Exploration des approches pangénomiques en
amélioration variétale chez l’orge à six rangs dans l’Est
du Canada
Thèse
Amina Abed
Doctorat en biologie végétale
Philosophiæ doctor (Ph. D.)
Exploration des approches pangénomiques en
amélioration variétale chez l’orge à six rangs dans
l’Est du Canada
Thèse
Amina Abed
Sous la direction de :
Résumé
L’émergence du génotypage à haut débit et le développement de méthodes statistiques reliant le génotype au phénotype ont donné lieu à des approches pangénomiques, c’est-à-dire à l’échelle du génome entier, exploitables en sélection des plantes. Ces approches ont d’abord permis d’examiner l'association entre génotype et phénotype via des analyses d'association pangénomique (GWAS en anglais) afin d’identifier des loci de caractères quantitatifs (« quantitative trait loci », QTL) utiles en sélection assistée par marqueurs (SAM). Plus récemment, ces approches ont été explorées pour la prédiction génomique, laquelle vise, d’une part, à identifier les croisements les plus prometteurs (la sélection des croisements), et d’autre part, à identifier les individus les plus prometteurs au sein d’une descendance (la sélection génomique). Dans les deux cas, ces prédictions reposent sur un modèle statistique reliant le génotype et le phénotype au sein d’une population de référence. Ces approches pangénomiques offrent un grand potentiel, mais sont encore émergentes et de nombreuses questions se posent encore chez l’orge. Notre étude s’intéresse à certaines de ces interrogations et elle est divisée en quatre volets de recherche.
Les approches pangénomiques nécessitent un nombre important de marqueurs moléculaires de type SNP (« single nucleotide polymorphism »). Ainsi dans le premier volet nous avons optimisé le protocole de génotypage par séquençage. Ce volet détaille tout le processus, de la préparation des librairies de séquençage jusqu’à la production d’un catalogue de SNP de haute qualité. À titre d’illustration, nous avons généré un catalogue de 30 000 SNP ayant une distribution chromosomique intéressante et une grande exactitude des génotypes.
Dans le deuxième volet, en utilisant les données phénotypiques et génotypiques d’une population d’amélioration, nous avons comparé l’efficacité de trois approches GWAS (Uni-SNP, Multi-SNP et Haplotypique) pour détecter des QTL pour des caractères agronomiques importants. Les approches Multi-SNP et Haplotypique ont identifié plus de QTL que l’approche Uni-SNP. Le chevauchement entre les approches était limité, chaque approche découvrant un sous-ensemble différent de QTL. Dans le cadre du troisième volet, nous avons étudié l’impact de trois facteurs sur la justesse de la sélection génomique : (1) la performance de différents modèles statistiques (incluant ou non l’épistasie), (2) le nombre de marqueurs employés ainsi que (3) leur localisation (génique/non-génique). Le modèle qui intègre les effets additifs et épistatiques a montré les meilleures performances même si les différences entre les modèles étaient modestes. Jusqu’à 2K SNP, la justesse de la sélection génomique est restée comparable à celle basée sur le catalogue entier (35K), mais une
diminution significative été observée à 500 SNP. Dans la plupart des cas, l’utilisation de SNP présents dans les régions géniques, voire codantes, n’a pas apporté une amélioration significative.
Enfin, dans le quatrième volet, nous avons exploré la sélection génomique et la sélection des croisements. En premier lieu, nous avons constitué une population de référence pour bâtir un modèle de sélection génomique et prédire les performances de 350 descendants développés dans un programme d’amélioration. A partir des prédictions, 35 lignées ont été sélectionnées et testées au champ afin d’examiner la corrélation entre les performances prédites et observées. Les corrélations étaient satisfaisantes pour la résistance à la fusariose et le rendement. Ensuite, sur la base de ce modèle, nous avons prédit la moyenne (𝜇) et la variance génétique (𝑉𝐺) de chacune des descendances
simulées issues de tous les croisements possibles (30 000). La validation de ces prédictions a été réalisée rétrospectivement sur un sous-ensemble de croisements précédemment réalisés, en examinant leur persistance dans le processus de sélection. Tel qu’attendu les croisements les plus persistants (>F9) ont présenté des 𝜇 supérieures, mais des 𝑉𝐺 modérées. Même si la résistance à la fusariose et
le rendement sont corrélés défavorablement, nous avons pu identifier des croisements (650) où cette corrélation était rompue. Parmi ces croisements, certains (40) auraient un réel potentiel avec des performances égales ou supérieures à des lignées témoins performantes.
Au terme de ce projet, nous avons démontré l'efficacité d'une procédure GWAS combinant des approches uni- et multi-locus à disséquer des caractères complexes et à détecter des QTL clés utilisables en SAM. Nous avons aussi démontré que la prédiction génomique peut être optimisée et efficacement intégrée en sélection génétique chez l’orge à six rangs pour identifier les meilleurs descendants, mais surtout pour identifier des croisements prometteurs.
Abstract
The emergence of high-throughput genotyping and the development of statistical methods linking genotype to phenotype have led to pangenomic approaches, performed on a genome-wide scale, exploitable in plant breeding. First, these approaches were used to examine the association between genotype and phenotype in genome-wide association studies (GWAS) in order to identify quantitative trait loci (QTLs) useful in marker-assisted selection (MAS). More recently, these approaches have been explored in genomic prediction which aims, on the one hand, to identify the most promising crosses (genomic mating), and on the other hand, to identify the most promising lines within a set of progeny (genomic selection). In both cases, these predictions are based on a statistical model linking genotype to phenotype in a training population. These genome-wide approaches offer great potential but are still emerging and many questions remain unanswered in barley. Our study focuses on some of these questions and is divided into four areas of research.
Genome-wide approaches require a large number of single nucleotide polymorphism (SNP) markers. Thus, in the first part of this project, we optimized the protocol of genotyping by sequencing (GBS). This part details the entire process, from the preparation of GBS libraries until the production of a high-quality SNP catalog. As an illustration, we generated a catalog of 30,000 SNPs with a broad chromosome distribution and high genotype accuracy.
In the second part, using phenotypic and genotypic data from a breeding population, we compared the effectiveness of three GWAS approaches (Single-SNP, Multi-SNP and Haplotype-based) to detect QTLs for important agronomic traits. The Multi-SNP and Haplotype-based approaches identified more QTLs than the Single-SNP approach. The overlap between the approaches was limited, as each approach uncovered a different subset of previously validated QTLs.
In the third part we studied the impact of three factors on the accuracy of genomic selection: (1) the performance of different statistical models (including epistasis or not), (2) the number of SNP markers included in the model as well as (3) their localization (genic/non-genic regions). The model that incorporates both the additive and epistatic effects of SNPs showed the best performance even though the differences between the models were modest. With as few as 2K SNP, the accuracy of genomic selection remained comparable to that based on the entire catalog (35K), while a significant decrease in accuracy was observed at 500 SNPs. In most cases, the use of SNPs located in genic regions, even coding regions, did not provide a significant improvement.
Finally, in the fourth part, we explored genomic selection and genomic mating in a breeding program. First, we established a training population to build a genomic selection model and to predict the performance of 350 progeny developed in a breeding program. Based on these predictions, 35 lines were selected and tested in the field to examine the correlation between predicted and observed performances. The correlations were satisfactory for Fusarium head blight (FHB) resistance and yield. Then, based on this model, we predicted the mean (µ) and the genetic variance (VG) of each simulated progeny from all possible crosses (n = 30,000) between lines of the training population. The validation of these predictions was carried out retrospectively on a subset of previously performed crosses by examining their persistence in the selection process. As expected, the most persistent crosses (> F9) displayed high µ but moderate VG. Although resistance to FHB and yield are unfavorably correlated, we could identify crosses (650) where this correlation was weakened. Among these crosses, some (40) are predicted to offer equal or better performance than current checks. Through this project, we demonstrated the efficiency of a GWAS procedure combining single- and multi-locus approaches to dissect complex characters and to detect key QTLs that can be used in MAS. We also demonstrated that genomic prediction can be optimized and efficiently integrated in genetic improvement of six-row barley to identify the best progeny but also to identify promising crosses.
Table des matières
Résumé ... II Abstract ... IV Table des matières ... VI Listes des tableaux ... X Liste des figures ... XI Liste des abreviations ... XIII Remerciements ... XVI Avant-propos ... XVII
Introduction générale ... 1
Chapitre 1 revue bibliographique ... 4
1.1. Culture de l’orge et ses contraintes ... 5
1.2. La caractérisation génotypique ... 5
1.3. Analyses d’association ... 9
1.3.1.PRINCIPE DES ANALYSES D’ASSOCIATION PANGENOMIQUE ... 9
1.3.2.LES FACTEURS INFLUENÇANT LE GWAS ... 9
1.3.2.1. Les approches statistiques ... 10
1.4. La prédiction génomique ... 12
1.4.1.LA SÉLECTION GÉNOMIQUE ... 13
1.4.1.1. Principe de la sélection génomique ... 13
1.4.1.2. Estimation de la justesse de la sélection génomique ... 13
1.4.1.3. Les facteurs qui influencent l’exactitude de la sélection génomique ... 15
1.4.1.3.1. Modèles statistiques ... 15
1.4.1.3.2. La taille et la composition de la population de référence ... 17
1.4.1.3.3. La densité des marqueurs moléculaires ... 18
1.4.1.3.4. L’héritabilité et l’architecture génétique ... 19
1.4.2.LA PRÉDICTION DES CROISEMENTS ... 19
1.4.2.1. Principe et avantages de la prédiction des croisements ... 19
1.4.2.2. Méthodes de prédiction des croisements ... 20
1.4.2.3. Avancée et limite de la prédiction des croisements ... 21
1.5. Projet de thèse de doctorat ... 21
Chapitre 2 genotyping-by-sequencing on the ion torrent platform in barley ... 24
2.1. Resume ... 25
2.2. Abstract ... 25
2.3. Introduction ... 25
2.4. Materials ... 26
2.4.1.DNA EXTRACTION ... 26
2.4.2.GBS LIBRARY PREPARATION AND SEQUENCING ... 27
2.5. Methods ... 29 2.5.1.DNA EXTRACTION ... 29 2.5.2.GBS LIBRARY PREPARATION ... 29 2.5.3.SEQUENCING ... 33 2.5.4.GBS DATA ANALYSIS ... 35 2.5.5.FURTHER SNP FILTERING ... 35 2.6. Example ... 36 2.7. Notes ... 40 2.8. References ... 42
Chapitre 3 comparing single-snp, multi-snp, and haplotype-based approaches in association studies
for major traits in barley ... 44
3.1. Resume ... 45
3.2. Abstract ... 45
3.3. Introduction ... 46
3.4. Materials and methods ... 49
3.4.1. GERMPLASM AND EXPERIMENTAL PHENOTYPIC DATA ... 49
3.4.2. PHENOTYPIC DATA ANALYSIS ... 50
3.4.3. GENOTYPIC DATA ... 51
3.4.4. SINGLE NUCLEOTIDE POLYMORPHISM CALLING AND FILTRATION PROCEDURE ... 51
3.4.5. HAPLOTYPE CATALOG CONSTRUCTION ... 52
3.4.6. POPULATION STRUCTURE ... 52
3.4.7. GENOME-WIDE ASSOCIATION APPROACHES ... 52
3.4.8. COMPARISON OF THE THREE GWAS APPROACHES ... 54
3.4.9. CANDIDATE GENE ANALYSIS AND VALIDATION ... 55
3.5. Results ... 55
3.5.1. PHENOTYPIC DATA ... 55
3.5.2. GENOTYPIC DATA ... 56
3.5.2.1. Genotyping and cataloguing of haplotypes ... 56
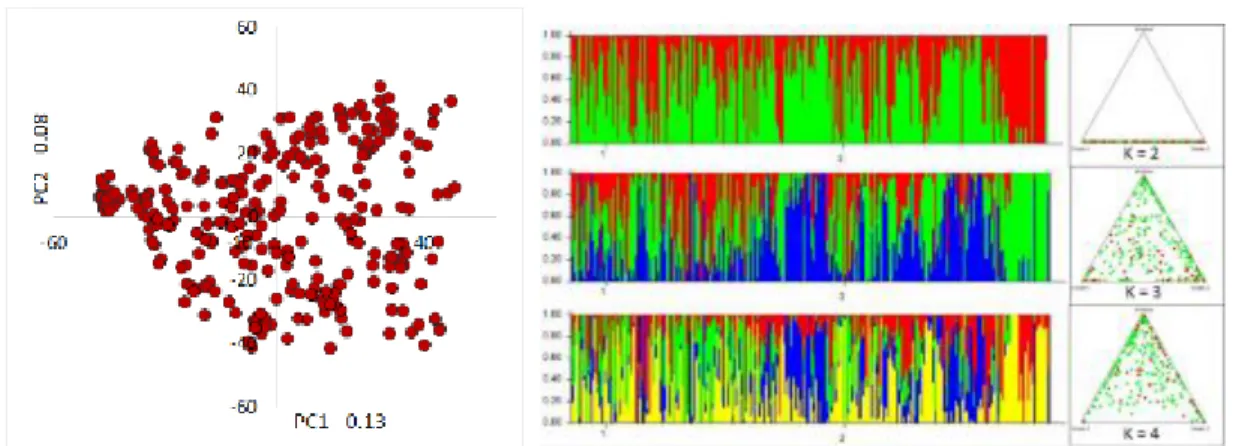
3.5.2.2. Population structure ... 57
3.5.3. GENOME-WIDE ASSOCIATION STUDY APPROACHES ... 57
3.5.3.1. Comparison of the three gwas approaches ... 57
3.5.3.2. Comparison with candidate genes and qtls ... 61
3.6. Discussion ... 62
3.6.1. WHICH APPROACH HAD THE HIGHEST CAPACITY TO DETECT QTLS? ... 62
3.6.2. COMPARISON WITH CANDIDATE GENES AND QTLS ... 64
3.6.3. IS THERE A CORRELATION BETWEEN YIELD AND THE QUALITY OF THE APPROACHES? 65 3.6.4. WHAT IS THE BEST APPROACH? ... 66
3.7. Supplemental information ... 67
3.8. Conflict of interest disclosure ... 68
3.9. Acknowledgments ... 68
3.10. Author contributions ... 68
3.11. References ... 68
Chapitre 4 when less can be better: how can we make genomic selection more cost-effective and accurate in barley? ... 76
4.1. Résumé ... 77
4.2. Abstract ... 77
4.3. Introduction ... 78
4.4. Materials and methods ... 81
4.4.1. BARLEY POPULATIONS ... 81
4.4.2. EXPERIMENTAL PHENOTYPIC DATA ... 81
4.4.3. PHENOTYPIC DATA ANALYSIS ... 83
4.4.4. GENOTYPIC DATA ... 83
4.4.5. SNP CALLING AND PROCEDURES FOR VARYING THE NUMBER OF SNPS OBTAINED ... 84
4.4.6. SNP DISTRIBUTION ON THE PHYSICAL AND GENETIC MAPS AND THEIR FUNCTIONAL IMPACT 84 4.4.7. METHODS FOR ESTIMATING THE ACCURACY OF PREDICTED PHENOTYPES ... 85
4.4.8. GENOMIC-ENABLED PREDICTION MODELS ... 85
4.4.9. POPULATION STRUCTURE AND RELATEDNESS BETWEEN LINES ... 87
4.4.10. IMPACT OF THE NUMBER OF SNPS ON PREDICTION ACCURACY ... 88
4.5. Results ... 88
4.5.1. IMPACT OF SEQUENCING DEPTH AND SNP FILTERING ON THE NUMBER OF SNPS AND THE ACCURACY OF GENOTYPE CALLS ... 88
4.5.2. SNP DISTRIBUTION AND FUNCTIONAL IMPACT ... 90
4.5.3. PHENOTYPIC EVALUATION ... 92
4.5.4. ACCURACY OF DIFFERENT GENOMIC SELECTION MODELS ... 92
4.5.5. IMPACT OF THE NUMBER OF SNPS ON THE ACCURACY OF PREDICTIONS ... 94
4.5.6. IMPACT OF THE LOCALIZATION OF SNPS ON PREDICTION ACCURACY ... 96
4.6. Discussion ... 97
4.6.1. CAN WE REDUCE THE NUMBER OF SNPS AND GENOTYPING COSTS? ... 97
4.6.2. TRADE-OFF BETWEEN MARKER DENSITY, GENOTYPING COST AND PREDICTION ACCURACY ... 98
4.6.3. IS THERE AN ADVANTAGE TO CAPTURING EPISTASIS IN GS MODELS? ... 101
4.6.4. DO SNPS IN GENIC REGIONS LEAD TO MORE ACCURATE PREDICTIONS? ... 101
4.7. Author contribution statement ... 102
4.8. Acknowledgements ... 102
4.9. Compliance with ethical standards ... 102
4.10. Supplementary material ... 102
4.11. References ... 103
Chapitre 5 exploring the realm of possibilities: trying to predict promising crosses and successful offspring through genomic mating in barley ... 111
5.1. Resume ... 112
5.2. Abstract ... 112
5.3. Introduction ... 113
5.4. Materials and methods ... 115
5.4.1. GERMPLASM AND EXPERIMENTAL DESIGN ... 115
5.4.2. PHENOTYPIC DATA AND ANALYSIS ... 115
5.4.3. GENOTYPIC DATA ... 116
5.4.3.1. Genotyping ... 116
5.4.3.2. Snp calling and filtration procedure ... 116
5.4.3.3. Assignment of snp genetic positions ... 117
5.4.4. GENOMIC PREDICTION APPROACHES ... 117
5.4.4.1. Genomic selection ... 117
5.4.4.1.1. Statistical model ... 117
5.4.4.1.2. Validation of genomic selection ... 118
5.4.4.2. Genomic mating ... 118
5.4.4.2.1. Validation of genomic mating ... 118
5.4.4.2.2. Selecting the best crosses ... 119
5.5. Results ... 119
5.5.1. GENOTYPING ... 119
5.5.2. TRAINING AND ASSESSMENT OF THE GENOMIC SELECTION MODEL ... 120
5.5.3. VARIANCE AND GEBV PREDICTIONS OF CROSSES ... 122
5.5.4. RETROSPECTIVE VALIDATION ... 123
5.5.5. CORRELATIONS BETWEEN PAIRWISE COMBINATIONS OF TRAITS ... 126
5.6. Discussion ... 127
5.6.1. GENOMIC SELECTION ACCURACY ... 127
5.6.2. ARE PREDICTIONS OF THE POTENTIAL PERFORMANCE OF CROSSES ACCURATE ENOUGH? 128 5.6.3. CORRELATIONS BETWEEN DON AND GYD ... 130
5.7. Conclusion ... 131
5.9. Author contributions ... 133
5.10. Conflicts of interest ... 133
5.11. Acknowledgments ... 133
5.12. References ... 133
Chapitre 6 Discussion générale ... 138
Conclusion... 148
Listes des tableaux
Table 2.1 Oligonucleotide sequences………..27
Table 2.2 Barcode sequences………..28
Table 2.3 Summary of genotyping by sequencing and SNP calling results……….38
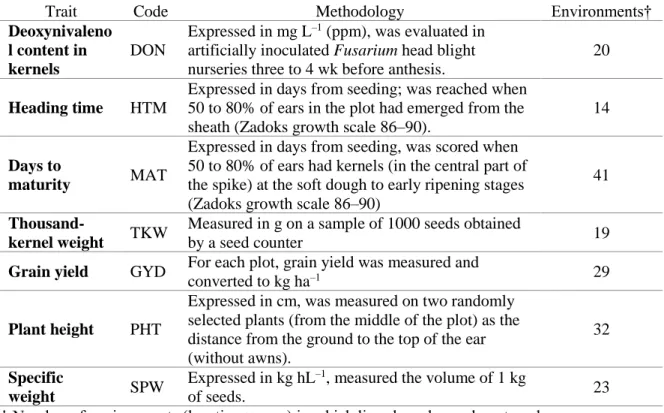
Table 3.1 Methodology of measurement for seven traits……….50
Table 3.2 Descriptive statistics of phenotypic data (blue) for each trait……….56
Table 4.1 Number of years, locations and environments from which phenotypic data were obtained for six traits for both the training and validation populations……..……….81
Table 4.2 Number, accuracy and reproducibility of SNP calls obtained at four depths of sequencing per line (multiplexing) and four levels of tolerance for missing data………..88
Table 4.3 Distribution of SNPs across the barley chromosomes in three highly contrasting sets of SNP markers (conditions)………...90
Liste des figures
Figure 1.1 Les étapes simplifiées d’une approche GBS depuis l’extraction d’ADN au séquençage…..7
Figure 1.2 Schéma représentant la procédure complète d’une approche GBS………..8
Figure 1.3 Schéma résumant une procédure de sélection génomique………..14
Figure 2.1 An example of high-quality library………34
Figure 2.2 A run summary for one chip ion torrent (Proton)………34
Figure 2.3 Read quality for one ion torrent chip computed with FastQC……….37
Figure 2.4 A sample of 30,000 SNP catalog obtained after filtration and imputation……….37
Figure 2.5 Distribution of 30,000 SNP loci across the IBSC physical map……….38
Figure 2.6 Manhattan plot for GWAS of heading time…...………39
Figure 2.7 Population structure analysis using two methods: (i) principal component analysis and (ii) clustering using structure………39
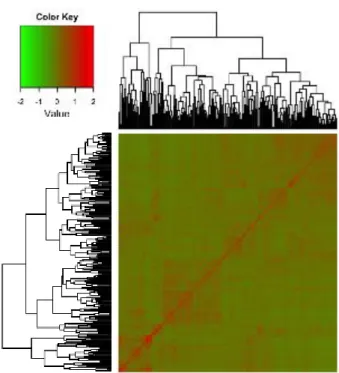
Figure 2.8 Heat map representing the realized genomic relationship (G matrix) among lines……….40
Figure 3.1 Manhattan plot of trait DON, HTM and TKW………..……….58
Figure 3.2 Phenotypic variance explained by significant SNPs (R2) for each GWAS approach.…….60
Figure 3.3 Cumulative phenotypic variance explained by significant QTLs (cumulative R2) for each GWAS approach……….………60
Figure 3.4 Heritability for each trait in each GWAS approach………61
Figure 3.5 Proximity of chromosomal regions detected by single-SNP, multi-SNP and haplotype-based approaches to QTL/genes reported by previous GWAS and linkage mapping studies on barley……….……….62
Figure 4.1 Distribution of SNP loci on the genetic map in three highly contrasted conditions………90
Figure 4.2 Pairwise scatterplot matrix of the accuracy of three prediction models assessed through cross-validation (80:20)………..…………92
Figure 4.3 Prediction accuracies for different sizes of SNP sets assessed through cross-validation (80:20)………....93
Figure 4.4 Principal component analysis (PCA) of the training population (TP) lines (circles) and validation population (VP) lines (triangles)………94
Figure 4.5 Prediction accuracies with different sizes of SNP sets for six traits assessed through inter-population validation………..………95
Figure 4.6 Prediction accuracies with the different categories of SNP sets for six traits assessed through inter-population validation………96 Figure 5.1 Prediction accuracies assessed through cross-validation (60:40)………119 Figure 5.2 Prediction accuracies assessed through a validation process………...120 Figure 5.3 Pattern of relationships between predicted progeny mean and variance for all 30,000 pairwise crosses between the 245 lines in the TP...………122 Figure 5.4 Persistence of crosses over the course of selection emphasizing grain yield (GYD) in the UL breeding program………123 Figure 5.5 Persistence of crosses over the course of selection emphasizing grain yield (GYD) in the Céréla breeding program…………...………124 Figure 5.6 Persistence of crosses over the course of selection emphasizing FHB resistance (low DON)………124 Figure 5.7 Correlations between GEBVs for three pairs of traits among the simulated progeny of all pairwise crosses between lines of the training set…….………....125
Liste des abréviations
ADN Acide DésoxyriboNucléiqueBOPA Barley Oligonucleotide Pool Assays DON désoxynivalénol
EBV estimated breeding values
FASTmrEMMA fast multi-locus random-SNP-effect efficientmixed model association GBS genotyping by sequencing
GEBV genomic estimated breeding value GWAS genome-wide association studies GYD le rendement
HTM l’épiaison
IGST IBIS Genotyping by Sequencing Tools
ISIS EM-BLASSO Iterative sure independence screening Expectation-Maximization (EM)-Bayesian least absolute shrinkage and selection operator
LASSO least absolute shrinkage and selection operator LD déséquilibre de liaison
MAT la maturité
MLMM multi-locus mixed-model
mrMLM a multi-locus random-SNP-effect mixed linear model NGS next-generation sequencing
PCR Polymerase chain reaction PHT la taille des plantes
pKWmEB Kruskal–Wallis test with empirical Bayes
pLARmEB polygenic-background-control-based least angle regression plus empirical Bayes QTL quantitative trait loci
RR-BLUP ridge regression best linear unbiased prediction SAM sélection assistée par marqueurs
SNP single nucleotide polymorphism SPW le poids à l’hectolitre
TKW le poids de mille grains
Remerciements
Mon directeur de thèse François BELZILE est la première personne que je veux remercier, non seulement pour la qualité exceptionnelle de son encadrement, mais aussi pour sa présence, son engagement et ses qualités humaines.
Je tiens à remercier les membres du jury : Lucia GUTIERREZ, Nathalie ISABEL et Charles GOULET d’avoir consacré une partie de leur temps précieux pour évaluer ma thèse.
Mes plus sincères remerciements vont à Suzanne MARCHAND, Martin LACROIX et Martine JEAN pour leur aide précieuse, leur disponibilité et les innombrables échanges, j’ai beaucoup appris à vos côtés.
Nos partenaires dans ce projet : Samuel OSTIGUY, Annie ARCHAMBAULT de Céréla et Nathalie LANOIE se sont démarqués par leur professionnalisme et l’intérêt, toujours grandissant, porté au projet. Ce fut un plaisir de collaborer avec eux.
J’ai eu la chance de collaborer avec José CROSSA et Paulino PEREZ-RODRIGUEZ au sein de l’unité biométrique et statistique de CIMMYT, je vous remercie de m’avoir donné cette opportunité très enrichissante. Par ailleurs, je remercie Juan BURGUEÑO de m’avoir invité à CIMMYT ainsi que toutes les personnes travaillant au sein de cette unité, des personnes brillantes et toujours disponibles.
Mes remerciements vont également à la Plate-forme d’analyse génomique, particulièrement à Brian BOYLE, ainsi que la Plateforme bio-informatique de l’IBIS pour l’excellent service offert par chacune des personnes travaillant au sein de ces plateformes.
Je tiens également à remercier Bachar CHEAIB et Sidiki MALLE, ainsi que tous les étudiants, stagiaires, personnels du laboratoire François Belzile, du département de phytologie et de l’IBIS (depuis janvier 2014) qui ont pu m’aider de prêt ou de loin.
Ma mère Fatma-Zohra et mon père Saïd ont joué un rôle très important dans cette aventure. Leur soutien inébranlable, leur foi en moi et leur fierté m’ont guidés tout le long de mon parcours. A mes sœurs et meilleures amies Leila et Khadidja, j’adresse mes plus sincères remerciements pour leur présence et leur soutien continus malgré la distance. A mes frères Adnane et particulièrement Hakim, merci de m’encourager à votre manière.
Mes petits trésors : Sofia, Narimene, Lylia et Yacine, qui ne comprenaient pas pourquoi ces études duraient aussi longtemps, je vous remercie d’avoir été compréhensifs lorsque je ratais un évènement important de votre vie et de m’encourager malgré tout.
A tous mes amis et proches, ici et ailleurs dans le monde, qui m’ont encouragés et soutenus, je vous dis merci.
Avant-propos
Notre étude se divise en quatre volets de recherche (chapitres 2, 3, 4 et 5), chacun prenant la forme d’un article scientifique publié ou d’un manuscrit pour fins de publication. Le chapitre 1 est dédié à une revue bibliographique qui met en lumière les principes des analyses d’association et de la prédiction génomique, les acquis réalisés chez l’orge, les interrogations non encore élucidés, ainsi que les limites de ces approches génomiques. Le chapitre 6 contient une discussion générale de l’étude dans laquelle les principaux résultats sont discutés et intégrés pour démonter la cohérence et la pertinence de la présente étude.
Le chapitre 2 a été publié en 2019, sa référence est : Abed A., Légaré G., Pomerleau S., St-Cyr J., Boyle B. and Belzile FJ. (2019). Genotyping-by-sequencing on the ion torrent platform in barley. In: W.A. Harwood, editor, Barley, Methods and protocols. Vol. 1900. Springer. New York. p. 233–252. AA et FB ont conçu l’étude. AA, GL, SP et JS ont réalisé et optimisé les protocoles expérimentaux au laboratoire, AA et BB ont effectué des analyses bio-informatiques et interprété les résultats. AA, BB et FB ont rédigé le manuscrit.
Le chapitre 3 a été publié en novembre 2019 dans le périodique « The Plant Genome ». Sa référence partielle est : Abed A. and Belzile F. (2019). Comparing Single-SNP, Multi-SNP, and Haplotype-Based Approaches in Association Studies for Major Traits in Barley. 12:180036.AA et FB ont mis au point l’étude. AA a réalisé des expériences sur le terrain et en laboratoire, effectué des analyses bio-informatiques et statistiques et interprété les résultats. AA et FB ont rédigé le manuscrit.
Le chapitre 4 a été publié en Juin 2018 dans le périodique « Theoretical and Applied Genetics ». Sa référence partielle est : Abed A., Pérez-Rodríguez P., Crossa J. and Belzile F. (2018). When Less Can Be Better: How Can We Make Genomic Selection More Cost-Effective and Accurate in Barley? 131(9):1873–1890. AA et FB ont conçu l'étude. JC et PP ont supervisé l'analyse statistique et examiné le manuscrit. AA a effectué des expériences sur le terrain et en laboratoire, effectué des analyses bio-informatiques et statistiques et interprété les résultats. AA et FB ont écrit le manuscrit.
Le chapitre 5 a été publié en Septembre 2019 dans la revue scientifique « Crop Breeding, Genetics and Genomics ». Sa référence partielle est : Abed A. and Belzile F. (2019). Exploring the realm of possibilities: trying to predict promising crosses and successful offspring through genomic mating in barley 1:e190019. AA et FB ont mis au point l’étude. AA a réalisé des expériences sur le terrain et au laboratoire, effectué des analyses bio-informatiques et statistiques et interprété les résultats. AA et FB ont rédigé le manuscrit.
Introduction générale
La volonté de rendre la sélection des plantes plus efficace a toujours été le moteur de développement de nouvelles procédures de sélection (Ortiz Ríos, 2015). Les procédures de génotypage à haut débit comme le génotypage par séquençage GBS (« genotyping by sequencing ») (Elshire et al., 2011; Poland and Rife, 2012) ont ouvert la voie au développement de méthodes statistiques cherchant à relier des milliers de génotypes, répartis sur tout le génome au phénotype, ouvrant la voie ainsi à des approches pangénomiques. Ces approches ont d’abord permis d'approfondir la dissection des caractères complexes via les analyses d'association pangénomique (GWAS pour « Genome-wide association studies ») (Lee et al., 2015). Plus récemment, elles sont explorées en prédiction génomique pour réaliser le choix des parents à croiser (sélection des croisements) (Neyhart and Smith, 2019) ou la sélection des descendants (sélection génomique) sur la base du seul génotype des lignées et du phénotype qui leur est prédit à l’aide d'un modèle statistique reliant le génotype et le phénotype au sein d’une population de référence (Crossa et al., 2010).
Les analyses d'association pangénomique examinent le degré d'association entre des milliers de polymorphismes mono-nucléotidiques (« single nucleotide polymorphism SNP ») et un caractère étudié (Wen et al., 2018). Elles identifient des régions chromosomiques ayant un effet sur celui-ci. L'approche GWAS la plus couramment utilisée repose sur un modèle où chaque SNP est testé indépendamment (Lee et al., 2015). Cependant, un modèle uni-locus n'est jamais adéquat si un caractère est contrôlé par un grand nombre de locus QTL (« quantitative trait loci »). Des modèles multi-locus et des modèles basés sur des haplotypes testent simultanément plusieurs SNP et, en théorie, devraient permettre de mieux saisir l'architecture sous-jacente des caractères quantitatifs complexes (Wang et al., 2016).
Par ailleurs, pour faire face à la complexité de caractères quantitatifs comme la résistance à la fusariose de l’épi (Kolb et al., 2001; Horsley et al., 2006; Liu et al., 2006; Steffenson et al., 2006) et le rendement (Wang et al., 2014), une stratégie prometteuse a été proposée: la sélection génomique. En sélection génomique, des marqueurs SNP uniformément distribués sont utilisés pour prédire les valeurs agronomiques d’une descendance réelle, pour identifier les individus les plus performants sans jamais avoir recours au phénotypage (Desta and Ortiz Ríos, 2014; Spindel et al., 2015). La sélection génomique s’appuie sur un modèle statistique, lequel établi un lien entre les informations phénotypiques et génotypiques au sein d’une population représentative, sans étape préalable de localisation des QTL et sans qu’il soit nécessaire de connaitre précisément les effets individuels des marqueurs ou des QTL (Endelman, 2011; Lorenz et al., 2011).
Une autre approche génomique prédictive s’appuie sur le même modèle utilisé en sélection génomique, mais vise cette fois à prédire les valeurs agronomiques de descendants simulés de plusieurs croisements. Au terme de ces simulations, un sélectionneur pourrait s’appuyer sur ces résultats afin de choisir les croisements les plus prometteurs (Bernardo, 2015); c’est la sélection des croisements. Le choix des parents à croiser est essentiel au succès du programme de sélection, mais il repose actuellement sur des données limitées. La sélection des croisements permet de discriminer les parents et d’identifier les plus prometteurs sans jamais avoir recours au croisement ni à l’évaluation au champ (Mohammadi et al., 2015).
D’après plusieurs auteurs, les procédures de la prédiction génomique offrent un grand potentiel pour augmenter le gain génétique réalisé au terme de chaque cycle de sélection (Bernardo, 2010; Pérez-Rodríguez et al., 2017). L’utilisation d’approches prédictives pour identifier les croisements les plus prometteurs se veut une première étape visant à améliorer la performance d’un programme de sélection. On peut ainsi identifier des croisements dont les descendants présenteront une performance moyenne élevée, mais également une variabilité supérieure. C’est au sein de telles descendances que l’on peut espérer identifier des ségrégants transgressifs, c’est-à-dire qui sont encore meilleurs que le meilleur des deux parents. De son côté, la sélection génomique vise à assurer une meilleure sélection des lignées au sein d’un croisement. En principe, en s’appuyant sur le génotype des individus, on espère prendre de meilleures décisions qu’en se fondant sur une mesure forcément imparfaite de la performance d’un génotype au champ.
Comme ces approches pangénomiques sont encore relativement nouvelles, du moins en amélioration génétique végétale, de nombreuses questions se posent encore. Peu d'études ont été menées pour déterminer l’efficacité de différents modèles GWAS chez l’orge et l’intégration de la prédiction génomique dans les programmes de sélection. Notre étude s’inscrit dans le cadre d’un projet de recherche appliqué faisant intervenir deux programmes d’amélioration de l’orge à six rangs dans l’Est du Canada: un programme public (Université Laval, UL) et un programme privé (Céréla). Cette étude se divise en quatre volets de recherche. Comme ces approches génomiques nécessitent le génotypage d’un nombre élevé de marqueurs SNP, en premier, nous avons mis au point un protocole optimisé de génotypage GBS chez l’orge. Nous avons ensuite comparé les modèles uni-locus vs multi-locus en analyse GWAS portant sur sept caractères agronomiques importants: la teneur en désoxynivalénol (DON) dans les grains, l’épiaison (HTM), la maturité (MAT), le poids de mille grains (TKW), le poids à l’hectolitre (SPW), le rendement (GYD), et la taille des plantes (PHT). Ensuite, nous avons exploré la performance de différents modèles statistiques reliant le génotype au phénotype ainsi que l’impact du nombre et de la localisation des SNP sur la justesse des prédictions phénotypiques en
sélection génomique. En dernier lieu, et profitant des acquis des précédents volets, nous avons exploré la sélection des croisements les plus prometteurs sur la base de progénitures simulées et de notre modèle statistique.
1.1. Culture de l’orge et ses contraintes
L'orge (Hordeum vulgare L.) est une culture importante sur le plan agronomique et économique (Malysheva-Otto et al., 2006; Purugganan and Fuller, 2009; Ullrich, 2011; Langridge, 2018). Le Canada occupait la 6ème place mondiale des producteurs d’orge en 2018, avec une production annuelle de 8.4 M de tonnes (Statistique Canada, 2018). Même si plus de 95 % de la superficie consacrée à l'orge au Canada se trouve dans l'Ouest du pays, l’orge reste encore une culture importante dans l'Est où les conditions de croissance sont très variables avec une multiplicité agro-écologique (Statistique Canada, 2018).
Au Canada, la production annuelle de l’orge a connu une diminution ces vingt dernières années (http://faostat.fao.org). Différentes contraintes biotiques et abiotiques sont à l’origine de cette tendance. La fusariose de l’orge est la maladie la plus dévastatrice car, en plus de causer des pertes de rendement, l’agent pathogène Fusarium graminearum produit différentes mycotoxines dont la plus importante est le désoxynivalénol (DON), une toxine dangereuse pour l’homme et les animaux (Kolb et al., 2001; Rudd et al., 2001; Boddu et al., 2006; Horsley et al., 2006; Steffenson et al., 2006). Pour réduire l’impact de cette maladie, la résistance génétique offre un potentiel énorme. De ce fait, plusieurs études (cartographie de QTL et analyse d’association pangénomique) ont été effectuées pour élucider la génétique de la résistance à la fusariose de l’épi chez l’orge. Elles ont révélé une nature quantitative et complexe de ce caractère qui est contrôlé par de nombreux gènes avec des effets relativement faibles répartis à travers tout le génome (Horsley et al., 2006; Massman et al., 2011; Ulrich, 2011). La complexité de cette architecture génétique a rendu difficilement praticable une sélection assistée par marqueurs (SAM). De plus l’existence d’une corrélation défavorable entre la résistance à la fusariose et le rendement (les lignées au plus haut rendement sont les plus sensibles à la fusariose) rend une sélection simultanée pour ces deux caractères très difficile et peu efficace (Mohammadi et al., 2015). Une alternative possible consiste à faire appel à des approches prometteuses, mais malheureusement encore peu éprouvées chez les plantes cultivées, soit la prédiction génomique.
1.2. La caractérisation génotypique
La caractérisation du polymorphisme génétique, au moyen de marqueurs SNP (« single nucleotide polymorphism »), est une étape essentielle pour des approches pangénomiques comme les analyses d’association pangénomique et la prédiction génomique. Elle permet de révéler les bases génétiques et la diversité allélique des caractères quantitatifs, d’estimer la structure de la population avec précision, d’éclairer les choix à l’étape de la composition des populations utilisées dans ces approches
et de mesurer le degré de similitude entre les lignées qui composent différents programmes de sélection.
Pour les génomes de grande taille et complexes, les approches de séquençage ciblant une représentation réduite de l’espace entier du génome sont nécessaires pour (i) séquencer de manière reproductible la même région chez plusieurs individus et (ii) caractériser à grande échelle les marqueurs SNP, réduisant ainsi drastiquement les couts du séquençage d’une population (Mascher et al., 2013a).
Le génotypage par séquençage (« genotyping by sequencing », GBS), développé par Elshire et al. (2011) chez le maïs (Zea mays), répond parfaitement à ces attentes. C’est une approche robuste, efficiente et simple dont l’objectif principal est de simultanément identifier les marqueurs SNP informatifs et appeler le génotype à chacun de ces marqueurs pour tous les individus analysés. Une autre force du GBS est que les séquences brutes obtenues sont dynamiques, c’est-à-dire qu’elles peuvent être analysées de nouveau afin de découvrir de plus amples informations au fur et à mesure que les méthodes bio-informatiques se raffinent (Elshire et al., 2011; Poland and Rife, 2012). Cette approche emploie deux stratégies avantageuses (i) une réduction de la complexité en ciblant un sous-ensemble du génome (ii) un multiplexage des individus, produisant une librairie d’ADN de plusieurs individus. Ces stratégies diminuent le coût et augmentent l'exactitude d’appel des SNP sans compromettre leur qualité (Elshire et al., 2011; Lu et al., 2013; Sonah et al., 2013).
La réduction de la complexité est illustrée dans la Figure 1.1-A. Elle est assurée par le choix approprié des enzymes de restriction qui évitent les régions répétées et méthylées du génome. Chez l’orge, Poland et al. (2012) ont exploré un protocole basé sur l’utilisation de deux enzymes de restriction : PstI (CTGCAG) à coupe rare et MspI (CCGG) à coupe fréquente. Ce protocole fournit une plus grande réduction de la complexité que le protocole original avec une seule enzyme ApeKI (Elshire et al., 2011). Dans l’étape du multiplexage, un système de codage des fragments de restriction est utilisé. Ainsi schématisé dans la Figure 1.1-B, des adaptateurs sont ligaturés aux extrémités des fragments de restriction et ces adaptateurs incluent des séquences spécifiques à chaque individu (code-barres) et des séquences communes à tous les individus. L’adaptateur à code-barres (forward) est conçu avec les extrémités cohésives de l’enzyme de restriction PstI alors que l’adaptateur commun (reverse) est complémentaire avec les extrémités cohésives de l’enzyme MspI (Poland et al., 2012). Un volume identique des produits de digestion/ligature de chaque individu est par la suite mélangé en une seule librairie GBS. Par la suite, comme indiqué à la Figure 1.1-C des amorces appropriées (de séquençage/de fixation sur support solide) sont additionnées et une PCR est effectuée pour amplifier
les fragments de restriction du mélange. Les produits PCR sont purifiés et les tailles des fragments de la librairie GBS sont vérifiées avant d’être séquencés.
Figure 1.1 Les étapes simplifiées d’une approche GBS depuis l’extraction d’ADN au séquençage. (Figure adaptée des travaux de Sartori, 2012)
L’étape qui suit la préparation des librairies GBS, détaillée plus haut, est celle du séquençage et de l’appel de SNP tel qu’illustré dans la Figure 1.2. Différentes technologies peuvent être utilisées pour le séquençage de la librairie GBS. L’approche du GBS a le plus souvent porté sur un séquençage avec la plateforme Illumina (HiSeq), mais récemment l’utilisation de la technologie Ion Torrent (Proton) a été démontrée. Ces deux technologies utilisent massivement un séquençage par synthèse, mais utilisent des chimies et des dispositifs de détection différents (Mascher et al., 2013a). Parallèlement aux avancées techniques dans le séquençage d’ADN, de nouveaux algorithmes et programmes computationnels pour l’analyse bio-informatique des données et la détection des SNP ont également été développés (Mascher et al., 2013a). Les logiciels communément utilisés pour l’appel de SNP à
partir de données GBS incluent deux classes de pipelines (i) les pipelines indépendants de la disponibilité d’un génome de référence, comme UNEAK (Lu et al., 2013) ou Stacks (Catchen et al., 2013) et (ii) les pipelines qui se basent sur un génome de référence pour la détection des SNP comme TASSEL-GBS (Glaubitz et al., 2014), IGST-GBS (Sonah et al., 2013) et Fast-GBS (Torkamaneh et al., 2017).
Figure 1.2 Schéma représentant la procédure complète d’une approche GBS (Figure adaptée des travaux d’Elshire et al., 2011 et Poland and Rife, 2012)
L'orge est une espèce diploïde autogame, son génome (2n = 2x = 14) est parmi les plus grands et complexes chez les plantes cultivées (5,1 gigabases) (Schulte et al., 2009). Le projet de séquençage du génome de l’orge, entrepris par le Consortium International de séquençage du génome de l’orge, a permis de produire une référence structurée de séquences physique, fonctionnelle et génétique qui décrit l'espace des gènes dans le contexte du génome entier du cultivar Morex (IBGSC, 2012; Mascher et al., 2017). Avec son génome complexe, l’orge est considérée comme un bon candidat pour une procédure GBS. Des cartes physique et génétique denses sont disponibles et pouvant servir comme une excellente ressource pour l’identification de marqueurs SNP de qualité supérieure (densité élevée, distribution satisfaisante et exactitude élevée). Celles-ci fournissent des informations nécessaires pour les approches pangénomiques et les travaux d’amélioration de cette culture céréalière importante (Muñoz-Amatriaín et al., 2011; Mascher et al., 2013b).
1.3. Analyses d’association
1.3.1. Principe des analyses d’association pangénomique
Les analyses d’association pangénomique ou GWAS (« Genome-wide association studies ») représentent une méthode largement utilisée pour disséquer des caractères quantitatifs complexes. Cette approche exploite la diversité génétique existante et les évènements de recombinaison historiques capturés dans une population de lignées distinctes (représentative de la diversité allélique pour un caractère) (Varshney and Tuberosa, 2007). La procédure GWAS examine le degré d’association entre des milliers de marqueurs moléculaires type SNP avec le caractère d’intérêt (Lee et al., 2015; Beilsmith et al., 2019). La détection d’une telle association repose sur le déséquilibre de liaison (« linkage disequilibrium », LD). Ce déséquilibre reflète le fait qu’une ségrégation non-aléatoire existe entre différents locus, que ce soit des SNP ou des QTL (« quantitative trait loci »). En effet, des loci proches sur un même chromosome auront tendance à être transmis ensemble, jusqu’à ce qu’un événement de recombinaison survienne pour briser cette association originelle. Au final, cela conduit à une association entre locus plus ou moins proches et constitue l’assise des analyses d’association (Hayes, 2013; Choi et al., 2015; Bartoli and Roux 2017).
Conçue à l'origine pour la génétique humaine, l’approche GWAS est devenue de plus en plus populaire et puissante dans la recherche en génétique des plantes au cours des dernières années; trois raisons principales peuvent expliquer cet intérêt. Premièrement, elle est pratique, car elle peut exploiter directement les abondantes données phénotypiques collectées au cours d’un programme de sélection; deuxièmement, comparée aux précédentes approches de cartographie de QTL, elle offre une résolution génétique accrue, car les populations de lignées fixées qui sont employées en GWAS capturent davantage d’événements de recombinaison et plus d’allèles que les populations biparentales (Waugh et al., 2014). Troisièmement, l'émergence d'approches de génotypage à haut débit et à faible coût (comme le GBS), produisant des milliers de SNP et fournissent ainsi au GWAS la densité de SNP requise (Biscarini et al., 2016).
1.3.2. Les facteurs influençant le GWAS
Bien que les approches de génotypage à haut débit rendent le GWAS pertinent pour de nombreuses espèces, plusieurs facteurs peuvent influer le pouvoir avec lequel de réelles associations SNP-QTL peuvent être détectées. Ces facteurs incluent la proportion de la variance phénotypique totale expliquée par le QTL, l’importance de l’effet du QTL, le nombre de locus affectant le caractère, la taille de la population, la fréquence de l’allèle rare du SNP associé et le niveau de signification défini par l’expérimentateur (Hamblin and Jannink, 2011; Uchiyama et al., 2013).
Une couverture en marqueurs SNP suffisamment exhaustive est également un facteur clé du succès du GWAS, car une couverture insuffisante réduit la puissance d'identification des QTL. La densité de marqueurs requise est fonction de l'étendue du LD dans une population (Waugh et al., 2014; N’Diaye et al., 2017). Si le LD décline rapidement dans une région génomique, un grand nombre de SNP sera nécessaire pour balayer cette région à la recherche de QTL sous-jacents et vice versa (Ersoz et al., 2007; Hayes, 2013; Uchiyama et al., 2013).
L'architecture génétique et l'héritabilité d'un caractère jouent un rôle important dans le succès de la détection, car une héritabilité réduite diminue le pouvoir statistique (Vaughn et al., 2014) et les différentes architectures sont susceptibles d'interagir avec les caractéristiques des marqueurs influençant la puissance statistique également (Hamblin and Jannink, 2011). Le succès du GWAS est également fortement influencé par le dispositif expérimental et l’évaluation précise du phénotype; pour une résolution plus élevée de la dissection allélique, une variation phénotypique plus importante est requise (Ersoz et al., 2007; Uchiyama et al., 2013).
L’un des problèmes majeurs en GWAS est que l’emploi d’une population de lignées faisant partie d’un programme d’amélioration implique un compromis entre la détection d’une variation phénotypique réelle et la confusion avec celle causée par la structure de la population (Pritchard et al., 2000; Ersoz et al., 2007; Zhao et al., 2007; Beilsmith et al., 2019). La structure de la population est en grande partie causée par l'origine et l'historique de la population. Les lignées faisant partie d’un programme d’amélioration génétique ont, par la force des choses, de nombreux liens tissés par le fait de partager un parent/ancêtre commun. Une telle situation entraîne des fréquences alléliques déséquilibrées parmi les sous-populations, un faux LD entre SNP et QTL et peut conduire à la détection d’associations significatives, mais fausses (des faux positifs) dues à des facteurs autres que le l’association génétique (Uchiyama et al., 2013; Waugh et al., 2014; Mihalyov et al., 2017). Afin de contrôler l’occurrence de faux positifs, il est primordial de corriger la structure de la population en incluant une matrice de similarité des lignées ou de parenté (Matrice K pour « kinship » en anglais) et une matrice décrivant la population (Matrice Q) dans le modèle linéaire mixte utilisé pour identifier les associations. (Kang et al., 2010; Bartoli and Roux, 2017; Beilsmith et al., 2019).
1.3.2.1. Les approches statistiques
Les approches statistiques utilisées pour identifier les associations SNP-QTL peuvent avoir un effet substantiel sur le succès du GWAS, car elles diffèrent par leur puissance et leur fiabilité (Yu et al., 2006; Stich et al., 2008; Zhang et al., 2010; Hayes, 2013; Gawenda et al., 2015; Visscher et al., 2017; Wen et al., 2018). L'approche la plus couramment utilisée est un modèle mixte à locus unique où
chaque SNP est testé indépendamment pour une association avec le phénotype (Bush and Moore, 2012; Gupta et al., 2014; Waugh et al., 2014; Wen et al., 2018). Cependant, un modèle uni-locus ne correspond pas à l’assise génétique d’un caractère quantitatif contrôlé simultanément par de multiples loci, ce qui est le cas de la plupart des caractères agronomiques (Gupta et al., 2014; Wang et al., 2016). De plus, sur le plan statistique, le nombre de tests impliqués est égal au nombre de SNP testés, une situation qui nécessite une correction pour les tests multiples. Si cette correction est trop conservatrice, il peut s’avérer impossible de détecter de nombreux locus importants (faux négatifs) (Wen et al., 2018). De plus, comme les SNP sont essentiellement bi-alléliques, un modèle uni-locus peut être moins approprié pour capturer la véritable diversité allélique présente dans une population (Lu et al., 2011; Contreras-Soto et al., 2017; N’Diaye et al., 2017). Une autre limitation des modèles uni-locus est que des interactions épistatiques au sein ou entre gènes proches peuvent exister, mais ne peuvent pas être détectées en évaluant chaque SNP séparément (Gawenda et al., 2015).
Pour surmonter certaines de ces limitations, des modèles multi-locus et des modèles basés sur les haplotypes ont récemment été proposés et appliqués chez certaines espèces. Tester simultanément plusieurs SNP avec un modèle multi-locus permet de mieux reproduire l'architecture sous-jacente de caractères quantitatifs complexes (Wang et al., 2016). Lorsque le nombre de marqueurs n'est pas grand, tous les effets des SNP peuvent être inclus dans un seul modèle d’association. Cependant, si le nombre de marqueurs est grand, l’approche est subdivisée en deux étapes. Dans une première étape, les SNP sont d’abord présélectionnés et uniquement ceux ayant une forte probabilité d'être associés au caractère sont retenus. Par la suite, ceux-ci sont inclus dans un seul modèle d’association multi-locus (Wen et al., 2018). Plusieurs modèles multi-multi-locus ont récemment été développés, tels que: MLMM (Segura et al., 2012), mrMLM (Wang et al., 2016), ISIS EM-BLASSO (Tamba et al., 2017), FASTmrEMMA (Wen et al., 2018), pLARmEB (Zhang et al., 2017) et pKWmEB (Ren et al., 2018). Alternativement, combiner des SNP adjacents ayant un fort LD en un haplotype unique multi-allélique et tester l'association entre ces haplotypes et le caractère d'intérêt peut être plus puissant et plus fiable pour la détection de QTL (Liu et al., 2008; Lorenz et al., 2010; Gupta et al., 2014; Gawenda et al., 2015; N’Diaye et al., 2017). L’utilisation d’une approche haplotypique peut surmonter la limitation bi-allélique inhérente aux marqueurs SNP et capturer avec précision la diversité allélique des gènes causals présents dans une population (Lou et al., 2003; Liu et al., 2008; Gawenda et al., 2015). Selon Lorenz et al. (2010), les haplotypes permettent d’augmenter le pouvoir de détection des interactions épistatiques entre les SNP d'un locus. Réduire le nombre de tests et éviter ainsi des ajustements trop conservateurs pour des tests multiples (pour l’erreur de type I) sont également
avancés comme avantages à l’utilisation des haplotypes en GWAS (Contreras-Soto et al., 2017; N’Diaye et al., 2017).
Des plates-formes de génotypage à haut débit et à coût relativement bas sont maintenant disponibles pour l'orge, permettant ainsi l'application de GWAS pour disséquer les mécanismes génétiques sous-jacents de caractères importants sur le plan agricole et pour identifier des marqueurs utiles pour les programmes de sélection (Bellucci et al., 2017). Plusieurs analyses d'association ont été rapportées chez l'orge et ont permis d’identifier des gènes majeurs (Ramsay et al., 2011), mais également des QTL pour des caractères agronomiques complexes (Cockram et al., 2010; Lorenz et al., 2010; Comadran et al., 2011; Pasam et al., 2012; Visioni et al., 2013; Pauli et al., 2014; Gawenda et al., 2015; Alqudah et al., 2016; Bellucci et al., 2017; Alqudah et al., 2018; Hu et al., 2018) ou de résistance à certains agents pathogènes importants (Roy et al., 2010; Massman et al., 2011; Mamo and Steffenson, 2015; Sallam et al., 2017). La majorité de ces études ont été menées avec un nombre relativement faible de SNP (1,5K - 9K), ce qui, probablement, n'a pas permis de révéler complètement les bases génétiques des caractères quantitatifs. De plus, parmi ces études, peu ont comparé les modèles à uni-locus avec ceux à haplotypes et aucune étude n’a encore porté sur la comparaison de modèles uni-locus et multi-locus pour des caractères complexes chez l’orge.
1.4. La prédiction génomique
La prédiction génomique permet l’augmentation du gain génétique pour des caractères quantitatifs (Pérez-Rodríguez et al., 2017; Neyhart and Smith, 2019). Selon l’objectif des prédictions, elle peut être divisée en deux approches; la sélection génomique (« genomic selection ») et la prédiction des croisements (« genomic mating »). En sélection génomique, des individus phénotypés et génotypés sont utilisés pour prédire les performances d'individus non phénotypés sur la base du seul génotype (Meuwissen et al., 2001). La sélection génomique porte principalement sur l’identification des descendants les plus prometteurs au sein d’une population. En prédiction des croisements, des individus phénotypés et génotypés sont utilisés pour prédire les performances d'individus à être utilisés comme futurs parents dans les croisements sans avoir recours à leurs phénotypes ni aux croisements (Bernardo, 2010). Idéalement, un croisement devrait donner une population avec une moyenne et une variance génétique élevées. C’est au sein de telles descendances qu’on pourra espérer trouver des individus qui dépassent les performances des deux parents, un phénomène que l’on nomme la ségrégation transgressive. (Mohammadi et al., 2015)
1.4.1. La sélection génomique
La capacité de prédire des caractères phénotypiques complexes à partir des seules données génotypiques (marqueurs moléculaires) est un objectif très souhaitable pour les programmes d’amélioration des plantes (Endelman, 2011; Heslot et al., 2015). Durant cette dernière décennie, il s’est fait beaucoup de travail dans le domaine de la modélisation de la sélection génomique chez les plantes, mais relativement peu de mise en pratique de ce concept (Crossa et al., 2010; Massman et al., 2013; Crossa et al., 2014; Guzman et al., 2016; Sallam and Smith, 2016).
1.4.1.1. Principe de la sélection génomique
Tel qu’illustré dans la Figure 1.3, la sélection génomique est typiquement basée sur deux populations: la population de référence, génotypée et phénotypée, et les candidats à la sélection, génotypés uniquement. Ces deux populations créent deux cycles complémentaires; celui de la calibration du modèle statistique et celui du développement des lignées. La population de référence joue un rôle essentiel puisqu’elle permet la calibration d’un modèle de prédiction (soigneusement choisi) qui à son tour estime les relations statistiques entre les génotypes et les phénotypes des caractères étudiés. Une fois la relation génotype-phénotype établie avec justesse, il est alors possible de prédire, pour chaque caractère et pour chaque lignée, une valeur génomique (« genomic estimated breeding value » ou GEBV) des candidats à la sélection (uniquement sur la base de leur génotype) (de los Campos et al., 2009b; Crossa et al., 2010). La sélection est ainsi effectuée sur la base uniquement des prédictions et nous affranchit de devoir réaliser une longue et laborieuse caractérisation phénotypique de chaque lignée (Meuwissen et al., 2001; Guillaume et al., 2009; Endelman, 2011; Lorenz et al., 2011; Lorenz et al., 2012; Desta and Ortiz Ríos, 2014; Sallam, 2015; Spindel et al., 2015).
Par rapport à la sélection conventionnelle (phénotypique), on espère que la sélection génomique apportera un raccourcissement potentiel du cycle de sélection, une augmentation de l’intensité de la sélection pour des caractères complexes, ainsi qu’un gain en précision des valeurs génétiques estimées des individus sélectionnés (Heffner et al., 2009; Heffner et al., 2010; Crossa et al., 2011; Hickey et al., 2012; Daetwyler et al., 2013; Sallam and Smith, 2016; Pérez-Rodríguez et al., 2017)
1.4.1.2. Estimation de la justesse de la sélection génomique
Comme il existe plusieurs modèles statistiques pour capturer la relation génotype-phénotype, il est nécessaire de pouvoir comparer ces différents modèles en termes d’efficacité à prédire la valeur génomique. Le « meilleur » modèle peut être choisi soit en se basant sur des critères statistiques de sélection de modèle, mais c’est l’utilisation de données réelles qui apportent la réponse la plus fiable (Guillaume et al., 2009; Lorenz et al., 2011; Mallick and Li, 2013). Il existe trois méthodes générales
pour évaluer la justesse des prédictions à l'aide de données réelles: (1) la validation croisée, (2) la validation inter-population et (3) la validation avec des descendants (Sallam et al., 2015).
Figure 1.3 Schéma résumant une procédure de sélection génomique (Figure adaptée des travaux de Heffner et al., 2009)
La validation croisée est utilisée pour calibrer, développer et évaluer un modèle à travers sa capacité à prédire un phénotype. Ensuite, le meilleur modèle, celui qui est le mieux ajusté, peut être utilisé afin de prédire les performances pour chacune des lignées qui composent les candidats à la sélection (Guillaume et al., 2009; Desta and Ortiz Ríos, 2014). La validation croisée consiste à diviser aléatoirement les données de la population de référence en deux sous-populations: (1) une majorité des lignées (60 à 90 %) de la population de référence est utilisée pour estimer les effets des marqueurs et (2) le reste de la population (40 à 10 %) est utilisé pour tester le modèle de prédiction choisi. Un modèle statistique est développé en utilisant les données de la sous-population de référence et les prédictions sont calculés en utilisant seulement les génotypes de la sous-population de validation (Lorenz et al., 2011). L’exactitude de la prédiction du modèle est estimée en calculant la corrélation entre les performances prédites (GEBV) et les performances observées au champ (« estimated breeding values » ou EBV) de la sous-population de validation (Desta and Ortiz Ríos, 2014).
La validation inter-population consiste à utiliser une population autre que la population de référence pour vérifier si le modèle permet de prédire avec justesse la performance de lignées sur la seule base de leur génotype. La population de validation peut être formée de descendants ou de lignées proches génétiquement de la population de référence, pour lesquelles on dispose de données génotypiques et phénotypiques. En utilisant un modèle de prédiction bâti à partir de la population de référence, les prédictions de la population de validation sont calculées. À nouveau, l’exactitude de la sélection génomique est évaluée en mesurant la corrélation entre les performances prédites et celles observées au champ pour la population de validation (Guillaume et al., 2009; Lorenz et al., 2011).
La validation avec des descendants implique que la population de référence comprenne les parents (ou grands-parents, etc.) des descendants qui composent la population de validation. Ainsi, en réalité cette méthode de validation reconstitue un scénario d’intégration de la sélection génomique dans un programme de sélection où des parents/grands-parents sont utilisés pour calibrer le modèle de prédiction et estimer les performances des descendants. La décroissance du LD entre les SNP et les QTL résultant de recombinaisons dans la descendance (Habier et al., 2007) est associée à une diminution de la justesse des prédictions. Par conséquent, une évaluation fiable d’une approche de sélection génomique devrait inclure la validation avec des descendants. Chez les plantes très peu d’étude ont évalué la sélection génomique à l'aide d'informations phénotypiques et génotypiques empiriques (Hofheinz et al., 2012; Sallam et al., 2015).
1.4.1.3. Les facteurs qui influencent l’exactitude de la sélection génomique
L’efficacité de la sélection génomique est directement liée à l’exactitude des prédictions, elle-même fortement dépendante des effets des gènes, de la complexité du caractère, de la taille de la population de référence, de la structure de la population, de la proximité génétique entre les lignées, ainsi que de la densité des marqueur SNP (de los Campos et al., 2009b; Crossa et al., 2010; Crossa et al., 2011; González-Camacho et al., 2012; Hickey et al., 2012; Lorenz et al., 2012; Pérez-Rodríguez et al., 2012; Riedelsheimer et al., 2012; Sallam el al., 2015; Pérez-Rodríguez et al., 2017). Par ailleurs, elle dépend moins de la méthode adoptée pour calculer la contribution du marqueur (Lorenz et al., 2012).
1.4.1.3.1. Modèles statistiques
Les modèles de prédiction utilisés dans la sélection génomique sont des modèles de régression linéaire et ceux-ci peuvent être composés de modèles paramétriques ou non-paramétriques. Parmi les modèles paramétriques, les plus largement utilisés chez les plantes sont les approches pénalisées et les approches bayésiennes (Lorenz et al., 2011; Desta and Ortiz Ríos, 2014; Heslot et al., 2015). Ces modèles ont différentes caractéristiques de calcul dépendamment des hypothèses retenues pour les
effets des marqueurs considérés dans le modèle (Lorenzana and Bernardo, 2009; Asoro et al., 2011; Desta and Ortiz Ríos, 2014).
Les méthodes de régression pénalisée régressent vers zéro l’effet des marqueurs de telle sorte que seuls ceux qui ont un effet suffisamment important influencent l’estimation de la valeur génétique. Ainsi, les autres marqueurs ayant un effet faible perturberont moins l’estimation des effets des marqueurs les plus prometteurs (Guillaume et al., 2009; Robert-Granié et al., 2011; Mallick and Li, 2013). Différentes approches de régression pénalisée ont été décrites: RR-BLUP (« ridge regression best linear unbiased prediction »), LASSO (« least absolute shrinkage and selection operator ») et « Elastic net » (une combinaison du LASSO et du RR-BLUP) (Robert-Granié et al., 2011; Mallick and Li, 2013). La méthode basée sur le RR-BLUP mesure l’effet de tout le catalogue de marqueurs à la fois sous l’hypothèse que les variances génomiques de tous les loci sont égales et suivent une loi normale, ceci implique une connaissance préalable que le caractère étudié est contrôlé par de nombreux locus à petits effets. Cette méthode fournit des résultats précis ainsi qu’une facilité de mise en œuvre et de calcul (Lorenz et al., 2011; Robert-Granié et al., 2011). Différentes méthodes utilisent le LASSO, une approche qui stipule qu’un nombre réduit de marqueurs sont en lien avec le caractère. Cette méthode permet ainsi une sélection simultanée des marqueurs et l’estimation de leur effet (Guillaume et al., 2009; Robert-Granié et al., 2011; Mallick and Li, 2013).
L'hypothèse faite par le RR-BLUP que les effets génétiques sont également répartis sur tout le génome a été jugée peu réaliste par Meuwissen et al. (2001). Ces chercheurs ont donc utilisé une approche bayésienne pour y remédier (Lorenz et al., 2011). Les méthodes bayésiennes lèvent la restriction imposée par le RR-BLUP en permettant l’introduction de distributions plus adaptées à la variabilité des effets des SNP. Ces méthodes combinent régulièrement une distribution a priori et un ensemble de données à travers lequel les effets des QTL inconnus sont capturés avec une distribution a posteriori. Les méthodes bayésiennes supposent effectivement que le caractère est contrôlé par relativement peu de locus qui varient en importance de l'effet (Lorenz et al., 2011). Les différentes méthodes bayésiennes utilisées en sélection génomique se distinguent par les hypothèses faites concernant la distribution des effets des SNP (Robert-Granié et al., 2011). Par exemple, BayesA stipule que les effets des marqueurs sont différents. BayesB suppose qu’une proportion donnée des SNP a un effet nul (variance nulle) et ne contribue donc pas au caractère. LASSO Bayésien fait l’hypothèse que les effets des marqueurs suivent une loi de Laplace (double exponentielle) reflétant qu’un grand nombre de SNP sont supposés avoir un effet pratiquement nul et que seuls quelques marqueurs ont un grand effet (Robert-Granié et al., 2011; Mallick and Li, 2013).
L'efficacité relative des modèles statistiques dépend souvent de l'architecture génétique d'un caractère (Howard et al., 2014). Ainsi, lorsqu’un caractère se compose de nombreux locus à petit effet, le RR-BLUP et les modèles exploitant les informations sur la relation génétique globale entre les lignées fonctionnent bien. Lorsque des QTL à large effet expliquent une plus grande variation génétique, des modèles tels que BayesB devraient être favorisés (Lorenz et al., 2011). De plus, des méthodes non paramétriques, telles que la RKHS « reproducing kernel Hilbert spaces regression (RKHS) », conviendraient mieux à l’architecture génétique non additive (dominance et épistasie), car elle tient compte de l’effet additif et épistatique dans la prédiction des GEBV (Gianola, 2006; Gianola and van Kaam, 2008; de Campos et al., 2009a; de Campos et al., 2010; Pérez-Rodríguez et al., 2012). Cependant, comme le suggèrent plusieurs études, les méthodes paramétriques basées sur les effets additifs peuvent être meilleures que les méthodes non paramétriques dans le cas d'architectures génétiques additives, de sorte que l'utilisation de modèles non paramétriques peut ne pas donner l’exactitude attendue (Desta and Ortiz Ríos, 2014; Howard et al., 2014).
1.4.1.3.2. La taille et la composition de la population de référence
Les caractéristiques de la population de référence (sa taille et sa composition) influencent grandement la précision de l’estimation des effets des marqueurs. De manière générale, la précision de la prédiction est proportionnelle à la taille de la population de référence (Lorenz et al., 2011; Robert-Granié et al., 2011; Nakaya and Isobe, 2012). Différentes études ont confirmé cette tendance. En effet, dans une population d’orges à six rangs, des populations de référence comptant 100, 200 ou 300 lignées ont produit des prédictions affichant des corrélations respectives de 0,25, 0,30 et 0,35 pour le rendement et de 0,65, 0,75 et 0,80 pour la taille des plants (Lorenz et al., 2011).
La composition génétique de la population de référence est un facteur important à considérer. En effet, il est possible de maximiser l'efficacité de la population de référence à travers sa conception en échantillonnant uniformément la diversité génétique d’un programme de sélection sans formation d’une structure dans la population (Asoro et al., 2011; Lorenz et al., 2011; Crossa et al., 2014). Selon Desta and Ortiz Ríos (2014), une considération particulière doit être accordée à la conception et à la composition de la population de référence en fonction des candidats à la sélection pour maintenir un degré élevé de précision. En plus, si la population de référence d’un programme est petite, il est avantageux de la compléter avec des individus d'autres populations en minimisant les distances génétiques entre les lignées afin de conserver au maximum l’association entre marqueurs et QTL (Hamblin and Jannink, 2011).