol binding protein 4
RI Récepteur à l'insuline
SAPK Stress-activated protein kinases
SFK Src Family Kinase
SH2 Src homolgy 2
SH3 Src homology 3
TNF-α Tumor necrosis factor-α
TACE Tumor necrosis α-converting enzyme TPKII Tau protein kinase II
YK Tyrosine kinases
Autres:
Ach Acétylcholine
ADI Alzheimer's disease international ADN Acide désoxyribonucléique
ANOVA Analyse de la variance, analysis of variance
ARN Acide ribonucléique
CAA Cerebral amyloid angiopathy DCB Dégénérescence cortico-basale
DFT Démences fronto-temporales
DFTP-17 Démences fronto-temporales avec syndrome parkinsonien liées au chr.17 DNF Dégénérescence neurofibrillaire
DRG Diète riche en gras
DT1 Diabète de type 1
DT2 Diabète de type 2
DT3 Diabète de type 3
DTT Dithiothreitol
DWML Deep white matter lesions
ECL Enhanced chemiluminescence
EDTA Acide éthylène diamine tétra acétique EGTA Acide éthylène glycol tétra acétique ELISA ENzyme-linked immunosorbent assay FAD Familial Alzheimer's disease
FDG 18F-fluoro-déoxyglucose GTT Glucose tolerance test
HCl Chlorure d'hydrogène
HDL High-density lipoprotein
HOMA-IR Homeostatic model of assessment-Insulin resistance i.c.v. Intracérébroventriculaire
IMC Indice de masse corporelle
i.p. Intrapéritonéale
IRM Imagerie par résonnace magnétique
IRMf Imagerie par résonnance magnétique fonctionnelle
ITT Insulin tolerance test
LCR Liquide céphalorachidien
LiCl Chlorure de lithium
LTD Long-term depression
LTP Long-term potentiation
MA Maladie d'Alzheimer
MCI Mild cognitive impairment
MT Microtubules
NaCl Chlorure de sodium
NaF Fluorure de sodium
NFD Neurofibrillary degeneration NFT Neurofibrillary tangles
NINCDS-ADRDA National Institute of Neuroloical Disorders and Stroke and the Alzheimer Disease Related Disorders Association
NMDA N-Methyl-D-Aspartate
NP-40 Nodinet P-40
OA Okadaic acid
PSP Paralysie supranucléaire progressive PTB Phospho tyrosine binding
RIP Regulated intramembrane proteolysis RIPA Radio immunoprecipitation assay
ROS Reactive oxygen species
SDS Sodium dodecyl sulfate
SDS-PAGE Sodium dodecyl-sulfate polyacrylamide gel electrophoresis
SH2 Src homology 2
STZ Streptozotocine
TEMP Tomographie par émission de photon unique TEP Tomographie par émission de positons UPR Unfolded protein response
Chapitre I
Introduction
1 La maladie d’Alzheimer
Le 25 Novembre 1901, Auguste D. (Fig. 1-1), une allemande âgée de 51 ans, se présente à l‟Hôpital des malades mentaux et épileptiques de Francfort souffrant d‟une perte de mémoire, des troubles de langage ainsi que d‟autres troubles comportementaux (épisodes d‟hallucinations, anxiété, etc) (Maurer et al, 1997). Elle sera examinée par le Dr. Alois Alzheimer (Fig. 1-2), qui, lors de la 37e conférence de la société des Neuropsychiatres du Sud de l‟Allemagne à Tübingen en 1906, décrira sa condition comme étant une « maladie particulière du cortex cérébral». En 1907, Alzheimer publiait la première description histologique post-mortem des caractéristiques pathologiques de la démence qui portera désormais son nom (Alzheimer et al, 1995).
La maladie d‟Alzheimer (MA) est une maladie neurodégénérative irréversible qui se caractérise par une atteinte progressive de la mémoire (amnésie) et des fonctions cognitives (aphasie, apraxie, agnosie). Ceci se traduit par des troubles comportementaux ayant un impact profond, non seulement sur les personnes atteintes, mais aussi sur leurs proches et leur entourage.
La progression des signes cliniques de la MA est accompagnée de nombreux changements structuraux et métaboliques dans les cerveaux des patients. En effet, au moyen des techniques d‟imprégnation argentique, Alois Alzheimer a décrit deux types de lésions qui constituent jusqu‟à nos jours les deux marqueurs neuropathologiques majeurs de la maladie : les plaques amyloïdes et la dégénérescence neurofibrillaire (DNF ; en anglais NFD pour neurofibrillary degeneration). Les plaques amyloïdes résultent de l‟accumulation extracellulaire du peptide β amyloïde (Aβ), qui lui-même provient du clivage protéolytique d‟une protéine précurseur nommée APP (amyloid precursor protein). La DNF, quant à elle, correspond à l‟accumulation intra-neuronale d‟enchevêtrements de neurofibrilles (NFT pour neurofibrillary tangles), composés d‟une protéine nommée Tau qui se trouve dans un état anormalement hyperphosphorylé et s‟accumule sous forme de filaments appariés en hélice ou PHF (paired helical filaments). Notre laboratoire s‟intéresse
mieux comprendre la pathophysiologie de la MA et aider par la suite à développer des nouvelles cibles thérapeutiques visant à contrôler sa progression.
Dans ce chapitre, nous rappellerons tout d‟abord les bases de la neuropathologie de la MA, pour ensuite aborder la structure et les fonctions connues de Tau ainsi que les types de phosphorylation existants et leurs rôles putatifs. Nous examinerons ensuite la régulation de la phosphorylation de Tau et discuterons des mécanismes possibles de l‟hyperphosphorylation de Tau dans la MA. Enfin, nous aborderons les données émergentes qui supportent un lien entre la MA et la dysfonction de l‟insuline.
Figure 1-1. Éléments du dossier médical original de la patiente Auguste D (Deter).
Perdu depuis 1910, ce dossier a été retrouvé en 1995 dans les archives de l‟Université de Johann Wolfgang Goethe, à Francfort, Allemagne. A. Une photo de la patiente, prise en 1902 et montrant son impuissance. B. La couverture du dossier : admise le 25 Novembre 1901 et décédée le 8 Avril 1906. C. Écriture d‟Auguste D. Ce papier a été commenté par Alois Alzheimer : « Quand on lui demande d‟écrire, elle prend le livre d‟une telle façon qu‟on a l‟impression qu‟elle a perdu une partie du champ visuel droit. Pour écrire Auguste D., elle essaie d‟écrire Mme mais oublie tout le reste. Il est nécessaire de répéter chaque mot. Désordre de l‟écriture d‟origine amnésique». Adaptée de (O‟Brien, 1996) et (Maurer et al, 1997).
Figure 1-2. Dr. Alois Alzheimer. Neuropathologiste et psychiatre Allemand, né le 14 Juin
1864 à Markbeit, petit village bavarois au Sud de l‟Allemagne. En 1988, il commence sa carrière comme médecin assistant à l‟hôpital spécialisé dans les maladies mentales et psychiques de Francfort. Il s‟intéressait particulièrement aux démences d‟origine dégénérative ou vasculaire. Son intérêt pour la neuropathologie des troubles de la démence était partagé par son ami et collègue Franz Nissl qui le rejoignit à Francfort en mars 1889. C‟est Nissl qui initia Alzheimer aux techniques histopathologiques cérébrales, notamment les imprégnations argentiques. En 1912, Alzheimer est nommé directeur de la clinique psychiatrique de l‟université Freidreich-Wilhelm de Breslau (aujourd‟hui Wroclaw, en Pologne). Il est alors à l‟apogée de sa carrière. Mais le neuropsychiatre est bientôt touché par une maladie rénale dont il meurt le 15 décembre 1915, à Breslau. Adaptée de :
1.1 Prévalence
Dans leur rapport « démence : une priorité de santé publique », l‟organisation mondiale de la santé (OMS) et l’Alzheimer Disease International (ADI), affirment qu‟un nouveau cas de MA ou d‟une maladie apparentée est diagnostiqué toutes les 4 secondes dans le monde ; ce qui représente un équivalent de 7.7 millions nouveaux cas par année. Selon l‟expert mondial en matière de santé, le Dr. Peter Piot, il s‟agit en effet d‟une « bombe à retardement» (http ://www.alzheimer.ca/fr/Get-involved/Raise-your-voice/WHO-report-dementia-2012).
Au Canada, plus de 700,000 personnes souffrent d‟une forme de démence, et la MA représente environ 65% de ces cas. On estime que, si rien ne change, ce nombre dépasserait un million de personnes d‟ici 2031. Les femmes sont plus touchées par la MA que les hommes. Le vieillissement de la population des pays occidentaux impose la MA comme l‟un des problèmes de santé publique majeurs à l‟heure actuelle, chiffré au Canada à presque 33 Milliards de dollars par an (http ://www.alzheimer.ca/fr/Get-involved/Raise-your-voice/A-new-way-of-looking-at-dementia).
1.2 Signes cliniques et diagnostic
À l‟heure actuelle, le diagnostic de la MA ne peut être confirmé d‟une manière définitive que par analyse neuropathologique et histologique post-mortem des cerveaux de patients. De plus, l‟absence de marqueurs biologiques périphériques fiables ainsi que la grande variabilité des symptômes entre les individus rendent plus difficile le diagnostic ante-mortem de la maladie.
Les critères définis par le NINCDS-ADRDA (National Institute of Neuroloical Disorders and Stroke and the Alzheimer Disease Related Disorders Association) sont actuellement utilisés pour la classification de la maladie (McKhann et al, 1984; McKhann et al, 2011). Ainsi, on distingue entre une MA définitive (confirmée à l‟autopsie), probable (toutes les autres causes de démence ont été exclues et la progression des symptômes est typique de la
démence avec une probabilité supérieure à 80%, mais la possibilité de distinction entre la MA et les autres formes de démence est moins précise (23-88%) (Ballard & Bannister, 2005).
Le diagnostic clinique peut être appuyé par les résultats d‟imagerie cérébrale structurelle ou fonctionnelle. En effet, l‟imagerie par résonnance magnétique (IRM) structurelle semi-quantitative est capable de détecter l‟atrophie du lobe temporal médian (notamment l‟hippocampe) observée dans la MA (voir plus loin, paragraphe 1.4.1, page 22), avec une probabilité supérieure à 85% (Waldemar et al, 2007).
D‟autres techniques d‟imagerie quantitatives plus sophistiquées, telles que l‟imagerie volumétrique, la cartographie cérébrale tridimensionnelle et la mesure de l‟épaisseur corticale représentent des outils plus prometteurs pour le diagnostic de la MA (Querbes et al, 2009; Scheltens et al, 2002; Wahlund et al, 2000).
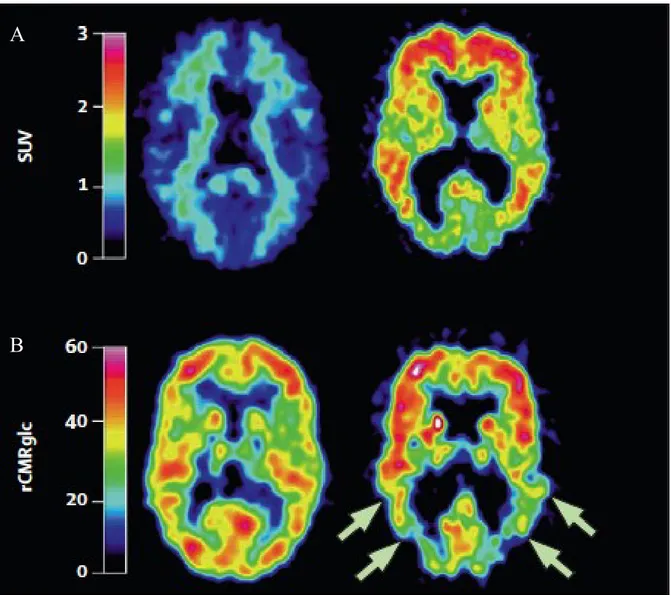
En plus des techniques d‟IRM structurelles, différentes techniques d‟imagerie fonctionnelles ont été développées pour le diagnostic de la maladie. Ces techniques comprennent principalement la TEP (tomographie par émission de positons), la TEMP (tomographie par émission de photon unique) et l‟IRMf (IRM fonctionnelle). De plus, le développement de radiotraceurs a permis l‟évaluation du métabolisme de différentes molécules dans le cerveau. Par exemple, la PET avec le 18F-fluoro-déoxyglucose (FDG) permet de mesurer le métabolisme du glucose qui se trouve significativement réduit lors de la MA (Devanand et al, 2010; Minoshima et al, 1995). De plus, le PIB (pour 11C-labelled Pittsburgh compound), un ligand d‟Aβ peut être utilisé pour visualiser les plaques amyloïdes in vivo (Nordberg, 2004), et plusieurs études ont montré une corrélation entre la rétention du PIB et les dépôts fibrillaires d‟Aβ ainsi que la distribution des plaques en post-mortem chez la majorité des patients atteints de la MA (Kadir et al, 2011; Svedberg et al, 2009) (Fig. 1-3).
Figure 1-3. Comparaison de la rétention du PIB et du métabolisme de glucose dans un cerveau normal et un cerveau de patient atteint de la MA. A. Rétention du Pittsburgh
compound B marqué avec du Carbone-11 (11C-PIB) chez un individu sain (67 ans, gauche) ou atteint de la maladie d‟Alzheimer (79 ans, droite). Noter la rétention importante du PIB dans les cortices frontal et tempo-pariétal. B. Images du métabolisme du glucose cérébral marqué par le 18F-fluoro-déoxyglucose (FDG) (μmol/min/100 mL) chez un individu sain (gauche) ou atteint de la MA (droite). Noter la diminution du métabolisme cérébral dans les régions tempo-pariétales (flèches). Adaptée de (Ballard et al, 2011).
A
Enfin, l‟analyse du liquide céphalorachidien (LCR) pourrait aussi confirmer un diagnostic clinique. On suggère que la MA pourrait être caractérisée par des niveaux réduits d‟Aβ42 (Fagan et al, 2007; Motter et al, 1995; Mulder et al, 2002), et des niveaux élevés de Tau et de Tau phosphorylée (Formichi et al, 2006; Hu et al, 2002; Vandermeeren et al, 1993). En revanche, il existe une grande variabilité dans les résultats des études du LCR. De plus, les kits d‟analyse disponibles sur le marché ne semblent pas être encore suffisamment fiables pour un usage en diagnostic.
1.3 Génétique
Alors que l‟étiologie de la majorité des cas de la MA (~99%) est d‟origine sporadique et à apparition tardive (plus que 65 ans), il existe des formes familiales (FAD, pour familial Alzheimer’s disease) (moins que 65 ans) qui sont transmises selon un mode autosomique dominant (Folstein, 1989). Ces mutations se trouvent sur 3 gènes situés sur les chromosomes 1, 14, et 21. De plus, il existe un facteur de risque pour les formes sporadiques de la maladie, dont le gène se trouve sur le chromosome 19 (Tableau 1-1). Il faut noter que des mutations sur l‟ADN mitochondrial ont été aussi associées avec la MA, mais il a été démontré par la suite que ces résultats étaient dus à des erreurs expérimentales (Orth & Schapira, 2001).
Symbole du gène
Locus Protéine Mode de
transmission Age d’apparition (ans) Contribution aux FAD APP 21q21.2 Amyloïde beta A4 Autosomal
dominant 40-60 ≤5%
PSEN1 14q24.3 Préséniline-1 Autosomal
dominant 30-58 ≥50-75%
PSEN2 1q31-q42 Préséniline-2 Autosomal
dominant 45-88 ≤1%
APOE 19q13.2 Apolipoprotéine E Facteur de
risque 40-90 Facteur de risque Tableau 1-1. Gènes impliqués dans les FAD. Adapté de (Alonso Vilatela et al, 2012).
1.3.1 APP
En 1984, Glenner et Wong ont isolé un polypeptide de 4.2 kDa contenu dans les plaques amyloïdes caractéristiques de la MA (Glenner & Wong, 1984). Il a été nommé amyloïde β à cause de sa structure en feuillets β. En 1987, le gène codant pour la séquence d‟Aβ a été
cloné et localisé sur le chromosome 21 (Goldgaber et al, 1987). Le clonage a pu aussi révéler que le peptide Aβ est synthétisé à partir d‟une protéine précurseur plus large dénommée APP (amyloid precursor protein). La première mutation dans le gène de l‟APP chez des familles atteintes de la MA a été reportée en 1991 par Goate et ses collaborateurs (Goate et al, 1991).
Le gène codant pour l‟APP est constitué de 18 exons qui, par épissage alternatif, donnent naissance à 10 isoformes comprenant de 563 à 770 acides aminés (a.a.) (Yoshikai et al, 1991). Les neurones expriment principalement l‟isoforme APP695 (Coulson et al, 2000).
L‟APP est un membre de la famille des APLP (amyloid precursor-like proteins), des glycoprotéines comportant un seul domaine transmembranaire, un domaine N-Terminal extracellulaire large et un domaine C-Terminal intracellulaire plus court (Fig. 1-4.A.). Ces protéines sont hautement conservées, régulées durant le développement, et exprimées dans tous les tissus mais à des niveaux élevés dans le cerveau.
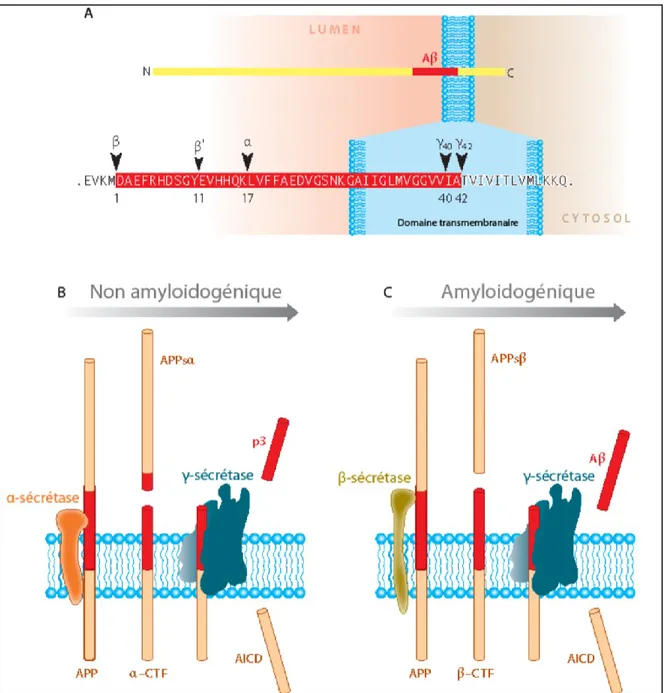
La protéine APP est clivée par une famille d‟enzymes appelées sécrétases, incluant les , , et sécrétases. Le clivage séquentiel par les , puis les sécrétases, qui libère respectivement les extrémités N et C-terminales du peptide A est caractéristique de la voie amyloïdogénique et entraine la production de différentes formes solubles de l‟A, principalement les formes A40 et A42, formées de 40 et 42 a.a., respectivement (Gouras et al, 1998; Vassar et al, 1999) (Fig. 1-4.C.).
Une coupure alternative de l‟APP par l‟-sécrétase intervient au milieu de la séquence A, prévenant ainsi la formation de l‟A et engendrant la sécrétion d‟un fragment appelé APP. La maturation de l‟APP faisant intervenir l‟-sécrétase est appelée voie non amyloïdogénique (Evin & Weidemann, 2002; Price et al, 1995) (Fig. 1-4.B.).
Figure 1-4. Structure et métabolisme de la protéine précurseur du peptide β-amyloïde (APP). A. La famille de l‟APP possède un domaine N-Terminal extracellulaire large et un
domaine C-Terminal intracellulaire plus court. La séquence du peptide Aβ commence à partir du domaine C-Terminal et s‟étend jusqu‟à la région transmembranaire (en rouge). B. Voie non amyloïdogénique impliquant le clivage de l‟APP par les α et γ-sécrétases. C. Voie amyloïdogénique montrant le clivage successif de l‟APP par les β et γ-sécrétases. Les 2 voies génèrent des domaines solubles (APPsα et APPsβ), ainsi qu‟un fragment C-Terminal intracellulaire identique (AICD, pour APP intracellular domain). CTF : C-Terminal fragment. Adaptée de (O‟Brien & Wong, 2011).
Les différentes sécrétases qui clivent l‟APP ne sont pas encore toutes identifiées. Les -sécrétases seraient des membres de la famille ADAM (α disintegrin and metalloprotease), telles que ADAM 9, ADAM 10 (Lammich et al, 1999; Lopez-Perez et al, 2001), et ADAM 17 (aussi appelée TACE pour tumor necrosis -converting enzyme) (Buxbaum et al, 1998). Les -sécrétases seraient BACE -1 et -2 (-site APP cleaving enzyme) (Vassar & Citron, 2000). Ce sont des protéases transmembranaires qui clivent l‟APP directement au niveau des sites +1 (qui suit le premier a.a.) et +11, par rapport à la séquence d‟A. Les souris BACE1-/- ne produisent pas d‟A, suggérant que BACE-1 serait la -sécrétase neuronale (Cai et al, 2001).
Les -sécrétases sont un complexe multiprotéique composé de présénilines 1 ou 2 ; la nicastrine (Nct), une glycoprotéine membranaire de type I, Aph-1 et Pen-2, deux protéines transmembranaires (pour revue, voir (Bergmans & De Strooper, 2010)). Les fonctions des différentes -sécrétases ainsi que leurs interactions au sein du complexe ne sont pas bien définies.
Les fonctions physiologiques de l‟APP ne sont pas bien comprises. La suppression du gène chez la souris ne produit pas de phénotype particulier jusqu‟à 12 semaines, ce qui suggère une possible substitution des fonctions de l‟APP par d‟autres membres de la famille des APLP (Heber et al, 2000; Zheng et al, 1996). Il a été montré que le domaine extracellulaire pourrait avoir un rôle dans l‟adhérence cellulaire, la croissance neuritique et la synaptogenèse ; alors que le domaine intracellulaire joue des rôles importants dans le transport axonal, la signalisation cellulaire et l‟apoptose (pour revue, voir (Zheng & Koo, 2006)).
À l‟heure actuelle, il existe plus qu‟une trentaine de mutations sur le gène de l‟APP (http ://www.alzforum.org/res/com/mut/app/default.asp), et ce sont les mutations proches des sites de clivage des et sécrétases qui entrainent l‟apparition des FAD
(Chartier-sécrétase entraine une augmentation du ratio A42/A40 (Suzuki et al, 1994), ce qui accélère la progression de la maladie, du fait que la forme A42 est plus hydrophobe et amyloïdogénique que la forme A40 (Jarrett et al, 1993). En revanche, la mutation Swedish de l‟APP (identifiée chez une famille Suédoise atteinte d‟une FAD), proche du site du clivage de la -sécrétase (Mullan et al, 1992), entraine une augmentation de la production des formes A40 et A42 sans toutefois en changer le ratio (Citron et al, 1994; Scheuner et al, 1996). D‟autres mutations situées au niveau de la région A elle-même, ne résultent pas en une augmentation de la production d‟A, mais augmente sa capacité d‟oligomérisation et d‟agrégation, ce qui entraine des accumulations massives de l‟amyloïde sur les parois des vaisseaux sanguins et des hémorragies cérébrales (Joachim et al, 1988). Ces mutations sont associées non seulement aux FAD, mais aussi à l‟angiopathie amyloïde cérébrale (CAA, pour cerebral amyloid angiopathy) (Grabowski et al, 2001; Nilsberth et al, 2001; Van Nostrand et al, 2001).
1.3.2 Présénilines
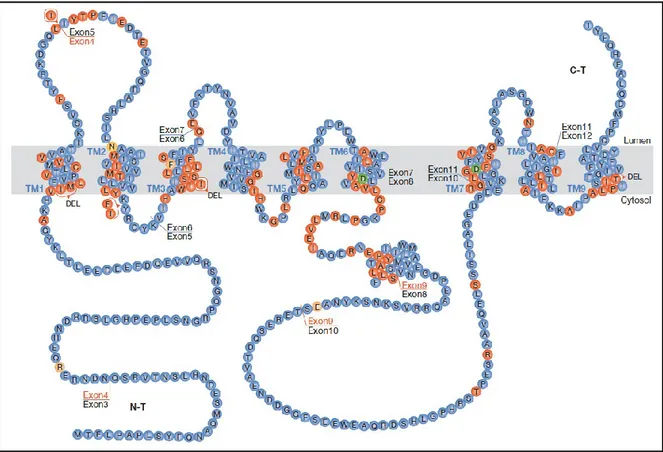
Les présénilines 1 et 2 (PS1 et PS2) sont deux protéines, de 467 et 441 a.a., respectivement, qui partagent 67% d‟homologie mais dont les gènes sont localisés sur deux chromosomes différents ; le gène codant pour PS1 est situé sur le chromosome 14 alors que le gène codant pour PS2 se trouve sur le chromosome 1 (Rogaev et al, 1995; Sherrington et al, 1995; St George-Hyslop et al, 1992). Ce sont des protéines membranaires à traversées multiples (multipass) possédant 8-9 segments transmembranaires hydrophobes, des domaines N et C-Terminaux cytosoliques ainsi qu‟une large boucle cytoplasmique hydrophile située entre les domaines VI et VII (Doan et al, 1996; Li & Greenwald, 1996; Li & Greenwald, 1998) (Fig. 1-5). Dans le système nerveux central (SNC), elles sont essentiellement exprimées au niveau des neurones (Huynh et al, 1997; Kovacs et al, 1996b; Moussaoui et al, 1996). À l‟échelle subcellulaire, les présénilines sont localisées majoritairement au niveau du réticulum endoplasmique et de l‟appareil de Golgi, mais aussi en faible quantité au niveau de la membrane plasmique (Annaert et al, 1999; Kovacs et al, 1996a; Lah et al, 1997).
Les PS sont la cible de plusieurs protéases, essentiellement une « présinilase » (Mercken et al, 1996; Thinakaran et al, 1996), et des caspases (Kim et al, 1997; Loetscher et al, 1997).
Le clivage par la présinilase entraine la production de fragments N-Terminal et C-Terminal de poids moléculaires respectifs de ~30 et ~20 kDa, qui forment des hétérodimères stables correspondant au complexe biologiquement actif des présénilines (Capell et al, 1998; Thinakaran et al, 1998).
Les PS ne sont ni acétylées, ni glycosylées, ni sulfatées, mais peuvent subir des phosphorylations au niveau de différents résidus, ce qui semblerait moduler leur fonction de contrôle de mort cellulaire (pour revue, voir (Checler, 1999)).
Figure 1-5. Structures des présénilines et mutations liées aux FAD. La structure
montrée est à 9 domaines transmembranaires. Les mutations en rouge sont pathogènes, celles en vert sont non pathogènes et celles en orange sont de nature pathogène non claire. N-T : N-Terminal; C-T : C-Terminal; TM : Transmembranaire; DEL : délétion. Adaptée de
(Hardy, 2007) et de
(http ://www.molgen.ua.ac.be/ADMutations/default.cfm?MT=1&ML=1&Page=MutByQue ry&Query=tblContexts.GeneSymbol%20In%20(„PSEN1‟)&Selection=Gene%20In%20(PS EN1).
Les fonctions des PS ne sont pas bien connues. On suggère que ce sont des aspartyl protéases qui ont un rôle clé dans le clivage intramembranaire de l‟APP ainsi que plusieurs autres substrats protéiques (Wolfe et al, 1999a; Wolfe et al, 1999b). En effet, L‟expression des PS au niveau de la membrane plasmique leur permet de participer à de nombreuses fonctions à la surface de la cellule, notamment la protéolyse intramembranaire régulée (RIP pour regulated intramembrane proteolysis). La RIP est un mécanisme conservé au cours de l‟évolution qui consiste à cliver des protéines transmembranaires au niveau de la membrane, permettant ainsi la libération des fragments cytosoliques qui pénètrent dans le noyau et contrôlent la transcription des gènes (Brown et al, 2000). Il a été établi que les PS participent dans la RIP ce qui leur permet par conséquent d‟influencer des processus vitaux, tels que la différentiation cellulaire, le métabolisme lipidique, ainsi que l‟UPR (unfolded protein response), (pour revue, voir (Brown et al, 2000; Fortini, 2002)). De plus, les PS contribuent à la maturation de l‟APP. Cette hypothèse est renforcée par les observations que les mutations dans les PS se manifestent par une augmentation de la production d‟A42 dans les cerveaux de patients atteints de la MA. Toutefois, le mécanisme par lequel les PS pourraient contrôler le métabolisme de l‟APP n‟est pas bien connu.
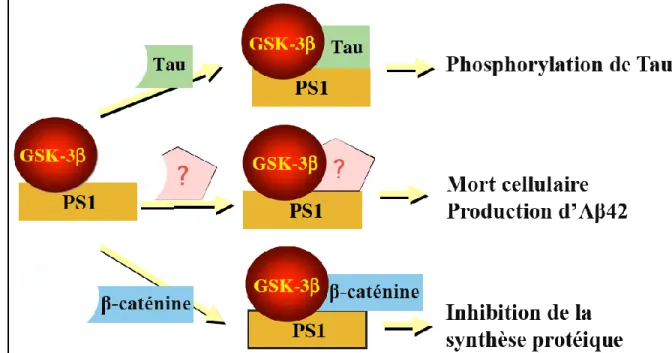
À l‟heure actuelle, plus de 180 mutations ponctuelles ont été identifiées sur les gènes de la PS1 et la PS2 (http ://www.alzforum.org/res/com/mut/pre/default.asp), ce nombre ne cessant pas de croître. Les mutations PS1 causent les formes les plus sévères des FAD et on pense que ceci se fait en modifiant le métabolisme de l‟APP. En effet, il a été établi que les mutations PS1 et PS2 entrainent une production massive du peptide « pathogène » A42, qui est plus amyloïdogénique qu‟A40 (Cruts & Van Broeckhoven, 1998; Fraser et al, 2000). D‟une manière intéressante, ceci ne pourrait être qu‟un aspect des effets délétères de ces mutations car PS1 est connue pour interagir avec de nombreuses autres protéines, notamment Tau et GSK-3 (Glycogène synthase kinase-3β), deux protéines impliquées dans la pathologie de la MA (voir plus loin, paragraphe 4.1.1, page 63) (Takashima et al, 1998b). En effet, GSK-3 montre une plus grande affinité pour les PS1 mutées, ce qui entraine une augmentation de l‟association de GSK-3 avec Tau, et favorise ainsi l‟hyperphosphorylation de Tau et la formation de NFT (Fig. 1-6) (Takashima et al, 1999).
Figure 1-6. Rôle du complexe PS1/GSK-3β dans la MA. La PS1 peut agir comme une
protéine qui lie la GSK-3β avec des substrats protéiques importants. Le complexe PS1/ GSK-3β peut se lier sur Tau, menant ainsi à sa phosphorylation. De plus, il a été établi que la β-caténine peut se lier avec la PS1, qui est un substrat pour la GSK-3β. La liaison PS1/β-caténine affecte les niveaux de la β-PS1/β-caténine soluble, et entraine une diminution de l‟activité de transcription de Tcf-4, résultant en une réduction de la synthèse des protéines induite par Tcf. Il pourrait aussi exister d‟autres substrats protéiques pouvant s‟associer à la PS1 et avoir un rôle dans l‟apoptose et la production d‟Aβ42. PS1 : Préséniline-1 ; GSK-3β : Glycogène synthase kinase-GSK-3β, Aβ : Amyloïde-β. Adaptée de (Takashima et al, 1999).
1.3.3 Apolipoprotéine E
L‟apolipoprotéine E (ApoE ) est une glycoprotéine de 299 a.a. et d‟un poids moléculaire de ~34 kDa (Rall & Mahley, 1992). Chez l‟Homme, le gène codant pour l‟ApoE est situé sur le bras long du chromosome 19 (Olaisen et al, 1982), et existe sous forme de 3 allèles différents (ε2, ε3 et ε4) (Zannis & Breslow, 1980; Zannis et al, 1982). Ces variations sont le résultat d‟une substitution d‟a.a. (Cystéine ou Arginine) aux positions 112 et 158 : ApoE2 (Cys112, Cys158), ApoE3 (Cys112, Arg158) et ApoE4 (Arg112, Arg158) (Mahley, 1988; Weisgraber et al, 1981) (Fig.1-7). Cette substitution affecte la structure des différentes isoformes de l‟ApoE et influence leur capacité à lier leurs récepteurs, les lipides, ainsi que le peptide A (Chen et al, 2011a; Frieden & Garai, 2012; Zhong & Weisgraber, 2009).
Figure 1-7. Structure et isoforme de l’Apolipoprotéine E. Les isoformes E2,
Apo-E3 et Apo-E4, codées respectivement par les allèles ε2, ε3 et ε4 du gène de l‟ApoE, diffèrent les unes des autres par les résidus d‟acides aminés en position 112/et ou 158 (cercles violets). L‟ApoE possède deux domaines structuraux: un domaine N-Terminal contenant une région de liaison au récepteur (résidus 136-150) et un domaine C-Terminal contenant une région de liaison aux lipides (résidus 244-272); les deux domaines étant liés par un coude. ApoE: Apolipoprotéine. Adaptée de (Liu et al, 2013).
Différence d’acides aminés
isoforme-spécifique Fréquence de l’allèle (%)
112 158 Population
générale
Maladie d‟Alzheimer
Apo-E2 Cys Cys 8.4 3.9
Apo-E3 Cys Arg 77.9 59.4
Dans les tissus périphériques, l‟ApoE est synthétisée majoritairement par le foie et les macrophages. Son rôle est de réguler l‟homéostasie lipidique en transportant les lipides soit d‟un tissu à l‟autre, soit d‟un type cellulaire à l‟autre (Mahley & Rall, 2000).
Dans le SNC, l‟ApoE est principalement produit par les astrocytes (Blain et al, 1996; Pitas et al, 1987; Uchihara et al, 1995), et transporte le cholestérol vers les neurones via les récepteurs ApoE, qui sont des membres de la famille des LDLR (low-density lipoprotein receptor) (Herz & Bock, 2002). Le cholestérol cérébral est un acteur important pour différents processus neuronaux, tels que la croissance axonale, la formation et le remodelage des synapses, un événement crucial requis pour la mémoire et l‟apprentissage (Mauch et al, 2001; Pfrieger, 2003).
Les fréquences des allèles ε2, ε3 et ε4 dans la population mondiale sont de 8.4%, 77.9% et 13.7%, respectivement (Farrer et al, 1997). En revanche, la fréquence de l‟allèle ε4 est dramatiquement augmentée (environ 40%) chez les patients atteints de la MA (Farrer et al, 1997), suggérant qu‟il représente un facteur de risque important pour les formes sporadiques de la maladie. En effet, il a été montré, que, dans les populations caucasiennes, les porteurs hétérozygotes de l‟allèle ε4 ont un risque 3 fois plus élevé, alors que les homozygotes 15 fois plus élevé de développer la maladie, en comparaison avec les individus ayant un génotype non-ε4 (Chartier-Harlin et al, 2002; Farrer et al, 1997). De plus, l‟âge d‟apparition des symptômes cliniques de la maladie semble être plus précoce chez les homozygotes (68 ans), en comparaison avec les hétérozygotes (76 ans), ou les non porteurs (84 ans) (Corder et al, 1993; Rebeck et al, 1993) (Tableau 1-2). Ces données suggèrent que l‟ApoE4 entraine une augmentation dramatique du risque de la MA à un âge plus précoce, d‟une manière proportionnelle au nombre d‟allèles ε4 portés.
En revanche, l‟allèle ε2 semble être protecteur : les porteurs homozygotes (ε2/ ε2) ou hétérozygotes (ε2/ ε3) ont moins de risque d‟avoir la MA, par rapport aux porteurs ε3 (Benjamin et al, 1994; Corder et al, 1994; Farrer et al, 1997; Talbot et al, 1994).
Génotype Non porteurs Apoε4 Hétérozygotes Apoε4 Homozygotes Apoε4 Fréquence dans la MA (%) 20 47 91
Age moyen d’apparition clinique de la maladie (ans)
84 76 68
Tableau 1-2. Effet de l’allèle Apoε4 sur la fréquence et l’âge d’apparition de la MA.
Adapté de (Liu et al, 2013).
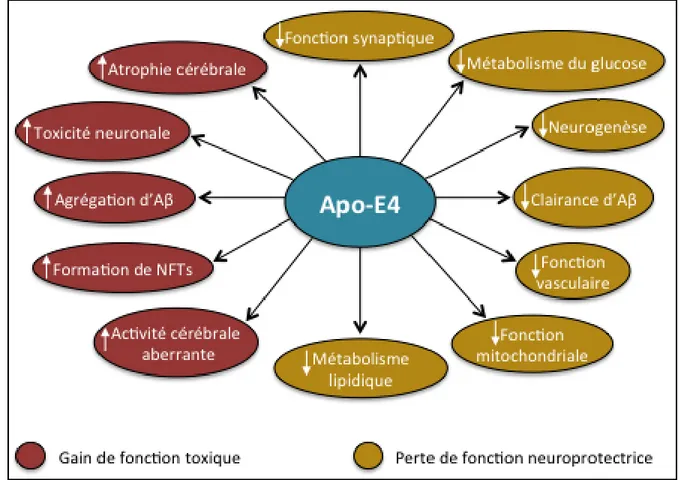
La contribution des différentes isoformes de l‟ApoE dans la MA n‟est pas bien connue. Des études ont montré que les niveaux d‟A et la formation des plaques amyloïdes sont régulées par l‟ApoE d‟une manière dépendante de l‟isoforme (ε4>ε3>ε2), suggérant un rôle important de la protéine dans le métabolisme et l‟agrégation d‟A (Bales et al, 2009; Castellano et al, 2011; Reiman et al, 2009). Les mécanismes par lesquels l‟ApoE4 contribue à la MA ne sont pas tous établis. On suggère que l‟ApoE4 confère un gain de fonction toxique et/ou une perte de fonction neuroprotectrice au sein du neurone. Par exemple, il a été établi que cette isoforme est moins efficace que ces homologues dans le maintien de nombreuses fonctions vitales, telles que la plasticité synaptique, le métabolisme du glucose, la neurogenèse, la stabilité du cytosquelette ainsi que la fonction mitochondriale. En parallèle, sa présence est associée à de nombreuses anomalies fonctionnelles, telles que l‟atrophie cérébrale, l‟inflammation, la toxicité neuronale, l‟agrégation d‟A ainsi que l‟hyperphosphorylation de Tau (Fig. 1-8) (pour revue, voir (Liu et al, 2013)).
Figure 1-8. Rôle de l’apolipoprotéine-E4 dans la MA. Apo-E4 confère un gain de
fonction toxique et/ou une perte des fonctions neuroprotectrices au cours de la pathogenèse de la MA. Les différences fonctionnelles majeures entre Apo-E4 et Apo-E3 sont illustrées. Abréviations : Aβ : Amyloïde-β ; ApoE : Apolipoprotéine. Adaptée de (Liu et al, 2013).
1.4 Neuropathologie
1.4.1 Atrophie cérébrale
Au niveau macroscopique, les patients atteints de la MA présentent une atrophie massive du tissu cérébral, une augmentation importante du volume des sillons et des ventricules cérébraux, ainsi qu‟un élargissement des espaces périvasculaires (Fig. 1-9). Cette atrophie serait le résultat d‟une perte neuronale importante menant à une réduction du ruban cortical, et par conséquent des circonvolutions cérébrales (Dhenain et al, 2002; Duyckaerts et al, 1985).
Figure 1-9. Atrophie cérébrale observée au cours de la MA. Comparaison de deux
coupes coronales d‟un cerveau normal (droite) et d‟un cerveau de patient atteint de la MA (gauche) à échelle identique. Noter l‟atrophie corticale importante et l‟élargissement des ventricules. Adaptée de (Holtzman et al, 2011).
1.4.2 Les plaques amyloïdes
Les plaques amyloïdes sont des dépôts extracellulaires sphériques (Fig. 1-10. A.) constitués d‟un peptide de 4.2 kDa nommé peptide amyloïde (A) (Glenner & Wong, 1984). Ces dépôts sont agrégés sous forme de feuillets -plissés à caractère argyrophile et peuvent être visualisés par différentes techniques, telles que les imprégnations argentiques (coloration Gallyas, Bodian ou Bielchowski), le marquage avec des intercalants des feuillets , tels que le congo Red et la thioflavine S, ou encore par immunohistochimie en utilisant des anticorps dirigés contre le peptide A (Fig. 1-10. C, E).
Figure 1-10. Marqueurs neuropathologiques de la MA et techniques de marquage. A.
Imprégnation argentique des plaques amyloïdes extracellulaires par la méthode de Bielschowski. B. Enchevêtrements neurofibrillaires intraneuronaux marqués par la méthode de Bielschowski. C. Marquage immunohistochimique des plaques amyloïdes au moyen d‟anticorps dirigés contre le peptide β-amyloïde (Aβ) montrant des plaques diffuses (grande
neurones (grande flèche) et au niveau des processus neuronaux appelés neuropils (petite flèche; X200). E. Marquage des lésions par la Thioflavine-S, un intercalant des feuillets β plissés, montrant les plaques amyloïdes (grande flèche) et les enchevêtrements neurofibrillaires (petite flèche, x200). Adaptée du site internet du VCU (Virginia Commonwealth University) medical center (department of patholgy) et de (Holtzman et al, 2011).
1.4.2.1 Classification et extension des plaques amyloïdes
Les plaques amyloïdes peuvent être classées en 5 sous-catégories en fonction de leur morphologie et leur réactivité aux colorants spécifiques aux feuillets -plissés : (1) les dépôts stéllaires, (2) les plaques diffuses, (3) les plaques primitives, (4) les plaques classiques et (5) les plaques compactes. Les dépôts stéllaires et les plaques diffuses (aussi nommées pré-amyloïdes, ou pré-plaques) ne présentent pas des composés neuritiques et peuvent être marquées par l‟imprégnation argentique mais non pas avec la Thioflavine S ou le Rouge Congo. En revanche, les plaques primitives (sans cœur dense mais avec un contour bien défini), les plaques classiques (avec un cœur dense de nature fibrillaire et une couronne de neurites dystrophiques) et les plaques compactes (avec un cœur dense mais sans couronne de neurites dystrophiques) contiennent des neurites dystrophiques ainsi que des fibres amyloïdes denses pouvant être marquées par la Thioflavine S ou le Rouge Congo (Delaere et al, 1991; Giannakopoulos et al, 2001). Certaines de ces neurites peuvent contenir des éléments synaptiques en dégénérescence, des mitochondries et des lysosomes, alors que d‟autres peuvent contenir des PHF similaires à ceux observés dans les NFT. Les plaques neuritiques immuno-réactives aux PHF se trouvent en prédominance dans la MA et corrèlent positivement avec la sévérité de la maladie (Giannakopoulos et al, 2001). Les plaques séniles classiques et notamment leur cœur dense, sont composées principalement d‟A40 alors que les plaques diffuses sont composées d‟A42 (Iwatsubo et al, 1994). Les plaques amyloïdes affectent principalement les régions néocorticales ; les régions temporales et occipitales étant les plus atteintes. En revanche, l‟hippocampe et le cortex entorhinal sont moins affectés (Arnold et al, 1991; Price et al, 1991). On distingue 3 stades de dépôts amyloïdes (Fig. 1-11) (Braak & Braak, 1991; Braak & Braak, 1997) :
- Stade A : les dépôts se trouvent en faible quantité dans les régions basales du cortex frontal, temporal et occipital. Le territoire hippocampique est épargné.
- Stade B : les dépôts amyloïdes affectent les aires associatives de presque l‟ensemble de l‟isocortex, avec une densité modérée. Le territoire hippocampique est légèrement touché.
- Stade C : l‟ensemble de l‟isocortex est atteint. Le territoire hippocampique reste modérément touché.
Figure 1-11. Distribution des plaques amyloïdes au cours de la MA. Au stade A, les
plaques sont présentes dans les aires basales de l‟isocortex. Le stade B montre la présence des dépôts amyloïdes dans presque toutes les aires associatives de l‟isocortex. Au stade final C, les dépôts affectent la totalité de l‟isocortex, incluant les parties sensorielles et motrices. L‟augmentation de l‟intensité de la couleur reflète l‟augmentation du nombre de plaques. Adaptée de (Braak & Braak, 1991).
1.4.2.2 L’hypothèse de la cascade amyloïde
La découverte des mutations dans les gènes de l‟APP, la PS1 et la PS2 dans les FAD, et la cytotoxicité des agrégats d‟A (Selkoe, 2001) ont conduit certains chercheurs à formuler « l‟hypothèse de la cascade amyloïde » (amyloid cascade hypothesis) (Hardy & Higgins, 1992). Selon l‟hypothèse originale, le peptide A (plus spécifiquement la forme A42) est l‟élément déclencheur de la neuropathologie de la MA, et provoque secondairement un stress cellulaire responsable d‟une cascade d‟évènements moléculaires menant à la dérégulation de la phosphorylation de Tau et la formation des NFT, qui ne seraient par conséquent qu‟un épiphénomène (Fig. 1-12).
Figure 1-12. Illustration de la cascade d’évènements reliant la pathologie amyloïde à la dégénérescence neurofibrillaire. Adaptée de (Hardy & Higgins, 1992).
En revanche, cette hypothèse est controversée par de nombreuses études, aussi bien chez l‟Homme que chez la souris, qui montrent que la formation des dépôts amyloïdes ne corrèle pas avec différents paramètres de la MA, tels que les déficits cognitifs, la toxicité neuronale, la perte synaptique et les altérations du cytosquelette (Delaere et al, 1990; Dickson et al, 1992; Morris et al, 1996; Neve & Robakis, 1998).
Ceci est renforcé par des observations montrant que la neurodégénérescence est observée dans certaines régions du cerveau où il n‟y a pas de dépôts A. De plus, les plaques amyloïdes ne sont pas spécifiques à la MA, et se développent aussi durant le vieillissement normal (Davies et al, 1988).
Ces résultats pris ensemble suggèrent que les agrégats d‟A, à eux seuls, ne sont pas capables d‟expliquer la corrélation entre le métabolisme de l‟APP et l‟évolution de la MA. En effet, dans la formulation plus récente de cette hypothèse, les oligomères solubles d‟A remplaceraient les agrégats (Palop & Mucke, 2010) (voir paragraphe 1.4.2.3, page 28). Il serait aussi possible que le facteur critique ne soit pas l‟A en lui-même, mais plutôt les fragments C-Terminaux de l‟APP (APP-CTFs pour APP-C-terminal fragments) (Cao & Sudhof, 2001), puisque leur diminution lors de l‟évolution de la MA corrèle fortement avec le développement des NFT (Sergeant et al, 2002).
L‟hypothèse de la cascade amyloïde a servi de base au développement de nombreuses stratégies thérapeutiques visant à réduire la quantité d‟A ou sa toxicité (vaccins contre l‟A42, inhibiteurs de la -sécrétases, etc.) (Hardy & Selkoe, 2002; Sommer, 2002). En revanche, un grand nombre d‟approches en essai clinique ont été arrêtées (Karran et al, 2011), (http ://www.alzforum.org/dis/tre/drc/default.asp), ce qui devrait donner raisons aux chercheurs qui proclament depuis des années que les fonctions physiologiques de l‟APP et les produits de son métabolisme doivent être mieux comprises avant de développer tout traitement dirigé contre eux.