ARTICLE IN PRESS
+ModelANDO-931; No.ofPages6
Disponibleenlignesur
ScienceDirect
www.sciencedirect.com
Annalesd’Endocrinologiexxx(2017)xxx–xxx
Klotz
communications
2017:
From
the
shortest
to
the
tallest
X-LAG:
How
did
they
grow
so
tall?
X-LAG
ou
comment
sont-ils
devenus
si
grands
?
Albert
Beckers
a,∗,
Liliya
Rostomyan
a,
Iulia
Potorac
a,
Pablo
Beckers
b,
Adrian
F.
Daly
aaDepartmentofEndocrinology,CentreHospitalierUniversitairedeLiège,UniversityofLiège,4000Liège,Belgium bDepartmentofClinicalBiochemistry,CentreHospitalierUniversitairedeLiège,UniversityofLiège,4000Liège,Belgium
Abstract
X-linkedacrogigantism(XLAG)isanew,pediatric-onsetgeneticsyndrome,duetoXq26.3microduplicationsencompassingtheGPR101gene. XLAGhasaremarkablydistinctphenotypewithdiseaseonsetoccurringbeforetheageof5inallcasesdescribedtodate,whichissignificantly youngerthaninotherformsofpituitarygigantism.ThesepatientshavemixedGHandprolactinpositiveadenomasand/ormixed-cellhyperplasia andhighlyelevatedlevelsofGH/IGF-1andprolactin.Giventheirparticularlyyoungageofonset,thesignificantGHhypersecretioncanleadto aphenotypeofseveregigantismwithveryadvancedage-specificheightZ-scores.Ifnotadequatelytreatedinchildhood,thisconditionresultsin extremefinaladultheight.XLAGhasaclinicalcoursethatishighlysimilartosomeofthetallestpeoplewithgigantisminhistory.
©2017ElsevierMassonSAS.Allrightsreserved.
Keywords: Gigantism;Pituitaryadenoma;X-linkedacrogigantism(X-LAG);GPR101gene;Familialisolatedpituitaryadenoma(FIPA) Résumé
«X-linkedacrogigantism»(XLAG)estun syndromepédiatriquerécemmentdécrit, liéàdesmicroduplications duchromosome Xq26.3, englobantlegèneGPR101,responsabledel’affection.LespatientsXLAGprésententunphénotyperemarquablementdistinctdesautrescasde gigantismehypophysaire.Danstouslescasdécrits,lamaladies’exprimeavant5anssoitbeaucoupplustôtquedanslesautresformes.Lespatients onthabituellementungrosadénomeouunehyperplasiemixtepourlaGHetlaprolactineetdestauxtrèsélevésdeGH/IGF1etprolactine.Enraison desondébuttrèsprécoce,l’hypersécrétionimportantedeGHpeutconduireàungigantismeextrêmementsévèreavecunZ-scoretrèsimportant pourl’âge.Sicetteconditionn’estpastraitéependantl’enfance,ellepeutconduireàunetaillefinaleextrême.XLAGmontreuneévolutionclinique similaireàcelleobservéechezlesgéantslesplusgrandsdel’histoire.
©2017ElsevierMassonSAS.Tousdroitsr´eserv´es.
Motsclés :Legigantisme;L’adénomehypophysaire;L’acro-gigantismeliéauchromosomeX(X-LAG);LegèneGPR101;L’adénomehypophysairefamilial isolé(FIPA)
1. Introduction
Acromegaly and gigantism result from chronic excessive
productionandsecretionofgrowthhormone(GH),usuallyby
apituitaryadenoma,andare consideredveryrare conditions.
GH-secreting pituitary tumors are predominantly sporadic
∗Correspondingauthor.
E-mailaddress:[email protected](A.Beckers).
lesionsthatoccurinadults,althoughrarepediatricorfamilial formscanoccurandthesecanhaveaggressivecharacteristics.
The molecular genetics of these aggressive forms of
soma-totropinomas is of interestfor research andclinical practice.
Known genetic syndromes associated with the occurrence
of somatotropinomas include multiple endocrine neoplasia
(MEN)type1andMEN4,Carneycomplex,McCune–Albright
syndrome(MAS),andpheochromocytoma/paragangliomaand
pituitaryadenomasyndrome(3PAs).Themostfrequentfamilial
formofpituitaryadenomaisfamilialisolatedpituitaryadenoma
http://dx.doi.org/10.1016/j.ando.2017.04.013
ARTICLE IN PRESS
+ModelANDO-931; No.ofPages6
2 A.Beckersetal./Annalesd’Endocrinologiexxx(2017)xxx–xxx
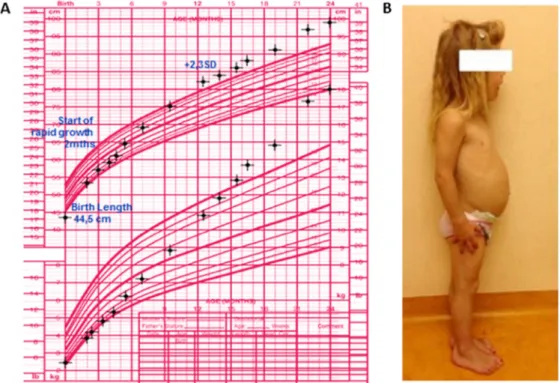
Fig.1.Growthpattern(panelA)andphysicalcharacteristics(panelB)ofapatientwithXq26.3duplication.Shewasborntonormally-sizedparentsinafamily withoutahistoryofgigantismandhadalreadyestablishedabnormallineargrowthandacralovergrowthbytheageof12months,causedbyaGH-and prolactin-secretingpituitarytumor[4].
(FIPA) syndrome [1]. Approximatively 15–20% of general
FIPAkindredsandabout50%ofhomogeneousFIPAfamilies
withacromegaly-gigantismare causedbymutations/deletions
inthearylhydrocarbonreceptorinteractingprotein(AIP)gene
[2,3].
In Trevellin et al., we recently described an international cohortof patientswithanewgeneticformofpituitary gigan-tismbeginninginearlychildhoodduetoamicroduplicationon
Xq26.3,thatwetermedX-linkedacrogigantism(XLAG)[4].In
alargepituitarygigantism-specificcohortstudyof208patients,
we found that AIP mutations andXLAG accountedfor 29%
and 10%, respectively, of geneticallytested patients, making
them the two most common causes of pituitary gigantism.
Thepreviouslyknowngeneticcausesofsomatotropinomasare
responsible for a much lower percentage of pituitary
gigan-tismcases:MAS(5%),MEN1(1%),andCarneycomplex(1%)
[5].To date,32 patients withXLAG duplications have been
publishedinthescientificliterature[4,6–11].Notably,XLAG explainsthemajorityofprepubertalcases(80%)andalmostall ofthetallestcasesinour208-patientcohort;theseXLAGcases
manifestedwitheitherexceedinglyexcessivegrowthrateora
veryincreasedfinaladultheight(heightZ-score>+4.5SD).
2. Clinicalprofile
XLAGisapediatric syndromeofpituitarygigantismwith
a very young ageof disease onset. Usually increased length
andweightareseenbeforetheageof1yearandmostpatients
are diagnosedby the age of three with already substantially
advanced overgrowth [4,9,12]. This young age at first
mani-festations renders this particular form of gigantism different
fromotherformsofgigantismandacromegaly.Geneticformsof
somatotropinomas,particularlyAIP-related,aremorefrequent
in younger patients than in the general sporadic acromegaly
population [5]. AIP-related cases, however, usually occur in
adolescentsand young adults, whereas childrenwith XLAG,
whoareusuallyborn withnormalanthropometric parameters
afteruncomplicatedpregnancies,begintheirabnormalgrowth
ininfancy(Fig.1A)[4].ThismeansthatpatientswithXLAG
canhavehadalongdurationof overgrowthbythe timethey
reachpuberty,whichtranslatesintoincreased heightZ-scores ascomparedwithotherformsofpituitarygigantism[5].
Besidestheearlyexcessiveaccelerationoflineargrowthand bodysize,theseyoungpatientsfrequentlypresentsomeclinical
signsandsymptomsofGH/IGF-1excessthataremoretypical
of adultacromegaly (Fig. 1B).They maydevelopsoft tissue
swellingandcoarsefacialfeaturesandanincreasedinterdental
space,aswellas markedenlargementof handsandfeet [12].
In somecases, increased appetite (25%) andsigns of insulin
resistance (such as acanthosis nigricans) are also noticed at
diagnosis[12].XLAGhasafemalepredominance(71%);this
feature differentiates it from AIP-mutation related gigantism
casesandgigantismpatientswithoutaknowngeneticcausein
whichcasesmostoftheaffectedsubjectsaremales(95%and
78%,respectively)[5].
In both male and female XLAG cases, the extraordinary
growth velocity and prolonged linear growth is underpinned
bytheaggressivebehaviorofthepituitarylesion[4,12].
Chil-drenwithXLAG developrelatively largepituitary adenomas
for their young age (Fig. 2), which can be accompanied by
pituitaryhyperplasia.Insomecases,diffusehyperplasiacanbe presentaloneanditisunknownwhetherthemixedGH-prolactin
ARTICLE IN PRESS
+ModelANDO-931; No.ofPages6
A.Beckersetal./Annalesd’Endocrinologiexxx(2017)xxx–xxx 3
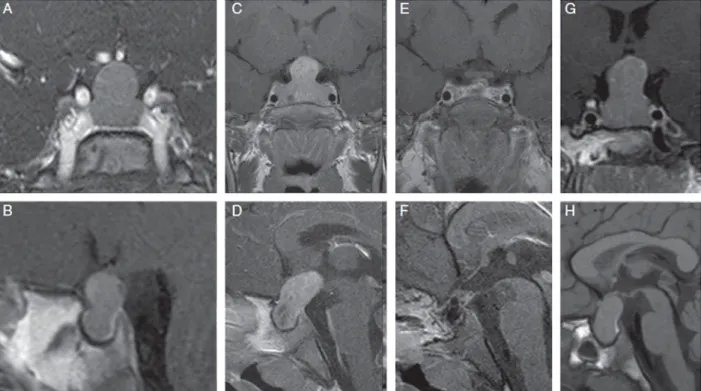
Fig.2.CoronalandsagittalMRIimagesofpatientswithXLAG.PanelsAandB.Alargepituitarymasswithimportantsuprasellarextensioninafemalepatient wasrevealedbygadolinium-enhancedMRimagingatdiagnosis(2years11months).PanelsCandD.Gadolinium-enhancedMRimagesofafemalepatient,who wasdiagnosedatage3yearswithalargepituitarymasswithmarkedsuprasellarandposteriorextensionandareasofdegenerativechanges.PanelsEandFshow postoperativeimagesfromthesamepatientwithresectedadenoma.However,theadministrationofSSAandpegvisomantwererequiredaftersurgeryinthispatient forhormonalandgrowthcontrol.PanelsGandHshowMRimagesofafemalepatientatdiagnosis(aged3years)presentingwithalargepituitarymasswith extrasellarexpansion[12].
secretingmacroadenomasthatareseeninmostXLAGpatients
beginashyperplasiabeforetransformingintoadenoma.
Pathol-ogy studies have demonstrated foci of transformation from
hyperplasia to adenoma, which further supports the concept
thatthe adenomasinXLAGdevelopagainstabackgroundof
hyperplasia[4,7,12,13](Table1).
WerecentlyshowedthatmodestlyelevatedcirculatingGH
releasing hormone (GHRH) levels can occur in XLAG[13].
GHRH levels in XLAG are not as high as those usually
encountered inectopicGHRH secretionfromneuroendocrine
carcinomas. Moreover, intense GHRH-receptor (GHRH-R)
stainingwasfoundinresectedpituitarytumortissueinXLAG
Table1
Historicalcasesofextremegigantisminwhichgrowthbeganinearlychildhood[12].
Name Country Sex Yearofbirth Birth
weight(kg) Normal family history Ageatwhich abnormalgrowth noted(years) Finalheight(cm)
MartinVanBurenBatesa USA M 1837 Normal Y 6 222
AnnaHainingSwana Canada F 1846 8.1 Y <4 227
EllaKateEwing USA F 1872 3.4 Y 7 225
FedorAndreevichMachnow Russia M 1878 NA Y 5 239
EdouardBeaupréb Canada M 1881 4.1 Y 3 251
Joh(a)nAasenb USA M 1890 NA No <8 218
AlbertJohanKramerb TheNetherlands M 1897 8.5 Y <7 238
RobertPershingWadlowb USA M 1918 4.1 Y <3 272
CecilBoling USA M 1920 Normal Y <7 235
RigardusRijnhoutb TheNetherlands M 1922 Normal Y >3 238
DoloresAnnPullardb USA F 1946 Normal Y 4 227
SandraElaine(Sandy)Allenb USA F 1955 2.95 Y 3 232
ZengJinlian China F 1965 Normal Y <1 249
YaoDefenb China F 1972 2.8 Y <3 234
aSwanandBatesweremarriedandhadtwopregnancies;onefemalechildwasstillborn(weighed8.1kg)andlaterasonwhodiedinearlyinfancywas10.8kg and76cminlengthwhenborn.
ARTICLE IN PRESS
+ModelANDO-931; No.ofPages6
4 A.Beckersetal./Annalesd’Endocrinologiexxx(2017)xxx–xxx
Fig.3.Photographsofsomeofthetallesthistoricalpatientspresentedintheorderoftheirheight.Theyappearedfrommedicalrecordsashavingearlychildhood-onset pituitarygigantism,whichisanimportantfeatureoftheXLAGsyndrome.Twogreysilhouettesillustratetheaveragehumanheight(175cm).
[4,12,13];whichsuggeststhatthe cellpopulationinvolvedin
hyperplasiaandadenomatoustransformationmaybesensitive
toGHRH.Furthermore,invitrocellculturestudiesofanXLAG
pituitarylesion revealedthat a GHRH-R antagonist inhibited
both GHand prolactin and antagonised the effect of GHRH
stimulation.Thesedatasupporthypothalamicinvolvementvia
GHRH and GHRH-R dysregulation in the pathogenesis of
XLAG.TheGHRH-relatedGHhypersecretioninXLAGcould
potentiallybetargetedbytherapeuticblockadeoftheGHRH-R
[13].
Intermsoftreatment,XLAGpatientshaveparticularly dif-ficult to treat pituitary lesions, not only due to their young ageandrelativelylargetumorsize.They oftenrequire multi-modaltreatment,includingpituitarysurgery,medicaltreatment
andradiotherapy.AlmostallXLAGcaseshavepartialor
com-pleteresistancetooctreotide/lanreotide,despitearelativelyhigh expressionofsomatostatinreceptortype2[12].Prolactin
hyper-secretion demonstrates variable responsiveness to dopamine
agonist therapy. In ordertocontrol GHhypersecretion,some
patients have required aggressive surgical resection
(includ-ing anterior hypophysectomywhen extensivehyperplasiahas
been present), incombination with radiotherapy. The
conse-quences of these multiple lines of treatment is the frequent
occurrence of partial or complete hypopituitarism, requiring
lifelong replacement therapystartingfrom avery youngage.
Adjuvant use of the GH receptor antagonist, pegivsomant,
has proven successful in controlling GH secretion in some
XLAGpatients[12].Promptandeffectivehormonalcontrolis
akeyrequisiteinhaltinglineargrowthbeforepatientsreacha markedlyincreasedadultheight[5].Intheabsenceofeffective
measurestocontroltumorgrowth,markedtumorprogression
cancontinue inXLAG,withexceptionallyseverephenotypes
of pituitary gigantism being established before puberty [7].
A number of patients with extremelygigantism described in
historical andmedical records had clinical presentationsthat
correspondedtothe growthpattern of XLAGpatients [4,12].
In particular, the tallestman, RobertWadlow, andthe tallest
woman,ZengJinlian,inrecordedhumanhistorypresentedthe
characteristicsofXLAGpatientswithrapidovergrowthinearly childhood(Fig.3).
3. Genetics
XLAGhasaspecificgeneticetiology.Unliketheother
pitu-itaryadenoma-associatedgeneticsyndromesusuallycausedby
inactivatingmutationsordeletionsintumorsuppressorgenesor
activatingmutationsinanoncogene,XLAGisduetoa
dupli-cation on chromosome Xq26.3. Array comparative genomic
hybridization(aCGH)isusually usedfordetectingsuchcopy
numbervariation(CNV)abnormalities.All18XLAGpatientsin
ourinitialcohortwerefoundtohavemicroduplicationswithtwo
smallestregionsofoverlap,encompassingfourgenes(CD40LG,
ARHGEF6,RBMXandGPR101)[4].Amongtheinitialfour
can-didategenes,GPR101,an orphanG-protein coupledreceptor
gene,wasshowntobetheonlyonesignificantlyoverexpressed inthepituitarytissueofpatientswithXq26.3microduplications.
Thecausativepathogenicroleof the GPR101gene inXLAG
wasthereaftersupportedbyaXLAGpatientwithduplicationof
GPR101alone[8].
GPR101 is anorphan Gsprotein-coupledreceptor, whose
endogenousligandandexactfunctionsareunknown.GPR101
isnormallyexpressedatparticularlyhighlevels inthemouse hypothalamusandinthepituitaryoftherhesusmacaqueandthe
rat.Inhumans,GPR101mRNAispredominantinthenucleus
accumbens anditsexpression islow inadultpituitarygland.
However,GPR101mighthavearoleduringpituitary
ontoge-nesis.GPR101 expressionwasfoundinthepituitaryglandof
humanembryosstartingfrom19weeksofgestationalagewith
thehighestlevelsofGPR101staining(65%)at38weeks[4,14].
GPR101duplicationmightsupportmixedGHandprolactin pos-itivecellularhyperplasiainXLAGstartingfromthefetalperiod withfurtheradenomaformationintheearlyperiodafterbirth.
However,theroleofGPR101inpituitarydevelopment,
tumori-genesisandstimulationofGH/prolactinhypersecretionremains
unclear.Sofar,inactivatingmutationsoftheGPR101genedo
ARTICLE IN PRESS
+ModelANDO-931; No.ofPages6
A.Beckersetal./Annalesd’Endocrinologiexxx(2017)xxx–xxx 5
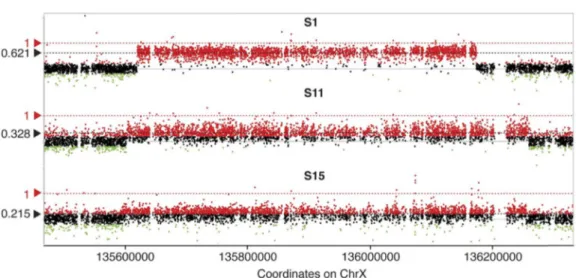
Fig.4.GenomicgainsonHD-aCGHinthreesporadicmales.ThetheoreticalLogratio(LR)valueofheterozygousduplicationonXchromosomeinmalesismarked astheredline.TheLRvaluesoftheXLAGduplicationsidentifiedineachsubject(blacklines)werefoundtobelowerthanthetheoreticalLRinallthreecases[9].
MostXLAGcasesaresporadicandduplicationsinall
spo-radiccasesappeartobenon-recurrentwithuniqueboundaries.
ThesearegenerallyduetoaDNAreplicationerrorbecauseof
microhomologiesbetweenproximatefragmentsatXq26.3[4].
Infamilialcases,atransmissionofthe sameXLAG
microdu-plication from affected mother to affected son with 100%
penetrancehasbeenreportedinthreehomogeneousFIPA
kin-dreds withearlyonset acrogigantism[4,11].Along withAIP
mutation/deletion,XLAGisthesecondknowngeneticcauseof
FIPAinAIP-negativekindredswithfamilialacrogigantism.
While the GPR101 duplication has been found in all the
XLAGcasesreportedsofar,insporadicmalestheaCGHresult
demonstratesthepresenceofsomaticmosaicismthatleadstoa variableproportionofaffectedcells(Fig.4)[9].Relativelylow levelsofGPR101duplicatedcells(aslowas16–17%)havebeen
confirmedusingdigitaldropletPCR(ddPCR),whichallowsthe
detectionof quitesmallrates of mutationsor CNV–deletions
or duplications (Fig. 5). The clinical presentation of XLAG
inallmosaiccasesissimilartothatof patientswith constitu-tionalduplication,intermsofovergrowthandpituitarydisease
severity. Thus, sporadic males could exhibit dramatic forms
of pituitary gigantism even when only a low proportion of
cellscarrytheGPR101duplication.Wescreenedalargegroup
of patients with acromegaly andgigantismusing quantitative
ddPCRforGPR101duplications.Thismethodallowsthe
iden-tificationofnewXLAGcasesbymeansofincreasedGPR101
copynumber,whichcanlaterbeconfirmedonaCGH[9].
Recently,wefurtherappliedthisscreeningddPCRmethod
toancientDNAretrievedfromskeletalremainsofahistorical
caseofextremeacrogigantism(giantConstantin;259cm).The
individualdiedofsepticemiafollowingsurgeryforgangreneas ayoungadultin1902,andhisautopsydemonstratedamassively enlargedpituitaryglandandpituitaryfossa[10].Accordingto
published sourcesand original medical documentation
avail-abletous,theabnormalgrowthstartedearlyandextremelytall stature(1.94m)wasalreadypresentattheageof14.
Paleonto-logicalDNAextractiontechniquespermittedtheextractionof
DNAfrombonepowderandsubsequentddPCRrevealed
ele-vatedcopynumberofGPR101indicatingaprobablediagnosis
ofXLAG(Fig.6).
Fig.5.Mosaicism-levelquantificationbyHD-aCGH(panelA)andddPCR(panelB)insporadicfemales(averagemosaicismlevelin11cases),familialfemale (1case),familialmales(3cases)andsporadicmales(3cases)withXLAGsyndrome[9].
ARTICLE IN PRESS
+ModelANDO-931; No.ofPages6
6 A.Beckersetal./Annalesd’Endocrinologiexxx(2017)xxx–xxx
Fig.6. Ahistoricalcaseofdramaticgigantism,thegiantConstantin,whomeasured259cm.HewasphotographedforaposterwhilevisitingBelgiumin Novem-ber1901,atwhichtimehehadextremelytallstatureandacromegalicfeatures(panelA).Aphotographofhisskullfrom1904(panelB)showsmarkedprognathism andenlargementofthemalarbones.AnenlargedsellaturcicaismarkedbytheopenarrowonpanelCshowinganinteriorviewofthesubject’scranium.DNAwas obtainedbydrillingasmallamountofbonepowder(filledarrow)[10].
4. Conclusion
XLAGsyndromeischaracterizedbyaveryspecific
pheno-typeofinfant-onsetgigantismwithmarkedlyincreasedheight
andweightz-scores.It iscausedbymixed GHandprolactin
secretingpituitaryadenomas and/orhyperplasiathat can
pro-duceexceptionallyhighlevelsofGHandprolactin.Thisnew
genetic from of pituitarygigantism is due tothe duplication
of the GPR101 gene andcan present either sporadically (as constitutional or mosaicduplication) orinaFIPA setting.To
date,32XLAGpatientshavebeenreported,mostofwhomare
females.XLAGisaclinicalentitythat,whilerare,leadstoan exceptionallyseverepituitarygigantismphenotypethatrequires earlyinterventiontocontrolovergrowth.
Disclosureofinterest
Theauthorsdeclarethattheyhavenocompetinginterest.
References
[1]RostomyanL,BeckersA.Screeningforgeneticcausesofgrowthhormone hypersecretion.GrowthHormIGFRes2016;30–31:52–7.
[2]BeckersA,etal.Familialisolatedpituitaryadenomas(FIPA)andthe pitu-itaryadenomapredispositionduetomutationsinthearylhydrocarbon receptorinteractingprotein(AIP)gene.EndocrRev2013;34:239–77.
[3]DalyAF,etal.Clinicalcharacterizationoffamilialisolatedpituitary ade-nomas.JClinEndocrinolMetab2006;91:3316–23.
[4]TrivellinG,etal.GigantismandacromegalyduetoXq26microduplications andGPR101mutation.NEnglJMed2014;371:2363–74.
[5]RostomyanL,etal.Clinicalandgeneticcharacterizationofpituitary gigan-tism:aninternationalcollaborativestudyin208patients.EndocrRelat Cancer2015;22:745–57.
[6]RoddC,etal.Somatic GPR101 duplicationcausingX-linked acrogi-gantism(XLAG)-diagnosisand management.JClinEndocrinolMetab 2016;101:1927–30.
[7]NavesLA,etal.Aggressivetumorgrowthandclinicalevolutioninapatient withX-linkedacro-gigantismsyndrome.Endocrine2016;51:236–44.
[8]IacovazzoD,etal.Germline orsomaticGPR101duplicationleadsto X-linkedacrogigantism:aclinicopathologicalandgeneticstudy.Acta Neu-ropatholCommun2016;4:56.
[9]DalyAF,etal.SomaticmosaicismunderliesX-linkedacrogigantism syn-dromeinsporadicmalesubjects.EndocrRelatCancer2016;23:221–33.
[10]BeckersA,etal.PaleogeneticstudyofancientDNAsuggestiveofX-linked acrogigantism.EndocrRelatCancer2017;24:L17–20.
[11]GordonRJ,etal.ChildhoodacromegalyduetoX-linkedacrogigantism: long-termfollow-up.Pituitary2016;19:560–4.
[12]BeckersA,etal.X-linkedacrogigantismsyndrome:clinicalprofileand therapeuticresponses.EndocrRelatCancer2015;22:353–67.
[13]DalyAF,etal.GHRHexcessandblockadeinX-LAGsyndrome.Endocr RelatCancer2016;23:161–70.
[14]TrivellinG,etal.CharacterizationofGPR101transcriptstructureand expressionpatterns.JMolEndocrinol2016;57:97–111.
[15]CastinettiF,etal.GPR101mutationsarenotafrequentcauseof con-genitalisolatedgrowthhormonedeficiency.HormMetabRes2016;48: 389–93.