Université de Montréal
Impact d'un traumatisme crânio-cérébral léger sur l’architecture du sommeil et le transcriptome dans un modèle murin
Présenté Par Meriem SABIR Dirigé par Dre. Valérie MONGRAIN
Mémoire présenté à la Faculté de médecine en vue de l’obtention du grade de maîtrise
en sciences biomédicales option sciences psychiatriques
Février 2015
Université de Montréal
Faculté des études supérieures et postdoctorales
Ce mémoire intitulé :
Impact d'un traumatisme crânio-cérébral léger sur l’architecture du sommeil et le transcriptome dans un modèle murin
Présenté Par Meriem SABIR
est évalué par un jury composé des personnes suivantes :
Valérie MONGRAIN, directeur de recherche (Faculté de médecine. Université de Montréal)
Pierre-Paul ROMPRÉ, président rapporteur (Faculté de médecine. Université de Montréal)
Roger GODBOUT, membre du jury (Faculté de médecine. Université de Montréal)
2 Remerciements
En tout premier lieu, je remercie le bon Dieu, tout puissant, de m’avoir donné la force pour survivre, ainsi que l’audace pour dépasser toutes les difficultés.
Le travail présenté dans cette thèse a été réalisé sous la direction de Dre. Valérie Mongrain. Ma plus grande gratitude va à mon encadreur, pour sa disponibilité et la confiance en mes capacités qu’elle m’a accordée. J’ai profité du savoir-faire dont j’ai pu bénéficier au cours de ces deux années. J’aimerais aussi la remercier pour l’autonomie qu’elle m’a accordée, pour ses orientations qui m’ont permis de mener à bien ce travail.
J’exprime toute ma reconnaissance à Dr. Pierre-Paul Rompré, vous m’avez fait un honneur en acceptant de présider ce mémoire. Aussi, je remercie vivement Dr Roger Godbout pour avoir bien voulu juger ce travail.
Merci à tous mes collègues de laboratoire, particulièrement à Érika Belanger Nelson, la coordinatrice de laboratoire et amie. Quand je suis arrivée au labo, tu étais en congé maternité, ton arrivée a fait une différence pour moi. Merci pour ton soutien, pour ton aide et surtout pour ton écoute.
Merci également à mon collègue de laboratoire à Marlène Freyburguer pour ton aide à mon arrivée au labo pour l’apprentissage des techniques et de répondre à mes questions.
Une spéciale dédicace à ma famille qui m’a supportée pendant ces deux années d’études… Spéciale dédicace à ma mère, à mon père, et à ma tante Saadia: Je vous remercie pour votre amour, votre affection votre soutien inconditionnel qui ne cesse point, et surtout merci pour les valeurs que vous m’avez transmises. Je vous aime et vous m’avez manqué énormément.
Un grand merci à mon mari, pour ses conseils et surtout merci pour ta patience, en particulier pendant la période de rédaction. Tu m’as gâté, c’est le terme…... Merci !!!
Merci aussi à ma sœur Soumia pour tout le soutien et l’aide, et surtout pour l’amour et la tendresse que t’as donné à Sidine quand moi j’étais pas la…!
Une pensée particulière à mon ange, ma petite Sidine!
Merci à tous mes amis, surtout à ceux qui étaient si compréhensifs envers ma préoccupation, sans avoir besoin d’expliquer pourquoi je ne suis pas là, qui m’ont soutenue aux moments difficile, et qui sont encore des chers amis.
3
Table des matières
Remerciements ... 2
Résumé ... 5
Abstract ... 6
Liste de tableaux ... 7
Liste des figures ... 8
Liste des abréviations ... 12
Chapitre 1: Introduction ... 14 1.1 Mise en contexte : ... 15 1. 2. Le sommeil ... 16 1.2.1 Définitions ... 16 1.2.1.1 Électroencéphalographie ... 16 1.2.1.2 Architecture du sommeil ... 17
1.2.2 Processus de régulation du sommeil ... 19
1.2.2.1 Processus circadien ... 19
1.2.2.2 Processus homéostatique ... 20
1.2.3 Effets de la privation de sommeil (PS) sur la cognition ... 21
1.2.4 Les mécanismes moléculaires impliqués dans l’homéostasie du sommeil ... 23
1.3 Le traumatisme crânien ... 28
1.3.1 Définition ... 28
1.3.2 Le traumatisme crânien léger (TCL) ... 28
1.3.3 Dommages dus aux TCL ... 30
1.3.4 Trouble de sommeil chez les TCL : ... 31
1.3.5 Implication de la génétique suite aux traumatismes crâniens ... 33
1.3.6 Réponses cellulaires, facteurs moléculaires et excitoxicité ... 34
Chapitre 2 : Objectifs, hypothèses et contribution spécifique ... 39
2.1 Objectifs ... 40
2.2 Hypothèses ... 40
2.3 Contribution spécifique de l’étudiante ... 40
2.4 Contribution spécifique des co-auteurs de l’article ... 40
Chapitre 3. Méthodologies et résultats - Article de recherche ... 43
Article publié dans Brain, Behavior and immunity ... 43
Chapitre 4 : Discussion ... 83
4
4.2 Effet du TCL sur le sommeil ... 85
4.2.1 Sommeil chez rongeurs TCL. Similarités et différences ... 85
4.2.2 Sommeil des patients TCL. Similarités et différences ... 87
4.2.3 Des plaintes de sommeil chez les patients trauma ... 90
4.2.4 La neurophysiologie des circuits régulateurs du sommeil. ... 90
4.2.5 Changements secondaires pouvant induire des changements neurophysiologiques : ... 91
4.3 Effet de la privation de sommeil après TCL ... 92
4.3.1 La privation de sommeil affecte l’expression des gènes de plasticité dans le cortex ... 93
4.3.2 La privation de sommeil affecte les gènes de l’horloge ... 95
4.3.3 TCL pourrait affecter l’homéostasie du calcium ... 96
4.3.4 Changement de l’expression des chimiokines et gènes gliaux après TCL ... 97
4.4 Limites de l’étude et perspectives ... 98
4.4.1 Limites de l’étude ... 99
4.4.2 Perspectives à venir ... 99
4.5 Conclusion ... 101
5 Références ... 102
5 Résumé
Le traumatisme crânien léger (TCL) est l'un des troubles neurologiques les plus courants affectant la santé publique. Aussi, les troubles du sommeil sont fréquents chez les patients atteints de TCL. Les études chez les rongeurs montrent que certains marqueurs de plasticité synaptique diminuent après le TCL, ce qui pourrait nuire à la plasticité du cerveau. Nous suggérons que la perte de sommeil intensifie l'effet négatif de TCL, qui peut refléter les changements des marqueurs de plasticité synaptique ou des changements des voies physiologiques qui régulent le sommeil. En utilisant un modèle de traumatisme crânien sur crâne fermé (closed head injury), nous avons étudié la relation bidirectionnelle entre le TCL et le sommeil en évaluant les effets de TCL sur l’activité électrique du cerveau par électroencéphalographie (EEG), et ceux de la privation de sommeil (PS) sur l'expression génique post-TCL. Premièrement, l'activité EEG a été enregistrée pour voir si l'architecture du sommeil est altérée suite au TCL. Nous avons ensuite voulu tester si la PS suite TCL induit des changements dans l'expression des gènes : Arc, Homer1a, Hif1a, Bdnf, Fos et éphrines, qui ont été liés à la plasticité synaptique et à la régulation du sommeil. Nous avons également étudié l'effet de la PS post-TCL sur le génome complet dans les régions cibles (cortex et l'hippocampe). Les principaux résultats obtenus dans cette étude confirment que TCL modifie de manière significative l'activité spectrale pendant l'éveil, le sommeil Rapid Eye Movement (REM) et le sommeil non-REM dans le deuxième 24 heures post-TCL. Fait intéressant, la capacité de maintenir de longues périodes d'éveil a été altérée immédiatement après TCL (première 24h post-TCL). La dynamique de l'activité delta pendant l'éveil a été modifié par le TCL. Parallèlement à ces modifications, des changements dans l'expression des gènes ont été observés dans le cortex et l'hippocampe. Seulement Arc et EfnA3 ont montré une interaction TCL / PS et ce dans l’hippocampe, tandis que l'expression de tous les autres gènes semblait être affectée par la PS ou TCL indépendamment. Nos résultats montrent pour la première fois que le TCL induit l'expression de deux chimiokines (Ccl3 et Cxcl5) à la fois dans le cortex cérébral et l'hippocampe 2,5 jours post-TCL. Également, nous avons observé que le TCL induit une diminution de l'expression de Lgals3 et S100A8 dans le cortex, et une augmentation d’Olig2 dans l'hippocampe. Les résultats concernant les effets de la PS sur le génome complet du cortex et de l'hippocampe montrent des changements significatifs dans les gènes impliqués dans diverses fonctions physiologiques, telles que les rythmes circadiens, la réponse inflammatoire, ainsi que de l'activation des cellules gliales. En général, nos résultats précisent les changements dans la qualité de l’éveil ainsi que dans l'expression de divers gènes après TCL.
6 Abstract
Mild traumatic brain injury (mTBI) is one of the most common neurological disorders affecting public health. Sleep disorders are common in patients with mTBI. Studies in rodents show that some synaptic plasticity markers decreased after mTBI which could impair brain plasticity. We suggest that sleep loss intensifies the negative effect of mTBI, which may reflect changes of synaptic plasticity markers or changes of different physiological pathway that regulates the sleep process. Using a "closed head injury" model, we have studied the bidirectional relationship between mTBI and sleep by investigating the effects of mTBI on sleep structure, and that of sleep deprivation (SD) on gene expression post-mTBI. First, EEG activity was monitored to investigate if sleep architecture is altered following mTBI. We then tested if SD, following mTBI, induces changes in gene expression of plasticity markers (Arc, Homer1a, Hif1a, Bdnf, Fos, and Ephrins), which have also been linked to sleep regulation. We also investigated the effect of SD post-mTBI on genome wide gene expression in target regions. The main results obtained in this study confirm that mTBI affects wakefulness, and significantly changes spectral activity during wakefulness, rapid eye movement (REM) sleep, and non-REM sleep on the second 24 hours post-TCL. Interestingly, the capacity to sustain long bouts of wakefulness was impaired immediately after mTBI. In addition, delta activity time course was altered by mTBI during wakefulness. In parallel to these alterations, changes in gene expression were observed. Only Arc and EfnA3 showed a mTBI/SD interaction in the hippocampus specifically, whereas expression of all other genes seemed to be affected by SD or mTBI independently. Our results indicate for the first time that the TCL induced the expression of two chemokines (Ccl3 and Cxcl5) in the cerebral cortex and hippocampus 2.5 days post-TCL. Also, we observed that the TCL induces a decrease in the expression of Lgals3 and S100A8 in the cortex, and an increase of Olig2 in the hippocampus.Results of SD effects on genome wide gene expression in the cortex and hippocampus show significant changes in genes involved in various physiological functions, such as circadian rhythms, inflammation, and also glial cell activation. In general, our results precise changes in wakefulness as well as in expression of various genes after mTBI.
7 Liste des tableaux
Chapitre 3. Méthodologies et résultats - Article de recherche
Table S1: Sequences of primers or probes and product numbers of probe sets used for qPCR.
Table S2: 24h mean (± SEM) duration (in sec) of individual bouts of wakefulness, non-rapid eye movement sleep [NREMS] and non-rapid eye movement sleep [REMS] in mTBI and Sham mice averaged separately for the first and the second recorded days.
Table S3: Genes with differential expression between mTBI-Control and Sham-Control mice in the cerebral cortex (p < 0.01).
Table S4: Genes with differential expression between mTBI-Control and Sham-Control mice in the hippocampus (p < 0.01).
Table S5: Biological functions and predicted regulators for which a significant enrichment was observed for genes differentially expressed with mTBI and SD in the cerebral cortex (p values computed with Fisher's Exact Tests using IPA).
Table S6: Biological functions and predicted regulators for which a significant
enrichment was observed for genes differentially expressed with mTBI and SD in the hippocampus (p values computed with Fisher's Exact Tests using IPA).
Table S7: Genes with differential expression in the cerebral cortex after SD in mTBI mice (FDR < 0.05).
Table S8: Genes with differential expression in the cerebral cortex after SD in Sham mice (FDR < 0.05).
Liste des figures
Chapitre 1. Introduction
Figure 1: Architecture du sommeil et de l'activité corticale. Mesurée par l’électroencéphalogramme (EEG) pendant l'éveil, le sommeil lent et le sommeil paradoxal chez un rat (Electrophysiological correlates of sleep homeostasis in freely behaving rats. Vyazovskiy et al., 2011).
Figure 2: L’effet de l’interleukine-1 bêta sur le sommeil chez la souris. (Involvement of cytokines in slow wave sleep. Krueger 2011).
Figure 3: Les effets à court et à long terme d’un TCL (The Young Brain and Concussion: Imaging as a Biomarker for Diagnosis and Prognosis. Toledo et al., 2012).
8
Figure 4: Schéma descriptif des changements métaboliques et anatomiques en réponse au TCL. (A): Changements neurologiques: schéma descriptif de la réponse cellulaire post-TCL. B: changements axonales (The Young Brain and Concussion: Imaging as a
Biomarker for Diagnosis and Prognosis. Toledo et al., 2012). Chapitre 3. Méthodologies et résultats - Article de recherche
Figure 1. (A) Schematic view of the ECoG/EMG recording protocol. mTBI was performed between ZT8 and ZT11 and was immediately followed by an electrode implantation surgery. The morning of the following day, mice were cabled around 15 min before ZT0 and ECoG/EMG were recorded continuously for 48 h starting at light onset. Sham control mice were recorded in parallel, and recordings were performed in groups of 4 to 6 mice. Gray bars indicate undisturbed/spontaneous wakefulness and sleep behavior (same in B), and red bars indicate ECoG/EMG recorded undisturbed/spontaneous wakefulness and sleep behavior. Light gray areas indicate dark periods (same in B). (B) Schematic view of the gene expression protocol. Mice were initially divided into two different groups in which animals were either submitted to mTBI or Sham surgery between ZT8 and ZT11. For each of these conditions, half of the animals were submitted to two consecutive days of 6 h sleep deprivation (SD), taking place between ZT0 and ZT6. All animals were then sacrificed on the morning of the third day for brain area sampling. (C) Neurological Severity Scale (NSS) score from an initial cohort of animals (n = 10 mTBI and 8 Sham). Only mTBI mice showed a significant increase in NSS score 6 h after injury in comparison to before injury (t = 3.2, p < 0.01 indicated by the star).
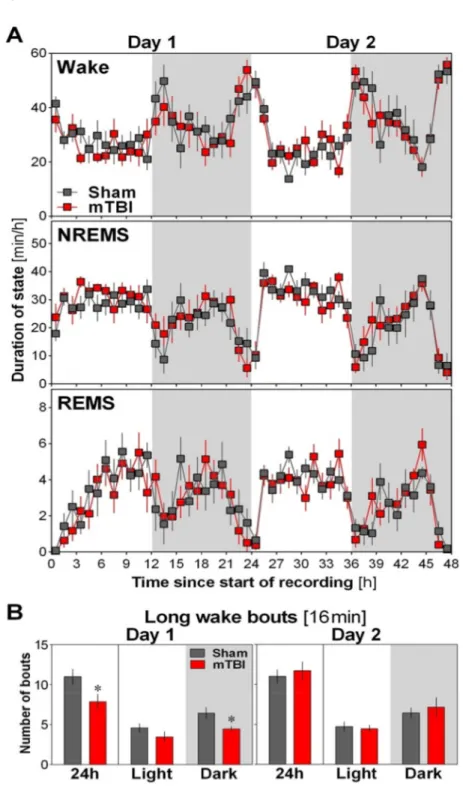
Figure 2. (A) Forty-eight-hour time course of vigilance state duration in mTBI and Sham mice measured using ECoG/EMG recording. The upper panel shows the time course of wakefulness, the middle panel that of NREMS and the lower panel that of REMS. The time course of the three vigilance states did not significantly differ between mTBI and Sham mice on both Day 1 (interaction: wakefulness F23,276 = 0.7, p = 0.9; NREMS F23,276 = 0.7, p = 0.9; REMS F23,276 = 0.8, p = 0.8) and Day 2 (interaction: wakefulness F23,276 = 0.7, p = 0.8; NREMS F23,276 = 0.7, p = 0.8; REMS F23,276 = 1.0, p = 0.5). The total duration of vigilance states was also similar between mTBI and Sham mice (Condition effect wakefulness: F1,12 < 1.3, p > 0.2; NREMS: F1,12 < 1.9, p
9
> 0.2; REMS: F1,12 < 0.7,p > 0.4). Grey areas indicate dark periods (same in B). (B) Number of long bouts of wakefulness (16min) in mTBI and Sham mice averaged for 24h, 12-h light, and 12-h dark periods separately for the first and the second recorded days. A significant Condition effect (mTBI vs. Sham) was found for 24h and 12-h dark period only for the first recorded day (t > 2.3, p < 0.05 indicated by stars; Day 1 12-h light period: t = 1.3, p = 0.2; Day 2: t < 0.8, p > 0.4).
Figure 3. (A) Wakefulness (upper panels), NREMS (middle panels) and REMS (bottom panels) relative spectral power averaged for 24 h per 0.25 Hz-bin between 0.75 to 25 Hz separately for Day 1 and Day 2 in mTBI and Sham mice. Significant group differences were observed only in NREMS for Day 1 for three bins around 5 Hz (t > 2.2, p < 0.05). For Day 2, significant group effects (t > 2.2,p < 0.05) were observed for wakefulness for Hz-bins around 5 Hz and at 16.25 and 20.25 Hz, for NREMS between 3.5 and 6.75 Hz and between 14.75 and 25 Hz, and for REMS between 3.25 and 5 Hz and between 21.75 and 23 Hz. Significant differences between mTBI and Sham mice are represented by black bars above the x-axis (thin bars: p < 0.05; thick bars: p < 0.01). Inserts show magnifications of indicated spectra with significant differences. (B) Forty-eight-hour time courses of relative delta activity during wakefulness (upper panel) and NREMS (bottom panel) in mTBI and Sham mice. A Condition (mTBI vs. Sham) by Interval interaction was found for relative delta activity during wakefulness (F35,420 = 1.8, p = 0.02). Stars indicate significant differences between mTBI and Sham mice for indicated points (p < 0.05, simple effect analysis). No significant Condition by Interval interaction or Condition effect was found for relative delta activity during NREMS (interaction: F35,420 = 1.0, p = 0.5; condition: F1,12 = 1.8,p = 0.2). Gray areas indicate dark periods.
Figure 4. Effect of mTBI and two consecutive days of 6 h sleep deprivation (SD) on the expression of plasticity and other target genes measured by qPCR on the third morning after surgery in the cerebral cortex and hippocampus. In the cerebral cortex, no significant interaction between Condition (mTBI vs. Sham) and Treatment (SD vs. Control (Ctrl)) and no significant Condition effect was found. Significant Treatment (SD vs. Control) effect was found for Arc, Bdnf, DnajB5, Fos, Homer1a, EfnA3 and EphA4 (F1,33 > 4.3,p < 0.05, indicated by +). In the hippocampus, a significant Condition by Treatment interaction was found for Arc and EfnA3(F1,33 > 4.6, p < 0.04, indicated by stars), and a tendency for
10
Condition by Treatment interaction was observed for Fgf1 (F1,33 = 2.6,p = 0.1). No significant Condition effect was found, but a significant Treatment (SD vs. Control) effect was found for EfnB3 (F1,33 = 8.6, p < 0.01 indicated by +), and a tendency for a significant Treatment effect was observed for Fos (F1,33 = 3.4, p = 0.07).
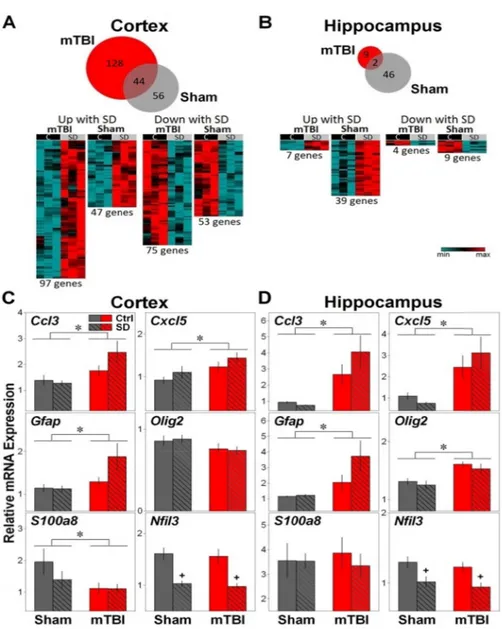
Figure 5. Venn diagrams (upper panels) and heatmap representations (lower panels) of genes differentially expressed in mTBI and Sham mice after two days of 6 h sleep deprivation (SD) in the cerebral cortex (A) and hippocampus (B) as measured using high-throughput sequencing (RNA-Seq). Venn diagrams show the overlap between genes differentially expressed (FDR < 0.05) in mTBI and Sham mice. In the cerebral cortex, 25.6% of genes differentially expressed after SD in mTBI mice overlap with those differentially expressed in Sham mice, whereas 44% of those in Sham mice overlap with those of mTBI mice. For the hippocampus, 18.2% of genes differentially expressed after SD in mTBI mice overlap with those differentially expressed in Sham mice, while only 4.2% of those in Sham mice overlap with those of mTBI mice. Heatmaps represent the expression of the same transcripts with significant differential expression after SD in mTBI and Sham mice, separately sorted according to the direction of the change with SD. For each condition (mTBI and Sham), columns refer to three pools of RNA of three control (C) and three SD mice (total nine per group). Transcripts were ordered by hierarchical clustering (complete linkage). (C and D) qPCR validations of the effect of mTBI and SD on the expression of selected transcripts on the third morning after surgery in the cerebral cortex and hippocampus. (C) In the cerebral cortex, significant Condition effects (mTBI vs. Sham, indicated by stars) were observed for Ccl3,Cxcl5, Gfap and S100a8 (F1,33 > 4.3, p < 0.05). A significant Treatment effect (SD vs. Control (Ctrl), indicated by +) was observed for Nfil3 (F1,33 > 34.6, p < 0.001). A trend for Condition by Treatment interaction was found for Gfap (F1,33 = 2.6, p = 0.1).
(D) In the hippocampus, significant Condition effects (mTBI vs. Sham, indicated by stars) were observed for Ccl3, Cxcl5, Olig2 and Gfap(F1,33 > 8.2, p < 0.05). A significant Treatment effect (SD vs. Control, indicated by +) was observed for of Nfil3 (F1,33 = 15.1,p < 0.001). No Condition by Treatment interaction was found for qPCR validation in the hippocampus.
11 Supplementary information
Figure S1. RNA-Seq quantification of the effect of mTBI and of two consecutive days of 6h sleep deprivation (SD) on the expression of Bhlhe41 on the third morning after surgery in the cerebral cortex and hippocampus.
12
Liste des abréviations
AMPA: α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid Arc: Activity-mediated cytoskeleton-associated
Bdnf: Brain-derived neurotrophic factor BCAA: Branched Chain Amino Acids
Bhlhe41: Basic helix-loop-helix family, member e41
Bmal1
:
Brain and muscle Aryl hydrocarbon receptor nuclear translocator like 1 Ca2+: Ion calciumCaMKII: Ca2+/calmodulin-dependent protein kinase II CCI : Impact cortical contrôlé
Ccl3: C-C motif ligand 3
Clock: Circadian Locomotor Output Cycles Kaput
Creb: Cyclic adenosine monophosphate Response Element-binding protein Cry: Cryptochrome
Cxcl5: Chemokine (C-X-C motif) ligand 5
DnajB5: DnaJ (Hsp40) Homolog Subfamily B, Member 5 EEG: Électroencéphalogramme
ECoG: Électrocorticoencéphalogramme Eph: Ephrin receptor family
Efn: Ephrin
EMG: Électromyogramme Fgf1: Fibroblast growth factor 1
Fos: Facteur de transcription synthétisé à la suite d'un signal activateur GABA: Gamma-aminobutyric acid
GFAP: Glial fibrillary acidic protein GCS: Glasgow coma scale
Homer1a: Homer protein homolog 1 a Hz: Hertz
Hif1a: Hypoxia inducible factor 1, alpha subunit IL : Interleukine
IRM: Imagerie par résonance magnétique KO: Knockout
Lgals3 : lectine galactoside contraignant soluble 3 ou galectine-3 MAPK I: Mitogen-Activated Protein Kinase
mGluR: Récepteurs métabotropiques du glutamate N1, N2, N3: non-rapid eye movement sleep stage 1, 2, 3
Nfil3 : Nuclear factor interleukin-3-regulated protein NGF: Nerve growth factor
NMDAR: N-methyl-D-aspartate receptor NREM: Non-Rapid Eye Movement NSS: Neurological severity scale
13 Olig2: Oligodendrocyte transcription factor 2 Per: Period
PKC : Protein kinase C
Processus S : Processus homéostatique de régulation du sommeil Processus C : Processus circadien de régulation du sommeil PS : Privation de sommeil
qPCR: Quantitative polymerase chain reaction REM: Rapid Eye Movement
ROS: Reactive oxygen species SD: Sleep deprivation
S100a8: Calcium binding protein family
TCC: Traumatisme crâniocérébral TCL: Traumatisme crânien léger TDM: TomodensitométrieTNF: Tumor Necrosis Factor TS: Trouble de sommeil ZT: Zeitgeber time
14
15 1.1 Mise en contexte :
Chez toutes les espèces animales, le sommeil est l'une des activités les plus fondamentales pour le bon fonctionnement de l’organisme et du cerveau. Il joue un rôle central dans la plasticité cérébrale, l'apprentissage, la consolidation de la mémoire, ainsi que dans la récupération des fonctions physiologiques. Le sommeil NREM, en particulier, exerce un rôle récupérateur lié à son intensité, surtout pour la récupération des fonctions cognitives (Aricò et al., 2010). Cela dit, une perturbation chronique de sommeil peut être assez nocive pour bouleverser la santé et la qualité de vie de l’individu, aussi, elle peut avoir des effets néfastes et considérables sur la performance des individus dans la vie quotidienne.
Selon le concept de l’homéostasie du sommeil, le besoin de dormir augmente durant l’éveil et se dissipe durant le sommeil, de façon exponentielle. Aussi, l’éveil prolongé augmente le besoin du sommeil. Certains marqueurs mesurés à partir de l’activité électroencéphalographique (EEG) aident à caractériser la dynamique de la régulation homéostatique du sommeil. Grâce à cette approche, nous avons évalué dans le présent travail la relation bidirectionnelle entre le trauma crânien léger (TCL) et le sommeil. Les traumas crâniens, particulièrement les TCL, sont associés à des déficits cognitifs à long terme (Hicks et al., 1993; Milman et al., 2005). Or, ils sont les moins diagnostiqués car ils ne montrent pas des lésions tissulaires avec les techniques de neuro-imageries. Autant chez l’humain que chez les rongeurs, les données indiquent que le cycle éveil-sommeil ainsi que la qualité de éveil-sommeil sont altérés suite aux TCL (Baumann et al., 2007). En effet, les patients ayant subi un trauma crânien rapportent souvent une prévalence élevée d’insomnie, des symptômes de fatigue et de somnolence (Baumann et al., 2007 ; 2010). Ceci suggère qu’il pourrait y avoir des changements au niveau de l’architecture de sommeil chez ces patients. Ces troubles du sommeil peuvent avoir des conséquences néfastes sur le fonctionnement des individus en question, et les dommages neuronaux post-TCL peuvent induire une atteinte au niveau de la plasticité synaptique, qui génère un besoin de récupération. De manière intéressante, la restauration d’un cycle éveil-sommeil normal semble être proportionnelle avec la récupération de certaines fonctions cognitives après avoir subi un tel dommage (Wiseman-Hakes et al., 2013). Cependant, aucune étude n’a permis de distinguer les
16
différents stades de sommeil ni d’évaluer son intensité par l’analyse spectrale dans les premiers jours post-TCL.
De tout cela s’inspire notre étude dont l’objectif est d’étudier en détail la relation sommeil-trauma crânio-cérébral. Le but de la présente étude était d’explorer la relation bidirectionnelle entre le TCL et le sommeil : comment le TCL peut affecter l’architecture de sommeil, et dans un sens inverse, comment le manque de sommeil peut affecter l’expression des gènes associés à la plasticité cérébrale et à la récupération post-TCL. Dans cette introduction, nous allons présenter les éléments de régulations de sommeil, les caractéristiques des TCC notamment les TCL, et les troubles de sommeil qui y sont associés, et enfin décrire comment ces deux aspects (sommeil et TCC) sont liés à une réponse immunitaire.
1. 2. Le sommeil 1.2.1 Définitions
Le sommeil n’est pas un processus passif, c’est un état périodique et réversible qui comporte plusieurs niveaux d’activité cérébrale. Les recherches ont prouvé que le sommeil est un comportement essentiel à la santé et occupe généralement le tiers de notre vie (Ohlmann et O'Sullivan., 2009). Il est caractérisé par la perte de conscience et la perte de l'activité sensorielle. En outre, il implique des processus physiologiques complexes allant de l’expression des gènes et mécanismes intracellulaires jusqu’aux différents circuits neuronaux (Hobson et al., 2002). Aussi, le sommeil joue un rôle primordial dans la plasticité synaptique et dans la récupération.
1.2.1.1 Électroencéphalographie
L’électroencéphalographie (EEG) est une technique très importante qui a permis de mesurer et de caractériser l’activité électrique synchronisée et désynchronisée des neurones corticaux. Des électrodes collées sur le crâne sont reliées par des fils à un amplificateur qui enregistre l'activité électrique des neurones situés au cortex. L'activité électrique se traduit par un potentiel de champ qui chevauche la surface du cerveau et qui est mesuré sur le cuir chevelu. Il est supposé que la contribution principale aux enregistrements EEG provient de nombreux neurones situés dans les couches
17
corticales et en particulier de grands neurones pyramidaux dirigés vers la surface corticale, avec arborisations dendritiques étendues dans les couches superficielles parallèles à la surface corticale (Coenen., 1997; Pedley et Traub., 1990). La fréquence et l'amplitude sont les deux caractéristiques de l'EEG qui permettent l'analyse spécifique de différents stades de sommeil.
Généralement, le sommeil NREM se distingue par trois stades de sommeil appelés N1 (sommeil léger), N2 (sommeil lent) et N3 (sommeil lent profond). Les phases de sommeil lent sont caractérisées par différents types d'ondes cérébrales. En effet, les ondes delta (fréquence de 1 à 5 Hz) qui sont associés à la récupération des fonctions physique apparaissent pendant le stade N3 et y représente plus de 20% de l'EEG de sommeil (Jouvet et al., 1967; Keenan et Hirshkowitz 2011). Généralement, le stade N3 est prédominant dans le premier tiers de la nuit, tandis que le sommeil paradoxal est plus présent dans le dernier tiers de la nuit. La fréquence de l’EEG durant le sommeil NREM est généralement associée à des ondes alpha lentes (8Hz), à des ondes thêta (4-7Hz) prédominantes durant le stade N1, et des ondes delta (1-4Hz) avec l’apparition du stade N3. Généralement, les rongeurs suivent la même succession des phases de sommeil observée chez l’humain, sauf qu’il y a moins de stades chez les rongeurs comparés aux humains et la durée des stades est moins courtes, par exemple chez la souris, le sommeil REM se manifeste toutes les 4 minutes de sommeil comparé a 90 minutes chez l’humain.
1.2.1.2 Architecture du sommeil
Le début de sommeil est précédé par une période de 10 à 20 minutes pendant laquelle la sensation de fatigue et le besoin de dormir s’accroient. Le sommeil chez l’humain suit une périodicité régulière qui comprend deux états de sommeil tel que décrit brièvement ci-haut (sommeil lent ou NREM et paradoxal ou REM) suivis par l’éveil formant ainsi l’architecture du sommeil.
Le sommeil lent (NREM : Non Rapid Eye Movement). D’une manière générale, le sommeil lent est le plus réparateur autant sur le plan physique qu’au plan psychique. Il est caractérisé par une baisse globale du débit sanguin cérébral et une diminution de l’activité des structures du tronc cérébral (Maquet et al., 1997). Le sommeil NREM
comm lent e cerve Le so parad pend minut répèt Au co somm ampl l’aton ocula L’éve produ yeux les ré les h impliq proce proce Figur et hyp mence par le et le stade eau à passer ommeil REM doxal, est la ant la journ tes chez l’hu tent dans de ours d'une meil lent, le itude sembla nie muscula aires rapides eil. Générale
uit une désy généraleme éflexes sont humains prè quées dans essus circad essus et leur re 1: Architec pnogramme c e stade N1, o N3 du somm r au somme M (Rapid E période des ée. Ce stad umain. Dans es périodes d nuit 3 à 7 c e sommeil R able à l’EEG aire. Aussi, s, et une res ement, l’éve ynchronisatio ent ouverts, vifs. Penda ès des deux la régulatio dien et homé r importance cture du somm correspondan ou le somme meil lent pro il REM (Fig. Eye Moveme s rêves et de de de somm s le sommei de temps ré cycles de so REM a une G de l’éveil le sommei piration irrég eil résulte on corticale. le temps de nt cette péri x tiers du t on de l’éveil éostatique. e dans la rég meil et de l'ac nt à 24h de ba 18 eil est léger, ofond, cette . 1). ent). Le som e la mise en meil se prése l humain, le gulières et s ommeil peuv e fréquence et une activ l REM est gulière. d’une augm Au cours d e réaction à iode l'activité temps. Plus et du somm Nous allons gulation du s ctivité corticale aseline (référ , suivi du sta dernière ph mmeil REM n mémoire d ente réguliè s états de s selon des cy vent se suc EEG plus vité muscula caractérisé entation de de l'éveil, la toutes stim é EEG est r sieurs zones meil qui son s présenter sommeil dan e. (A) L'activi rence) chez u ade N2 ou le hase semble M, aussi app es informati èrement tout sommeil (NR ycles de 60 à ccéder. Cont élevée ave aire EMG trè é par des e l’activité d vigilance es mulations est rapide. Il rep s dans le c nt aussi cont avec plus d ns la section ité à ondes le un rat. Les ba e sommeil es e préparer l pelé somme ons acquise tes les 60-9 REM/REM) s à 90 minutes trairement a ec une faib ès faible dû mouvement du cortex qu st élevée, le t très court e présente che cerveau son trôlés par le de détails ce suivante. entes de l’EE arres verticale st le eil es 90 se s. au le à ts ui es et ez nt es es G es
19
représentent valeurs consécutifs de 4-s de l’activité à ondes lentes (AOL). L’AOL diminue au cours de la période de lumière de 12 h, et augmentent après les long épisodes de l’éveil. (B) L’électroencéphalogramme (EEG) pendant l'éveil, le sommeil lent et le sommeil paradoxal chez un rat (Reproduit de Vyazovskiy, et al., 2011, Electrophysiological correlates of sleep homeostasis in freely behaving rats).
1.2.2 Processus de régulation du sommeil
Il est connu que le sommeil est régulé par deux processus, impliquant que les transitions entre le sommeil et l'éveil sont médiées par l'interaction d'un processus homéostatique (processus S) et un processus circadien (Processus C) (Borbely, 1982; Daan et al., 1984). La pression contrôlé par le processus S augmente au cours de l’éveil et diminue durant le sommeil, et c’est ainsi qu’il induit la sensation du besoin de dormir et l’intensité du sommeil (Daan et al., 1984; Borbély et al., 1982 ; Franken et al., 1991). Quant au processus C, il est généré par notre horloge biologique interne située dans les noyaux suprachiasmatiques (NSC) de l’hypothalamus. En effet, l’horloge biologique serait responsable de la rythmicité du cycle éveil-sommeil (Ralph et al., 1990; Daan et al., 1984), elle contrôle un grand nombre de fonctions physiologiques sur un rythme d’environ 24 heures, appelée rythme circadien.
1.2.2.1 Processus circadien
Le cycle éveil-sommeil est l’un des processus biologiques contrôlé par le rythme circadien (Aschoff et al., 1965; Hur et al., 1998; Lavie, 2001). Toutes les cellules du corps humain sont capables de générer ces rythmes circadiens, et la variation génétique est en partie responsable de la différence des horloges circadiennes internes et la rythmicité entre les individus (Harano et Miyatake 2010). Cette variabilité génétique peut aussi être responsable de la différence de cycle éveil-sommeil entre les individus et du chronotype qui définit les préférences pour l’horaire de sommeil (individus dits matinaux qui se couchent et se lèvent tôt ou vespéraux qui se couchent et se lèvent tard). La rétine, l’hormone mélatonine et la température corporelle jouent un rôle essentiel dans le système circadien et sont impliquées dans le contrôle des variations endogènes importantes au cours de la journée et/ou en réponse aux stimuli du cycle lumière/obscurité.
20
La rythmicité circadienne origine d’un ensemble de mécanismes moléculaires impliquant des gènes nommés gènes de l’horloge (ex : Brain and muscle ARNT like 1 (Bmal1), Circadian Locomotor Output Cycles Kaput (Clock), Period (Per)) (King et al., 1997). Le premier gène de l’horloge isolé est appelé Period (per), pour lequel différentes mutations ont été identifiées. Ce sont des gènes qui composent une boucle de rétroaction où l'activation initiale d'un gène est régulée par ce dernier selon un cycle qui dure environ 24 h. L'horloge circadienne chez les mammifères est contrôlée par deux facteurs de transcription principaux BMAL1 et CLOCK qui forment un hétérodimère et se lient aux séquences E-box (séquence d'ADN trouvé dans certaines régions de promoteur qui agit comme un site de liaison de protéine) dans l’ADN des gènes Period (Per1, Per2) et Cryptochrome (Cry1, Cry2). Le fonctionnement en boucle de rétroaction fait en sorte qu’après la traduction de Per et Cry en protéines, elles interagissent ensuite pour inhiber l'activité de BMAL1/CLOCK (Young et al., 2001; Ko et Takahashi 2006).
L’isolement des mutants est une approche assez prometteuse pour comprendre les fonctions de certains gènes. En effet, des souris dont le gène Per1 est muté montrent des actogrammes ayant une période de l'horloge interne plus courte que la normale. Aussi, des souris dont le gène Clock est inactivé ont perdu leur rythmicité lorsqu’elles ont été plongées dans l’obscurité constante. Le gène Clock, isolé et séquencé. Les animaux portant la mutation Clock présentent un actogramme anormal.
1.2.2.2 Processus homéostatique
La dynamique de l’homéostasie du sommeil est mesurée par l'activité à ondes lentes de l’EEG (Fig. 1). La pression homéostatique pour le sommeil augmente pendant l’éveil et diminue pendant le sommeil. Ainsi, un manque de sommeil provoque une augmentation compensatoire de l'intensité du sommeil, tel que mesurée par l’activité à ondes lentes. Le processus S définit donc la récupération pendant le sommeil en fonction de la période d’éveil. Cette fonction récupératrice a été proposée en se basant sur le fait que l'activité à ondes lentes augmente durant le sommeil proportionnellement à la durée précédente de l’éveil.
Plusieurs hypothèses ont été développées pour répondre à la question fondamentale de la fonction du sommeil et de l’intensité du sommeil qui reste toujours un mystère. Dans les dernières décennies, de nombreuses hypothèses sur la fonction de l’homéostasie du
21
sommeil ont été proposées, en mettant l'accent sur le maintien de l'homéostasie cellulaire de l'énergie, la plasticité synaptique, la consolidation de la mémoire, la thermorégulation, la biosynthèse de macromolécule et sur d’autres fonctions (Abel et al., 2013; Cirelli et Tononi 2008; Diekelmann et Born 2010; Frank 2006; Mackiewicz et al 2007; Varshavsky., 2012; Vyazovskiy et Harris., 2013). Ces hypothèses ne sont pas nécessairement exclusives, il est possible et probable que différents processus moléculaires se produisent en parallèle durant le sommeil.
Dans la plupart des espèces de mammifères étudiées, la quantité de sommeil est élevée au cours de la période néonatale, la phase de la vie qui se caractérise par le développement du cerveau et beaucoup de plasticité synaptique (Frank et Heller 1997; Frank., 1997; Roffwarg et al., 1966). Ceci suggère un lien étroit entre le sommeil et la plasticité synaptique. Ainsi, l’une des principales hypothèses qui ont été abordées les dernières années est l’hypothèse de l’homéostasie synaptique proposé par Tononi et Cirelli (2003; 2006; 2013). Cette hypothèse suggère que le sommeil reflète des changements liés à la plasticité synaptique. Plus précisément, elle suggère que pendant l’éveil, les connexions entre les neurones sont augmentées en induisant ainsi le besoin de sommeil. Ceci serait lié à la régulation homéostatique du sommeil et à la récupération.
Il est établi que l’état d’éveil est associé à certains changements moléculaires, structurels et fonctionnels dans le cerveau qui se produisent progressivement. Il s’avère ainsi logique de conclure que des difficultés de sommeil en réponse à une certaine perturbation peuvent avoir un impact négatif sur la récupération, et ils peuvent également exacerber d’autres troubles. Une étude effectuée sur des patients vacciné, ayant subi ensuite une privation de sommeil ou non, a rapporté que ceux qui ont été privés de sommeil avaient des niveaux d'anticorps plus faibles quelques semaines plus tard, ce qui démontre que le manque de sommeil peut, après la vaccination, nuire à la formation des acteurs de défenses immunitaires spécifiques de l'antigène (Spiegel et al., 2002). Ceci montre qu’outre la cognition, l’effet de manque du sommeil pour s’étendre et affecter d’autres processus physiologiques tel que le système immunitaire. Afin de mieux comprendre les conséquences de manque de sommeil, nous allons présenter les effets de la privation de sommeil dans les sections suivantes.
22
1.2.3 Effets de la privation de sommeil (PS) sur la cognition
La privation de sommeil expérimentale est une excellente méthode qui permet d’élucider l’importance du sommeil dans les processus physiologiques et comportementaux. De nombreuses études ont rapportés que la privation chronique de sommeil peut avoir des effets néfastes et considérables sur la performance, l’apprentissage, la mémoire et la récupération des fonctions physiques ou psychologiques (Killgore 2010; Diekelmann et Born., 2010; Palagini et Rosenlicht., 2011; Krueger et al., 1999). En général, plus la durée de l’éveil est prolongée, plus la performance est altérée. Aussi, il a été observé que le temps de réaction à des tâches de vigilance psychomotrice était plus élevé après une privation de sommeil total, partielle ou même après un sommeil fragmenté (Dinges., 1997; Belenky et al., 2003), ce qui indique que la PS entraine un ralentissement cognitif considérable. Cette détérioration de la performance peut être restaurée quand le tiers du sommeil perdu est récupéré (Horne., 1988; 1993; Dinges et Kribbs., 1991).
Les résultats d'une étude de neuro-imagerie, ont montré qu'une seule nuit sans sommeil altère la connexion entre l’amygdale et le cortex préfrontal. Plus précisément, la PS génère une perte de contrôle et empêche l’inhibition de l’activation de l’amygdale (Yoo et al., 2007). Les auteurs de la même étude ont suggéré que le sommeil semble effectuer une remise à la base (reset) du cerveau et de la réactivité émotionnelle, en assurant le maintien de l'intégrité du circuit préfrontal. Des études chez les rongeurs montrent que la mémoire et l’apprentissage dépendent du sommeil. Des souris soumises à 6h de privation de sommeil immédiatement après une tâche de reconnaissance d'objet ont eu des troubles de mémoire, tandis que la mémoire reste intacte si la privation de sommeil n’est effectuée qu’après 6h suite à la même tâche (Palchykova et al., 2006). Ces données montrent une relation étroite entre le sommeil, la plasticité et la récupération des fonctions cognitives.
La privation de sommeil affecte la cognition, l'attention, et les comportements émotionnels contrôlés par les régions du cerveau comme le néocortex et l'hippocampe (Nilsson et al., 2005; Smith et al., 2002; Yoo et al., 2007). Dans le cortex préfrontal : Les structures corticales sont impliquées dans le maintien de l'attention et la cognition. Des données de traitement du signal électroencéphalographique (EEG) suggèrent que pendant la privation de sommeil, les performances lors des tâches de mémoire et
23
d'attention sont associés à une baisse de l’amplitude des caractéristiques spectrales associées à la vigilance (Gevins et al., 1997). Aussi, l’effet de l'éveil prolongé au-delà de 15 heures sur la capacité de mémoire de travail a montré une association fonctionnelle avec des déficits neuroanatomique, tel que les structures du lobe pariétal et frontal connues pour être impliquées dans la mémoire de travail et d'attention. Des données montrent que la privation de sommeil est liée à l'augmentation de l'activation du cortex préfrontal et du lobe pariétal lors de tâches de l'apprentissage. Cette information suggère que les tâches d'attention partagée nécessitent plus de ressources attentionnelles que normalement requis par une personne privée de sommeil (Drummond et al., 2001) .
Dans l’hippocampe : Des études antérieures ont montré que la perte de sommeil induit une diminution de l’activités de l'hippocampe (Yoo et al., 2007). Ceci est également suggéré par le fait que la privation de sommeil altère l’apprentissage dépendant de l'hippocampe et la mémoire, ceci est associés à des changements des molécules liés à la mémoire et à la plasticité synaptique telles que P-CaMKII, BDNF (Youngblood et al., 1997; Alhaider et al., 2010; McDermott et al., 2003). En outre, le BDNF est réduit après la PS dans l'hippocampe. Il est bien connu que le déficit d'expression hippocampique de BDNF affecte la cognition comme la reconnaissance d'objet et la mémoire spatiale (Heldt et al., 2007)
1.2.4 Les mécanismes moléculaires impliqués dans l’homéostasie du sommeil De nombreuses voies moléculaires sont associées au sommeil/éveil. Nous allons présenter dans ce chapitre la relation entre le sommeil et les marqueurs de plasticité, les cytokines et les gènes de l'horloge biologique, et comment ces éléments peuvent être impliqués dans la régulation de sommeil. Essentiellement, le sommeil est régulé via deux voies moléculaires principales :
1.2.4.1 Voies moléculaires liées à la plasticité synaptique
Suite à une privation de sommeil, l’expression des gènes de plasticité augmente immédiatement d’une manière importante au niveau de l’hippocampe et le cortex cérébral. Cela comprend les gènes de réponse immédiate (Arc, c-Fos, NgfI-A,
24
Homer1a), les facteurs de transcription (Creb), les gènes liés au métabolisme, des facteurs de croissance neuronale (Bdnf) et les protéines de choc thermique (Sheng et Greenberg., 1990; Cirelli and Tononi., 2000; Chen et al., 2006; Maret et al., 2007; Aton et al., 2009; Curie et al., 2013; El Helou et al., 2013). Aussi, la privation de sommeil induit des changement sur l’expression des protienes d’adhesion synaptique telle que Nlgn1, mais ces changement varient selon la souche de l’animal et le variant transcriptionnel étudiés (El Helou et al., 2013). Le sommeil semble être régulé par ces éléments dépendant de la plasticité synaptique. Il est important de souligner que, indépendamment de la catégorie dans laquelle ils sont répertoriés, plusieurs de ces gènes qui augmentent durant l'éveil ou après la PS ont été impliqués dans les mécanismes moléculaires de la plasticité neuronale. Aussi, plusieurs de ces gènes ont été associés à l'apprentissage, la mémoire et à la cognition. La PS induit une régulation différentielle des gènes à réponse immédiate dans le noyau suprachiasmatique et le néocortex (Thompson et al., 2010). Il y a eu des évidences que les gènes d'horloge peuvent être impliqués dans l'homéostasie du sommeil dans le cortex cérébral, en particulier Per1, Per2 et Dbp. En effet, la privation de sommeil induit l’augmentation de Per2 principalement dans le cortex cérébral, tandis que son niveau dans le noyaux suprachiasmatiques (NSC) reste inchangé (Franken et al., 2007; Curie et al., 2015) Les gènes de plasticité sont affectés différemment dans le cortex et dans l’hippocampe. Des études ont permis d’examiner l'expression des gènes de plasticité après la privation de sommeil. Cela a conduit à l'identification des classes de gènes dont les expressions sont modifiées par la privation de sommeil dans le cortex et l'hippocampe (Mackiewicz et al 2009; Wang et al., 2010).
Dans le cortex : Après des périodes de courte durée de la privation de sommeil, il y a une augmentation observée des gènes de plasticité tels que Bdnf, Arc, c-fos et NGFI-A dans le cortex cérébral. Quand la privation de sommeil s’allonge, des changements d'expression des gènes dans d'autres classes de gènes (tels que les gènes exprimant pour : des protéines de choc de chaleur, des protéines chaperons, des facteurs de croissance, des molécules d'adhésion, et des composants de neurotransmission présynaptique et postsynaptique) sont observés dans le cortex (Cirelli et Tononi 2000; Tononi et Cirelli 2001; Cirelli 2002; Cirelli et al 2006; Mackiewicz et al 2009; Wang et al 2010; Curie et al 2013).
25
Dans l’hippocampe: Comme l’hippocampe est lié directement à l'apprentissage et la mémoire, il n’est pas surprenant que la privation de sommeil perturbe la plasticité synaptique dans l'hippocampe. Les changements dans l'expression des gènes liés à la plasticité synaptique dans l'hippocampe après PS diffèrent de ce qui a été observé dans le cortex. Les chercheurs ont même observé des résultats contradictoires concernant l'expression des gènes après la privation de sommeil. Taishi et al. (2001) ont constaté que, après 8 h de la privation de sommeil, le gène à réponse immédiate Arc augmente dans l'hippocampe, tandis que Bdnf reste inchangé. Cependant, d’autres chercheurs ont observé plus tard que le Bdnf, ainsi que CREB, et l’expression de CAMKII ont été réduites après privation de sommeil (Guzman-Marin et al., 2006). Ces changements dans l'expression des gènes au niveau de l'hippocampe sont inversés après 2,5 h de sommeil suite à la PS (Vecsey et al. 2012).
L’état de sommeil et l'éveil diffèrent sur plusieurs volets, que ce soit en matière de comportement, du métabolisme, d’activité neuronale, des médiateurs de l'inflammation ou au niveau de l'expression des gènes. Il est évident que la transition entre ces deux états engendre d’autres changements physiologiques tels que la synthèse des protéines, les neurotransmetteurs et le métabolisme qui peuvent également être associés à la plasticité synaptique. Effectivement, des données récentes montrent que le besoin accru de sommeil induit des changement de l'expression des gènes dans le cerveau impliquant l'ensemble du génome (voir Rachalski et al., 2014 pour revue de littérature).
Les gènes de plasticité et la régulation de sommeil:
L'activité à ondes lentes (0-4 Hz) pendant le sommeil NREM est un indicateur du besoin de sommeil, car elle augmente avec la durée de l'éveil préalable et diminue pendant le sommeil. Dans une étude, Huber et ses collaborateurs (2007) ont voulu déterminer chez des rats, si la plasticité neuronale sous-tend le lien entre l'éveil et l’activité à ondes lentes. À l’aide d’une expermientation qui combine l’EEG et la stimulation magnétique transcrânienne (SMT), ils ont mesurés la quantité d'activité à ondes lentes (AOL) durant le sommeil suivant une tâche d’apprentissage. Plus precisemment, les sujets de l'étude ont subi une potentialisation par SMT et les réponses corticales ainsi que les changements locaux en AOL ont été surveillés à l'aide l’EEG. Les résultats montrent que
26
la SMT produit une potentialisation suivie par une augmentation locale des AOL durant le sommeil. Ensuite ils ont mesuré l’expression corticale de gènes liés à la plasticité (Bdnf, Arc, Homer, NGFI-A). Leur étude suggère un lien direct entre la plasticité synaptique déclenché durant l'éveil et identifie BDNF comme un médiateur majeur de ce lien au niveau moléculaire. En effet, après une privation de sommeil par manipulation douce chez les jeunes rats, les niveaux corticaux de Bdnf ont été augmentés (Hairston et al., 2004). De plus, suite à une cartographie du génome, Thompson et al. (2010) ont montrés que l’expression d’Arc est augmenté dans le cortex et l'hippocampe des souris ayant subi 6h de privation de sommeil comparés aux contrôles.
1.2.4.2 Voies moléculaires liées à l’inflammation
Il a été démontré que des perturbations du cycle veille sommeil peuvent affecter de manière significative la fonction immunitaire. Par exemple, la privation de sommeil augmente les taux circulants de marqueurs inflammatoires telles que l'IL-6, le facteur de nécrose tumorale (TNF) α, et la protéine C réactive (Vgontzas et al., 2004; Meier-Ewert et al., 2004; Irwin et al., 2004). Aussi, la perte de sommeil induit l’altération de la réponse innée de système immunitaire. Ceci est rapporté par une étude ayant observé après une nuit de privation de sommeil une augmentation significative de cytokines et de TNF-α au niveau des monocytes périphériques comparés aux contrôles (Ackermann et al., 2012; Zielinski et al., 2014). En outre, la perte de sommeil a un impact sur l'expression de gènes de cytokines pro-inflammatoires (Irwin et al., 2006). Dans une étude sur de jeunes adultes portant sur la durée/qualité du sommeil et la sécrétion d'IL, la privation de sommeil a augmenté les niveaux diurne d’IL-6 et provoqué de la somnolence et de la fatigue pendant la journée suivante (Vgontzas et al., 1999). Ceci suggère que la perte de sommeil est assez nocive pour troubler le système immunitaire et pour créer un véritable stress pour l'organisme en général et pour le cerveau en particulier (Ackermann et al., 2012).
Les cytokines sont des molécules sécrétées par les cellules du système immunitaire qui agissent comme médiateurs et régulateurs de processus immunitaires, l'inflammation et l'hématopoïèse. Des cytokines, telles que IL-1 ou de TNF-α, NGF, IL-10 et IL-4 semblent tous faire partie d’un réseau de régulation biochimique du sommeil, et modulent plusieurs fonctions telles que la mémoire, la cognition, la fatigue et la somnolence (Pugh
27
et al., 2001; McAfoose et al., 2009; Kurzrock., 2001; Vgontzas et al., 1997). Il est établi que le sommeil à un rôle important dans le renforcement du système immunitaire et que celui-ci change tout au long de la journée en même temps que le cycle veille-sommeil, d’où l’existence probable d’une communication bidirectionnelle entre le sommeil et le système immunitaire (Krueger et al., 1995; Krueger et Toth., 1994). D’ailleurs, un grand nombre de variables qui influent sur le sommeil ont été liées à des mécanismes hormonaux et humoraux, qui sont régulés par une cascade biochimique impliquant des éléments du système immunitaire.
Ainsi, dans les conditions physiologiques de sommeil normal, des cytokines comme l’IL-6 et l’IL-2, sont impliquées à la régulation de sommeil. Une étude chez l’humain, a démontré que les concentrations circulantes d'IL-2 changent dans la période nocturne, avec des niveaux élevés des IL-2 dans les premières heures de sommeil (Irwin et al., 1999). Pareillement, des données suggèrent une interaction entre le sommeil, les rythmes circadiens, et les cytokines (Keller et al., 2009; Yoshida et al., 2014). Comme par exemple, la concentrations de TNF et d'IL-6 chez des patients atteints de troubles du sommeil montrent souvent des différence entre la journée et la nuit (Vgontzas et al., 1997; 1999; Bauer et al 1994). Toutes ces observations suggèrent que le sommeil est modulé par l'inflammation.
Des études au cours des dernières années ont permis d'élucider l'influence de cytokines inflammatoires sur le système circadien. En effet, des observations indiquent que les deux cytokines IL-1 et TNF inhibent l'expression des gènes de l’horloge (Clock, Bmal1, Per) via la perturbation d'activation des éléments de régulation E-box (Cavadini et al., 2007; Krueger et al 2007; Krueger., 2008; Petrzilka et al., 2009). De plus, le niveau de ces cytokines dans le sang est élevé en début de soirée, et plus bas dans la matinée. Par ailleurs, dans les conditions physiologiques normales, les taux plasmatiques de TNF varient parallèlement avec l’activité EEG à ondes lentes et avec la propension à s'endormir. Aussi, l’injection des deux cytokines TNF- α ou IL-1β induit de fortes augmentations du sommeil NREM, et l'inhibition de ces mêmes cytokines réduit le sommeil NREM (Fig. 2). De plus, des observations chez des souris dépourvues de récepteurs TNF ou IL-1β rapportent qu’ils dorment moins comparés aux contrôles. Ceci porte à penser que des changements des acteurs du système immunitaire peuvent assumer un rôle important dans la régulation homéostatique du sommeil (Redwine et al., 2000; Hublin et al., 2007; Papanicolaou et al., 1998).
Figur pour grand (cercl in slow Dans IL-1 hypot 2011 en lie l'expr 2005 est m cellul libéra produ reflèt Krueg somm re 2: L’effet d les 24h suiv de après IL-1β es pleins) pe w wave sleep s le même c dans le fo thèses sur l ; Tononi et C en directe a ression des ). D’autre médiée en p es, l’ATP s ation de cyto uit de la dég te aussi le ger., 2011). meil et l’évei de l’interleuki vantes l’inject β (cercles vid ndant environ p). contexte, Kru onctionneme la fonction d Cirelli, 2006) avec la pla récepteurs g part, la re partie par l se trouve au okines est re gradation de niveau d’a Également l, de la régu ne-1 bêta su tion d’IL-1β. es) que celle n 6h. (Reprod ueger et al. ent synaptiq des oscillatio ). Ces chang sticité syna glutaminergi elation entre ’adénosine-5 ussi dans le enforcée pa l’ATP (adén activité des , l'adénosin lation homéo 28 ur le sommei La durée du observée pe duit de Krueg (2011) prop que ‘Scalin ons lentes p gements aux ptique, surt iques et la p e le proces 5'triphospha es cellules ar l'activité n nosine tripho s neurones e e a été pro ostatique (D l chez la sou u sommeil N endant les enr
er et al., 201 posent un rô ng’, qui es pendant le s x niveaux de tout que le plasticité syn ssus inflam ate (ATP). C gliales (Bur neuronale via osphate). La et des cellu oposé comm Dworak et al. uris. Le somm NREM était b registrements 1; Involveme
ôle des cyto t l'une des sommeil (So es cytokines TNF est im naptique (Pic mmatoire et Comme dan rnstock et a a l’adénosin a production les gliales me marqueu ., 2010). meil enregistr beaucoup plu s des contrôle ent of cytokine okines TNF e s principale omeren et al s peuvent êtr mpliqué dan ckering et al le somme ns toutes le al., 2007). L ne, qui est u n d’adénosin (Zielinski e r, pendant l ré us es es et es l., re ns l., eil es La un ne et le
29
1.3
Le traumatisme crânien
1.3.1 Définition
Le TCC est un problème de santé publique rencontré dans la communauté générale. Il entraîne un large éventail de séquelles à court et à long terme, qui peuvent persister pendant une longue période de temps. Cliniquement, les types de TCC sont classés en fonction de l'échelle de Glasgow (Glasgow coma scale (GCS)), une méthode qui permet de mesurer la sévérité d’un coma, et ce par l’évaluation de 4 critères cliniques: l’ouverture des yeux, la réponse verbale, la perte de conscience et la réponse motrice. Un TCC sévère est définit par un coma avec échelle de Glasgow < 9, et le TCC modéré se définit par un Glascow de 9 à 12. Quant au TCL, il est traduit par un score de Glasgow de 13 à 15, avec un ou plusieurs des éléments suivants : confusion, désorientation, amnésie post-trauma inférieure à 24 heures, perte de conscience inférieure à 30 minutes (Teasdale et Jennett., 1974, Prins et al., 2013). Le présent projet portera spécifiquement sur les TCL.
1.3.2 Le traumatisme crânien léger (TCL)
Selon les données épidémiologiques, plus de 1,5 millions de personnes subissent un TCC chaque année aux États-Unis, dont 75 % sont considéré comme TCL. Ces blessures peuvent provoquer des séquelles à long terme. De plus, beaucoup de patients avec TCL ont des difficultés pour la reprise des activités quotidiennes, et peuvent devenir incapables de retourner au travail pendant plusieurs semaines voir des mois (Bazarian et al., 2005; Macciocchiet al., 1993). Outre les conséquences sur la santé humaine, le TCL entraine des coûts élevés au budget de la santé. Aux États-Unis par exemple, le coût des TCL est de 17 milliards de dollars chaque année selon le centre national de prévention et de contrôle des traumatismes (Rapport au Congrès sur TCL aux États-Unis, 2003).
Malgré la nature légère du TCL, la prévalence des séquelles post-TCL est non négligeable. Ainsi, le TCL est fréquemment associée à un dysfonctionnement cognitif et psychologique, qui comprend des plaintes de fatigue, des maux de tête, de l’anxiété, de la dépression et bien évidemment des troubles de sommeil comme étant les plus courants (Fig. 3). Ces séquelles entraînent des conséquences socio-économiques importantes tel que le chômage et l’invalidité (Dikman et al., 2009; Englander et al.,
1992 patie Petch La va ainsi récup perm bless répon la gr rappo 2011 Figur comm et à lo as a B 1.3 La pl TCL o comm ). En géné nts TCL ap hprapai et W ariabilité du que le man pération diff ettrait de c sures, du pr ndre de man rande major ortera des s ). re 3: Les effe motionnels et d ong terme (Re
Biomarker fo 3.3 Domm upart des ét ont reçu mo motion céré éral, ces sy rès un mois Winkelman., 2 diagnostic nque de bio ficile (Ellenb catégoriser ronostic de nière approp rité des per symptômes ets à court et des éléments eproduit de T or Diagnosis mages dus a tudes ont po ins d'attentio brale avec ymptômes p s, et entre 2007). associée à omarqueurs berg et al., l’offre des récupération priée. En ou rsonnes atte persistants à long terme s qui servent p Toledo et al., and Progno aux TCL orté sur les T
on comparés un impact 30 post-commo 15 à 25% a une définitio fiables rend 2009). Un soins en f n et du nive tre, si le pro eintes de T au-delà de e d’un TCL. S pour le diagn 2012; The Y osis.)
TCC modéré s aux problè moins sévè otionnel affe après un an on plus ou dent l’évalu ne meilleure fonction du eau de serv onostic de ré TCL, un cer trois mois Schéma desc ostic des com
Young Brain a ées et sévèr èmes qui en ère tend à ectent jusqu n (Bazarian moins unifo ation du pro e identificati niveau de vices nécess écupération rtain nombr
(Wiseman-criptif des sym mmotions céré and Concus res. En cons découlent. P affecter be u'à 50% de et al., 1999 orme du TCL onostic de l ion des TC gravité de saires pour est bon pou re d’individu Hakes et al mptômes pos ébrales à cou sion: Imagin séquence, le Pourtant, un eaucoup plu es 9; L, la CL es y ur us l., st-urt ng es ne us
31
fréquemment les patients. C’est ainsi que les TCL sont décrits comme une épidémie silencieuse (Feinstein et Rapoport 2000). On sait que le cerveau est protégé contre les chocs par le crâne, mais les méninges et le liquide céphalo-rachidien demeurent toutefois très fragiles. Suite à la lésion, la force de l’impact externe induit une atteinte du flux sanguin et de la pression intracrânienne (Fig. 3). Cet impact cause souvent des dommages et des lésions aux tissus, appelés dommages primaires. Cependant, les lésions dues aux TCL sont souvent invisibles en neuro-imagerie (IRM, TDM), mais ils entraînent toutefois des altérations au niveau neuronal, telle que l’atteinte de la substance blanche, la dégénérescence des dendrites, ainsi que la réduction du nombre des synapses (Gao et al., 2011). Effectivement, une étude chez les patients TCC montre que la concentration de la matière grise a diminué dans plusieurs régions du cerveau y compris le cortex frontal et temporal comparés aux contrôles (Gale et al., 2005). Les données actuelles soulignent également que les changements de la substance blanche existent chez la population TCC à différentes sévérités, y compris les TCL. Aussi la morphologie des épines dendritiques est affectée de manière significative suite aux lésions du cortex ipsilatéral (Winston et al., 2013; Gao et al., 2011; Tay et al., 2010; Kraus et al., 2007). Ces altérations synaptiques et dendritiques peuvent nuire considérablement au fonctionnement du système nerveux central et en conséquence nuire à la mémoire, la cognition et au sommeil (Tada et Sheng., 2006).
Les changements qui succèdent aux dommages primaires donnent naissance aux dommages secondaires. Il a été montré que le TCC déclenche une réponse cellulaire (Fig. 4A) dans les différents circuits neuronaux que nous allons présenter dans les sections suivantes. Cette réponse cellulaire aboutie à la neuro-dégénérescence corticale et hippocampique, entraînant une perte majeure de tissu dans le cortex, qui s’étend souvent dans l'hippocampe. Aussi, des données indiquent que le TCC modéré déclenche une mort nécrotiques des neurones immatures dans l'hippocampe chez la souris (Colicos et al., 1996; Fox et al., 1998; Kochanek et al., 2006; Mtchedlishvili et al., 2010; Yang et al., 2010; Zhou et al., 2012).
Figur (A): C dépol métab huma immu as a B 1.3 Les t préva peuve somm somn somn Baum collec sugg (Ducl altéré l’altér troub assoc al., 2 re 4: Schéma Certaines cha arisation neu boliques et l’a ain suite au nohistochimie Biomarker for 3.4 Troub roubles de s alence varie ent prendre meil), l’insom nolence diur nolence diur mann et al., cte des don
érant que l los et al., 20 é présente u ration de so bles d’attenti cié à la dou 009; Wilde e a descriptif de angements p uronale, la li altération de TCL identif e. (Reproduit Diagnosis and le de somm sommeil (TS selon les é plusieurs fo mnie (difficu rne et le déla rne excessiv 2007). Dan nées pertine es TS se d 013; 2014; T un impact n ommeil peu on et de la leur post-TC et al., 2007; es changeme hysiologiques ibération de débit sanguin fiés par APP de Toledo et d Prognosis.)
meil chez les S) sont les p études de 30 ormes, telles ulté d’endo ai de phase ve semble ns les derniè entes sur le développent Tham et al., égatif sur la ut exacerbe vitesse de t CL (Castriott Bloomfield e 32 ents cellulaire s surviennent neurotransm n cérébral. (B P (immunore t al., 2012; Th . s TCL : roblèmes les 0 à 80% (Co s que l’hyper rmissement e (se couche être la plus ères années s TS à la s à travers 2012). Les a récupératio r les troubl traitement d ta et al., 200 et al., 2010; es et métabo t au cerveau metteurs exci B): Changem eactivity to a he Young Brai s plus rappo ohen et al., rsomnie (allo ou de ma er plus tard e s répandue s, la recherc uite de diffé un large sp données su on neuronal les de l'hum de l'informati 09; Mahmoo Khoury et a liques en rép u suite au TC tateurs, des ents axonaux amyloid prec in and Concu ortés suite à 1992). Les ongement d aintien du et se lever p (Baumann che cliniques érentes form pectre des uggèrent qu le et cogniti meur, de m ion, et il peu od et al., 200 al., 2013). ponse au TC CL, tels que changemen x dans le tiss cursor protein ussion: Imagin à un TCL, leu TS post-TC e la durée d sommeil), l plus tard). L et al., 2012 s a permis l mes des TCC patients TC ue le somme ve. En outre mémoire, de ut même êtr 04; Makley e L. la ts su n) ng ur CL de la La 2; la C, CL eil e, es re et
33
Ces troubles sont suffisamment prévalents pour qu’ils soient pris en considération. Toutefois, quelques difficultés nuancent l’identification des TS post-TCL comme l’absence d’homogénéité dans la blessure puisqu’on ne retrouve pas les mêmes symptômes et les mêmes TS chez les différents patients. Plusieurs facteurs augmentent la variabilité, comme la capacité du patient à s’autoévaluer et à rapporter son vécu, à cause du choc subi qui affecte souvent la cognition (Baumann et al., 2007; Castriotta et al., 2001).
Au cours des dernières décennies, des modèles animaux ont été développés pour reproduire les différents aspects de TCC humain, afin de mieux comprendre la physiopathologie sous-jacente, et pour explorer des traitements pertinents. Pareillement pour les TCL, les modèles animaux sont nécessaires pour élucider la physiopathologie des troubles du sommeil post-TCL. Récemment, une étude utilisant un système de cage piézoélectrique pour l’enregistrement de sommeil, a montré que les souris avec un TCC modéré ou léger avaient une augmentation importante du sommeil dans les premières heures après le TCC et indépendamment du moment de la journée où la chirurgie TCC a eu lieu (Rowe et al., 2014). Cependant, les limites de cette étude n’ont pas permis l’identification des différents états de sommeil post-TCL (sommeil NREM et REM), étant donné que l’enregistrement avait été fait par un système qui n’enregistre pas l’EEG. Une autre étude menée chez des souris TCL/Sham par percussion de fluide (Fluid percussion injury (FPI)), et utilisant l’EEG, rapporte plusieurs épisodes d'éveil de courte durée une semaine après un TCL, concluant que les souris TCL étaient incapables de rester éveillées pendant de longues périodes de temps une semaine post-TCL (Lim et al., 2013). De plus les épisodes de sommeil étaient fragmentés (épisodes individuels plus court) et la durée totale du sommeil NREM était augmentée (Lim et al., 2013). Une augmentation générale de la fragmentation des épisodes d’éveils et de sommeil a aussi été observée pendant la période d’activité des souris, et ce dans les trois premiers jours après un TCC modéré à sévère (Willie et al., 2012). Autre donnée intéressante, les neurones orexinergiques qui aident à maintenir l’éveil sont affectés suite au TCL, spécifiquement, le TCL réduit le marquage pour l’Orexine-A dans l'hypothalamus latéral (Chemelli et al., 1999; Skopin et al., 2014). Par ailleurs, ces données concordent bien avec des études menées chez l’humain, montrant une diminution des niveaux d'orexine dans le liquide céphalo-rachidien après la lésion traumatique (Nishino et al., 2001; Strawn et al., 2010). Ces données ensembles indiquent l’altération de l’éveil et des systèmes qui y sont reliés après TCL. Toutefois, d’autres études sont nécessaires pour élucider le développement et la physiopathologie des troubles du sommeil post-TCL et surtout pour comprendre avec plus de détail la progression du sommeil et son rôle dans la récupération suite au TCL.