DOCTORAT THESIS
Presented by:
Yassine SOUILMI
Discipline: Biology
Specialty: Bioinformatics
HIGH-THROUGHPUT NEXT-GENERATION SEQUENCING DATA
ANALYSIS FOR PRECISION MEDICINE
Defended May 10
th, 2016
In front of the jury
President
Saaïd AMZAZI:
PES, School of Sciences, Mohammed V University, Rabat
Examinators:
Youssef BAKRI:
PES, School of Sciences, Mohammed V University, Rabat
Ali IDRI:
PES, National School of Informatics and Systems Analysis, Mohammed V University, Rabat
Hassan GHAZAL:
PH, Multidisciplinary School, Mohammed I University, Oujda
COPYRIGHTS
The following work is following the general guidelines of the Creative Common License for the sections that doesn't have a specific license belonging to a journal publisher. For the sections free from in a journal publisher or a university domain's you are free to:
- Share -- copy and redistribute the material in any medium or format
- Adapt -- remix, transform, and build upon the material for any purpose, even commercially. The licensor cannot revoke these freedoms as long as you follow the license terms.
Under the following terms:
- Attribution -- You must give appropriate credit, provide a link to the license, and indicate if changes were made. You may do so in any reasonable manner, but not in any way that suggests the licensor endorses you or your use.
- ShareAlike -- If you remix, transform, or build upon the material, you must distribute your contributions under the same license as the original.
- No additional restrictions -- You may not apply legal terms or technological measures that legally restrict others from doing anything the license permits.
- These three aforementioned terms are valid where to all the following material except what belongs to a journal publisher or a university domain.
Notices:
- You must comply with the license for the elements of this material in a journal publisher or a university domain.
- You do not have to comply with the license for elements of the material in the public domain or where your use is permitted by an applicable exception or limitation.
- No warranties are given. The license may not give you all of the permissions necessary for your intended use. For example, other rights such as publicity, privacy, or moral rights may limit how
DECLARATION
I hereby declare that except where specific reference is made to the work of others, the contents of this dissertation are original and have not been submitted in whole or in part for consideration for any other degree or qualification in this, or any other university. This dissertation is my own work and contains nothing that is the outcome of work done in collaboration with others, except as specified in the text and Acknowledgements. This dissertation complies with Mohamed V University in Rabat guidelines and respects its code of ethics.
DEDICATION
I dedicate this work to whom I hold dearest:
My parents
My brother and sister
My dear nieces
My friends
My mentors and advisors
ACKNOWLEDGEMENT
First of all I would like to thank my supervisory team Pr. Saaid Amzazi, Pr. Hassan Ghazal, Pr. Peter J. Tonelllato and Pr. Dennis P. Wall for their support and guidance. I am grateful to Pr. Amzazi for his unconditional support and wise advices throughout my masters and Ph.D. research. Pr. Ghazal, I am thankful for all the mentorship, technical help and scientific and philosophic supervision. I will be forever grateful to Pr. Tonellato for his supervision, empowerment and mentorship; he pushed my level of expertise to the bleeding edge, as well as providing me with the necessary funding to pursue my research in the best conditions possible. I am also very grateful to Pr. Wall for his supervision and his visionary advice.
I would like to thank my colleagues Jared B. Hawkins for making my adaptation to my host lab easy, and providing me with technical support. Jae-Yoon Jung (the Wall lab) and Erik Gafni (Invitae) for their continuous technical support. Alex Lancaster (the Wall lab) for his technical support, and scientific valuable and mentorship. I thank Angel Pizarro and Leo Zhadanovsky from Amazon Web Services for advice and assistance. Gratitude goes also to Justin Riley for his continuous help and support with his StarCluster cloud tool. In addition, I would like to address my gratitude to David L. Osterbur and the NCBI helpdesk for their help designing the custom search queries.
I would also like to extend my thanks to Dr. James Miller the director of the Moroccan Fulbright commission and his team, for the funding of my research scholarship at Harvard Medical School. I also thank Aimee Doe and Katherine Flannery from the Department for Biomedical Informatics, Harvard University as well as John Orak and Matthew Guckenberg from the Washington DC AMIDEAST office for all of their administrative support.
The research in this thesis project received the following funding: Amazon Web Services, National Institutes of Health [1R01LM011566 to P.J.T., 5T15LM007092 to P.J.T. and J.B.H., 1R01MH090611-01A1 to D.P.W.]; and a Fulbright Fellowship [to Y.S.]. Funding from the Hartwell Foundation and the Hartwell Autism Research and Technology Initiative directed by D.P.W.
PUBLICATIONS
1- Souilmi, Yassine, Alex K Lancaster, Jae-Yoon Jung, Ettore Rizzo, Jared B Hawkins, Ryan Powles, Saaid Amzazi, Hassan Ghazal, Peter J Tonellato, and Dennis P Wall. 2015. “Scalable and Cost-Effective NGS Genotyping in the Cloud.” BMC Medical Genomics 8 (1): 64. doi:10.1186/s12920-015-0134-9.
2- Souilmi, Yassine, Jae-Yoon Jung, Alex Lancaster, Erik Gafni, Saaid Amzazi, Hassan Ghazal, Dennis Wall, and Peter Tonellato. 2015. “COSMOS: Cloud Enabled NGS Analysis.” BMC Bioinformatics 16 (2). BioMed Central: 1. doi:10.1186/1471-2105-16-S2-A2.
3- Gafni, Erik, Lovelace J Luquette, Alex K Lancaster, Jared B Hawkins, Jae-Yoon Jung, Yassine Souilmi, Dennis P Wall, and Peter J Tonellato. 2014. “COSMOS: Python Library for Massively Parallel Workflows.” Bioinformatics 30 (20): 2956–58. doi:10.1093/bioinformatics/btu385.
TALKS
1- Souilmi, Y., Jung, J.-Y., Lancaster, A., Gafni, E., Amzazi, S., Ghazal, H., . . . Tonellato, P. (2015, October). High throughput NGS variant calling in the cloud. Paper presented at the ISMB Students Council Symposium, Boston, MA
2- Souilmi, Y., Jung, J.-Y., Lancaster, A., Gafni, E., Amzazi, S., Ghazal, H., . . . Tonellato, P. (2014, July). COSMOS: cloud enabled NGS analysis. Paper presented at the ISMB Students Council Symposium, Boston, MA
3- Hawkins, J., Souilmi, Y., Powles, R., Jung, J., Lancaster, A., . . . Tonellato, P. (2014, November). COSMOS: NGS Analysis in the Cloud. Paper presented at the AMIA 2014 Joint Summits on Translational Science, San Francisco, CA
POSTERS
1- Yassine Souilmi, Alex K. Lancaster, Jae-Yoon Jung, Ettore Rizzo, Ryan Powles, Peter J. Tonellato, Dennis P (2015, July). Wall. High-throughput Clinical NGS Data Analysis on the Cloud. Poster presented at ISMB Students Council Symposium, Dublin, Ireland
2- Souilmi, Y., Jung, J.-Y., Lancaster, A., Gafni, E., Amzazi, S., Ghazal, H., . . . Tonellato, P. (2015, April). COSMOS: cloud enabled NGS analysis. Poster presented at the New England Science Symposium, Boston, MA
3- Souilmi, Y., Jung, J.-Y., Lancaster, A., Gafni, E., Amzazi, S., Ghazal, H., . . . Tonellato, P. (2014, July). COSMOS: cloud enabled NGS analysis. Poster presented at the ISMB Students Council Symposium, Boston, MA
ABSTRACT
Since the discovery of the first sequencing technologies, biomedical research took a very early interest in the role played by the human genome in the development of diseases. However, the increasing discovery rate started by the launch of the human genome project, pushed towards the development of high-throughput sequencing platforms, which accelerated the genomic data generation. However, the new sequencing technology routinely generates petabyte scale data, a data deluge and a computational bottleneck.
Genomic data analysis is a multi-step processing operation, and the importance of the data size requires the development of a new generation of workflow management systems. We developed COSMOS; a workflow management system designed with genomic data specifics in consideration. COSMOS offers high levels of parallelization yet easy to implement, and is cloud-ready. To create a golden standard timings and quality reference for genomic data analysis, we created GenomeKey, a “GATK best practices” COSMOS workflow. We performed a systematic benchmarking of timings, cost and quality control of the variant calls generated by the pipeline, creating a road-map to routine, cost effective, clinical-turnaround, whole genome analyses.
The high-throughput data generation of new sequencing platforms, and the cost-effective and fast data processing of GenomeKey/COSMOS running on cloud platforms opens the door to a new era of genomic discovery and evidence-bases medical genomics practices. Thus, we explore the feasibility of a Precision Medicine project in Morocco, by assessing what are the country’s challenges and opportunities.
RESUME
Dès l’introduction des premières technologies de séquençage, la recherche biomédicale a très tôt prêté attention au rôle du génome humain dans le développement des maladies. Par contre, la cadence des découvertes s’est accélérée après le lancement du projet du génome humain, poussant vers le développement de plateformes de séquençage à haut débit. C’est plateformes génèrent régulièrement des données à l’échelle du pétaoctet (106GO) de à grande échelle, ceci représente un volume très important de données et un obstacle dans l’analyse des données.
L'analyse des données génomique est un processus de traitement des données a étapes multiples, et l'importance du volume des données nécessite le développement d'une nouvelle génération de systèmes de gestion de flux de travail. Nous avons développé COSMOS ; un système de gestion de flux de travail conçu en considérant les spécificités des données génomiques. COSMOS offre des niveaux élevés de parallélisation, facile à opérer, et compatible avec le cloud. Pour créer une référence du temps de fonctionnement et une référence de qualité pour l'analyse des données génomiques, nous avons créé GenomeKey, un flux de travail de COSMOS pour analyser les données génomiques basé sur “GATK best practices”. Nous avons effectué une analyse comparative et systématique des temps de calculs, le coût et de la qualité des mutations génétiques identifiées par le flux de travail. Ceci offre une vision claire pour rendre ce type d’analyse génomique une routine, avec un coûts et un temps de fonctionnement cliniquement acceptable.
La génération de données à haut débit sur les plateformes de séquençage nouvelle génération, et le traitement abordable et rapide des données qu’offre GenomeKey / COSMOS fonctionnant dans le cloud ouvre la porte à une nouvelle ère de découverte génomique, et rend possible la pratiques de la génomique médicale. Ainsi, nous explorons la faisabilité d'un projet de médecine de précision au Maroc, en évaluant les défis et les opportunités qu’offre le pays.
LIST OF ABBREVIATIONS
AWS: Amazon Web Services
BAM: Binary Alignment/Mapping
BQSR: Base Quality Score Recalibration
BWA: Burrows-Wheeler Aligner
Ch: Chromosome
CPU: Central Processing Unit
DNA: Deoxyribonucleic acid
Ex: Exome
GATK: Genome Analysis Tool kit
Ge: Genome
hg18/19: Human Genome build 18/19
Indel: Insertion/deletion
MAQ: Mapping and Assembly with Qualities
NGS: Next-generation Sequencing
RG: Read group
S3: Simple Storage Service
SNP: Single Nucleotide Polymorphism
VCF: Variant Call Format
LIST OF FIGURES
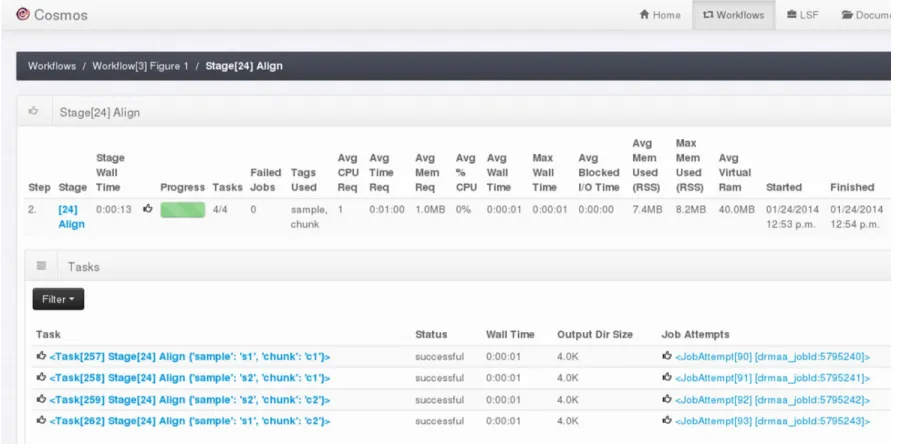
Figure 2.1 Human male karyotype ... 12 Figure 3.1 (a) Tools are defined in COSMOS by specifying input and output types, not files, and a cmd() function returning a string to be executed in a shell. cpu_req and other parameters may be inspected by a programmer-defined Python function to set DRM parameters or redirect jobs to queues (b) Workflows are defined using map-reduce primitives: sequence_, map_ (execute the align tool from (a) on each ‘chunk’ in parallel) and reduce_ (group the aligned outputs by sample tag) (c) Directed acyclic graph of jobs generated by the workflow in (b) to be executed via the DRM for four input FASTQ files (with sample tags s1 and s2, and chunk tags of 1 and 2). ... 23 Figure S3.3. Screenshot showing the completed four stages in the workflow from Figure 1
along with runtime statistics. Note the number of tasks/job for each stage corresponds to the number of jobs in Figure 1(c). ... 27
Figure S3.5. Sample in-progress COSMOS workflow of next-generation sequencing (NGS)
analysis of three exomes, from loading of BAM files, through alignment, variant calling via the Genome Analysis Toolkit (GATK 3.x) to variant quality scoring of output of VCF files. ... 29
Figure S3.6. (A) Python code from the Pegasus tutorial
(http://pegasus.isi.edu/wms/docs/latest/tutorial.php#idp10013792) implementing a
“diamond” DAG with (B) roughly equivalent COSMOS code. Pegasus requires the programmer to manage the file names of inputs and outputs, while COSMOS handles this automatically. ... 30
Figure S3.6. (A) Python code from the Pegasus tutorial
(http://pegasus.isi.edu/wms/docs/latest/tutorial.php#idp10013792) implementing a
“diamond”DAG with (B) roughly equivalent COSMOS code. Pegasus requires the programmer to manage the file names of inputs and outputs, while COSMOS handles this
Each arrow represents a stage of the workflow, and the level of parallelization for each stage is described in the Methods section under “Workflow”. b Deployment of the workflow on the Amazon Web Services Elastic Compute Cloud (EC2) infrastructure using the COSMOS workflow management engine. ... 42
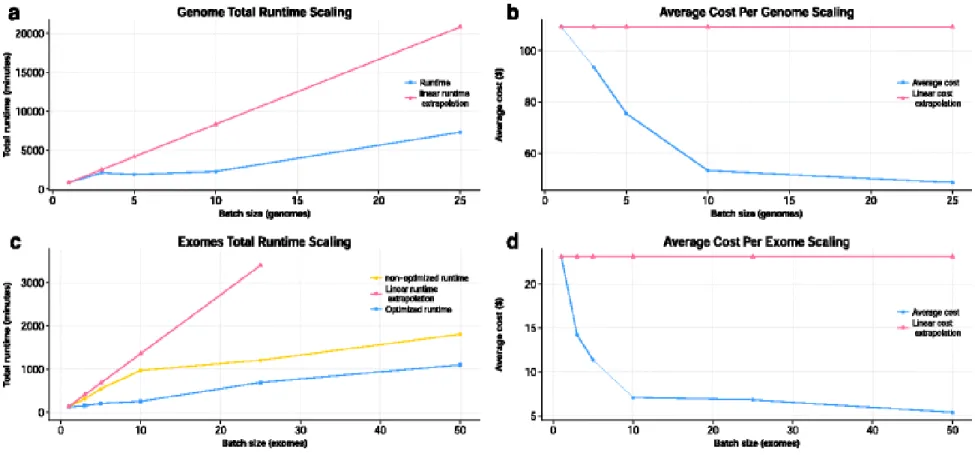
Figure 4.2 GenomeKey scalability. GenomeKey workflow efficiently scales with increasing
number of genomes. a Wall time and b cost as a function of number of genomes compared to a linear extrapolation single genome. GenomeKey workflow scales efficiently with increasing number of exomes compared on different GlusterFS configurations. The blue curve represents the 1, 3, 5 and 10 exomes runs performed on a cluster with one GlusterFS brick; the yellow curve represents the scalability on a cluster with four GlusterFS bricks. c Wall time and d cost as a function of exome and size for as compared to a linear extrapolation of a single exome ... 49
Figure 4.3 Cluster Resources Usage. Cluster resources are utilized more efficiently as batch
size increases. When the number of exomes increases from (a) 5 exomes to (b) 10 exomes, overall cluster CPU usage (shown as the brown “Total” line) is higher across the entire runtime. Percent CPU usage for each job across the entire 20-node was summed within 5-min “wall time” windows and then scaled by the total number of cores (20 nodes × 32 cores/node = 1920 cores) to quantify the overall system utilization. CPU usage for jobs not fully contained within each 5 min’ window was pro-rated according to how much they overlapped. The contribution of each stage to the entire total (brown line) as a function of time further illustrates the parallelization. ... 51
Figure S4.1. Exomes total runtime scaling. Replicated exomes runs, shows highly
reproducible runtimes. Error bars represent variance. ... 56
Figure S4.2. Automation script. Organogram of the automation script that downloads the
data, run the pipeline, save all the steps timestamps and backup the data. ... 57
Figure 5.1 Number of publications in medical genomics in Morocco since the launch of the
HGP. ... 72
Figure 5.2. Map of Morocco showing the major University Hospitals, and the regional
LIST OF TABLES
Table S3.1. Feature comparison between COSMOS and other workflow tools discussed in
main text with respect to use in next-generation sequencing analysis ... 32
Table 4.1 GlusterFS configurations used to increase shared disk ... 43
Table 4.2 Comparison of variant calls results ... 48
Table S4.1 Input datasets per run ... 58
Table S4.2. FastQC quality control table. ... 61
Table S4.3. Comparison with existing implementations of the GATK best practices, features. ... 62
Table S4.4. Runs summary with detailed cluster configuration, mean size, coverage, splitting strategy (chr: chromosome, RG: read-group), total runtime, total cost, average runtime and average cost. ... 63
Table S4.5 Comparison with previous benchmarks for specific 1000 g exomes/genomes. .. 65
Table S5.1 PubMed search terms used to collect the number of publications in the filed of genomics in Morocco. The search term include also the publications conducted in collaboration with foreign institutions, or completed partially at foreign laboratories. ... 78
TABLE OF CONTENT
COPYRIGHTS ... i DECLARATION ... ii DEDICATION ... iii ABSTRACT ... vi RESUME... viiLIST OF ABBREVIATIONS ... viii
LIST OF FIGURES ... ix
LIST OF TABLES ... xi
TABLE OF CONTENT... xii
FOREWORD ... 1
CHAPTER 1: GENERAL INTRODUCTION ... 2
1.1 CHALLENGE ... 4
1.2 RESEARCH AIMS & OBJECTIVES ... 5
1.3 ORGANIZATION AND CONTRIBUTIONS ... 5
CHAPTER 2: HUMAN GENOME AND BIOINFORMATICS ... 9
2.1. HUMAN GENOME ... 10
2.2 SEQUENCING ... 15
2.3 BIOINFORMATICS ... 17
2.4 CLOUD-COMPUTING ... 18
CHAPTER 3 : MASSIVELY PARALLEL WORKFLOW MANAGEMENT SYSTEM .. 19
Souilmi, Yassine, Alex K Lancaster, Jae-Yoon Jung, Ettore Rizzo, Jared B Hawkins, Ryan Powles, Saaid Amzazi, Hassan Ghazal, Peter J Tonellato, and Dennis P Wall. 2015. “Scalable and Cost-Effective NGS Genotyping in the Cloud.” BMC Medical Genomics 8 (1): 64. doi:10.1186/s12920-015-0134-9.
CHAPTER 4: SCALABLE AND COST-EFFECTIVE NGS GENOTYPING IN THE
CLOUD ... 33
Souilmi, Yassine, Alex K Lancaster, Jae-Yoon Jung, Ettore Rizzo, Jared B Hawkins, Ryan Powles, Saaid Amzazi, Hassan Ghazal, Peter J Tonellato, and Dennis P Wall. 2015. “Scalable and Cost-Effective NGS Genotyping in the Cloud.” BMC Medical Genomics 8 (1): 64. doi:10.1186/s12920-015-0134-9. Souilmi, Yassine, Alex K Lancaster, Jae-Yoon Jung, Ettore Rizzo, Jared B Hawkins, Ryan Powles, Saaid Amzazi, Hassan Ghazal, Peter J Tonellato, and Dennis P Wall. 2015. “Scalable and Cost-Effective NGS Genotyping in the Cloud.” BMC Medical Genomics 8 (1): 64. doi:10.1186/s12920-015-0134-9. CHAPTER 5: PRECISION MEDICINE IN MOROCCO ... 66
CONCLUSION ... 79
REFERENCES ... 82
ANNEXES... 91
ANNEXE 1: COSMOS DOCUMENTATION ... 92
FOREWORD
The current work was accomplished at the Laboratory of Biochemistry and Immunology at the School of Sciences, Mohamed V University in Rabat.
An important part of this work was performed at the Laboratory for Personalized Medicine, Department for Biomedical Informatics, Harvard Medical School, Harvard University, Boston, MA. In addition, this research project benefited from the collaboration, and the mentorship of the Wall Laboratory, Department of Pediatrics and Psychiatry (by courtesy), Division of Systems Medicine & Program in Biomedical Informatics, Stanford University, Stanford, CA.
I would like to express my gratitude to Professor Saaïd Amzazi for his valuable support and supervision and for accepting to preside the jury of my thesis defense. I also extend my gratitude Professor Peter J. Tonellato for hosting me in his laboratory and providing me with the technical knowledge and tools to drive this project to success.
I am thankful to Professor Youssef Bakri (Professeur d’Enseignement Supérieur) and Professor Badaoui (Professeur Assistant), School of Sciences, Mohamed V University in Rabat, as well as Professor Hassan Ghazal, Polydisciplinary Faculty, for his scientific and methodological help and advise, as well as for accepting to evaluate my research work and provide their valuable feedback. I also thank Professor Ali Idri (Professeur d’Enseignement Supérieur), National School of Informatics and Systems Analysis, Mohamed V University in Rabat for accepting to be part of the jury of my defense, and provide this work with his valuable comments and constructive critiques.
CHAPTER 1:
Medicine is a slow evolving science due to its nature, and strong ethical barriers that hold medical doctors and researchers from experimenting with unorthodox methods. The developments of advanced diagnosis tools (radiology, molecular diagnosis), led to the discovery of more complex diseases, such as neurodegenerative diseases and cancers. However, classic medical approaches could not fully provide satisfying understanding nor efficiently cure these diseases.
It was the work of the Gregor Mendel in the 19th century that started a series of scientific discoveries that will launch a new era of Medicine. Mendel’s work combined with the discovery of the nuclein by Friedrich Miescher in 1869, and the groundbreaking discoveries of the functional role of the Deoxyribonucleic acid (DNA) molecule by Nikolaj Koltzoff (N. K. Koltzoff 1927; N. Koltzoff 1934). However, it was the discovery of the three-dimensional structure of the DNA molecule by the chemistry Nobel prizes Watson and Crick in the mid-50s that started a molecular biology revolution.
The discovery of the DNA molecule three-dimensional structure, followed by the discovery of the twenty amino acids and their sixty corresponding messenger RNA codons that started the scientific community to find efficient methods to read the sequence of amino acids encoded that form the DNA string, and discover how it encodes for genes and all the different molecules that govern a living organism. The first technologic breakthrough in this direction was the development of the “rapid DNA sequencing” (AKA: Sanger sequencing). Sequencing technology reads the series of bases (sequence) forming a given DNA strand fragment. Frederick Sanger’s is of high importance, and mark the beginning of an era of genomic discovery, and he received a chemistry Nobel Prize for his work in 1980.
However, the effort to fully decipher the human genome started with the Human Genome Project (HGP) in 1990, an international project that led by the U.S. National Center for Human Genome Research (now the National Human Genome Research Institute; NHGRI). Sequencing the human genome took 13-year and $100 million. The project shed the light on a new perspective of the human health and disease understanding (E. D. Green et al. 2015). The end of the HGP was marked with excitement and disappointments, as the project brought far more questions than answers on the human genome.
world (Genomes Project et al. 2010), the HapMap project that generated genome-wide data that fully examine less common alleles in populations with a wide range of ancestry {TheInterna-tionalHapMapConsortium:2010en}, and The Cancer Genome Atlas (TCGA) project to accelerate the understanding of the molecular basis of cancer (Tomczak et al. 2015).
1.1 CHALLENGE
The fast discovery rate following the HGP, and the better understanding the aforementioned projects brought accelerated the development of high-throughput sequencing technology, commonly referred to as next-generation sequencing (NGS). NGS technology is unique as brought the sequencing cost down in a faster pace than Moor’s law predicts. This rapid decline in brought the cost of sequencing of patient’s genomes within reimbursable cost ($1,000) (Desai & Jere 2012; Sboner et al. 2011). As a result, NGS popularity both increased for research and clinical application, thus, producing a data creating a computational bottleneck in term of data storage and data analysis (Schatz & Langmead 2013). To take on the computational challenges, it required computational biologist and bioinformaticians to use novel computation techniques, such as MapReduce programming (Gafni et al. 2014), advanced parallelization, and harnessing the power of cloud computing (Marx 2013; Xing et al. 2015; Noor et al. 2015).
Next-generation sequencing and the computational advances in data analysis for genomic data such as the ones presented in this work leads to the development of a more modern and novel approach in medicine to take on complex diseases called “Precision Medicine.” Precision medicine takes environmental, lifestyle and genetic variations into consideration for treatment discovery and prevention strategies design (Kass-Hout & Litwack 2015). Aware of its importance in providing quality healthcare, the US President Barack Obama in 2015 launched the U.S. Precision Medicine Initiative, to generate the scientific evidence needed to move the concept of precision medicine into clinical practices (Collins & Varmus 2015; Kass-Hout & Litwack 2015). Morocco represents a leading nation in Africa in healthcare and medicine. The country’s healthcare system is based on a single payer model, and the most complex cases are redirected to the regional university hospitals, which provide a special and unique opportunity to develop an
1.2 RESEARCH AIMS & OBJECTIVES
The objective of this research work is to facilitate the development and adoption of modern medical genomic practices in Morocco to improve healthcare response and outcomes. However, to accomplish the results we seek our response was divided into three major.
From a bioinformatics perspective the most important area that needs solving is the genomic data analysis bottlenecks, for which we proposed a two-fold solution that represents the two first aims of this thesis:
Aim 1: Solve the genomic data analysis complexity by developing a novel workflow
management system with advanced capabilities, higher computational efficiency, and features rich while using modern computational technics.
Aim 2: Develop an accurate and cost-effective NGS data analysis pipeline with cloud
capabilities. A pipeline within clinical turnaround time, and reimbursable cost point by leveraging the advanced workflow management system capabilities developed in Aim 1, and the computational power, availability and cost effectiveness of cloud computing. Also, providing accuracy and timings benchmarking reference.
Aim 3: To take advantage of recent medical genomic advances, NGS technologies reaching
affordable pricing, and the computational efficiency of the tools developed in Aim 1 and 2 in clinical applications in Morocco, we assess the challenges and opportunities facing the adoption of the Precision Medicine in Morocco, and offer actionable points for the development of effective pilot projects.
1.3 ORGANIZATION AND CONTRIBUTIONS
In Chapter 2 I introduce the background concept that is use thought this entire thesis work; from the biomedical concepts from the molecule of DNA, Human Genome, and the sequencing technologies developed to identify the series of nucleotides that compose it. The bioinformatic tools used to analyze genomic data, as well as the informatics technics used for the
In Chapter 3, I present the first building block of this research, in the form of the first step to solving the data analysis computational challenges, through the development of an efficient, workflow management system for massively parallelized workflows. While multiple workflow management systems were either adapted for bioinformatics applications or designed for it, most were developed for research purposes or user friendliness and were not optimized for large-scale clinical applications. Thus, we developed COSMOS, a python based library for highly parallelized workflow definition, monitoring, and management. COSMOS offers functionalities unavailable in other solutions. For instance, COSMOS uses the MapReduce programming paradigm to implement, modern parallelization or its workflows while offering at the same time an easy to use definition language of the different workflow stages. Also, COSMOS uses an advanced tagging system that reads or generates ‘tags’ from the input data that will be used to compute the number of tasks to be generated for each stage of the defined workflow, as well as to create dynamically a tasks decencies direct acyclic graph (DAG). COSMOS’ tagging system is more advanced than other alternatives (Figure S3.1) since it uses upon definition of the workflow an input and output, not specific files, which allow the workflow to dynamically read the input or inputs and generate the tags, which allows the system to create the DAG.
COSMOS also offers a powerful interaction with the most common distributed resource management systems (DRM), which allows easy jobs submission, tracking, and resubmission of each job. The inputs/outputs, DAG, generated jobs data, and metadata is stored in a MySQL (or PostgreSQL) database, which data is visualized through a web interface. The web interface also offers the ability to monitor in real-time the progress of the workflow and each of its jobs, as well as all the needed information for efficient debugging (Figures S3.2-S3.5).
In Chapter 4, we present a scalable and cost-effective whole genome and whole exome data analysis pipeline, the pipeline when combined with the powerful features of COSMOS and taking advantage of the scalability, and availability of cloud computing resources offers a very rapid data analysis, that is accurate and cost-effective. Thus, solving the large-scale data analysis
To tackle the computational challenge, we used COSMOS powerful workflow management system capabilities presented in chapter 3, and we have constructed an NGS analysis pipeline implementing the Genome Analysis Toolkit - GATK v3.1 - best practice protocol (DePristo et al. 2011; Van der Auwera et al. 2013), a set of tools and best practices for whole genome and exome data analysis considered and the golden standard in the industry. The pipeline named Genomekey, compute a thorough data analysis, which includes quality control steps, alignment of the sequence reads or realignment if needed, variant calling steps and an unprecedented level of annotation using a custom extension of ANNOVAR that we have built.
With GenomeKey pushing the parallelization to the limits without compromising the quality of the analysis, and COSMOS taking advantage the resources of a high-performance on-demand compute cluster, it becomes possible to process genomic data at the petabyte scale, generated nowadays, routinely at NGS core facilities. Also, our implementation drops the cost of whole genome analysis under the $100 barrier, placing the analysis within reimbursable insurance cost point, as well as placing the turnaround time within clinically acceptable limits. Such result allows for an easy clinical setting adoption and solves one of the most challenging obstacles facing the development of efficient Precision Medicine programs.
While in Chapters 3 and 4 I take on the computational data analysis bottleneck limiting the adoption of clinical genomics and the launch of efficient Precision Medicine, in Chapter 5 I present an assessment of the challenges facing the development of a Precision Medicine strategy as well as the opportunities that Morocco, as a country can offer, and what will be the eventual benefits from such advanced programs.
While we start with a limited data, it is clear that the challenges could be summarized in three major blocks: 1) lack of trained scientists and medical doctors with mostly poor infrastructures; 2) economic limitations, and 3) poor or absent policies, providing clear regulations for patients data privacy, intellectual propriety and other medical trials and applications. Whereas 3 is a limitation, it is also an opportunity for the research community, private actors, and policymakers to propose new policies that learn from other countries mistakes, and take advantage from their experience.
Morocco is also developing a modern medical infrastructure around seven major public university hospitals, covering each one of the geographic regions; such system offers the advantage of the resources sharing, which optimizes the equipment and training investment. Furthermore, these regional centers important ‘hubs’ for rare diseases and complex disease care, and thus, solving the patients data collection for later processing.
The tight relations between different Moroccan, European and American universities as well as being part of large consortiums such as the Health Humans and Heredity in Africa Project (H3Africa) offers a unique opportunity for capacity building in the major areas of a modern healthcare system. Which will not only further the research contributions of the Moroccan university but also empowers researchers and technicians, improve significantly the healthcare outcomes and investments and improves the economic investment in the health sciences field.
CHAPTER 2:
HUMAN GENOME AND
BIOINFORMATICS
In the following chapter, we cover the required biologic and computational concepts needed to fully understand the presented work.
2.1. HUMAN GENOME
The human genome is the ensemble of the human organism’s set of genetic code necessary to build, develop and grow a human organism (The ENCODE Project Consortium 2012). The same genetic code is available to each cell of the human body, with the exception of the gametes. The set of genetic code of the human body is stored in the DNA molecule principally stored in the nucleus of the cell, with a small molecule stored in the mitochondria. The DNA is a macromolecule composed of two complementary strands, anti-parallel, running in opposite directions to each other. The two molecules are called strands (The ENCODE Project Consortium 2012). The two strands are coiled around each other forming the double helix form discovered by Watson and Crick. DNA is where all the hereditary biologic information of an organism is stored and transmitted from one generation of cells to the following.
In 1869, Friedrich Miescher discovered the “nuclein”, a new molecule isolated from white cells extracts collected from bandages. The molecule has a unique chemical composition (Anon 2008). While Miescher studied extensively the molecule, he didn’t realize its importance in regard to inherited traits. It was Nikolaj Koltzoff, in 1927, that predicted that inherited traits are passed from a generation to the next through a "giant hereditary molecule" made up of "two mirror strands that would replicate in a semi-conservative fashion using each strand as a template" (N. Koltzoff 1934). In 1934, Koltzoff contended that the proteins that contain a cell's genetic information replicate.
"I think that the size of the chromosomes in the salivary glands [of Drosophila] is determined through the multiplication of genonemes. By this term I designate the axial thread of the chromosome, in which the geneticists locate the linear combination of genes; … In the normal
The nuclein as described by Miescher, or the double stranded “giant hereditary molecule” that Koltzoff described is the Deoxyribonucleic acid, mostly known as DNA. The DNA molecule was extensively studied by chemists, and in 1952 Rosalind Franklin used X-ray crystallography technique to capture two sets of high-resolution photos of crystallized DNA fibers and determine the dimensions of DNA strands, the structure seemed helical. Here work was published in the same issue of Nature as Watson and Crick’s paper introducing their 3-D model of DNA structure in 1953. However, Watson and Crick’s work was more extensive and their model represented the DNA molecule as a double helix, with sugars and phosphates forming the outer strands of the helix and the bases pointing into the center. Hydrogen bonds connect the bases, pairing A–T and C–G; and the two strands of the helix are parallel but oriented in opposite directions. Their 1953 paper notes that the model “immediately suggests a possible copying mechanism for the genetic material”, thus, confirming Koltzoff finding in 1927.
About a decade later, Marshall Nirenberg and his team at the U.S. National Institute of Health (NIH) identified the process of transcription, and the role of messenger Ribonucleic acid (RNA). They also successfully identified all the 20 amino acids and their 60 mRNA codons.
2.1.1. Chromosomes
The DNA molecule within each cell’s nucleus is organized in mostly ‘X’ shaped structure called chromosomes. Within each chromosome, the DNA is compacted and organized around histones and other chromatin proteins. Such organization plays and important role in the DNA-protein interaction, for instance it contributes to the DNA transcription process.
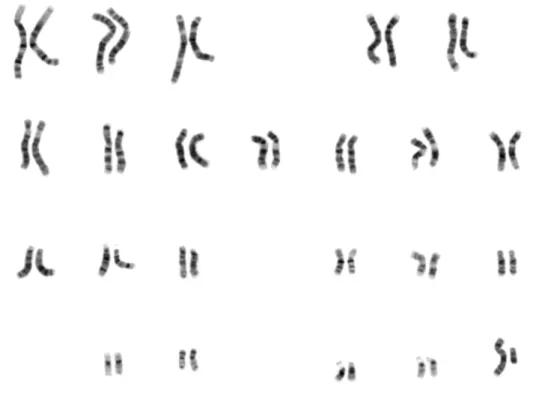
Figure 2.1 Human male karyotype
Human genome is a diploid and is made of a total of 46 chromosomes, 23 of them are provided by the female gamete and 23 by the male gametes. Human chromosomes are classified in two groups, the autosomal chromosomes (1-22) and allosomal (X/Y) or sex chromosomes. The allosomal chromosomes combination in humans determine sex of the offspring, the females carries two X chromosomes one from the father and the second from the mother, and males carries one copy of the X chromosome inherited from the mother and a copy of the Y chromosome from the father.
2.1.2. Gene
A gene is a segment of the DNA molecule, also named locus that encodes a functional RNA or protein. Genes are considered the molecular unit of heredity (Slack 2014). The genes are transmitted from a one-generation to the following (offspring) when the DNA molecule is replicated. It is the basis of inheritance genetic traits, however, this classic understanding “gene = protein = function” called the central dogma of molecular biology was demonstrated wrong. Most biological traits are under the influence of polygenes as well as the gene–environment interactions.
2.1.3. Variants
In the process of replication of DNA, genes can acquire mutations in their sequence of basis. The mutations lead to a different version of the gene called alleles. An allele encodes for different versions of the protein, resulting in a different phenotype trait. Genetic variants are the engine of significant biological phenomena such as the evolution, cancers and immunologic system development. Variants can have no effect, so-called silent mutations, or have a dramatic effect, such as silencing a gene, or damaging the produced protein (cancer). There are two major types of variations discussed bellow.
2.1.3.1 Small-scale sequence variation 2.1.3.1.1. Indels
Small insertions and deletions, and are commonly named indels (Gonzalez et al. 2007). Indels results in a slight change in the total count of nucleotides in a given sequence. Indels can have a dramatic impact on a coding regions of the genome (gene) the number of inserted/deleted nucleotides is not a multiple of 3; in this case the result is a frameshift mutation leading to a totally different protein or silencing the gene, as a result of the reading of a premature stop codon. A very known example of frameshift mutation in the BLM gene caused by indels, is Dwarfism or the Bloom syndrome characterized by a short stature but more importantly by an immune system deficiency and a higher susceptibility to leukemia and lymphoma (Wechsler et al. 2011). Indels represents 16% and 25% of all sequence polymorphisms in humans (Mills 2006).
2.1.3.1.2. Single Nucleotide Polymorphisms
Single Nucleotide Polymorphism (SNPs), is a single nucleotide substitution (variation), also referred to as point mutation or Single Nucleotide Variation (SNV). As example in a specific position in the human genome, an A appears in the majority of the population but for a minority a T occupy that position. However, to consider a single nucleotide substitution a SNP it should be occurring in at least in over 1% of the studied population (The ENCODE Project Consortium 2012). Human SNPs are catalogued and referenced in the U.S. National Center for Biotechnology Information (NCBI) database dbSNP. A reference number called an rsID identifies each known and confirmed SNP.
SNPs are widely used as markers for a wide large number of diseases. For instance a single SNPs could be the cause of a high risk of disease, e.g. rs429358 SNP (Anon n.d.) in the gene APOE on Chromosome 19 (NM_000041.3(APOE):c.388T>C (p.Cys130Arg)) is associated with a higher risk to develop Alzheimer disease (Wolf et al. 2013). SNPs also in cases determines the way a patient responds to a given treatment, e.g. patients with specific SNPs alleles in the genes VKORC1 and CYP2C9 genes, determines the organisms response to the Warfarin, an anticoagulation drug (Johnson et al. 2011), thus, impacting the dosing.
2.1.3.2 Large-scale Structural variation 2.1.3.2.1 Copy-number variations
Copy-number variations (CNVs) are deletion or duplications of sections of the DNA ranging between 1 Kilobase (1,000 bases) to multiple Megabases, and they account for approximately 13% of human mutations (Stankiewicz & Lupski 2010). For a given gene made of four sections A-B-B-C-C-C-D-D, we might have copy number loss of the section B for example and the sequence will be A-B-C-C-C-D-D, or copy number gain in the section D and have for example A-B-B-C-C-C-D-D-D-D. CNVs are the result of crossover events responsible that occur during the production of sperm and eggs, resulting in offspring carrying additional, or lost genetic information that were present in either of their parents’ genomes. CNVs are strongly associated
2.1.3.2.2. Structural Variants
Structural variation (SVs) are alterations in the structure of the chromosomes themselves, while the classic definition includes CNVs, in most cases CNVs and SVs are reported separately and are detected by different tools. In addition to CNVs, SVs consist of large (1 Kilobase-3 Megabases) deletions, duplications, copy-number variants, insertions, inversions and translocations (Feuk et al. 2006). The molecular and phenotypic effects of SVs are not very well studied, however, recently SVs were associated with diseases such as in the triple-negative breast cancer (TNBC) (Craig et al. 2013).
2.2 SEQUENCING
Sequencing the DNA molecule is the chemical process of determining the nucleotides sequence of a given fragment. Sequencing became fundamental in the study of new organism, understanding the molecular processes underlying a studied disease, and finding new treatments for a pandemic, e.g. the latest Zika virus and Ebola virus outbreaks used heavily sequencing to study the different strands and to develop vaccines. We can identify two major types of sequencing in use today, Sanger sequencing and high-throughput sequencing.
2.2.1 Sanger
The Frederick Sanger sequencing method developed in the mid-70s (Sanger & Coulson 1975) is one of the most widely used and validated method of sequencing. The method is a golden standard and still in use to validate the results of alternative sequencing methods. The technique uses a single strand of DNA of about 500 bases in length, to this strand a DNA primer, a short and marked small DNA strand of known sequence, this primer will hybridize with the complementary matching short segment of the studied DNA strand and serve as a known start point of the sequence, then the DNA polymerase will be used to replicate the remaining of the strand using the deoxynucleosidetriphosphates (dNTPs), and di-deoxynucleosidetriphosphates (ddNTPs) nucleotides lacking the 3’-OH group which impeaches phosphodiester bond between two nucleotides, thus, stopping the DNA extension by the DNA polymerase and determining the ‘strand elongation’. Modern Sanger sequencers uses florescent ddNTPs to automatically detect the DNA strand sequence.
2.2.3 Next-Generation Sequencing
The increasing demand for high-throughput sequencing techniques, pushed towards the development of the Next-generation sequencing (NGS) platforms. NGS platforms enabled large scale genomic project such as the 1000 Genome Project (Consortium 2015), the HapMap Project (The International HapMap Consortium et al. 2010), and The Cancer Genome Atlas (TCGA) (Network 2008). These project shed revealed unprecedented knowledge about the human genome and furthered our understanding of inheritance of multiple diseases such as cancers, neurodegenerative diseases and hereditary traits such as type 2 Diabetes to cite a few.
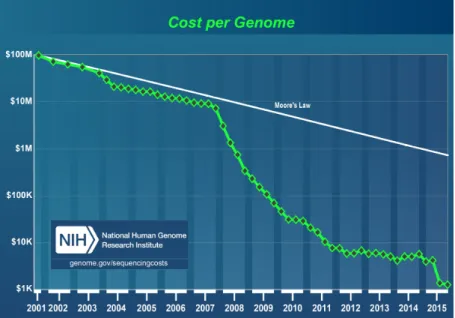
In addition, the fast development and adoption of NGS platform resulted in a dramatic drop in the cost of sequencing. A decrease that surpassed the benchmarking models that is Moore’s law. Nowadays, the sequencing of a whole human genome at clinically acceptable quality and will cost $1,000, placing sequencing within reimbursable price point (Figure 2.2).
This rapid cost decrease, and the importance of genomic information in the understanding of multiple diseases both for research purposes and recent medical applications, created a data deluge (Schatz & Langmead 2013). Thus, creating a data analysis and data storage bottlenecks, and advanced bioinformatics and computational technics are needed to process NGS data.
2.3 BIOINFORMATICS
Bioinformatics is a new field in the biomedical sciences; its emersion is traced back to the period after the development of the first sequencing technologies and the need for automated means to align sequences. Bioinformatics uses computer science approach to manage and analyze biological and genetic data. After the HGP, bioinformatics expanded beyond sequence alignment, to include protein modeling, genomic databases, integrative data analysis, genome-wide association studies and other techniques used to store, sort and data mine genomic and biological information.
In contrast with other science disciplines, bioinformatics benefited from the public aspect of the HGP and most of the golden standard widely used bioinformatics tools are open-source, or free for research and academic purposes. For instance the most used genomic short reads aligner BWA (Borrows-Wheeler Alignment tool) (Li & Durbin 2009; Li & Durbin 2010), and the Broad Institute’s golden standard variant-calling genome analysis toolkit (GATK) (Van der Auwera et al. 2013). In this work, we use a series of open-source or free for academic and research purposes bioinformatics data analysis tools, these tools are details and referenced in the methods section of the chapter 4 of this thesis.
2.4 CLOUD-COMPUTING
The genomic data deluge generated by the democratization of NGS platforms is a big data issue (a single whole genome is >150GB). While many universities own a high-performance computing platform, low and middle-income countries universities (such as morocco) have access to only nationally shared computational resources, medical centers also lack adapted computational resources to analyze on-demand patient’s genomic data. However, on one hand the wealth of data generated by these platforms need important computational resources, and on the other hand the sensitivity of the data (patients data) and the privacy requirements makes the use of a shared infrastructure impossible in many cases.
Cloud computing is a paradigm-shift where computing networking, CPU cycles, storage and memory are provided as utility (L. Wang et al. 2010; Cusumano 2010; Wilhite 2012). It provides adapted computational power and storage on-demand, which corresponds to the bioinformatics and medical genomic applications where the computational load is discrete. Bioinformaticians took very early interest in cloud computing and it was adopted as early as 2010 for multiple research and medical projects (Liu et al. 2014; Karthikeyan & Sukanesh 2012; Fusaro et al. 2011; Souilmi et al. 2015).
CHAPTER 3 :
MASSIVELY PARALLEL
WORKFLOW MANAGEMENT
SYSTEM
3.1 GENERAL INTRODUCTION
Next-generation sequencing platforms brought high-throughput genomic sequencing to the clinical field. However, these platforms generate massive data on a daily basis, introducing a data analysis bottleneck. In addition, the NGS data analysis needs several data conversion, cleaning, processing steps. Multiple solutions were proposed in the past to take on the workflow analysis, with solutions focusing on high performance but complicated to use such as Pegasus (Deelman et al. 2005), or user friendly solutions but sacrificing the high-throughput and high levels of parallelization such as Galaxy (Goecks et al 2010ea).
Herein, we present COSMOS a workflow management system written in Python, and adopting the MapReduce programming paradigm. COSMOS focuses on an easy and reusable workflow definition, as well as a powerful tagging system that enables dynamically very high-levels of parallelization. In addition, COSMOS is cloud-enabled and error tolerant, offering the user the ability to take advantage of the very low cost of the transient computation cost model offered by certain cloud-computing providers (e.g. spot instances on AWS
3.2 PUBLICATION TECHNICAL PRESENTATION
Title: COSMOS: Python Library for Massively Parallel Workflows Journal: Oxford Bioinformatics, Vol. 30 no. 20 2014, pages 2956–2958 DOI: 10.1093/bioinformatics/btu385
Authors: Erik Gafni1, Lovelace J. Luquette1, Alex K. Lancaster1,2, Jared B. Hawkins1, Jae-Yoon Jung1, Yassine Souilmi1,3, Dennis P. Wall1,2, and Peter J. Tonellato1,2
Authors Affiliations:
1
Center for Biomedical Informatics, Harvard Medical School, 10 Shattuck Street, Boston, MA 02115.
2
Available Online: June 30, 2014
License: Published by Oxford University Press. This is an Open Access article distributed under
the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/ by-nc/3.0/), which permits non-commercial re-use, distribution, and reproduction in any medium, provided the original work is properly cited. For commercial re-use, please contact [email protected]
Availability and implementation: Source code is available for non-commercial purposes, in
addition to documentation. Both are provided at http://lpm.hms.harvard.edu/.
Funding: This work was supported by the National Institutes of Health [1R01MH090611-01A1
to D.P.W]; a Fulbright Fellowship [to Y.S.].
Conflict of interest: LJ Luquette is also an employee with Claritas Genomics Inc., a licensee of
a previous version of COSMOS.
3.3 ABSTRACT
Summary: Efficient workflows to shepherd clinically-generated genomic data through
the multiple stages of a next-generation sequencing (NGS) pipeline is of critical importance in translational biomedical science. Here we present COSMOS, a Python library for workflow management that allows formal description of pipelines and partitioning of jobs. In addition, it includes a user-interface for tracking the progress of jobs, abstraction of the queuing system and fine-grained control over the workflow. Workflows can be created on traditional computing clusters as well as cloud-based services.
Availability and implementation: Source code is available for non-commercial
purposes, in addition to documentation. Both are provided at http://lpm.hms.harvard.edu/. Contact erik_ [email protected], [email protected],
3.4 INTRODUCTION
The growing deluge of data from next-generation sequencers leads to analyses lasting hundreds or thousands of compute hours per specimen, requiring massive computing clusters or cloud infrastructure. Existing computational tools like Pegasus (Deelman et al. 2005) and more recent efforts like Galaxy (Goecks et al. 2010) and Bpipe (Sadedin et al. 2012) allow the creation and execution of complex workflows. However, few projects have succeeded in describing complicated workflows in a simple but powerful language that generalizes to thou-sands of input files; fewer still are able to deploy workflows onto distributed resource management systems (DRMs) such as LSF or Sun Grid Engine that stitch together clusters of thou-sands of compute cores. Here we describe COSMOS, a Python library developed to address these and other needs.
3.5 FEATURES AND METHODS
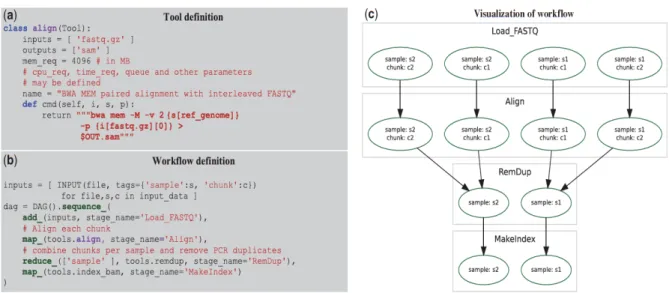
An essential challenge for a workflow definition language is to separate the definition of tools (which represent individual analyses) from the definition of the dependencies between them. Several workflow libraries require each tool to expect specifically named input files and produce similarly specific output files; however, in COSMOS, tool I/O is instead con-trolled by specifying file types. For example, the BWA alignment tool (Fig. 1a) can expect FASTQ-typed inputs and produce a SAM-typed output, but does not depend on any specific file names or locations. Additionally, tool definitions do not re-quire knowledge of the controlling DRM.
Figure 3.1 (a) Tools are defined in COSMOS by specifying input and output types, not files, and a cmd() function
returning a string to be executed in a shell. cpu_req and other parameters may be inspected by a programmer-defined Python function to set DRM parameters or redirect jobs to queues (b) Workflows are defined using map-reduce primitives: sequence_, map_ (execute the align tool from (a) on each ‘chunk’ in parallel) and reduce_ (group the aligned outputs by sample tag) (c) Directed acyclic graph of jobs generated by the workflow in (b) to be executed via the DRM for four input FASTQ files (with sample tags s1 and s2, and chunk tags of 1 and 2).
Once tools have been defined, their dependencies can be formalized via a COSMOS workflow, which is defined using Python functions that support the map-reduce paradigm (Dean and Ghemawat, 2004) (Fig. 3.1b). Sequential workflows are de-fined primarily by the sequence_ primitive, which runs tools in series. The apply_ primitive is provided to describe work-flows with potentially unrelated branching by executing tools in parallel. To facilitate map-reduce in large and branching workflows, COSMOS introduces a tagging system that associates a set of key-value tags (e.g., a sample ID, chunk ID, sequencer ID or other job parameter) with specific job instances. This tagging feature enables users to formalize reductions over existing tag sets or to split by creating new combinations of tags (Fig. S 3.1). To execute a workflow, COSMOS creates a directed acyclic graph (DAG) of tool dependencies at runtime (Fig. 3.1c) and automatically links the inputs and outputs be-tween tools by recognizing file extensions as types. All file paths generated by tool connections are managed by COSMOS, automatically assigning intermediate file names.
Another major challenge in workflow management is execution on large compute clusters, where transient errors are commonplace and must be handled gracefully. If errors cannot be automatically resolved, the framework should record exactly which jobs have failed and allow the restart of an analysis mid-way after error resolution. COSMOS uses the DRMAA library (Tröger et al. 2007) to manage job submission, status checking and error handling. DRMAA supports most DRM platforms, including Condor, although our efforts utilized LSF and Sun Grid Engine. Users may control DRM submission parameters by overriding a Python function that is called on every job control event. COSMOS’ internal data structures are stored in an SQL database using the Django framework (https://djangoproject.com) and is distributed with a web application for monitoring the state of both running and completed workflows, querying individual job states, visualizing DAGs and debugging failed jobs (Figs S3.2-S3.5).
Each COSMOS job is continuously monitored for resource usage, and a summary of these statistics and standard output and error streams are stored in the database. This allows users to fine tune estimated DRM parameters such as CPU and memory usage for more efficient cluster utilization. Pipeline restarts are also facilitated by the persistent database, as it records both success and failure using job exit codes.
3.6 COMPARISON AND DISCUSSION
Projects such as Galaxy and Taverna (Wolstencroft et al. 2013) are aimed at users without programming expertise and offer GUIs to create workflows, but come at the expense of power. For example, it is straightforward to describe task dependencies in Galaxy’s drag-and-drop workflow creator; however, to parallelize alignment by breaking the input FASTQ into several smaller chunks to be aligned independently, input stages must be manually created for each chunk or the workflow must be applied to each chunk by hand. In addition, the user must fix the number of input chunks a priori. COSMOS resolves this tedious process for the programmer by building its DAG at runtime.
Such limitations may not be a major concern for small-scale experiments where massive parallelization to reduce runtime is not critical; however, when regularly analyzing terabytes of raw data, the logistics of parallelization and job management play a central role. Snakemake (Köster and Rahmann, 2012) looks to the proven design of GNU make to describe DAGs for complicated workflows while the Ruffus project (Goodstadt, 2010) aims to create a DAG by providing a library of Python decorators. However, neither of these projects directly support integration with a DRM. The Pegasus system offers excellent integration with DRMs and even the assembly of several independent DRMs using the Globus software, however, the description of some simple workflows can require considerably more code than the equivalent COSMOS code (Fig. S6) and the DAG is not determined at runtime, so it cannot depend on the input. Bpipe offers an elegant syntax for defining the DAG, but does not include a GUI for monitoring and runtime statistics. Additionally, COSMOS’ persistent database and web-frontend allow rapid diagnosis of errors in data input or workflow execution (see Table S3.1 for a detailed feature comparison). COSMOS has been tested on the Ubuntu, Debian and Fedora Linux distributions. The only dependency is Python 2.6 or newer and the ability to install Python packages; we recommend a DRMAA-compatible DRM for intensive workloads.
Figure S3.2 Screenshot of the COSMOS web server displaying the current list of workflows. Here we depict just the workflow “Figure 1” corresponding
to the completed workflow in Figure 1 from the main
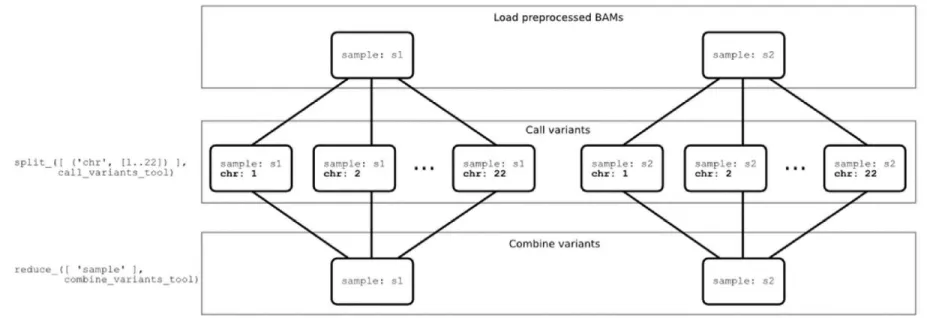
COSMOS introduces a tagging system that associates a set of key-value tags (e.g., a sample ID, chunk ID, sequencer ID or other job parameter) to each task in the stage. These tags provide a way to easily locate a particular task in the web interface, API and database, but more importantly are the parameters used by COSMOS’ formalized workflow description primitives. For example, if we wish to parallelize variant calling by processing each chromosome independently, we would issue a split_ over a new tag, say, chr with values 1…22, as shown below. The split_ call below will propagate all parent tags (sample) and create the new chr tag with values 1…22 and launch a job for each chr value. For each unique sample tag value v, the reduce_ call below will create a new job whose input is the set of output files from jobs with sample=v. Note that the split and reduce calls do not depend on the number of input samples.
Figure S3.3 Screenshot showing the completed four stages in the workflow from Figure 1 along with runtime statistics. Note the number of tasks/job for each
Figure S3.5 Sample in-progress COSMOS workflow of next-generation sequencing (NGS) analysis of three exomes, from loading of BAM files, through
Figure S3.6 (A) Python code from the Pegasus tutorial (http://pegasus.isi.edu/wms/docs/latest/tutorial.php#idp10013792) implementing a “diamond”DAG with (B) roughly equivalent COSMOS code. Pegasus requires the programmer to manage the file names of inputs and outputs, while COSMOS handles this automatically.
# Import the Python DAX library
os.sys.path.insert(0, "/usr/lib64/pegasus/python") from Pegasus.DAX3 import *
# The name of the DAX file is the first argument if len(sys.argv) != 2:
sys.stderr.write("Usage: %s DAXFILE\n" % (sys.argv[0])) sys.exit(1)
daxfile = sys.argv[1] # Create a abstract dag print "Creating ADAG..." diamond = ADAG("diamond") # Add a preprocess job print "Adding preprocess job..." preprocess = Job(name="preprocess") a = File("f.a") b1 = File("f.b1") b2 = File("f.b2") preprocess.addArguments("-i",a,"-o",b1,"-o",b2) preprocess.uses(a, link=Link.INPUT)
preprocess.uses(b1, link=Link.OUTPUT, transfer=False, register=False) preprocess.uses(b2, link=Link.OUTPUT, transfer=False, register=False) diamond.addJob(preprocess)
# Add left Findrange job print "Adding left Findrange job..." frl = Job(name="findrange") c1 = File("f.c1")
frl.addArguments("-i",b1,"-o",c1) frl.uses(b1, link=Link.INPUT)
frl.uses(c1, link=Link.OUTPUT, transfer=False, register=False) diamond.addJob(frl)
# Add right Findrange job print "Adding right Findrange job..." frr = Job(name="findrange") c2 = File("f.c2")
frr.addArguments("-i",b2,"-o",c2) frr.uses(b2, link=Link.INPUT)
frr.uses(c2, link=Link.OUTPUT, transfer=False, register=False) diamond.addJob(frr)
# Add Analyze job print "Adding Analyze job..." analyze = Job(name="analyze") d = File("f.d")
analyze.addArguments("-i",c1,"-i",c2,"-o",d) analyze.uses(c1, link=Link.INPUT) analyze.uses(c2, link=Link.INPUT)
analyze.uses(d, link=Link.OUTPUT, transfer=True, register=False) diamond.addJob(analyze)
# Add control-flow dependencies print "Adding control flow dependencies..."
diamond.addDependency(Dependency(parent=preprocess, child=frl)) diamond.addDependency(Dependency(parent=preprocess, child=frr)) diamond.addDependency(Dependency(parent=frl, child=analyze)) diamond.addDependency(Dependency(parent=frr, child=analyze)) # Write the DAX to stdout
print "Writing %s" % daxfile f = open(daxfile, "w") diamond.writeXML(f) f.close()
from cosmos.contrib.ezflow.tool import INPUT
in_list = [ INPUT("f.a", tags={'sample': 1}) ]
dag = DAG().sequence_( add_(in_list), map_(preprocess),
apply_(findrange_b1, findrange_b2, combine=True), map_(analyze
Table S3.1 Feature comparison between COSMOS and other workflow tools discussed
in main text with respect to use in next-generation sequencing analysis
COSMOS Bpipe Ruffus Galaxy Taverna Pegasus
Workflow language Python Groovy Python GUI GUI Java, Python, Perl
Workflow abstraction syntax Yes Yes Yes No No No[1]
DAG constructed at run-time Yes Yes Yes No No No
Support for complex
parallelization Yes Yes Yes No No Yes
Realtime monitoring dashboard Yes No No Yes Yes Yes
SQL-like persistent database Yes No No Yes No No
Uses process exit codes[2] Yes No No Yes Yes Yes
Combine subworkflows Yes Yes No No ? Yes
Support for >10k jobs[3] Yes No No No No Yes
Built-in DRM support Yes Yes No Yes No Yes
DRMAA[4] support Yes No No Yes via
plugin No
Task DAG visualizer Yes No No Yes Yes Yes
Stage DAG visualizer Yes Yes Yes No No Yes
Topological execution[5] Yes No No Yes Yes Yes
Auto-delete intermediate files Yes No No No No No
[1]
Pegasus provides an API for constructing DAGs, but the workflow is not abstracted from the DAG itself; in effect, the Pegasus programmer must construct the DAG directly.
[2]
COSMOS’ persistent database provides the ability to store task’s exit codes, this solves some issues with Ruffus and Bpipe’s reliance on output file timestamps is that failed jobs can be misinterpreted as successful when resuming a failed workflow.
[3]
Because Ruffus and Bpipe both create a thread per job, they may require more RAM than is available on modern machines for massive workflows. COSMOS was able to perform the GATK joint calling workflow with a dataset of 99 whole genomes.
[4]
Although Bpipe supports many popular DRMs, DRMAA has support for almost every widely used DRM including GridEngine, Condor, PBS/Torque, GridWay, PBS, and LSF.
[5]
COSMOS (and Galaxy and Taverna) execute a task as soon as its dependencies have successfully completed. Ruffus and Bpipe wait for an entire stage to complete before advancing.
CHAPTER 4:
SCALABLE AND COST-EFFECTIVE
NGS GENOTYPING IN THE CLOUD
4.1 GENERAL INTRODUCTION
The dramatic fall of next generation sequencing (NGS) cost in recent years positions the price in range of typical medical testing, and thus whole genome analysis (WGA) may be a viable clinical diagnostic tool. Modern sequencing platforms routinely generate petabyte data. The current challenge lies in calling and analyzing this large-scale data, which has become the new time and cost rate-limiting step.
To address the computational limitations and optimize the cost, we have developed COSMOS (http://cosmos.hms.harvard.edu), a scalable, parallelizable workflow management system running on clouds (e.g., Amazon Web Services or Google Clouds). Using COSMOS (Gafni et al. 2014), we have constructed a NGS analysis pipeline implementing the Genome Analysis Toolkit - GATK v3.1 - best practice protocol (DePristo et al. 2011; Van der Auwera et al. 2013), a widely accepted industry standard developed by the Broad Institute. COSMOS performs a thorough sequence analysis, including quality control, alignment, variant calling and an unprecedented level of annotation using a custom extension of ANNOVAR. COSMOS takes advantage of parallelization and the resources of a high-performance compute cluster, either local or in the cloud, to process datasets of up to the petabyte scale, which is becoming standard in NGS.
This approach enables the timely and cost-effective implementation of NGS analysis, allowing for it to be used in a clinical setting and translational medicine. With COSMOS we reduced the whole genome data analysis cost under the $100 barrier, placing it within a reimbursable cost point and in clinical time, providing a significant change to the landscape of genomic analysis and cement the utility of cloud environment as a resource for Petabyte-scale genomic research.