Étude des fonctions de la protéine FANCI
via la caractérisation d’un nouveau modèle murin
de l’anémie de Fanconi
Thèse
Émilie Dubois
Doctorat en Biologie Cellulaire et Moléculaire
Philosophiæ Doctor (Ph.D.)
Québec, Canada
© Emilie Dubois, 2018
Étude des fonctions de la protéine FANCI
via la caractérisation d’un nouveau modèle murin
de l’anémie de Fanconi
Thèse
Émilie Dubois
Sous la direction de :
Résumé
On compte aujourd’hui plus de 2000 patients dans le monde affectés par l’anémie de Fanconi (AF). Cette maladie rare est causée au niveau cellulaire par un défaut de réparation d’un type très particulier de lésions de l’ADN : les pontages inter-brins (ou ICLs, pour Interstrand CrossLink). Ceux-ci peuvent compromettre la transcription et la réplication, et donc à long terme la survie cellulaire. Pour y faire face, plus d’une vingtaine de protéines sont impliquées dans la voie FA-BRCA, chargée de détecter et d’activer la réparation de ces lésions dangereuses pour la cellule. Au niveau macroscopique, les manifestations de l’AF sont vastes et pas systématiquement présentes, ce qui en complique le diagnostic : pancytonémie, malformations développementales congénitales et prédisposition à certains cancers.
Comme la plupart des symptômes ne sont pas spécifiques à l’AF, les enjeux et l’intérêt de son étude deviennent multiples. L’anémie de Fanconi offre un modèle d’étude particulièrement intéressant, qui permet d’explorer des mécanismes aussi variés que la cancérogenèse, l’hématopoïèse, la gamétogenèse et la réparation de l’ADN. Les connaissances sur le mécanisme moléculaire à proprement parler sont grandissantes, même si de nombreuses interrogations persistent. Parmi elles, le rôle précis du complexe ID2, formé des protéines FANCI et FANCD2, reste nébuleux. De plus en plus d’études décrivent des fonctions exclusives à FANCD2, laissant formuler l’hypothèse que la protéine FANCI pourrait elle aussi posséder ses propres rôles. Cependant, les études biochimiques ont certaines limites, comme par exemple l’impossibilité d’étudier in vitro des processus biologiques trop complexes. La création de modèles animaux peut être une solution.
Pour répondre aux interrogations portées sur les fonctions de la protéine FANCI, nous avons décidé de caractériser un nouveau modèle murin Fanci-/- et de le comparer à la souris Fancd2-/-, déjà
décrite dans la littérature. Nos résultats indiquent d’une part plusieurs rôles épistatiques avec FANCD2 et d’autre part un rôle propre dans le cadre de la gamétogenèse.
En conclusion, cette thèse apporte un tout nouveau modèle animal pour étudier l’AF et plus spécifiquement FANCI. Elle apporte aussi les preuves physiologiques d’un rôle de FANCI indépendant au complexe ID2, ce qui viendrait nuancer le mécanisme biochimique actuellement établi.
Abstract
Nowadays, about 2000 patients have been diagnosed with Fanconi Anemia (FA). At a cellular level, this rare disease is caused by a defect in reparing a specific type of DNA damage: Interstrand CrossLinks (ICLs). Both transcription and replication can be compromised by ICLs, and therefore long term cell survival. The FA-BRCA pathway is in charge of detecting DNA lesions caused by ICLs and activating their repair, and counts now more than twenty proteins. At a macroscopic level, FA diagnosis is complex due to various and not systematic symptoms: pancytonemia, congenital and developmental defects, and cancer predisposition.
As most of FA symptoms are not exclusive to the disease, concerns and interests are multiple. As a consequence, Fanconi Anemia represents an interesting model of study, when it comes to work on oncogenesis, hematopoiesis, gametogenesis, and DNA repair. The description of the molecular mechanism is improving, although many questions remain. Among them, the exact role of the ID2 complex, formed by FANCI and FANCD2 proteins, remains nebulous. More and more studies have been describing unique functions for FANCD2, leading to the hypothesis of independent functions for FANCI. As complex physiological processes can not be studied in vitro, creating and studying animal models represents an interesting alternative.
In order to answer the questions raised on FANCI functions, we have decided to create and characterize the new Fanci-/- mouse model, with a possible comparison with the already described
Fancd2-/- mouse. Our results show epistatic roles with FANCD2 on one hand, and independent
functions of FANCI during gametogenesis on the other hand.
To conclude, this thesis brings a strong and new mouse model to study Fanconi Anemia and more specifically the roles of FANCI. It also gives some physiological proofs that FANCI can act independently of the ID2 complex, which could slightly nuance the actual molecular pathway.
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des figures ... viii
Liste des tableaux ... x
Liste des figures supplémentaires ... xi
Liste des abbréviations ... xii
Remerciements ... xviii
Avant-propos ... xxi
Chapitre 1 : Introduction ... 1
1.1 La cellule face à l’instabilité génomique ... 1
1.1.1 La réparation de l’ADN ... 1
1.1.1.1 Généralités ... 1
1.1.1.2 La formation des ICLs ... 2
1.1.1.2.1 Généralités ... 2
1.1.1.2.2 Les aldéhydes : présentation, incidence et prise en charge ... 2
1.1.1.3 La réparation des ICLs et des cassures double-brin ... 4
1.1.2 Les maladies associées à l’instabilité génomique ... 7
1.2 L’anémie de Fanconi ... 7
1.2.1 Découverte et incidence ... 8
1.2.2 Manifestations cliniques et diagnostic ... 8
1.2.3 Groupes de complémentation ... 12
1.2.4 Les traitements de l’anémie de Fanconi ... 14
1.2.4.1 La prise en charge hématologique ... 14
1.2.4.2 La thérapie génique sur cellules souches hématopoïétiques ... 14
1.2.4.3 Le traitement des symptômes non spécifiques à l’AF ... 15
1.2.4.4 Les stratégies anticancéreuses ... 16
1.2.4.4.1 Les défis ... 16
1.2.4.4.2 L’importance d’un dépistage plus précoce ... 16
1.2.4.4.3 Les traitements anti-tumoraux en cours de développement ... 16
1.2.4.5 L’utilisation des aldéhydes, un futur prometteur ... 18
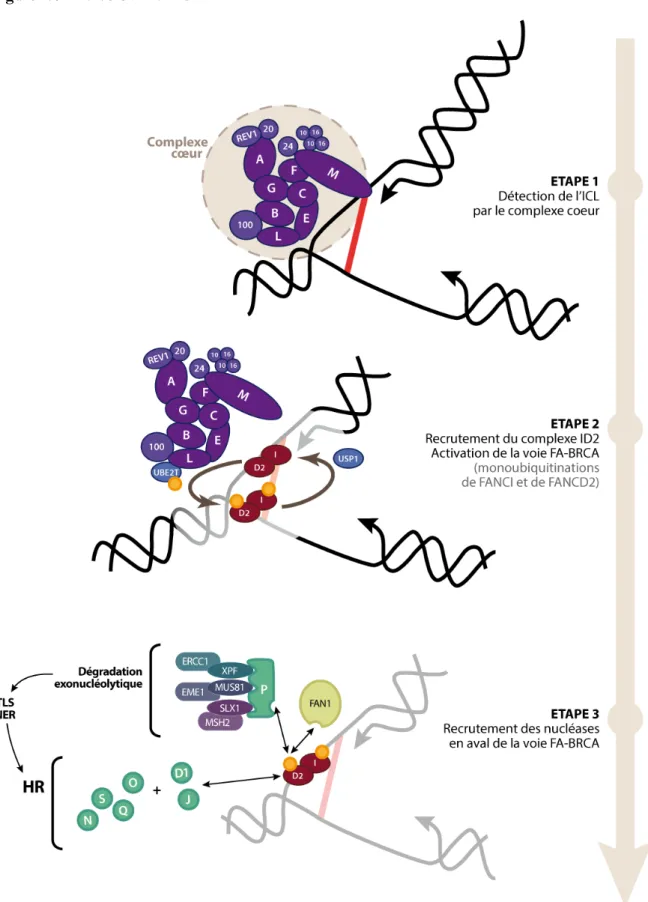
1.2.5 La voie FA-BRCA ... 19
1.2.5.1 Généralités ... 19
1.2.5.2 Mécanisme biochimique ... 19
1.2.5.3 Les fonctions annexes des protéines FANC ... 22
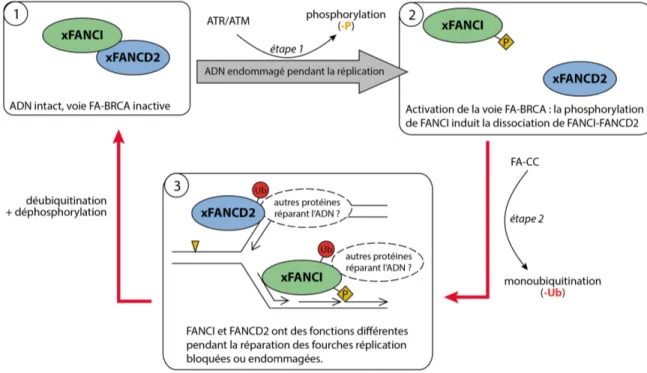
1.2.5.6 FANCI et FANCD2, jamais l’un sans l’autre ... 27
1.2.5.7 Controverses actuelles sur des partenaires pas toujours si proches ... 27
OBJECTIFS ... 31
Chapitre 2 : Les modèles animaux de l’anémie de Fanconi Ou comment les différences
peuvent être aussi précieuses que les similitudes… ... 32
2.1 Avant-propos ... 33
2.2 Résumé ... 34
2.3 Abstract ... 35
2.4 Introduction ... 36
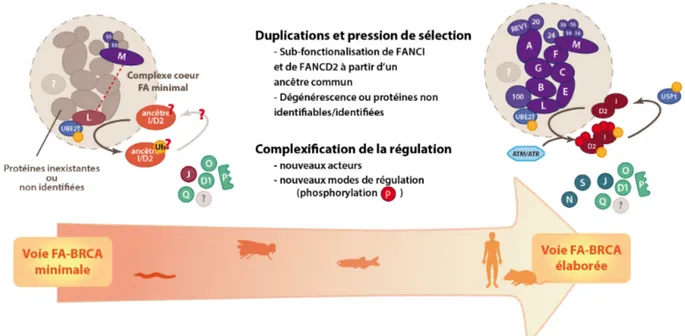
2.5 La voie FA-BRCA au cours du temps ... 39
2.6 Les découvertes majeures apportées par les modèles animaux pour l’AF ... 40
2.6.1 Le nématode ... 40
2.6.2 La drosophile ... 41
2.6.3 Le poisson zèbre ... 41
2.6.4 La souris ... 42
2.7 Perspectives ... 44
Chapitre 3 : A Fanci knock-out mouse model reveals common and distinct functions for
FANCI and FANCD2 ... 47
3.1 Avant-propos ... 48 3.2 Résumé ... 49 3.3 Abstract ... 50 3.4 Significance statement ... 51 3.5 Introduction ... 52 3.6 Results ... 53
3.6.1 Generation of a FANCI knock-out mouse ... 53
3.6.2 Fanci-/- mice display phenotypes similar to FA ... 54
3.6.3 Hematological status in Fanci-/- mice ... 57
3.6.4 Interbreeding FANCD2 and FANCI deficient mice and epistatic relationships for hematopoietic defects ... 59
3.6.5 Skeletal defects in the Fanci-/- mouse ... 61
3.6.6 Sterility and differential meiotic localization for FANCD2 and FANCI ... 63
3.6.7 FANCI, unlike FANCD2, stimulates RAD51-mediated D-loop formation ... 66
3.7 Discussion ... 66
3.8 Material and methods ... 69
3.9 Supplemental data ... 72
Chapitre 4 : Discussion ... 78
4.1 Dernières avancées sur la biochimie de FANCI ... 78
4.1.1 Identification de la protéine UBL5 et impact sur FANCI ... 78
4.1.3 FANCI et les origines de réplication ... 80
4.2 Conclusions sur les modèles animaux ... 82
4.2.1 Les modèles animaux spontanés pour l’AF ... 82
4.2.2 Les limites de notre modèle murin ... 83
4.2.2.1 La gamétogenèse ... 83
4.2.2.2 La souche C57BL/6 et ses phénotypes aggravés ... 83
4.2.3 Localisation et fonction de FANCI dans les souris de type sauvage ... 84
4.2.3.1 FANCI et la réparation des ICLs pendant la gamétogenèse ... 84
4.2.3.2 Contribution maternelle ... 88
4.2.4 Conclusions sur la souris Fanci-/- ... 90
4.2.4.1 Une spermatogenèse perturbée ... 90
4.2.4.2 Un rôle potentiel dans la morphogenèse ... 91
4.2.4.3 Exploration du lien entre FANCI et la transcription dans les MEFs Fanci-/- ... 93
4.2.5 Potentiel expérimental des souris Fanci-/- ... 94
4.2.5.1 Étude de l’impact des aldéhydes sur les souris pour l’AF ... 94
4.2.5.2 Quel lien entre FANCI et le cancer chez la souris ? ... 96
4.3 Les patients AF : futur et potentiels ... 96
4.3.1 Une vaste source d’informations utiles à tous ... 96
4.3.2 FANCI : un marqueur prédictif pour le cancer ovarien au même titre que BRCA1 et BRCA2 ? ... 97
4.3.3 Des traitements qui impliqueraient FANCI ? ... 98
Chapitre 5 : Conclusion ... 99
Annexe 1 ... 100
Liste des figures
Chapitre 1 – Introduction
Figure 1.1 – Mécanismes réparant les ICLs, selon l'avancée dans le cycle cellulaire ... 5
Figure 1.2 – Principales manifestations des patients atteints de l'anémie de Fanconi ... 9
Figure 1.3 – Principe de complémentation par fusion cellulaire ... 13
Figure 1.4 – La voie FA-BRCA ... 21
Figure 1.5 – Les multiples fonctions des protéines FANC ... 23
Figure 1.6 – Les domaines protéiques de FANCI et FANCD2 ... 26
Figure 1.7 – Modèle moléculaire le plus récent ... 30
Chapitre 2 – Les modèles animaux de l’anémie de Fanconi
Figure 2.1 – Frise chronologique des grandes étapes de la recherche sur l'anémie de Fanconi . 37Figure 2.2 – La voie FA-BRCA au cours de l'évolution ... 38
Chapitre 3 – A Fanci knock-out mouse model reveals common and distinct functions
for FANCI and FANCD2
Figure 3.1 – Fanci-/- animals are smaller ... 55Figure 3.2 – Fanci-/- mouse embryonic fibroblasts display FA hallmarks ... 56
Figure 3.3 – Fanci-/- mice show bone marrow failure ... 58
Figure 3.4 – Combined inactivation of both FANCI and FANCD2 is epistatic for bone marrow failure and eye abnormalities ... 60
Figure 3.5 – Fanci-/- mice show squeletal malformations ... 62
Figure 3.6 – Fanci-/- mice are infertile ... 64
Figure 3.7 – Discrepancies in FANCI and FANCD2 localization during meiosis ... 65
Chapitre 4 – Discussion
Figure 4.1 – Mécanisme moléculaire hypothétique d’interaction entre FANCI, FANCD2 et UBL5 ... 79Figure 4.2 – Fonctions distinctes de FANCI et de FANCD2 au niveau des origines de réplication ... 81
Figure 4.3 – La réparation de l'ADN pendant la gamétogenèse murine ... 86
Figure 4.4 – Stratégie d'étude de la spermatogenèse chez les souris Fanci-/- ... 91
Figure 4.5 – Vue 3D des axes, des zones de régulation et des protéines assurant la croissance des membres chez la souris ... 93
Annexe 1
Figure 5.1 – Proportions variables de la forme des chromosomes dans les MEFs Fanci-/- ... 101
Figure 5.2 – La chromatine des cellules Fanci-/- interphasiques serait plus condensée ... 102
Figure 5.3 – Niveau d’expression de quelques protéines d’intérêt dans les MEFs Fanci+/+ et
Liste des tableaux
Chapitre 1 – Introduction
Table 1.1 – Fréquence d'apparition des différents symptômes chez les patients atteints de l'AF 9
Table 1.2 – Conservation des protéines FANC dans divers organismes ... 11
Liste des figures supplémentaires
Chapitre 3 – A Fanci knock-out mouse model reveals common and distinct functions
for FANCI and FANCD2
Supplemental Figure 3.1 – Targeted disruption of the murine Fanci locus ... 72
Supplemental Figure 3.2 – Pattern of expression of FANCI in mouse tissues, and antibody validation ... 73
Supplemental Figure 3.3 – Limited growth of Fanci-/- MEFs ... 74
Supplemental Figure 3.4 – Fanci-/- BMCs can not compete with wild-type BMCs ... 75
Supplemental Figure 3.5 – Fanci-/- males display a Sertoli-like phenotype ... 76
Supplemental Table 3.1 – Under-Mendelian ratios obtained while breeding Fanci+/-Fancd2 +/-mice ... 77
Liste des abbréviations
3D 3 dimensions4-HNE 4-Hydroxy-2NonEnal
4H-TTD 4H-Thiopyran-4-one, Tétrahydro-3,5-bis[(4-nitrophényl)méthylène]-,1,1-Dioxide
A. thaliana Arabidopsis thaliana
ADN Acide DésoxyriboNucléique
ADH5 Alcool DésHydrogénase 5
AER de l’anglais « Apical Ectodermic Ridge »
AF Anémie de Fanconi
Alt-NHEJ de l’anglais « Alternative-NHEJ »
ALDH2 ALdéhyde DésHydrogénase 2
APRIN de l’anglais « Androgen-induced PRoliferation INhibitor »
ARN Acide RiboNucléique
ARNm Acide RiboNucléique Messager
ATM de l’anglais « Ataxia Telangiectasia Mutated »
ATP Adénosine TriPhosphate
ATR de l’anglais « Ataxia- and Rad-3Related »
BACH1 de l’anglais « BRCA1-Associated C-terminal Helicase 1 »
BLM de l’anglais « BLoom syndrome Mutated protein »
BMCs de l’anglais « Bone Marrow Cells »
BRCA1/2 de l’anglais « BReast Cancer 1/2, early onset »
BRIP1 de l’anglais « BRCA1 Interacting Protein C-terminal helicase 1 »
BS de l’anglais « Brachyspina Syndrome »
C. elegans Caenorhabditis elegans
C-NHEJ voie classique du NHEJ
CDB(s) Cassure(s) Double-Brin
CDC2 de l’anglais « Cell Division Cycle 2 »
CFC de l’anglais « Colony Forming Cell assay »
CFU de l’anglais « Colony Forming Unit »
ChIP-Seq de l’anglais « CHromatin ImmunoPrecipitation Sequencing »
CHUL Centre Hospitalier Universitaire de l’université Laval
CRISPR de l’anglais « Clustered Regularly Interspaced Short Palindromic Repeats »
CUE de l’anglais « Coupling of Ubiquitin conjugation to Endoplasmic reticulum degradation »
D. Rerio Danio rerio
D. melanogater Drosophila melanogaster D3T 3H-1,2-dithiole-3-thione D1-D2-G-X3 Complexe FANCD1-FANCD2-FANCG-XRCC3 DAC 5-aza-2’-déoxycytidine DDN 2,3-dichloro-5,8-dihydroxy-1,4-naphthoquinone DEB DiÉpoxyButane DMBA DiMéthylBenz[a]Anthracène
DNA de l’anglais « DeoxyriboNucleic Acid »
DNA-PK de l’anglais « DNA-dependent Protein Kinase catalytic subunit »
Dr(e) Docteur(e)
DT40 lignée stable de cellules lymphocytaires B de poulet
DTT DiThioTréitole
EDTA de l’anglais « Ethylene Diamine Tetraacetic Acid »
EF24 diphényl difluorocétone, analogue du curcumin
EOC de l’anglais « Epithelial Ovarian Cancer »
et al. et autres, locution latine « et alter »
FA de l’anglais « Fanconi Anemia »
FA-BRCA de l’anglais « Fanconi Anemia - Breast Cancer »
FAAP de l’anglais « Fanconi Anemia-Associated Protein »
FACS de l’anglais « Fluorescence-Activated Cell Sorting »
FARF de l’anglais « Fanconi Anemia Research Fund »
FAZF de l’anglais « FA-Zinc Finger protein »
FDA de l’anglais « Food and Drug Administration »
FGF de l’anglais « Fibroblast Growth Factor »
FOXO3a de l’anglais « FOrkhead BoX O3 »
GG-NER de l’anglais « Global Genome NER »
GRP94 de l’anglais « Glucose-Regulated Protein 94 »
H. sapiens Homo sapiens
HES1 de l’anglais « Hairy and Enhancer of Split 1 protein »
HGSC de l’anglais « High-Grade Serous Carcinomas »
HJ de l’anglais « Holliday Junction »
HLA de l’anglais « Human Leukocyte Antigen test »
HMG de l’anglais « High Mobility Group protein »
HPV de l’anglais « Human Papilloma Virus »
HR de l’anglais « Homologous Recombination »
HSCs de l’anglais « Hematopoietic Stem Cells »
HSP70 de l’anglais « Heat Shock Protein 70kDa »
HSPCs de l’anglais « Hematopoietic Stem and Progenitor Cells »
HU HydroxyUrée
ICL de l’anglais « Interstrand CrossLink »
ID2 Complexe FANCI-FANCD2
IF ImmunoFluorescence
IFAR de l’anglais « International Fanconi Anemia Registry »
IFN-γ InterFéroN γ
IHC ImmunoHistoChimie
IR Radiations Ionisantes, de l’anglais « Ionising Radiation »
IRIC Institut de Recherche en Immunologie et en Cancérologie
K ou Lys Lysine
Kb Kilo Base
KD de l’anglais « Knock-Down »
kDa kiloDalton
KO de l’anglais « Knock-Out »
LAM Leucémie Aigüe Myéloïde
LIG de l’anglais « DNA ligase »
LK (cells) cellules Lin- (Sca1-) c-Kit+
LSK (cells) cellules Lin- Sca1+ c-Kit+
Lys Lysine
M. musculus Mus musculus
MCM de l’anglais « MiniChromosome Maintenance complex component 2 »
MDa MégaDalton
MGA de l’anglais « Mid-preimplantation Gene Activation »
MHF de l’anglais « FANCM interacting Histone-Fold protein »
MiTF de l’anglais « Microphtalmia Transcription Factor »
MLH1 de l’anglais « MutL Homolog protein 1 »
MMC MitoMycine C
MNCs de l’anglais « MonoNuclear blood Cells »
MRI de l’anglais « Magnetic Resonance Imaging »
MRN MRE11-RAD50-NSB1
NAD de l’anglais « Nicotinamide Adenine Dinucleotide »
NBS1 de l’anglais « Nijmegen Breakage Syndrome protein 1 »
ND Non Documenté
NER de l’anglais « Nucleotid Excision Repair »
NGS de l’anglais « Next-Generation Sequencing »
NHEJ de l’anglais « Non-Homologous End Joining »
NLS de l’anglais « Nuclear Localization Signal »
nM nanoMolaire
p-RPA forme phosphorylée de la protéine RPA
PALB2 de l’anglais « Partner and Localizer of BRCA2 »
PARP de l’anglais « Poly-ADP-Ribose Polymerase »
PARPi inihibteur de PARP
PBS de l’anglais « Phosphate Buffered Saline »
PCR de l’anglais « Polymerase Chain Reaction »
PFA ParaFormAldéhyde
PIP-box de l’anglais « PCNA-Interaction box »
PKR de l’anglais « Protein Kinase regulated by RNA »
PMSF de l’anglais « PhenylMethylSulfonyl Fluoride »
Pol Polymérase
RH Recombinaison Homologue
RNP complexe RiboNucléoProtéique
RPA de l’anglais « Replication Protein A »
S. cerevisiae Saccharomyces cerevisiae
SCC de l’anglais « Squamous Cellule Carcinoma »
SCO de l’anglais « Sertoli Cell-Only »
SDS DodécylSulfate de Sodium
Ser, S Sérine
SHH de l’anglais « Sonic HedgeHog protein »
SOD1 Superoxyde Dismutase 1
SSC de l’anglais « Saline-Sodium Citrate »
S/TQ résidus Sérine/Thréonine et Glutamine
STAT1 de l’anglais « Signal Transducer and Activator of Transcription 1 »
TAp63 Transcription-Associated protein 63kDa
TBE solution de Tris, Borate, EDTA
TC-NER de l’anglais « Transcription-Coupled NER »
TCGA de l’anglais « The Cancer Genome Atlas group »
TERT de l’anglais « TElomerase Reverse Transcriptase protein »
Thr, T Thréonine
TLS Synthèse TransLésionnelle, de l’anglais « TransLesion Synthesis »
TMA de l’anglais « Tissue MicroArray »
TNF-α de l’anglais « Tumor Necrosis Factor α »
TUNEL de l’anglais « Terminal deoxynucleotidyl transferase dUTP Nick End Labeling »
Ub Ubiquitine
UBD de l’anglais « Ubiquitin Binding Domain »
UBE2T de l’anglais « UBiquitin-conjugating Enzyme E2 T »
UBL5 de l’anglais « Ubiquitin like 5 »
UBZ de l’anglais « Ubiquitin-Binding Zinc finger motif »
USP1 de l’anglais « Ubiquitin Specific Peptidase 1 »
UV Ultra-Violet
V(D)J Variant Distance Junction
WBC de l’anglais « White Blood Cell »
WNT de l’anglais « WiNgless-Type family »
WT de l’anglais « Wild-Type »
WT1 de l’anglais « Wilms Tumor protein 1 »
XPC, XPF de l’anglais « Xeroderma Pigmentosum group C/F-complementing protein »
XRCC de l’anglais « X-ray Repair Cross Complementing »
WBC de l’anglais « White Blood Cell »
WES de l’anglais « Whole Exome Sequencing »
Je dédie cette thèse à la vie, à la rudesse de sa beauté,
et à ma famille, qui m’a toujours soutenue,
quel que soit leur niveau de folie de mes idées …
“What we do not see, what most of us never suspect of existing,
is the silent but irresistible power which comes to the rescue
of those who fight on in the face of discouragement.”
- Napoleon HillRemerciements
Je tiens tout d’abord à remercier mon directeur de thèse, le Dr J.-Y. Masson, pour m’avoir accueilli dans son laboratoire, donné un aussi beau projet et offert toutes les opportunités d’enrichir mon bagage scientifique via de belles collaborations et des congrès internationaux. Je vous suis particulièrement reconnaissante pour votre confiance et l’autonomie que vous m’avez laissée. Merci aussi pour votre soutien et vos encouragements, de m’avoir laissé le temps nécessaire à mieux définir mon avenir et d’avoir respecté mes choix de carrière.
En second lieu, je tiens à remercier très sincèrement les membres de mon jury, qui ont bien voulu réviser ma thèse et y apporter des commentaires constructifs. Merci également pour ces discussions captivantes lors de nos rencontres à des congrès ou à des séminaires, lorsque l’occasion s’est présentée.
J’ai un énorme merci à adresser au Dr Guy Sauvageau pour sa grande passion contagieuse pour la science mais aussi d’avoir accepté de collaborer avec nous sur le volet hématopoïétique, et en me confiant à Jalila. Jalila, tu resteras un exemple sans égal de passion, de motivation, de connaissances et de compétence ! Merci pour ces riches échanges, merci de m’avoir aussi bien encadrée et choyée lors de mes visites à Montréal. J’ai puisé dans ce que tu m’as appris pour encadrer de mon mieux lorsque l’occasion s’est présentée. Enfin dans la même équipe à l’IRIC, j’ai eu le plaisir de collaborer sur un tout autre sujet avec Céline. Elle suivait l’avancée du projet sur la souris depuis son bureau voisin, que j’occupais clandestinement pendant les longues soirées de manip.
Les voyages pour les congrès ont été l’occasion de rencontrer des personnalités scientifiques marquantes et inspirantes. Parmi elles, je voudrais remercier le Dr Meyns pour ses conseils et ses suggestions, ainsi que d’avoir sollicité son équipe pour nous aider, car une partie de mon travail en découle. À sa manière, la Dre Fradet-Turcotte m’a marquée par son dynamisme et a représenté un modèle de travail acharné. Plus globalement, merci à tous les chercheurs de transmettre sans retenue cet amour pour la science !
De mes débuts au laboratoire, je souhaite remercier Annie, la première à m’avoir guidée sur ce projet qui sortait totalement des sentiers battus du laboratoire. Merci également à Rémi de m’avoir initiée à un haut niveau d’exigence pour fournir le meilleur travail possible, ainsi qu’à une persévérance vitale à tout doctorant.
Toujours au sein du laboratoire, impossible de ne pas remercier du fond du cœur Yan, le « Couteau suisse » de l’équipe ou encore « The Cloning Machine ». Sa disponibilité, son savoir, son aide technique et sa patience ont été cruciaux à l’avancée du projet. Je tiens également à remercier Joris pour les discussions sur nos projets respectifs, sans doute les plus proches de l’ensemble des thématiques du laboratoire, merci également pour l’aide technique, la bonne humeur et la compagnie lors des congrès. Mariline, à ton tour de souffrir de mon merci ! Je n’aurais jamais espéré meilleure stagiaire en tous points ! Ta motivation, ta curiosité, ton intelligence et ta gentillesse donnent le goût d’avoir d’autres stagiaires à encadrer. Ce fût un réel plaisir de partager ce beau projet avec toi ! Ta volonté a nourri notre travail en collaboration. J’espère que tu aimeras la Science encore longtemps et qu’elle te le rendra bien.
Niraj, plus besoin de traduction en anglais ou en hindi, tu as d’une certaine manière complété un second doctorat en langue française ! Je n’oublierai jamais ton grand soutien amical tout au long de ce cheminement, et ce fût très plaisant de collaborer avec toi vers la fin de ma thèse. Merci également à Marie-Christine (que j’ai sans doute rebaptisée plus d’une fois MMC – pour MitoMycine C – sur le tableau du laboratoire, pardon…) d’avoir assuré la transition lors de mon départ du laboratoire. Ce fût un plaisir de travailler dans un contexte aussi calme et agréable, comme tu en as le don.
En continuant dans ma lancée « scientifique », je vais faire une parenthèse hors laboratoire pour adresser une grande pensée à Lucie ! Merci pour tout ce temps partagé, à débattre passionnément de Science, à se conseiller (je l’espère réciproquement) sur des points techniques pour mener nos travaux de manière constructive et créative.
Par la même occasion, je tiens à remercier le Dr Lebel pour sa disponibilité et son mentorat tout au long de ma thèse, pour m’avoir permis de travailler dans son laboratoire avec son aide technique, même après un déménagement au CHUL. Merci aussi à Chantal et à Serges d’avoir accueilli toujours avec bonne humeur et humour mes visites impromptues dans leur espace de travail.
Du fait du caractère très « animal » de mon projet, j’ai été amenée à envahir la salle à mouches pour collaborer avec l’équipe du Dr Laprise. J’ai eu la chance d’être formée et de travailler avec des personnes aussi compétentes et passionnées que Clémence et Emilie H. Cléclé, je te dois un gros merci pour ton amitié, ton soutien constant, surtout lors des passes difficiles au cours desquelles tu as su aller chercher mon sourire.
Enfin, je finis ma tournée hors laboratoire par les équipes des Dr Moss, Dr Charron et Dre Jeannotte, qui ont toujours accueillies avec bienveillance mes visites et qui ont répondu à des masses de questions techniques.
Au laboratoire, j’ai eu beaucoup de plaisir à côtoyer Stéphanie, pour sa gentillesse, sa curiosité et son professionnalisme. Je n’oublierai jamais à quel point sa franchise et sa force de caractère ont été inspirantes et motivantes ! Le congrès Fanconi à Bethesda a également été le fun à ses côtés !
Ranjan et Hemanta, mes deux « benchmates » préférés… la force tranquille de votre présence, votre assiduité et votre attention bienveillante sont inégalables et ont été grandement porteuses. Merci pour les belles personnes que vous êtes et pour ce que vous dégagez et donnez, sans même vous en rendre compte.
Enfin, je n’oublie pas les autres membres du laboratoire pour leurs conseils et leur aide, ou du moins leur sourire : Tony, Jana, Amélie, Denis, Kenny, Anne-Marie et Marie-Michèle.
J’ai également eu la chance de travailler avec une super équipe à l’animalerie, notamment Émilie, Julie, Mélissa et Karine, puis Andrée au CHUL. Toutes étaient mes yeux et mes mains à distance.
Merci également à d’autres collaborateurs plus discrets mais tout aussi essentiels : Carl St Pierre au service de microscopie et Jean Lagueux pour tous les scans des souris. La qualité des échanges et leur bonne humeur ont rendu ce travail encore plus plaisant, sans pour autant perdre en rigueur scientifique.
Merci à mes merveilleux et irremplaçables amis Nupur, Nikunj et Niraj, partenaires de voyages et de découvertes sur les routes d’Amérique du Nord et de l’Inde. Nous avons partagé des moments riches, colorés et inoubliables, rechargeant les batteries et remplissant les yeux de merveilles. Shukriya mera/maru indian dost ! And thank you for keeping your love for life, whatever happens !
Merci à d’autres irremplaçables, mes perles : « Ganou » et « Dreynou », à la compréhension et au soutien inconditionnels, ainsi que pour les divers voyages, sorties et aventures audacieux. Je n’oublie pas non plus le soutien de Marie-Gab’, Laurence, Françoise, Corinne, « Lapinou » et « Bella », qui ont tenu la distance, présents à chaque étape ! De vrais anges gardiens…
Je ne veux pas non plus oublier d’autres amis précieux : Joan, Chelsea, Andréanne, Mathieu et ses adorables monstres, Élodie, Caro, Maria, Rachel, Pauline, Nicolas, Simon, Hélène, Lydie, Mélanie, Anindita, Surya, Supreet et je m’excuse pour ceux que j’ai oubliés…
Merci à mes partenaires sportifs, qui ont entraîné mon endurance et ma volonté à me battre pour une équipe. Merci à la communauté Thèsez-vous ensemble, qui m’a fait aller chercher les dernières forces pour achever ce travail titanesque. Merci aussi à mes collègues entrepreneurs, qui ont vu et soutenu les débuts de ma transition professionnelle.
Le meilleur vient toujours à la fin. Source de motivation (même si pas toujours consciente, ni vocalisée), merci à ma famille de m’avoir soutenue et supportée, depuis ma traversée de l’Atlantique jusqu’aux derniers moments, et pendant toutes les mauvaises passes qui ont ponctué ce cheminement. J’ai un merci tout particulier à adresser à ma maman… mon papa, mon « Shrek », ma « Dindounette », mes grands parents maternels, pour leur écoute et leur soutien inconditionnel !
Merci enfin à mes deux amours, mes deux « J », mes deux irremplaçables et inégalables. Je n’en citerai pas un avant l’autre, pas de bagarre. Vous êtes des incarnations de la détermination dans un gant de velours et de la bonté dans un gant de fer. Je vous serai toujours reconnaissante pour m’avoir acceptée, accompagnée et choyée, à votre manière et sans rien demander en retour. Merci d’être le deuxième soleil de chaque jour et l’envie d’aller toujours plus loin…
Avant-propos
Le laboratoire du Dr Jean-Yves Masson a pour mission de mieux connaître et de décrire la réparation des cassures double-brin de l’ADN, une thématique qui rejoint l’étude de l’anémie de Fanconi (AF). Ces deux sujets ont été les piliers de mon doctorat, dont l’objectif était de déterminer si la protéine FANCI avait un rôle additionnel à ceux du complexe ID2, qu’elle forme avec FANCD2.
Dans ce contexte, le Chapitre 1 est une introduction qui aborde les notions de réparation de l’ADN et plus précisément des pontages covalents de l’ADN, qui sont problématiques dans le cas de l’AF et qui peuvent dériver en cassures double-brin. Ce chapitre présente également la maladie rare qu’est l’anémie de Fanconi, ses symptômes, ses fondements moléculaires, ses traitements et ses défis. Les sujets abordés étant très vastes, j’ai fait des choix dans les sections développées pour donner les informations les plus pertinentes en lien avec mon objectif, sans pour autant surcharger le manuscrit.
Ayant caractérisé et employé un nouveau type de souris pour répondre à mon objectif de thèse, le Chapitre 2 vient compléter l’introduction. Il est essentiel dans le sens où il aborde le thème des modèles animaux pour mieux étudier et comprendre cette maladie complexe. Il s’intitule « Les modèles
animaux de l’anémie de Fanconi, ou comment les différences peuvent être aussi précieuses que les similitudes » (Émilie L. Dubois, Mariline Béliveau et Jean-Yves Masson) et a été publié en juillet
2016 dans la revue francophone Médecine/Sciences.
Enfin, le Chapitre 3 renferme le cœur même de mes travaux, avec presque la totalité de ma contribution au laboratoire. Ces travaux sont présentés sous forme d’article (en anglais) car ils seront soumis à publication dans les prochaines semaines. L’article aura pour titre « A Fanci knock-out
mouse model reveals common and distinct functions for FANCI and FANCD2 » (Émilie L.
Dubois, Mariline Béliveau, Laure Guitton-Sert, Kalindi Parmar, Jalila Chagraoui, Julien Vignard, Joris Pauty, Marie-Christine Caron, Yan Coulombe, Rémi Buisson, Karine Jacquet, Clémence Gamblin, Patrick Laprise, Michel Lebel, Guy Sauvageau, Alan d’Andrea, and Jean-Yves Masson). Cette partie est absolument fondamentale dans la mesure où (i) elle décrit de nouveaux outils physiologiques pour étudier l’AF, et (ii) elle permet de répondre du mieux possible à mon hypothèse d’étude. De plus, elle contribue à une description plus précise et complète du mécanisme moléculaire de la voie FA-BRCA. Dans le futur, ce nouveau modèle animal pourra servir de référence aux autres laboratoires travaillant
J’ai également participé à d’autres projets, variés, qui ont concerné :
- l’étude de drosophiles dFanci-/- et l’impact de l’insertion de répétitions de type CTG dans leur
génome, en collaboration avec le laboratoire du Dr Patrick Laprise du Centre de Recherche. Malheureusement, quelques problèmes techniques n’ont pas permis au projet d’aboutir, mais je suis convaincue que les données préliminaires sont suffisamment intéressantes pour motiver la reprise du projet à temps plein.
- la caractérisation cellulaire de plusieurs mutations de la protéine humaine FANCI, dans le cadre d‘un consortium. Le projet, intitulé « gène Fanci, un gène de prédisposition au cancer de
l’ovaire émergeant », n’est pas encore terminé, mais ces résultats préliminaires auront un impact futur
non négligeable. En effet, selon les résultats de l’étude, un dépistage des mutations de FANCI pourrait être proposé aux patientes atteintes d’un cancer de l’ovaire. Un dépistage similaire est déjà pratiqué pour les célèbres gènes BRCA1 et BRCA2.
- l’étude de l’interaction entre le facteur de transcription E4F1 et sa cible CHK1, via la création et la purification de protéines recombinantes marquées. Ce petit travail a été réalisé dans le cadre d’une collaboration avec la post-doc Céline Moisan du Laboratoire du Dr Sauvageau à l’IRIC (Montréal), et avec l’aide de Yan Coulombe. Le papier qui en est issu est intitulé « E4F1 Is a Master Regulator of
CHK1-Mediated Functions » (David Grote, Céline Moison, Stéphanie Duhamel, Jalila Chagraoui,
Simon Girard, Jay Yang, Nadine Mayotte, Yan Coulombe, Jean-Yves Masson, Grant W. Brown, Sylvain Meloche et Guy Sauvageau), publié dans le journal Cell Reports en avril 2015.
1.
Chapitre 1 : Introduction
1.1 La cellule face à l’instabilité génomique
1.1.1 La réparation de l’ADN
1.1.1.1 Généralités
Afin d’assurer sa survie, tout organisme doit protéger le bagage génétique qu’il renferme. En effet, l’ADN peut être altéré par de multiples agents, de source exogène (i.e. radiations ionisantes (IR), Ultra-Violets (UV) et substances chimiques), ou endogènes (i.e. erreurs de réplication, produits du métabolisme). Plusieurs types de dommages en découlent : mésappariements, sites abasiques, mutations de bases, insertions ou délétions de nucléotides, pontages intra- ou inter-brin, cassures simple- ou double-brin (CDBs). Nombre de ces altérations du génome peuvent évoluer en CDBs. Il s’agit du type de dommage le plus dangereux auquel les cellules ne sont pas capables de survivre, ce qui déclenche leur mort programmée par apoptose. Si la réparation des CDBs est mauvaise et que la cellule ne meurt pas, des mutations ou des translocations de chromosomes compromettent la stabilité du génome et mènent à la tumorigenèse. Certains dommages à l’ADN peuvent cependant s’avérer bénéfiques au bon fonctionnement de la cellule, comme par exemple les CDBs générées pour la recombinaison V(D)J (pour Variant (Distance) Junction) des cellules immunitaires ou encore lors de l’entrée en méiose des cellules germinales.
De nombreux paramètres influencent le choix du mécanisme de réparation : la phase du cycle cellulaire dans laquelle la cellule se trouve, la balance entre l’énergie disponible et celle requise pour réparer [1], la disponibilité des acteurs de la réparation, la localisation de la lésion tant au sein du génome que dans le noyau, et le niveau d’organisation de la chromatine [2]. Dépendamment du type de dommage, la réponse cellulaire et le mécanisme de réparation de l’ADN varient [3]. Dans toutes les situations, la prise en charge des lésions suit un scénario commun : (1) des protéines senseurs du dommage (i.e. ATM/ATR) arrêtent le cycle cellulaire en phosphorylant leurs cibles, et recrutent (2) des médiateurs qui vont amplifier le signal d’altération de l’ADN, attirant (3) les transducteurs à la lésion où ils phosphorylent les effecteurs (i.e. p53) pour permettre (4) la réparation de l’ADN ainsi que toutes les réponses cellulaires associées ou qui en découlent [4].
Ma thèse a pour thème général l’Anémie de Fanconi (AF), caractérisée entre autres par une défaillance dans la réparation des pontages de l’ADN (désignés plus loin par ICLs, pour Interstrand CrossLinks). Les deux prochains paragraphes détaillent les enjeux cellulaires que les ICLs soulèvent, ainsi que les mécanismes classiques qui assurent leur réparation. Après une présentation des manifestations de l’AF, s’ensuit celle des acteurs principaux de cette thèse, et enfin mes hypothèses et objectifs.
1.1.1.2 La formation des ICLs
1.1.1.2.1 Généralités
Un pontage inter-brin (ou ICL) est une liaison covalente des deux brins complémentaires de l’ADN. La forte distorsion de l’hélice d’ADN qu’il génère perturbe les processus vitaux de la cellule. Par exemple, le blocage des polymérases – recrutées lors de la réplication ou de la transcription, génère une forte instabilité génomique qui peut aboutir à des CDBs létales, ou à l’inverse favoriser la mutagenèse et donc la tumorigenèse [5].
Les ICLs peuvent être générés spontanément par des molécules dites pontantes ou bialkylantes, car liant de manière covalente deux bases complémentaires. Bien que décrits tout récemment, les aldéhydes sont les plus connus des agents pontants endogènes et sont présentés dans la section
suivante. De l’autre côté, certaines drogues employées en chimiothérapie produisent des effets
similaires. Le DiÉpoxyButane (DEB), la MitoMycine C (MMC), le cisplatine et les psoralènes photoactivés sont les agents alkylants bifonctionnels les plus répandus. L’emploi des deux premières molécules dans le cadre de l’AF est détaillé dans les sections 1.2.2 et 1.2.3.
1.1.1.2.2 Les aldéhydes : présentation, incidence et prise en charge
Mes travaux n’ont pas employé ces molécules, mais un point de la discussion soulève ce sujet d’actualité et certaines perspectives expérimentales pourraient les intégrer. Pour ces raisons, j’en ferai une brève présentation dans cette section.
Les aldéhydes sont générés à partir de précurseurs ubiquitaires, tant endogènes qu’environnementaux [6].
D’un côté, les aldéhydes biogéniques, c’est à dire de source biologique endogène, sont des métabolites produits dans le noyau des cellules [7] et sont essentiels à certains processus biologiques tels que la vision, le développement embryonnaire et la neurotransmission [8]. Ils participent à la transduction de signaux, la régulation génique et la prolifération [8]. Ils sont issus de la peroxydation des lipides et du stress oxydatif (le malondialdéhyde (MDA), le monoxyde d’azote, l’acroléine, le 4-hydroxy-2nonenal
(4-HNE)), du métabolisme des acides aminés (l’acroléine), ou de la déméthylation des histones et des protéines (le formaldéhyde) [7, 9-11]. De l’autre côté, l’alcool, l’aspartame, le tabagisme et la pollution de l’air génèrent des précurseurs exogènes. Une fois anabolisés, ces précurseurs donnent l’acroléine, le formaldéhyde ou l’acétaldéhyde [6, 12].
Ainsi, des sources et des réactions différentes mènent aux mêmes aldéhydes. Parmi elles, le formaldéhyde est la molécule la plus simple et paradoxalement la plus dangereuse pour la cellule. Il est formé en plus grande quantité que les acétaldéhydes [7, 13], et pour ces raisons il est largement employé dans les tests cellulaires actuels.
Qu’elles soient produites dans nos cellules ou à l’extérieur, ces molécules renferment un fort potentiel cytotoxique et oncogénique. En effet, leur nature électrophile les rend très réactives et leur toxicité viendrait de plusieurs caractéristiques : une durée de vie plus longue que les radicaux libres, une capacité à atteindre des cibles éloignées (via leur capacité à diffuser ou à être transportées) et une interaction avec un large panel de molécules (phospholipides, protéines, ARN et ADN) [8]. Mécanistiquement parlant, les adduits qu’ils forment avec l’ADN (de préférence les guanines) évoluent en pontages ADN-protéine ou ADN-ADN (tels que les ICLs), ce qui induit notamment des bris génomiques [14]. Comme déjà mentionné, ces altérations ont par la suite une incidence sur la transcription et la réplication, et peuvent induire des mutations.
Étant donné que les aldéhydes sont essentiels au bon fonctionnement cellulaire mais qu’ils représentent en même temps un danger, les cellules ont développé des stratégies pour s’en protéger à différents niveaux.
Dans un premier temps, les enzymes de détoxification de phase I (oxydatives ou réductrices) viennent les neutraliser. Le nombre de familles et de protéines détoxifiantes est très vaste. Je ne présenterai que les enzymes ADH5 et ALDH2, car elles sont actuellement au centre des investigations en lien avec l’AF.
ADH5 appartient à la famille des déshydrogénases de l’alcool (ADH) et se retrouve très conservée des bactéries aux Vertébrés. Elle détoxifie par oxydation le formaldéhyde en acide formique, et l’acétaldéhyde en acétate en utilisant le NAD+ [7].
ALDH2 fait quant à elle partie d’une autre famille, celle des aldéhydes déshydrogénases (ALDH). Elle transforme exclusivement l’acétaldéhyde en acétate par un mécanisme distinct de celui employé par les
ADH [8]. Elle est exprimée dans de multiples tissus et organes et est localisée dans la matrice mitochondriale [8].
Ces deux enzymes portent aussi une activité de réduction des nitrates, ce qui leur permet de bioinactiver le monoxyde d’azote. Même si toute inhibition (naturelle ou thérapeutique) de ces enzymes est anticipée comme délétaire pour la cellule (à cause de ces doubles fonctions), il semblerait que l’absence de ADH5 soit plus néfaste, car ALDH2 n’est pas capable de compenser son absence dans les cellules souches hématopoïétiques [7].
Si cette première barrière n’est pas suffisante et que des dommages génomiques sont induits, la réponse cellulaire varie en fonction de la dose locale d’aldéhyde [15]. Lorsque celle-ci est faible, une activation forte et rapide de la kinase ATM est induite spécifiquement en phase S et ATR n’est pas impliquée. ATM et ATR sont deux kinases clés pour l’initiation de la réparation de l’ADN. Elles phosphorylent un très grand nombre de cibles pour arrêter le cycle cellulaire et permettre la réparation de l’ADN. À l’inverse, si la concentration en aldéhyde est forte, ATM est inhibée et on observe la formation de pontages de l’ADN plus larges [15]. Par la suite, la voie FA-BRCA est activée via la monoubiquitination de FANCD2 et la phosphorylation de BRCA1 [16]. Cette activation peut être considérée comme un indicateur général de la réponse cellulaire aux aldéhydes. Par contre, si la réparation des lésions génomiques n’est pas parfaitement accomplie et que la concentration en molécules pernicieuses est trop grande, soit l’activation de la voie FA-BRCA devient promutagénique, soit l’apoptose est activée [8, 12, 16].
1.1.1.3 La réparation des ICLs et des cassures double-brin
Les ICLs peuvent être détectés et réparés dans différents contextes cellulaires. La Figure 1.1 résume les mécanismes activés et les principales protéines qui en font partie. Selon les acteurs recrutés, différentes voies de réparation sont enclenchées : la Réparation par Excision de Nucléotide (NER pour Nucleotide Excision Repair), la Synthèse TransLésionnelle (TLS pour TransLesion Synthesis), la Recombinaison Homologue (RH) et les voies classiques ou alternatives de la Jonction des Extrémités Non Homologues (NHEJ pour Non-Homologous End Joining) [17-19]. Chacun de ces mécanismes est présenté brièvement à cette suite.
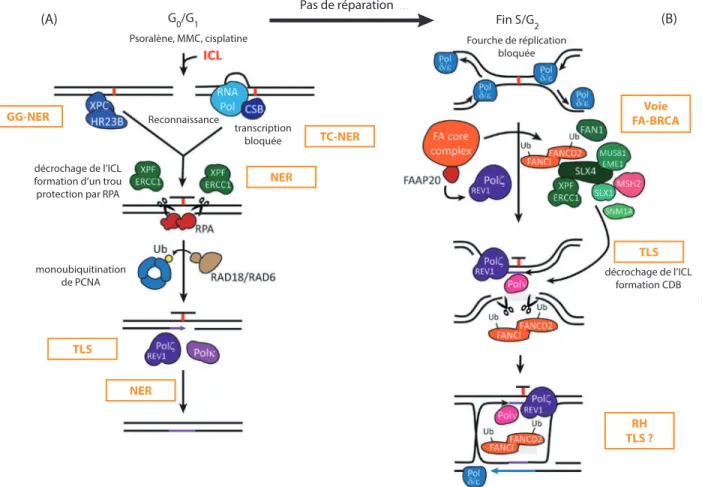
Figure 1.1 – Mécanismes réparant les ICLs, selon l'avancée dans le cycle cellulaire
Légende : (A) En phases G0 et G1, les agents bialkylants induisent un pontage covalent des deux brins de l’ADN.
Dans un contexte transcriptionnel, le blocage de l’ARN polymérase suivi du recrutement de la protéine CSB (du TC-NER) permet d’activer la réparation rapide de la lésion en recrutant les autres facteurs du NER. En dehors de la transcription, les protéines XPC et HR23B de la sous-voie GG-NER détectent le pontage inter-brin et conduisent à la réparation par le NER. Ainsi, le décrochage de l’ICL par les nucléases XPF et ERCC1 génère un espace dans l’ADN en face duquel l’ADN simple-brin restant est protégé par la protéine RPA. Puis PCNA est monoubiquitinée par RAD18/RAD6 et recrute les polymérases translésionnelles du TLS (Pol eta, Pol kappa et REV1). Ces dernières comblent l’absence de nucléotide, tandis que l’ICL resté sur le brin complémentaire est de nouveau pris en charge par les protéines du NER pour être excisé. (B) Si toutefois la cellule n’a pas détecté ou réparé à temps le pontage avant la réplication, elle va se retrouver arrêtée au point de contrôle entre les phases S et G2 par une fourche de réplication bloquée par l’ICL. Dans ce cas, les protéines de la voie FA-BRCA entrent en jeu
selon un mécanisme développé dans la section 1.2.5. Schéma adapté de [17].
Pas de réparation Reconnaissance G0/G1 Psoralène, MMC, cisplatine transcription bloquée décrochage de l’ICL
formation d’un trou protection par RPA
monoubiquitination de PCNA (A) Fin S/G2 Fourche de réplication bloquée décrochage de l’ICL formation CDB (B) GG-NER NER TC-NER TLS NER Voie FA-BRCA TLS RH TLS ?
Le NER a pour fonction de détecter et réparer les pontages entre deux nucléotides consécutifs, générés entre autres par une exposition aux UV (les dommages induits sont donc appelés des photoproduits). Deux sous mécanismes peuvent être activés, selon le contexte cellulaire et les acteurs recrutés. Dans un cas, les photoproduits sont détectés au cours de la transcription lorsque l’ARN polymérase s’y retrouve bloquée, avec l’aide des protéines CSA et CSB. Ces protéines constituent le sous mécanisme du TC-NER (Transcription-Coupled NER). Dans un contexte plus général de réparation du génome, les protéines XPC et HR23B sont recrutées et activent la Réparation Globale du Génome (ou GG-NER pour Global Genome NER). Même si le mode de détection est différent entre le TC-NER et le GG-NER, le mécanisme de réparation engagé par la suite est identique.
Le TLS permet de synthétiser une petite portion du brin d’ADN en face d’une lésion. Il emploie des polymérases dites translésionnelles, c’est-à-dire capables de passer outre un dommage pour insérer une base en face de la lésion ou du site abasique [5], contrairement aux polymérases processives qui y restent arrêtées. La faible processivité des polymérases translésionnelles a l’avantage de minimiser le risque d’insertion de mutations.
La Recombinaison Homologue est le mécanisme de réparation des CDBs le plus fidèle, mis en place uniquement au cours des phases S et G2. Suite à la résection de 5’ en 3’ des extrémités libres de l’ADN, limitée par le complexe MRN, les brins d’ADN protubérants sont couverts et protégés par RPA. Puis, RPA est chassée par la formation du filament de RAD51, qui va aller chercher l’homologie dans l’ADN de la chromatide-sœur avec l’aide de ses paralogues. Cet ADN homologue (présent uniquement en phase S) est pris comme modèle pour initier la synthèse du brin complémentaire et combler la lésion. BRCA1, BRCA2 et PALB2 sont également des acteurs majeurs qui participent activement à différentes étapes de ce processus [20-22].
La voie classique du NHEJ (C-NHEJ) est à l’inverse un mécanisme de réparation non fidèle de l’ADN. Il religue les brins situés de part et d’autres d’une CDB sans la présence de chromatide sœur comme guide. Il s’agit du mécanisme employé par défaut pour réparer les CDBs en dehors des phases S et G2, essentiellement en G0 et G1. Le C-NHEJ est activé très rapidement (dans les 30 minutes après la détection d’une CDB) et fait intervenir les protéines Ku70 et Ku80, LIG4/XRCC4/XLF, Artemis, CtIP et DNA-PKcs [19]. Cependant, le C-NHEJ peut induire l’ajout ou la perte de quelques paires de base au site de la CDB, et des translocations chromosomiques à plus large échelle. La grande instabilité chromosomique qu’il génère peut mener au cancer. Son impact serait limité par une vitesse de réparation lente [19].
Dans certaines situations, ces mécanismes de réparation peuvent être défaillants. La voie alternative du NHEJ (Alt-NHEJ) prend alors la relève de la RH (dans 20% des cas) ou du NHEJ [19]. Comme elle ne nécessite pas non plus de séquence homologue pour procéder, elle peut être mise en place tout au long du cycle cellulaire (avec malgré tout une plus forte activité observée en phases S et G2). Le Alt-NHEJ génère lui aussi de l’instabilité génomique. Même s’il est activé lorsque la réparation de l’ADN est déjà initiée, la présence des acteurs de la RH ou du NHEJ au site de la lésion ne freine pas son activation : la ligase LIG4 du NHEJ est remplacée par LIG1 et LIG3 – cette dernière œuvrant avec ses partenaires habituels XRCC1 et PARP1 issus d’autres voies de réparation. L’initiation de la résection par la RH fait ressortir des sites de microhomologie, ce qui favorise l’activation du Alt-NHEJ [19].
1.1.2 Les maladies associées à l’instabilité génomique
L’importance des multiples mécanismes de réparation de l’ADN se traduit par un large spectre de maladies génétiques qui leurs sont associées (syndrome de Bloom, syndrome de Cockayne, syndrome de Werner, maladie de Seckel, syndrome de Nijmegen, Xeroderma Pigmentosum et anémie de Fanconi). Symptômes et manifestations sont parfois communs : une instabilité génomique, des défauts neuro-dégénératifs, des déficiences immunitaires, une infertilité, un vieillissement prématuré, des problèmes métaboliques, malformatifs ou encore cardiovasculaires, et un dysfonctionnement des cellules souches [4]. La plupart de ces syndromes ont une transmission héréditaire de type récessive et se retrouvent également associés à une prédisposition à développer des cancers. L’anémie de Fanconi s’inscrit parmi ces syndromes d’instabilité génomique et sera présentée dans la section suivante.
1.2 L’anémie de Fanconi
Ma thèse a pour but d’étudier les fonctions de la protéine FANCI chez la souris, dont une mutation ou une absence chez l’homme induit l’anémie de Fanconi (AF). Cette section dresse l’étiologie de cette maladie rare, ses caractéristiques ainsi que les traitements actuellement proposés. Le Chapitre 2 complète cette partie en traitant de l’apport des modèles animaux pour mieux comprendre l’AF.
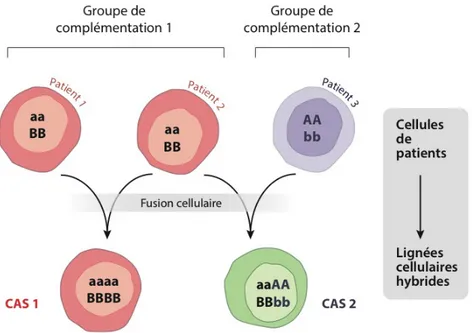
Pour assurer la clarté du propos, il est utile de préciser que les gènes impliqués dans l’AF sont nommés Fanc et les protéines FANC. Les patients sont regroupés dans des groupes de complémentation FA-X, X changeant à chaque groupe.
1.2.1 Découverte et incidence
L’anémie de Fanconi fait partie des maladies cassantes liées à l’instabilité du génome, mentionnées plus haut. Elle a été initialement décrite en 1927 par le pédiatre suisse Dr Guido Fanconi, puis nommée en 1931 par l’hématologue Dr Otto Naegeli [23]. Son découvreur a été un médecin hautement prolifique, qui a également donné son nom à une autre maladie – le Syndrome de Toni-Debré-Fanconi, à ne pas confondre avec l’AF. Le premier cas décrit par le Dr Fanconi a été l’anémie pernicieuse juvénile létale chez trois garçons d’une famille de cinq enfants [23]. Ces trois frères présentaient également d’autres manifestations de l’AF, présentées un peu plus loin.
Aujourd’hui, cette maladie autosomale récessive conserve une faible incidence (indice statistique correspondant à l’évaluation du risque de développer cette maladie, pour une personne ou une catégorie de personnes) : une naissance sur 100000 aux États-Unis [24], soit environ 31 nouveaux cas par an [12], et 1/300000 en France [25]. Ce risque est nettement augmenté chez les groupes des Juifs Ashkénazes, des gypsies espagnols ou des Afrikaners (1/22000) [24-27]. Parmi ces groupes ethniques, des mutations fondatrices ont été identifiées dans les gènes Fanca et Fancc [27-29]. Actuellement, on compte plus de 2000 personnes atteintes par la maladie dans le monde [30], mais ce nombre est sans doute sous-estimé pour de nombreuses raisons : hétérogénéité des symptômes donc diagnostic parfois incertain ; séquençage du génome, tests biologiques et hématologiques non systématiques ; accès aux soins et à la technologie limités dans certains pays.
Le mode de transmission de cette maladie génétique est de type récessif et la plupart des gènes impliqués sont portés par les chromosomes autosomaux. Mis à part Fancb qui est lié au chromosome X, seules les personnes homozygotes pour l’allèle muté – donc nées de parents hétérozygotes – vont développer la maladie. Dans la population mondiale, la fréquence des porteurs hétérozygotes est estimée à 1/300 et ces chiffres augmentent fortement à 1/77 pour les Afrikaners hétérozygotes et à 1/89 pour les populations juives ashkénazes hétérozygotes [25].
1.2.2 Manifestations cliniques et diagnostic
Malgré la grande hétérogénéité des symptômes de l’AF, il est possible de dresser la liste de ses signes caractéristiques (Table 1.1) [23, 25, 31]. Les patients peuvent développer une hypoplasie médullaire (ou défaillance de la moelle osseuse), un retard staturo-pondéral, des anomalies congénitales du squelette (par exemple une syndactylie (fusion des os de deux doigts consécutifs), un pouce surnuméraire ou au contraire absent), des malformations d’autres organes (microcéphalie, microphtalmie, hypogonadisme, infertilité partielle ou totale, défauts d’audition, malformation des voies urinaires, des reins et du cœur), et une prédisposition au cancer (Figure 1.2). L’âge médian de survie est de 29 ans [12].
Figure 1.2 – Principales manifestations des patients atteints de l'anémie de Fanconi
Légende : (A) Cas de malformation des membres de patients AF : duplication du pouce (haut) ou absence d’un doigt (bas).
(B) Frottis sanguin illustrant une anémie
(moitié de droite), par rapport à un patient sain (moitié de gauche). (C) Formes anormales typiques des
chromosomes de patients AF. De gauche à droite et de haut en bas : bris
chromosomiques, chromosomes triradiaux, chromosomes quadriradiaux, figures chromosomiques complexes, chromosomes dicentriques.
Illustrations tirées de
http://atlasgeneticsoncology.org/Kprone s/FA10001.html et [23, 25, 32, 33]
Table 1.1 – Fréquence d'apparition des différents symptômes chez les patients atteints de l'AF
Symptômes Fréquence d’apparition
Hypoplasie médullaire 90% (risque à vie)
Retard staturo-pondéral 63-90%
Malformations squelettiques 67-71%
Anomalies cutanées 40-64%
Microphtalmie 38%
Anomalies de la colone vertébrale 34%
Anomalies génitales 25%
Microcéphalie 25%
Malformations rénales et urinaires 20-34%
Anomalies oto-rhino-laryngologiques 10%
Anomalies du tube digestif 7-14%
Légende : Ce tableau illustre la grande hétérogénéité des symptômes de l’AF. À noter qu’environ 40% des patients ne présentent aucun de ces malformations congénitales. La gravité de ces signes pathologiques semble varier en intensité selon le groupe de complémentation. Données issues de [25, 31].
B A
La plupart des symptômes hématologiques sont présents dès la naissance. L’âge médian d’apparition de l’aplasie médullaire est de 7 ans et le diagnostic est généralement établi avant 10 ans [34]. À noter qu’il existe un mosaïcisme génétique, c’est à dire que certaines cellules souches hématopoïétiques acquièrent une mutation rectifiant la déficience de la protéine FANC en question [35]. Ainsi, les cellules de la moelle osseuse ayant conservé la mutation meurent par apoptose tandis que les cellules dites corrigées sont saines et colonisent le compartiment. Ces rares patients ne développent jamais d’anémie et ont même une moelle osseuse saine et fonctionnelle, ce qui peut biaiser le diagnostic. Chez les patients sans mosaïcisme, l’hypoplasie médullaire peut progresser en leucémie aigüe myéloïde (LAM), avec un risque 15000 fois supérieur à la population infantile normale [36].
La prédisposition à développer des masses solides concerne les carcinomes squameux de la tête et du cou, de l’œsophage, les carcinomes hépatocellulaires, les cancers de la peau, du cerveau, des reins ainsi que les cancers gynécologiques. Malheureusement, les traitements aux androgènes employés pour palier les retards de croissance de l’AF pourraient stimuler la progression tumorale.
Les médecins qui ont pris en charge des patients AF ont décrit que l’agressivité de la maladie varie selon le groupe de complémentation. Par exemple, (i) les patients FA-A développent des leucémies plus précocement, (ii) les symptômes des patients FA-N (mutés sur PALB2) et FA-D1 (BRCA2) sont habituellement plus forts [37]. On ne trouve d’ailleurs pas tous les groupes de complémentation dans les mêmes proportions au sein des patients AF : les groupes FA-A, -C et -G représentent à eux seuls plus de 85% des cas. Les autres groupes sont considérés comme rares lorsqu’ils représentent moins de 2% des patients. Pour plus de détails, se référer à la Table 1.2.
Table 1.2 – Conservation des protéines FANC dans divers organismes
Légende : Tableau récapitulant l’ensemble des protéines impliquées dans la voie FA-BRCA, les autres noms sous lesquels elles peuvent être retrouvées, et le nom des orthologues respectifs retrouvés chez la souris (Mus musculus), le poisson zèbre (Danio rerio), la mouche du vinaigre (Drosophila
melanogaster) et le nématode
(Caenorabditis elegans). Les pourcentages de similarité avec la protéine humaine sont indiqués entre parenthèses. La proportion de patients dans chacun des groupes est également indiquée.
ND : valeurs non renseignées. ? : orthologue putatif mais non identifié encore.
Données collectées sur www.genecards.org api en s A li as % p ati en t M . m u sc u lu s D . r er io D . m el an ogas te r C. e le gan s S c er ev is iae S . pom be A t h al ian a N C A -64% FA N CA (67,13%) FA N CA (37,93%) -N C B -2% FA N CB (50,42%) FA N CB (28,52%) -N C C -12% FA N CC (68,47%) FA N CC (33,84%) -N C D 1 BRCA 2 2% BRCA 2 (60,49%) BRCA 2 (27%) Brc a2 (N D ) BRC-2 (N D ) -N C D 2 -4% FA N CD 2 (75,3%) FA N CD 2 (54,71%) F D 2/ CG 17269 (26,17%) F CD -2 (N D ) -A T 4G 14970 (32,95%) N C E -1% FA N CE (66,28%) FA N CE (35,62%) -N C F -2% FA N CF (53,27%) FA N CF (31,37%) -N C G -8% FA N CG (73,56%) FA N CG (32,03%) -N C I -1% FA N CI (81,08%) FA N CI (61,86) FA N CI/ CG 13745 (25,62%) F N CI-1 (W 02D 3.10) (N D ) -FA N CI (N D ) N C J BRIP 1 2% BRIP 1 (75,37%) BRIP 1 (D r.15442) (75,4%) CG 4078 (39%) D O G -1 (31%) CH L 1 (23%) -N C L -0,4% FA N CL (79,41%) FA N CL (59,67%) CG 12812 (22%) m f-1 13 ? (N D ) -A T 5G 65740 (35,27%) N C M 0,1% FA N CM (66,92%) FA N CM (52-64%) CG 7922 (25%) F N CM -1 (drh-3) (N D ) M P H 1 (N D ) fm l1 (N D ) -N C N PA L B2 0,7% PA L B2 (63,69%) PA L B2 (18%) -N C O RA D 51C 0,1% RA D 51C (86,65%) D r.14386 (N D ) spn-D (28%) RF S -1 (N D ) D M C1 (26%) -RA D 51C (40,85%) N C P S L X 4, BT BD 12 0,5% S L X 4 (60,89%) S L X 4 (26-42%) m us 312 (N D ) hi m -18 (N D ) S L X 4 (N D ) S L X 4 (N D ) -N C Q E RCC4 0,1% E RCC4 (86,14%) w ufi 03a 05 (N D ) m ei -9 (44,87%) xpf-1 (C47D 12.8) (29%) RA D 1(Y P L 022W ) (37,12%) ra d16 (37,89%) U V H 1 (44,04%) N C R RA D 51 ? RA D 51 (98,82%) RA D 51 (91,04%) RA D 51 (68%) RA D -51 (47%) RA D 51 (51%) RH P 51 (75,08%) RA D 51 (71,3%) N C S BRCA 1 0,1% BRCA 1 (58,16%) brc a1 (N D ) -brc -1 (N D ) -N C T U BE 2T ? ? ube 2t (N D ) ? ube 2t (N D ) -? ? N C U X RCC2 ? X RCC2 (N D ) X rc c2 (47%) -N C V RE V 7 ? M A D 2L 2 (N D ) M A D 2L 2 (85%) RE V 7 (30%) -RE V 7 (N D ) A P 100 C17orf70 -? si :dke y-57h18.1 (N D ) -? A P 24 C19orf40 -? zgc :162267 (N D ) -? A P 20 C1orf86 -? si :c h73-70k4.1 (N D ) -? A P 16 A P IT D 1 (M H F 1, CE N P S ) -? api td1 (N D ) -? M H F 1 (N D ) m hf1 (N D ) ? A P 10 S T RA 13 (M H F 2, CE N P X ) -? st ra 13 (N D ) -F 35H 10.5 (N D ) M H F 2 (N D ) m hf2 (N D ) ? N 1 M T M R15 (K IA A 1018) -? fa n1 (N D ) -fa n-1 (N D ) -fa n1 (N D ) ? -? ? ? ? ? ? ?