Analyse
des
herbicides phénoxyacides par
chromatographie
en
phase
liquide
:
1er
essai
inter-laboratoires
HPLC
analysis
of
phenoxyacid
herbicides
:
1st
interlab
oratory
comparison
study
Alain
TURCANT»*,
Betty
DEHON(2)#,
Catherine
GANIÈRE-MONTEIL3*,
Sylvain
DULAURENT4*, Moustapha
MOULSMA(5),
Corinne
CHARLIER
m
(1) Phai-macologie-Toxicologie,CHUAngers (2) Biochimie UF Toxicologie, CHU
Lille
(3) Pharmacologie, CHU Nantes (4)Pharmacologie-Toxicologie, CHU Limoges (5) UF Pharmacotoxicologie, CHUE. Herriot Lyon (6) Toxicologie Clinique etMédicolégale,CHU Liège #Groupedetravail "Pesticides" delaSFTA
*Auteuràquiadresserlacorrespondance :
Alain TURCANT,
ServicedePharmacologie etToxicologie,CHU, 4,rueLarrey- 49933 ANGERS Cedex 9 - France Tel : 02 41 35 45 52 -Fax : 0241 3548 77 - E-mail : [email protected]
RESUME
Afindedocumenter au mieux,pardesdosagesplasmatiques,
les casd'intoxicationparlesherbicides phénoxyacides, six laboratoires ont évalué diverses approches analytiques de
type CLHPaprès purificationparsimpleprécipitation des
protéines plasmatiquesparVacétonitrileouaprèsextraction liquide-liquide en milieu acide ou encore après extraction liquide-solide. Un essai comparatifconcernant 4 plasmas surchargéset4casréelsd'intoxicationamontréunevaria¬
bilité correcte avec un CVlaplupart du temps inférieurà
20%.L'extractionliquide-solide, non totalement satisfaisan¬
te dans cetessai, nécessite une investigation pluspoussée
tandis que la simple précipitation protéique apporte une
(Reçule 16
février
2006; acceptéle19juin
2006)SUMMARY
The evaluation
of
acutephenoxyacid poisonings in.humans requirestheidentificationof
theherbicideand,if
possible, its quantification in plasma. Six laboratories tested different analyticalapproaches, using HPLC, aftersampletreatment either by simple protein precipitation or by liquid-liquid extraction under acidic pH conditions or by solid phase extraction. A comparative study on4 spiked plasmas and4real acutecasesshowedgood resultswith CV < 20%. Solid phase extraction needsfurther investigation while simple precipitation byacétonitrileprovidesa rapidandsufficient cleaning procedure toevaluate themajority
of
acutepoiso¬nings. Liquid-liquid extraction, providing lower limits
of
135
Analyse
des
herbicides phénoxyacides par
chromatographie
en
phase
liquide
:
1er
essai
inter-laboratoires
HPLC
analysis
of
phenoxyacid
herbicides
:
1st
interlab
oratory
comparison
study
Alain
TURCANT»*,
Betty
DEHON(2)#,
Catherine
GANIÈRE-MONTEIL3*,
Sylvain
DULAURENT4*, Moustapha
MOULSMA(5),
Corinne
CHARLIER
m
(1) Phai-macologie-Toxicologie,CHUAngers (2) Biochimie UF Toxicologie, CHU
Lille
(3) Pharmacologie, CHU Nantes (4)Pharmacologie-Toxicologie, CHU Limoges (5) UF Pharmacotoxicologie, CHUE. Herriot Lyon (6) Toxicologie Clinique etMédicolégale,CHU Liège #Groupedetravail "Pesticides" delaSFTA
*Auteuràquiadresserlacorrespondance :
Alain TURCANT,
ServicedePharmacologie etToxicologie,CHU, 4,rueLarrey- 49933 ANGERS Cedex 9 - France Tel : 02 41 35 45 52 -Fax : 0241 3548 77 - E-mail : [email protected]
RESUME
Afindedocumenter au mieux,pardesdosagesplasmatiques,
les casd'intoxicationparlesherbicides phénoxyacides, six laboratoires ont évalué diverses approches analytiques de
type CLHPaprès purificationparsimpleprécipitation des
protéines plasmatiquesparVacétonitrileouaprèsextraction liquide-liquide en milieu acide ou encore après extraction liquide-solide. Un essai comparatifconcernant 4 plasmas surchargéset4casréelsd'intoxicationamontréunevaria¬
bilité correcte avec un CVlaplupart du temps inférieurà
20%.L'extractionliquide-solide, non totalement satisfaisan¬
te dans cetessai, nécessite une investigation pluspoussée
tandis que la simple précipitation protéique apporte une
(Reçule 16
février
2006; acceptéle19juin
2006)SUMMARY
The evaluation
of
acutephenoxyacid poisonings in.humans requirestheidentificationof
theherbicideand,if
possible, its quantification in plasma. Six laboratories tested different analyticalapproaches, using HPLC, aftersampletreatment either by simple protein precipitation or by liquid-liquid extraction under acidic pH conditions or by solid phase extraction. A comparative study on4 spiked plasmas and4real acutecasesshowedgood resultswith CV < 20%. Solid phase extraction needsfurther investigation while simple precipitation byacétonitrileprovidesa rapidandsufficient cleaning procedure toevaluate themajority
of
acutepoiso¬nings. Liquid-liquid extraction, providing lower limits
of
AnnalesdeToxicologie Analytique, vol.
XVIII,
n° 2,2006bonne rapiditédepréparationdeséchantillonsetunesensi¬
bilité suffisante pour la majorité des cas d'intoxication
aiguë. L'extraction liquide-liquidepermetunemeilleuresen¬
sibilité, notammentavec unedétectionparspectrométriede masse en tandem et rendcetteapproche applicable auxcas
d'intoxicationfaible voireaux expositions professionnelles.
MOTS-CLES
Phénoxyacides, phytohormones, plasma, CLHP.
herbicides, intoxication,
detection especiallywhen tandem massspectrometryis used,
will be moreinteresting inminororprofessional exposition
to theseherbicides.
KEY-WORDS
Phenoxyacids, herbicides, acutepoisoning, plasma, HPLC.
Introduction
Les herbicides phénoxyacides (fig.
la)
constituent unefamille
de produits phytosanitaires agissant de façonsystémique par pénétration
foliaire
en stimulant de façon anarchique làcroissance de la plante. Sept sub¬stances sontcommercialisées en France : cinq sont de
type acide phénoxy-acétique, avecdifférentes substitu¬ tions par un ou plusieurs atomes de chlore (Cl) ou un groupement méthyle (CH3),deuxd'entre ellesprésen¬ tant un groupement méthyle sur la chaîne alcanoïque, d'où leur autre appellation d'acides phénoxy-propio-niques, et 2 sont des acides phénoxy-butyriques (Tableau I). Lefénopropou 2,4,5-TP, égalementappe¬
lé silvex, noncommercialisé en France mais référencé en Belgique comme devant être recherché dans les eaux derivières,complète cetteliste.Letriclopyr,où le cycle phényle est remplacé par un noyaupyridine, est apparenté à cette famille (fig.
lb).
Les pKa diffèrent légèrement d'une substance àl'autre en fonctionde la longueurdela chaîne acide ou delasubstitutionpar un ou plusieurs Cl ou un groupement méthyle. Ces pro¬ duits sont souvent commercialisés en association par deux ou encore avec d'autres molécules comme le dicamba (herbicidedelafamille
des dérivés del'acide benzoïque). Ces herbicides agissent commel'Acide
Indole Acétique
(IAA)
qui est une auxine naturelle et sont donc également appelés auxines de synthèse ou encorephytohormones (1). L'analogie structurale avecF
IAA
(fig.2) est basée sur 3 similitudes :- unepartie plane (cycles aromatiques),
- laprésence d'unefonction acide carboxylique
- l'existence d'une structuredipolaire entrele groupe¬ ment acide et une zone électrophile, azote du noyau indoleou carbone enposition2 du cycle. En raison de
l'effet
électro-attracteur des halogènes, la distanceentre lescharges estvoisine de5,5 angstroms (1).
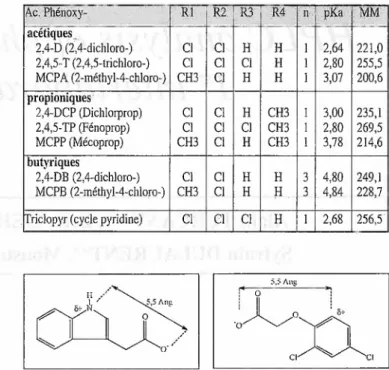
Tableau I : Dénomination, substituants, pKa et masses
molairesdes différents phénoxyacidesetapparentés(cffor¬ mulesfigure 1). (a) R, I R,
O
l3 0-(CH)n-COOH Ac. Phénoxy-acétiques, 2,4-D(2"4-dichloro-) 2,4,5-T(2,4,5-trichloro-) MCPA(2-méthyl-4-chloro-) propioniques 2,4-DCP(Dichlorprop) 2,4,5-TP (Fénoprop) MCPP(Mécoprop) butyriques 2,4-DB(2,4-dichloro-) MCPB(2-raéthyI-4-chloro-)riclopyr(cyclepyridine)
Ri Cl Cl CH3 Cl Cl CH3 Cl CH3 Cl R2 Cl Cl Cl Cl Cl Cl Cl Cl Cl R3 H Cl H H Cl H H H Cl R4 H H H CH3 CH3 CH3 H H. . H n pKa 1 2,64 1 2,80 1 3,07 1 3,00 1 2,80 1 3,78 3 4.80 3 4,84 1 2,68 MM 221,0 255,5 200,6 235,1 269,5 214,6 249,1 228,7 256,5 o-0
1
5,5Ally v /-°\ \/ V CI-' ' S*T
1
KA
N:!Figure 1 : Formules chimiques des phénoxyacides (a) et
triclopyr(b).
Figure 2 :Analogie géométrique entre l'acide indole acé¬
tiqueetl'acide2,4-D.
Si latoxicité par absorption cutanée est faible,
il
n'en est pas de même lors d'ingestion orale puisque les dosespotentiellement mortelles sont évaluées entre 80 et 800 mg/kg selon les substances, soit quelques grammes (2). Les principaux signes cliniques en casd'intoxication
aiguë sont les vomissements précoces,lestroublesdela conscience,l'hypotension artérielleet
Facidose métabolique.
Les méthodes de dosage plasmatique et/ou urinaire de
cesherbicides phénoxyacides, décritesdansla littératu¬
reencas
d'intoxication,
sontessentiellementbaséessurlachromatographieenphaseliquideavecdétection
UV
(3,4) plus facile à mettre en oeuvre que la chromato¬ graphie en phase gazeuse qui nécessite une étape de méthylation (5).
Le groupe de travail « Pesticides » de la Société Française de Toxicologie Analytique s'est intéressé au
suividesintoxicationsparherbicides phénoxyacidesen AnnalesdeToxicologie Analytique, vol.
XVIII,
n° 2,2006bonne rapiditédepréparationdeséchantillonsetunesensi¬
bilité suffisante pour la majorité des cas d'intoxication
aiguë. L'extraction liquide-liquidepermetunemeilleuresen¬
sibilité, notammentavec unedétectionparspectrométriede masse en tandem et rendcetteapproche applicable auxcas
d'intoxicationfaible voireaux expositions professionnelles.
MOTS-CLES
Phénoxyacides, phytohormones, plasma, CLHP.
herbicides, intoxication,
detection especiallywhen tandem massspectrometryis used,
will be moreinteresting inminororprofessional exposition
to theseherbicides.
KEY-WORDS
Phenoxyacids, herbicides, acutepoisoning, plasma, HPLC.
Introduction
Les herbicides phénoxyacides (fig.
la)
constituent unefamille
de produits phytosanitaires agissant de façonsystémique par pénétration
foliaire
en stimulant de façon anarchique làcroissance de la plante. Sept sub¬stances sontcommercialisées en France : cinq sont de
type acide phénoxy-acétique, avecdifférentes substitu¬ tions par un ou plusieurs atomes de chlore (Cl) ou un groupement méthyle (CH3),deuxd'entre ellesprésen¬ tant un groupement méthyle sur la chaîne alcanoïque, d'où leur autre appellation d'acides phénoxy-propio-niques, et 2 sont des acides phénoxy-butyriques (Tableau I). Lefénopropou 2,4,5-TP, égalementappe¬
lé silvex, noncommercialisé en France mais référencé en Belgique comme devant être recherché dans les eaux derivières,complète cetteliste.Letriclopyr,où le cycle phényle est remplacé par un noyaupyridine, est apparenté à cette famille (fig.
lb).
Les pKa diffèrent légèrement d'une substance àl'autre en fonctionde la longueurdela chaîne acide ou delasubstitutionpar un ou plusieurs Cl ou un groupement méthyle. Ces pro¬ duits sont souvent commercialisés en association par deux ou encore avec d'autres molécules comme le dicamba (herbicidedelafamille
des dérivés del'acide benzoïque). Ces herbicides agissent commel'Acide
Indole Acétique
(IAA)
qui est une auxine naturelle et sont donc également appelés auxines de synthèse ou encorephytohormones (1). L'analogie structurale avecF
IAA
(fig.2) est basée sur 3 similitudes :- unepartie plane (cycles aromatiques),
- laprésence d'unefonction acide carboxylique
- l'existence d'une structuredipolaire entrele groupe¬ ment acide et une zone électrophile, azote du noyau indoleou carbone enposition2 du cycle. En raison de
l'effet
électro-attracteur des halogènes, la distanceentre lescharges estvoisine de5,5 angstroms (1).
Tableau I : Dénomination, substituants, pKa et masses
molairesdes différents phénoxyacidesetapparentés(cffor¬ mulesfigure 1). (a) R, I R,
O
l3 0-(CH)n-COOH Ac. Phénoxy-acétiques, 2,4-D(2"4-dichloro-) 2,4,5-T(2,4,5-trichloro-) MCPA(2-méthyl-4-chloro-) propioniques 2,4-DCP(Dichlorprop) 2,4,5-TP (Fénoprop) MCPP(Mécoprop) butyriques 2,4-DB(2,4-dichloro-) MCPB(2-raéthyI-4-chloro-)riclopyr(cyclepyridine)
Ri Cl Cl CH3 Cl Cl CH3 Cl CH3 Cl R2 Cl Cl Cl Cl Cl Cl Cl Cl Cl R3 H Cl H H Cl H H H Cl R4 H H H CH3 CH3 CH3 H H. . H n pKa 1 2,64 1 2,80 1 3,07 1 3,00 1 2,80 1 3,78 3 4.80 3 4,84 1 2,68 MM 221,0 255,5 200,6 235,1 269,5 214,6 249,1 228,7 256,5 o-0
1
5,5Ally v /-°\ \/ V CI-' ' S*T
1
KA
N:!Figure 1 : Formules chimiques des phénoxyacides (a) et
triclopyr(b).
Figure 2 :Analogie géométrique entre l'acide indole acé¬
tiqueetl'acide2,4-D.
Si latoxicité par absorption cutanée est faible,
il
n'en est pas de même lors d'ingestion orale puisque les dosespotentiellement mortelles sont évaluées entre 80 et 800 mg/kg selon les substances, soit quelques grammes (2). Les principaux signes cliniques en casd'intoxication
aiguë sont les vomissements précoces,lestroublesdela conscience,l'hypotension artérielleet
Facidose métabolique.
Les méthodes de dosage plasmatique et/ou urinaire de
cesherbicides phénoxyacides, décritesdansla littératu¬
reencas
d'intoxication,
sontessentiellementbaséessurlachromatographieenphaseliquideavecdétection
UV
(3,4) plus facile à mettre en oeuvre que la chromato¬ graphie en phase gazeuse qui nécessite une étape de méthylation (5).
Le groupe de travail « Pesticides » de la Société Française de Toxicologie Analytique s'est intéressé au
évaluant les capacités analytiques de six laboratoires, tant
d'un
point de vuequalitatif
que quantitatif, soit par la miseenplaced'une méthodeanalytique nouvel¬ le soit par une réévaluation de sa propre méthode de dosage vis-à-visde 9 substances. Ladémarcheinitiale
aété, pourchaquelaboratoire,depréciserles limitesde détection de ces produits avec sa propre méthode de screening toxicologiqueparchromatographie enphase
liquide
avec détectionUV
à barrette de diodes(CLHP/BD)aprèsextractionalcaline, méthode miseen dans la plupart des laboratoires pour la recherchedes substances en cause au coursd'intoxica¬ tions aiguës médicamenteuses. L'extraction en
milieu
alcalinest a
priori
défavorablepourla miseenéviden¬ cedesherbicides phénoxyacides. Ensuite, chaquelabo¬ ratoireparticipantdevaitétablir des conditions optimi¬séesderecherche etdosage deces produitsens'aidant éventuellement d'une démarche analytique de type CLHP/BD, proposée parlelaboratoired'Angers, mais chacun disposaitdu libre choixtant pourl'aspectchro¬ matographiquequepourladétectionou lapréparation
de
l'échantillon
par extractionliquide/liquideou liqui¬de/solide ou encore par simple précipitation des pro¬ téines.Enfinunessai inter-laboratoires aétéréalisé sur
8 échantillonsplasmatiques correspondant à4 plasmas surchargés età4 casréels
d'intoxication.
Matériel
et
Méthodes
Les poudres, obtenues chez
Aldrich,
FlukaetRiedel de Haën(Sigma-Aldrich,SaintQuentin Fallavier,France), chez Cluzeau (Ste Foy la Grande, France) ou LGC Promochem (Molsheim, France), sontde pureté com¬ prise entre 95 et 99,9 % selon les produits. Les solu¬ tionsmères sont préparées, selonles substances, soità1, à 5 ou à 10
g/L
dansFacétonitrile (pouranalyses) et non le methanolpourminimiserle risque d'estérifica-tionpossible in vitro àlongterme.La
limite
dedétectionpourchacundecesproduits aétéestimée, pour chaque laboratoire, par l'obtention
d'un
spectre
UV
interprétable, en surchargeant des plasmas témoins àdesconcentrationsallantde 10 à 100mg/L
et en testant ses propres conditions d'extraction liquide-liquideenmilieu
alcalin.La méthode proposée pour
l'optimisation
du dosagedesherbicides
fait
appelàuneprise d'essaidelOOpl de plasma auquelestajouté unvolume équivalentd'acéto¬nitrile
contenant l'acide2-méthyl-4-chlorophénoxybu-tyrique (MCPB) utilisé comme étalon interne à 300 mg/L, cette substance n'ayant jamais été rencontrée
danslescas
d'intoxication
dans l'expérienceangevine.Lesurnageantdecentrifugationestensuitediluéau 1/2 dans l'eau puis analysé par chromatographie de parta
ge en phases inversées soit dans les conditions du screening classique de ce laboratoire (6), c'est-à-dire un gradienttampon phosphatepH6 etacétonitrile avec une détection à 3 longueurs d'onde (230, 285 et 210 nm), soit par une élution isocratique en tampon formiate2mMàpH3etacétonitrile67/33 v/v pourpal¬
lier
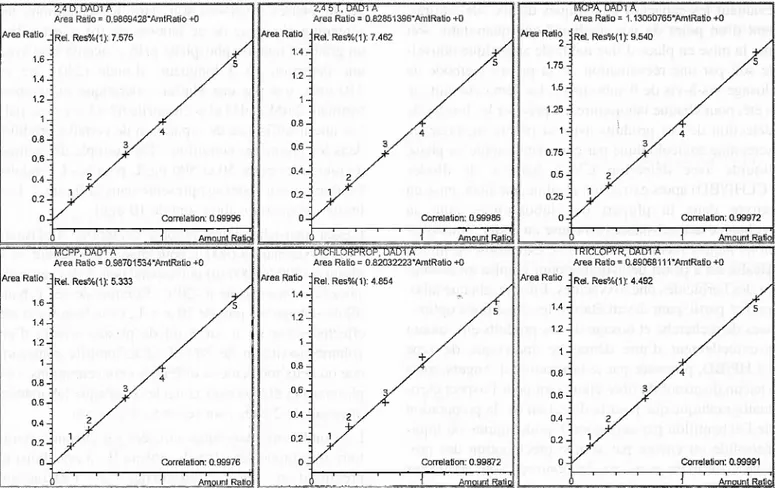
une insuffisance de séparation decertains produits dans les premières conditions. Un exemple de gamme d'étalonnage entre 50 et500mg/L
pour les6 produits habituellement testésest présenté dans lafigure3. Les limites de quantification sontde 10mg/L.L'essai inter-laboratoires proposé comportait 4 plasmas témoinssurchargés(800 ulpourchaquelaboratoire)et4 plasmas (300 à 1000 ul) correspondaientàdes casréels stockés en sérothèque à -20°C. Certains de ces échan¬
tillonsdataientdeplusde 10 ans.Les surchargesontété effectuées par ajout sur 5 ml de plasma témoin d'un volume maximum de 200 ul d'acétonitrile contenant une ou deuxmoléculesàdifférentes concentrations. Les plasmas ont été envoyés congelés et chaquelaboratoire disposaitde2moispourrépondreàcet essai.
Lesconditions analytiques utilisées par chaquelabora¬ toiresont rapportées dansle tableau
II
: 3 ontchoisilaprécipitation
par acétonitrile, 2l'extraction
liquide/liquide enmilieu trèsacide (pH <1) et un l'ex¬
traction
liquide/solide
sur cartouche échangeused' anions (OASIS©
MAX)
à partir de lOOpl de prise d'essai. La séparation chromatographique est réalisée surcolonne C8ouCl
8 éventuellement avecfaibledia¬mètre interne (2 voire
lmm)
avec des tampons phos¬ phate ou formiate de concentration et de pH variés, 4 laboratoires ayant des conditions de gradientet 2 desconditions isocratiques avec des temps de séparation comprisentre 15et30 min.Un laboratoire aidentifiéet dosé les phénoxyacides par spectrométrie de masse tandem, les autres utilisant
FUV
barrette de diodes avec une ou plusieurs longueurs d'onde. Cinq labora¬ toires ont quantifié avecl'aide
d'un étalon interne, 1par étalonnage externe, ce laboratoire ayant procédé à
l'identification initiale
parCPG/SMaprèsméthylation.Résultats
Les résultats des analyses dans les conditions de scree¬
ningmontrent évidemment lafaibledétectabilitédeces
molécules acides dans desconditions d'extractionalca¬
line même si, compte tenu des concentrations impor¬ tantes rencontrées en intoxication,
il
n'est pas impos¬ sibledelesmettreenévidence. SelonlepHetlesolvant d'extraction on observe une grande disparité de détec¬tion. Uneextraction deplasmaàpH 11 par le dichloro¬
méthane ne permet pas de détecter ces produits à la concentration de 100 mg/1. Uneextraction enprésence évaluant les capacités analytiques de six laboratoires,
tant
d'un
point de vuequalitatif
que quantitatif, soit par la miseenplaced'une méthodeanalytique nouvel¬ le soit par une réévaluation de sa propre méthode de dosage vis-à-visde 9 substances. Ladémarcheinitiale
aété, pourchaquelaboratoire,depréciserles limitesde détection de ces produits avec sa propre méthode de screening toxicologiqueparchromatographie enphase
liquide
avec détectionUV
à barrette de diodes(CLHP/BD)aprèsextractionalcaline, méthode miseen dans la plupart des laboratoires pour la recherchedes substances en cause au coursd'intoxica¬ tions aiguës médicamenteuses. L'extraction en
milieu
alcalinest a
priori
défavorablepourla miseenéviden¬ cedesherbicides phénoxyacides. Ensuite, chaquelabo¬ ratoireparticipantdevaitétablir des conditions optimi¬séesderecherche etdosage deces produitsens'aidant éventuellement d'une démarche analytique de type CLHP/BD, proposée parlelaboratoired'Angers, mais chacun disposaitdu libre choixtant pourl'aspectchro¬ matographiquequepourladétectionou lapréparation
de
l'échantillon
par extractionliquide/liquideou liqui¬de/solide ou encore par simple précipitation des pro¬ téines.Enfinunessai inter-laboratoires aétéréalisé sur
8 échantillonsplasmatiques correspondant à4 plasmas surchargés età4 casréels
d'intoxication.
Matériel
et
Méthodes
Les poudres, obtenues chez
Aldrich,
FlukaetRiedel de Haën(Sigma-Aldrich,SaintQuentin Fallavier,France), chez Cluzeau (Ste Foy la Grande, France) ou LGC Promochem (Molsheim, France), sontde pureté com¬ prise entre 95 et 99,9 % selon les produits. Les solu¬ tionsmères sont préparées, selonles substances, soità1, à 5 ou à 10
g/L
dansFacétonitrile (pouranalyses) et non le methanolpourminimiserle risque d'estérifica-tionpossible in vitro àlongterme.La
limite
dedétectionpourchacundecesproduits aétéestimée, pour chaque laboratoire, par l'obtention
d'un
spectre
UV
interprétable, en surchargeant des plasmas témoins àdesconcentrationsallantde 10 à 100mg/L
et en testant ses propres conditions d'extraction liquide-liquideenmilieu
alcalin.La méthode proposée pour
l'optimisation
du dosagedesherbicides
fait
appelàuneprise d'essaidelOOpl de plasma auquelestajouté unvolume équivalentd'acéto¬nitrile
contenant l'acide2-méthyl-4-chlorophénoxybu-tyrique (MCPB) utilisé comme étalon interne à 300 mg/L, cette substance n'ayant jamais été rencontrée
danslescas
d'intoxication
dans l'expérienceangevine.Lesurnageantdecentrifugationestensuitediluéau 1/2 dans l'eau puis analysé par chromatographie de parta
ge en phases inversées soit dans les conditions du screening classique de ce laboratoire (6), c'est-à-dire un gradienttampon phosphatepH6 etacétonitrile avec une détection à 3 longueurs d'onde (230, 285 et 210 nm), soit par une élution isocratique en tampon formiate2mMàpH3etacétonitrile67/33 v/v pourpal¬
lier
une insuffisance de séparation decertains produits dans les premières conditions. Un exemple de gamme d'étalonnage entre 50 et500mg/L
pour les6 produits habituellement testésest présenté dans lafigure3. Les limites de quantification sontde 10mg/L.L'essai inter-laboratoires proposé comportait 4 plasmas témoinssurchargés(800 ulpourchaquelaboratoire)et4 plasmas (300 à 1000 ul) correspondaientàdes casréels stockés en sérothèque à -20°C. Certains de ces échan¬
tillonsdataientdeplusde 10 ans.Les surchargesontété effectuées par ajout sur 5 ml de plasma témoin d'un volume maximum de 200 ul d'acétonitrile contenant une ou deuxmoléculesàdifférentes concentrations. Les plasmas ont été envoyés congelés et chaquelaboratoire disposaitde2moispourrépondreàcet essai.
Lesconditions analytiques utilisées par chaquelabora¬ toiresont rapportées dansle tableau
II
: 3 ontchoisilaprécipitation
par acétonitrile, 2l'extraction
liquide/liquide enmilieu trèsacide (pH <1) et un l'ex¬
traction
liquide/solide
sur cartouche échangeused' anions (OASIS©
MAX)
à partir de lOOpl de prise d'essai. La séparation chromatographique est réalisée surcolonne C8ouCl
8 éventuellement avecfaibledia¬mètre interne (2 voire
lmm)
avec des tampons phos¬ phate ou formiate de concentration et de pH variés, 4 laboratoires ayant des conditions de gradientet 2 desconditions isocratiques avec des temps de séparation comprisentre 15et30 min.Un laboratoire aidentifiéet dosé les phénoxyacides par spectrométrie de masse tandem, les autres utilisant
FUV
barrette de diodes avec une ou plusieurs longueurs d'onde. Cinq labora¬ toires ont quantifié avecl'aide
d'un étalon interne, 1par étalonnage externe, ce laboratoire ayant procédé à
l'identification initiale
parCPG/SMaprèsméthylation.Résultats
Les résultats des analyses dans les conditions de scree¬
ningmontrent évidemment lafaibledétectabilitédeces
molécules acides dans desconditions d'extractionalca¬
line même si, compte tenu des concentrations impor¬ tantes rencontrées en intoxication,
il
n'est pas impos¬ sibledelesmettreenévidence. SelonlepHetlesolvant d'extraction on observe une grande disparité de détec¬tion. Uneextraction deplasmaàpH 11 par le dichloro¬
méthane ne permet pas de détecter ces produits à la concentration de 100 mg/1. Uneextraction enprésence
Annales deToxicologie Analytique,vol.
XVIII,
n°2,2006 Area Ratio 1.6 1.4 1.2 1 0.8 0.6 0.4 0.2 0Area Ratio=0.9869428"AmtRatio-K!
Rel.Res%(1):7.575 4 3 2 1 Correlatio
Area Ratio=0.82851396-AmtRalio+0 Area Ratio=113050765'AmtRatio+0
5 4 Correlation: 0.99996 Amount Ratio AreaRatio 1.4 '1.2 1 0.8 0.6 0.4 0.2 0 Rel.Res%(1):7.462 3 2 1 5 4 Correlation: 0.99986 Amount Ratio Area Ratio 2 1.75 1.5 1.25 1 0.75 0.5 0.25 0 Rel Res%(1):9.540 3 2 Correlation:0.99972 Am n ti
Area Ratio=0.98701534'AmtRatio +0 Area Ratio=0.82032223*AmtRatio +0 Area Ratio 1.6 1.4 1.2 1 0.8 0.6 0.4 0.2 0 Rel Res%(1):5.333 3 2 1 4 Correlatior A 5 [j i :0.99976 mountR tî reaRatio 1.4 1.2 1 0.8 0.6 0.4 0.2 0 Correlation:0.99872
Area Ratio=0.89068111*AmtRatio+0 Area Ratio Re|.Res%(l):4.492
1.4 1.2 1 0.8 4 0.6 3 0.4 2 0.2 ! 0 Correlation: 0.99991
Figure3:Courbesd 'étalonnagede6phénoxyacidesentre50et500mg/L après simpleprécipitationdesprotéines plasmatiques (El =MCPB300 mg/L).
TableauII : Conditions analytiques utiliséesparles différentslaboratoires.
P. c. s. D. LQ Ll CH3CN 1/1 v/ve C18 Hypersil di2,1mm P20mM pH6 CH3CN G 18min UV-BD (230,285,210) El:MCPB 10mg/L L2 HC1 IN 1/2v/ve Ether 8/1 v/ve C8 Symmetry P44mM pH3,8 CH3CN G 30min UV-BD (230) El:Prazépam 0,2mg/L L3 OasisMAX L: MeOH E:MeOH/ac formique 98/2 C18 Eclipse F5mM pH3 CH3CN I70/30 15min UV-BD (230) El:MCPB 1à 10mg/L L4 HC16N 1/4v/ve AcEt 4/1 v/ve C18 Nucleosil di1mm F2mM pH3 CH3CN G 18min SM/SM MRM (2ions) El:Méphénytoïne 20 à 50fig/L L5 CH3CN 1/1 v/ve C18 Atlantis di 2,1mm F5mM pH6 CH3CN 167/33 30min UV-BD (230,200) EE L6 CH3CN 1/1v/ve C8 Symmetry P50mM pH3,6 CH3CN G 25min UV-BD (220) El:MCPB
(P.:préparationéchantillon;v/vevolumeréactif/volume échantillon;L.lavage;E.élution;C.colonne;didiamètreinterne;S.solvant;P.phos¬
phate;F.formiate;Ggradient;Iisocratique;D. détection;BDbarrette de diodes,longueursd'onde dequantificationentre parenthèses;SM/SM
spectrométriedemasse entandem;MRMmultiplereactionmonitoring;étalonnageinterneElouexterneEE;LQlimitedequantification).
de carbonate 1M (2/1 v/v) par un solvant quaternaire éther/dichlorométhane/hexane/alcool isoamylique 50/30/20/0,5
v/v/v/v
permet de détecter les herbicides entre 10 et 100 mg/L selon le pKa du produit. Uneextraction à pH 9,5 en milieu NH4C1 saturé par un mélange ternaire chloroforme/isopropanol/heptane 60/14/26
v/v/v
permetàdeuxlaboratoires une détection proche de 10 mg/L,quelles quesoientles molécules. Annales deToxicologie Analytique,vol.XVIII,
n°2,2006Area Ratio 1.6 1.4 1.2 1 0.8 0.6 0.4 0.2 0
Area Ratio=0.9869428"AmtRatio-K!
Rel.Res%(1):7.575 4 3 2 1 Correlatio
Area Ratio=0.82851396-AmtRalio+0 Area Ratio=113050765'AmtRatio+0
5 4 Correlation: 0.99996 Amount Ratio AreaRatio 1.4 '1.2 1 0.8 0.6 0.4 0.2 0 Rel.Res%(1):7.462 3 2 1 5 4 Correlation: 0.99986 Amount Ratio Area Ratio 2 1.75 1.5 1.25 1 0.75 0.5 0.25 0 Rel Res%(1):9.540 3 2 Correlation:0.99972 Am n ti
Area Ratio=0.98701534'AmtRatio +0 Area Ratio=0.82032223*AmtRatio +0 Area Ratio 1.6 1.4 1.2 1 0.8 0.6 0.4 0.2 0 Rel Res%(1):5.333 3 2 1 4 Correlatior A 5 [j i :0.99976 mountR tî reaRatio 1.4 1.2 1 0.8 0.6 0.4 0.2 0 Correlation:0.99872
Area Ratio=0.89068111*AmtRatio+0 Area Ratio Re|.Res%(l):4.492
1.4 1.2 1 0.8 4 0.6 3 0.4 2 0.2 ! 0 Correlation: 0.99991
Figure3:Courbesd 'étalonnagede6phénoxyacidesentre50et500mg/L après simpleprécipitationdesprotéines plasmatiques (El =MCPB300 mg/L).
TableauII : Conditions analytiques utiliséesparles différentslaboratoires.
P. c. s. D. LQ Ll CH3CN 1/1 v/ve C18 Hypersil di2,1mm P20mM pH6 CH3CN G 18min UV-BD (230,285,210) El:MCPB 10mg/L L2 HC1 IN 1/2v/ve Ether 8/1 v/ve C8 Symmetry P44mM pH3,8 CH3CN G 30min UV-BD (230) El:Prazépam 0,2mg/L L3 OasisMAX L: MeOH E:MeOH/ac formique 98/2 C18 Eclipse F5mM pH3 CH3CN I70/30 15min UV-BD (230) El:MCPB 1à 10mg/L L4 HC16N 1/4v/ve AcEt 4/1 v/ve C18 Nucleosil di1mm F2mM pH3 CH3CN G 18min SM/SM MRM (2ions) El:Méphénytoïne 20 à 50fig/L L5 CH3CN 1/1 v/ve C18 Atlantis di 2,1mm F5mM pH6 CH3CN 167/33 30min UV-BD (230,200) EE L6 CH3CN 1/1v/ve C8 Symmetry P50mM pH3,6 CH3CN G 25min UV-BD (220) El:MCPB
(P.:préparationéchantillon;v/vevolumeréactif/volume échantillon;L.lavage;E.élution;C.colonne;didiamètreinterne;S.solvant;P.phos¬
phate;F.formiate;Ggradient;Iisocratique;D. détection;BDbarrette de diodes,longueursd'onde dequantificationentre parenthèses;SM/SM
spectrométriedemasse entandem;MRMmultiplereactionmonitoring;étalonnageinterneElouexterneEE;LQlimitedequantification).
de carbonate 1M (2/1 v/v) par un solvant quaternaire éther/dichlorométhane/hexane/alcool isoamylique 50/30/20/0,5
v/v/v/v
permet de détecter les herbicides entre 10 et 100 mg/L selon le pKa du produit. Uneextraction à pH 9,5 en milieu NH4C1 saturé par un mélange ternaire chloroforme/isopropanol/heptane 60/14/26
v/v/v
permetàdeuxlaboratoires une détection proche de 10 mg/L,quelles quesoientles molécules.AnnalesdeToxicologie Analytique, vol.
XVHI,
n°2, 2006Les plasmas surchargés
(PI
àP4) contenaientsoit une seule molécule, letriclopyr
(PI)
ouun mélangede 2,4-D etDCP (P2), un mélange 2,4,5-Tet fénoprop (P3) ou encoreun mélangeMCPA etMCPP (P4). Le chro¬ matogramme obtenu parl'un
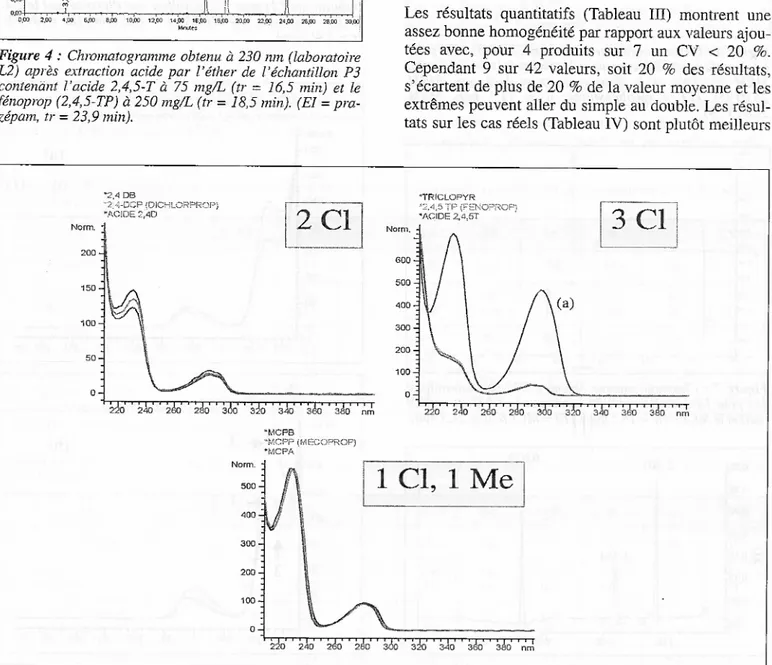
des laboratoires, après extractionparl'éther,est présentédanslafigure4.Les spectresUV
de ces différentes substances (fig.5) pré¬sentent des analogies spectrales avec, pourtous, une
1.20 1.10 1.00 0,80 0,80 0.70 30.60 0.50 0.40 0,30 050 0.10 0,00 o. 2.O0
3.716
4.00 6.00 8.00 10,00 12.00 CD O i 2 3 14.00 16.00 16.00 20.00 Minutes O Si R 22.00 24.00 26,00 28.00 30.00.Figure 4: Chromatogramme obtenuà230nm (laboratoire 12) après extraction acidepar l'éther de l'échantillon P3 contenant l'acide 2,4,5-Tà 75 mg/L (tr = 16,5 min) et le
fénoprop (2,4,5-TP)à250mg/L
(tr
= 18,5min). (El=pra-zépam, tr =23,9 min).
longueur d'onde maximale ou un épaulement à 230 nm, ainsi qu'un 2emaximum entre 280 à 300nm selon le nombre de Cl. Ces analogies sont plus importantes
lorsqu'il
y a unmême type de substitution (2 Cl, 3 Clou
lCl-lMe),
le spectre dutriclopyr
montrant une intensitédesmaxima très différenteenraisondu cycle pyridine.L'identification
nepourra donc complètementse faire surle simple spectre mais nécessite une sépa¬
ration chromatographique satisfaisante pour un maxi¬ mumde spécificité.
Les plasmas issus des cas réels P5, P6 et P7 (fig. 6-8) contenaient respectivement du triclopyr, du 2,4-D et MCPP ou du 2,4-D et 2,4,5-T. Le dernier échantillon P8 (fig.9) contenait un mélange 2,4-D etMCPA, non séparésdansles conditionsd' élutionàpH6,comme on peut le
voir
dans la superposition spectrale et dans la comparaison du spectre hybride etde celui de chacundesproduits (fig.10aetb).Mais ceux-ci sontséparés à
pH 3 (fig.11).
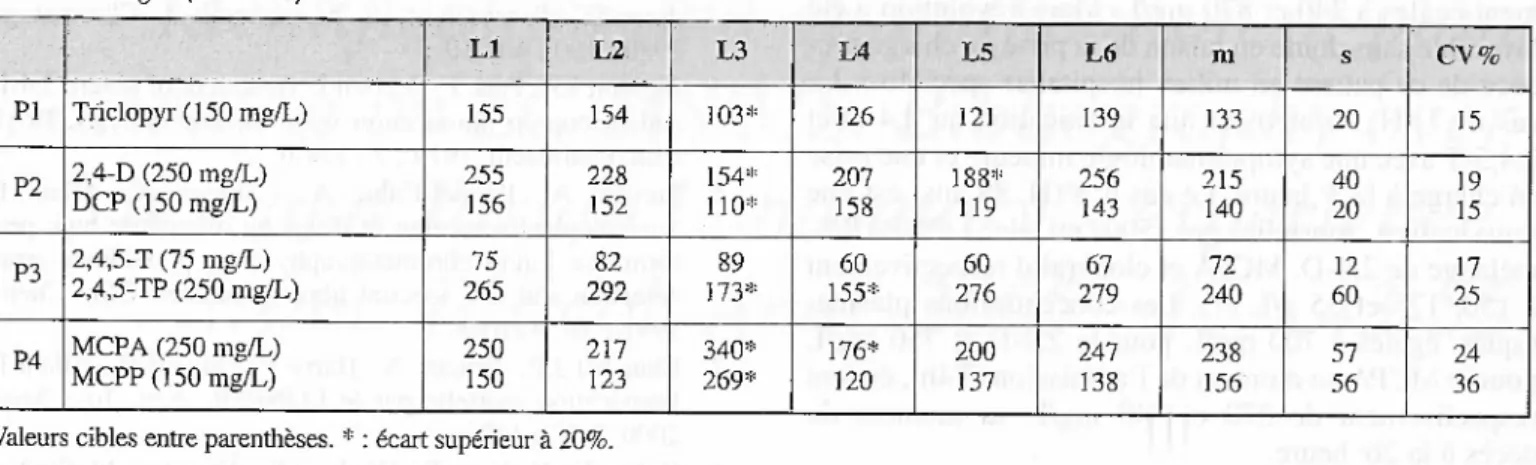
Les résultats quantitatifs (Tableau
III)
montrent une assezbonnehomogénéité parrapport auxvaleurs ajou¬ tées avec, pour 4 produits sur 7 un CV < 20 %. Cependant 9 sur 42 valeurs, soit 20 % des résultats, s'écartentdeplus de20 % de lavaleurmoyenne et les extrêmes peuventallerdu simpleau double.Les résul¬ tats surles cas réels(TableauIV)
sontplutôt meilleurs3.4 DB ~2«-DCP(DICHLORPROP! ACIDE S^D Norm. 200 150
2
Cl
20 20 20 20 30 30 30 30 30 "MCPB MCPP cMECOf-'ROF) MCPA Norm. 500 400 TRICLOPYR *2.1,5TP(FENOPROP! "ACIDE 2.3.5T3
Cl
600 500 400 300 200 100 0 (a) 20 20 20 20 30 30 340 30 301
CL
1
Me
20 20 20 20 30 30 30 30 30 nmFigure 5 : Spectres UVobtenus
àpH6
entre210et400nm desphénoxyacidesetapparentés:(2C1) dérivésdichlorés;(3C1) dérivéstrichlorésdontletriclopyr(a);(ICI,
IMe)dérivéschloro-etméthyl-.AnnalesdeToxicologie Analytique, vol.
XVHI,
n°2, 2006Les plasmas surchargés
(PI
àP4) contenaientsoit une seule molécule, letriclopyr
(PI)
ouun mélangede 2,4-D etDCP (P2), un mélange 2,4,5-Tet fénoprop (P3) ou encoreun mélangeMCPA etMCPP (P4). Le chro¬ matogramme obtenu parl'un
des laboratoires, après extractionparl'éther,est présentédanslafigure4.Les spectresUV
de ces différentes substances (fig.5) pré¬sentent des analogies spectrales avec, pourtous, une
1.20 1.10 1.00 0,80 0,80 0.70 30.60 0.50 0.40 0,30 050 0.10 0,00 o. 2.O0
3.716
4.00 6.00 8.00 10,00 12.00 CD O i 2 3 14.00 16.00 16.00 20.00 Minutes O Si R 22.00 24.00 26,00 28.00 30.00.Figure 4: Chromatogramme obtenuà230nm (laboratoire 12) après extraction acidepar l'éther de l'échantillon P3 contenant l'acide 2,4,5-Tà 75 mg/L (tr = 16,5 min) et le
fénoprop (2,4,5-TP)à250mg/L
(tr
= 18,5min). (El=pra-zépam, tr =23,9 min).
longueur d'onde maximale ou un épaulement à 230 nm, ainsi qu'un 2emaximum entre 280 à 300nm selon le nombre de Cl. Ces analogies sont plus importantes
lorsqu'il
y a unmême type de substitution (2 Cl, 3 Clou
lCl-lMe),
le spectre dutriclopyr
montrant une intensitédesmaxima très différenteenraisondu cycle pyridine.L'identification
nepourra donc complètementse faire surle simple spectre mais nécessite une sépa¬
ration chromatographique satisfaisante pour un maxi¬ mumde spécificité.
Les plasmas issus des cas réels P5, P6 et P7 (fig. 6-8) contenaient respectivement du triclopyr, du 2,4-D et MCPP ou du 2,4-D et 2,4,5-T. Le dernier échantillon P8 (fig.9) contenait un mélange 2,4-D etMCPA, non séparésdansles conditionsd' élutionàpH6,comme on peut le
voir
dans la superposition spectrale et dans la comparaison du spectre hybride etde celui de chacundesproduits (fig.10aetb).Mais ceux-ci sontséparés à
pH 3 (fig.11).
Les résultats quantitatifs (Tableau
III)
montrent une assezbonnehomogénéité parrapport auxvaleurs ajou¬ tées avec, pour 4 produits sur 7 un CV < 20 %. Cependant 9 sur 42 valeurs, soit 20 % des résultats, s'écartentdeplus de20 % de lavaleurmoyenne et les extrêmes peuventallerdu simpleau double.Les résul¬ tats surles cas réels(TableauIV)
sontplutôt meilleurs3.4 DB ~2«-DCP(DICHLORPROP! ACIDE S^D Norm. 200 150
2
Cl
20 20 20 20 30 30 30 30 30 "MCPB MCPP cMECOf-'ROF) MCPA Norm. 500 400 TRICLOPYR *2.1,5TP(FENOPROP! "ACIDE 2.3.5T3
Cl
600 500 400 300 200 100 0 (a) 20 20 20 20 30 30 340 30 301
CL
1
Me
20 20 20 20 30 30 30 30 30 nmFigure 5 : Spectres UVobtenus
àpH6
entre210et400nm desphénoxyacidesetapparentés:(2C1) dérivésdichlorés;(3C1) dérivéstrichlorésdontletriclopyr(a);(ICI,
IMe)dérivéschloro-etméthyl-.Annales deToxicologie Analytique, vol.
XVÏÏI,
n°2, 2006avec des
CV
tous inférieurs à 20 % et seulement 7 résultatsendehorsde±20%.Lesvaleurs trouvées sont voisines de celles initialement rendues par le labora¬ toireangevin,montrantunebonnestabilitéà-20°C.Le plasma6était un mélange de3 prélèvements avec des valeurs voisinesde 150à200mg/L
pourle2,4-D et de 600 à700mg/L pourle MCPP. Int 3.0e5 2.0e5 1.0e5 0 ensity, çps 2 4 Triclopyr Fluroxypyr N11.26 6 8 10 12 Time, min 13.48 | II... 14 16Figure6:Chromatogramme obtenuen CLHP/SM/SMpour l'échantillon réel P5 contenantle triclopyr(tr = 13,5min,
MRM 254/196) et le fluroxypyr (tr - 11,3 min, MRM 253/195). (El = méphénytoine, tr =11,1 min, MRM 217/137,9). 0*0 0.55 0» 0.45 0.40 u» 030 3 020-, O.l f'l1 ooo ; S3 s \ I . '-'A,
OOO EOO 1OO0 15.00
Miiutc jj n 1
*Ai
20 00 i \ 25m do'oo -t 35COFigure 7: Chromatogramme obtenu à 220nm (laboratoire L6)pour l'échantillonréelP6 contenantle2,4-D (tr = 15,9
min)et leMCPP(tr=18,5min).(El= MCPB,tr=21,3min).
0,35 0,30 0,2 0,20
^
0,15 0,10 0.0 0,00 2,303 2,00 3,364 4,00 6,653 6,00 8,00 10,00Figure 8 : Chromatogramme obtenu à 230nm (laboratoire L3)pour l'échantillonréelP7 contenantle2,4-D(tr = 2,30 min)et le2,4,5-T(tr=3,63min).(El= MCPB, tr=6,65min).
DAD1A.Sig=230.4Ref=650.100 (AOUT05\0810-Q21 rnAU i I 400-j 300! l 056 11! "* 200-! 100\ 0\ L
%h
-^
ïv 0 2 4 6 D) -7.396 j! 8 minFigure 9 : Chromatogramme obtenu à 230 nm et à
pH
6 (laboratoireLl) pourl'échantillonréelP8 contenantle 2,4-D(tr=4,05min)etleMCPA(tr= 4,05 min). (El=MCPB, tr= 7,40min).*DAD1.3.978(231mAU,Up2) Ref= 5.501of0810-021D
*OAD1.4053(37^SmAU.Apx) Refe5501of0810021.D
*DAD1.4.16S(327mAU.Dnl)Ref= 5.501of 0810-021.D
Norm. 350 300 250 200 150 100 50 0 (a) 220 240 260 280 300 320 340 360 380 nm Norm. 350 300 250 200 150 100 50 0 "MCPA ACIDE 2,*D
DAD1,4.056(374mAU.Apx) Ref=5.501 of0810-021.D
«-3
(b)
1
t
2
220 240 260 280 300 320 340 360 380 nm
Figure10 : (a) : Testdepuretédupic(tr=4,06min)dela figure9:(b):comparaisonentrespectrehybrideausommet
(1)etspectresdu2,4-D (2) etduMCPA(3).
Annales deToxicologie Analytique, vol.
XVÏÏI,
n°2, 2006avec des
CV
tous inférieurs à 20 % et seulement 7 résultatsendehorsde±20%.Lesvaleurs trouvées sont voisines de celles initialement rendues par le labora¬ toireangevin,montrantunebonnestabilitéà-20°C.Le plasma6était un mélange de3 prélèvements avec des valeurs voisinesde 150à200mg/L
pourle2,4-D et de 600 à700mg/L pourle MCPP. Int 3.0e5 2.0e5 1.0e5 0 ensity, çps 2 4 Triclopyr Fluroxypyr N11.26 6 8 10 12 Time, min 13.48 | II... 14 16Figure6:Chromatogramme obtenuen CLHP/SM/SMpour l'échantillon réel P5 contenantle triclopyr(tr = 13,5min,
MRM 254/196) et le fluroxypyr (tr - 11,3 min, MRM 253/195). (El = méphénytoine, tr =11,1 min, MRM 217/137,9). 0*0 0.55 0» 0.45 0.40 u» 030 3 020-, O.l f'l1 ooo ; S3 s \ I . '-'A,
OOO EOO 1OO0 15.00
Miiutc jj n 1
*Ai
20 00 i \ 25m do'oo -t 35COFigure 7: Chromatogramme obtenu à 220nm (laboratoire L6)pour l'échantillonréelP6 contenantle2,4-D (tr = 15,9
min)et leMCPP(tr=18,5min).(El= MCPB,tr=21,3min).
0,35 0,30 0,2 0,20
^
0,15 0,10 0.0 0,00 2,303 2,00 3,364 4,00 6,653 6,00 8,00 10,00Figure 8 : Chromatogramme obtenu à 230nm (laboratoire L3)pour l'échantillonréelP7 contenantle2,4-D(tr = 2,30 min)et le2,4,5-T(tr=3,63min).(El= MCPB, tr=6,65min).
DAD1A.Sig=230.4Ref=650.100 (AOUT05\0810-Q21 rnAU i I 400-j 300! l 056 11! "* 200-! 100\ 0\ L
%h
-^
ïv 0 2 4 6 D) -7.396 j! 8 minFigure 9 : Chromatogramme obtenu à 230 nm et à
pH
6 (laboratoireLl) pourl'échantillonréelP8 contenantle 2,4-D(tr=4,05min)etleMCPA(tr= 4,05 min). (El=MCPB, tr= 7,40min).*DAD1.3.978(231mAU,Up2) Ref= 5.501of0810-021D
*OAD1.4053(37^SmAU.Apx) Refe5501of0810021.D
*DAD1.4.16S(327mAU.Dnl)Ref= 5.501of 0810-021.D
Norm. 350 300 250 200 150 100 50 0 (a) 220 240 260 280 300 320 340 360 380 nm Norm. 350 300 250 200 150 100 50 0 "MCPA ACIDE 2,*D
DAD1,4.056(374mAU.Apx) Ref=5.501 of0810-021.D
«-3
(b)
1
t
2
220 240 260 280 300 320 340 360 380 nm
Figure10 : (a) : Testdepuretédupic(tr=4,06min)dela figure9:(b):comparaisonentrespectrehybrideausommet
Discussion
Enraison du caractère acide faibledes herbicides phé¬
noxyacides, les procédures classiques d'extraction en milieu alcalin, mises en Auvre dans les méthodes de screening toxicologique, ne sont pas complètement adaptéesàladétectiondeces substances,surtoutencas
Figure11 :Séparationchromatographiqueà
pH
3 (labora¬ toireLl)
del'
échantillonréelP8:2,4-D(tr
= 8,51 min)etMCPA
(tr
=8,56 min). (El=MCPB,tr
= 11,75 min).d'intoxication
mineure.Il
estdoncnécessaire de dispo¬serd'une méthode de dosage spécifiquepour ces her¬
bicides même si lesintoxications sévères, caractérisées pardes concentrations plasmatiques de quelques cen¬
taines de mg/L, sont généralement mises en évidence parces conditions descreeningtoxicologique.
Parmi les 7 molécules présentes dans l'essai, deux
n'ont
pas été correctement identifiées dans un premiertemps par deux laboratoires, car ces substances n'étaientpas référencées dans leur méthode : dans un
cas, le
triclopyr
a étéconfondu avec leMCPA malgréun spectre
UV
apriori
différentet dansl'autre cas, les transitions SM/SM du fénoprop n'avaient pas été inclusesdans la tabled'acquisition.Après actualisation desméthodes,chacunapuprocéderaudosage.Pour un laboratoire,l'identification
et le dosage du plasma P8 ontnécessitéunchangementdephasemobileenraison de lacoélution du 2,4-D etdu MCPA àpH6.Les résultats des 8 essais ont montréune bonnehomo¬ généitépourles laboratoires ayantutiliséla simple pré¬
cipitation des protéines ou l'extraction liquide-liquide. Cependant le laboratoire ayantutilisél'extraction liqui¬ de-solideaprésentéplusieurs valeurs endehors de ± 20%.Cetteapprocheserait-elle moinsperformantepour
cesproduitsou uneaméliorationdelaprocédure est-elle envisageable ? Plusieurs questions peuvent être
soule-Tableau
III
:Résultats(mg/L)parlaboratoire(Ll
à16)et moyenne, écart-typeetcoefficientdevariation(m, s, CV%)desplas¬ massurchargés(PI àP4).PI P2 P3 P4 Triclopyr(150mg/L) 2,4-D(250mg/L) DCP(150mg/L) 2,4,5-T(75mg/L) 2,4,5-TP (250mg/L) MCPA(250mg/L) MCPP (150mg/L) Ll 155 255 156 75 265 250 150 L2 154 228 152 82 292 217 123 L3 103* 154* 110* 89 173* 340* 269* L4 126 207 158 60 155* 176* 120 L5 121 188* 119 60 276 200 137 L6 139 256 143 67 279 247 138 m 133 215 140 72 240 238 156 s 20 40 20 12 60 57 56 " CV% 15 19 15 17 25 24 36
Valeurscibles entre parenthèses.*:écartsupérieurà20%.
Tableau
IV
: Résultats(mg/L)parlaboratoire(Ll
àL6)et moyenne, écart-typeetcoefficientdevariation (m, s, CV%) descasréels(P5àP8). P5 P6° P7 P8 Triclopyr(250mg/L) 2,4-D MCPP 2,4-D(155 mg/L) 2,4,5-T(92mg/L) 2,4-D (370mg/L) MCPA(340mg/L) Ll 260 202 760 170 95 375 400 L2 231 177 670 160 104 330 321 L3 255 133* 553 101* 61* 461* 442* L4 160* 196 680 173 96 328 306 L5 228 141 611 135 82 276* 300 L6 216 202 695 168 97 359 349 m 225 175 662 151 89 355 353 s 36 31 72 28 16 62 57 CV% 16 18 11 19 17 17 16
Valeursinitialesentre parenthèses.°poolde3plasmas.*:écartsupérieurà20%.
Discussion
Enraison du caractère acide faibledes herbicides phé¬
noxyacides, les procédures classiques d'extraction en milieu alcalin, mises en Auvre dans les méthodes de screening toxicologique, ne sont pas complètement adaptéesàladétectiondeces substances,surtoutencas
Figure11 :Séparationchromatographiqueà
pH
3 (labora¬ toireLl)
del'
échantillonréelP8:2,4-D(tr
= 8,51 min)etMCPA
(tr
=8,56 min). (El=MCPB,tr
= 11,75 min).d'intoxication
mineure.Il
estdoncnécessaire de dispo¬serd'une méthode de dosage spécifiquepour ces her¬
bicides même si lesintoxications sévères, caractérisées pardes concentrations plasmatiques de quelques cen¬
taines de mg/L, sont généralement mises en évidence parces conditions descreeningtoxicologique.
Parmi les 7 molécules présentes dans l'essai, deux
n'ont
pas été correctement identifiées dans un premiertemps par deux laboratoires, car ces substances n'étaientpas référencées dans leur méthode : dans un
cas, le
triclopyr
a étéconfondu avec leMCPA malgréun spectre
UV
apriori
différentet dansl'autre cas, les transitions SM/SM du fénoprop n'avaient pas été inclusesdans la tabled'acquisition.Après actualisation desméthodes,chacunapuprocéderaudosage.Pour un laboratoire,l'identification
et le dosage du plasma P8 ontnécessitéunchangementdephasemobileenraison de lacoélution du 2,4-D etdu MCPA àpH6.Les résultats des 8 essais ont montréune bonnehomo¬ généitépourles laboratoires ayantutiliséla simple pré¬
cipitation des protéines ou l'extraction liquide-liquide. Cependant le laboratoire ayantutilisél'extraction liqui¬ de-solideaprésentéplusieurs valeurs endehors de ± 20%.Cetteapprocheserait-elle moinsperformantepour
cesproduitsou uneaméliorationdelaprocédure est-elle envisageable ? Plusieurs questions peuvent être
soule-Tableau
III
:Résultats(mg/L)parlaboratoire(Ll
à16)et moyenne, écart-typeetcoefficientdevariation(m, s, CV%)desplas¬ massurchargés(PI àP4).PI P2 P3 P4 Triclopyr(150mg/L) 2,4-D(250mg/L) DCP(150mg/L) 2,4,5-T(75mg/L) 2,4,5-TP (250mg/L) MCPA(250mg/L) MCPP (150mg/L) Ll 155 255 156 75 265 250 150 L2 154 228 152 82 292 217 123 L3 103* 154* 110* 89 173* 340* 269* L4 126 207 158 60 155* 176* 120 L5 121 188* 119 60 276 200 137 L6 139 256 143 67 279 247 138 m 133 215 140 72 240 238 156 s 20 40 20 12 60 57 56 " CV% 15 19 15 17 25 24 36
Valeurscibles entre parenthèses.*:écartsupérieurà20%.
Tableau
IV
: Résultats(mg/L)parlaboratoire(Ll
àL6)et moyenne, écart-typeetcoefficientdevariation (m, s, CV%) descasréels(P5àP8). P5 P6° P7 P8 Triclopyr(250mg/L) 2,4-D MCPP 2,4-D(155 mg/L) 2,4,5-T(92mg/L) 2,4-D (370mg/L) MCPA(340mg/L) Ll 260 202 760 170 95 375 400 L2 231 177 670 160 104 330 321 L3 255 133* 553 101* 61* 461* 442* L4 160* 196 680 173 96 328 306 L5 228 141 611 135 82 276* 300 L6 216 202 695 168 97 359 349 m 225 175 662 151 89 355 353 s 36 31 72 28 16 62 57 CV% 16 18 11 19 17 17 16
Annales deToxicologie Analytique, vol.
XVIII,
n° 2,2006vées : une estérification de ces produits sur la colonne
lors del'étapedelavageau methanol est-elle possible ?
Le pourcentaged'acideformique(2%)danslemethanol est-il suffisantpour éluerdela même façontous lespro¬
duits ? Cependant celaboratoire ayantutilisé le MCPB comme étalon interne etpar ailleurs les gammes d'éta¬ lonnage' ayant étéfaites dans les mêmes conditions que les plasmas tests, ces différences restent difficiles à
expliquer. Un nouvel essai a été effectué par ce même laboratoire,5moisplus tard, sur les mêmeséchantillons, avec une colonne chromatographique neuve et des
gammes d'étalonnage obtenues à partir de nouvelles poudresstandards.Lesrésultatssontpluscohérents avec lesvaleurs attenduesmais 3 concentrations (1 surcharge et 2casréels) sontencoreendehors de± 20%. Laques¬
tionreste doncpourlemomentensuspens.
Deux laboratoires ont mentionnéla présence dans les
casréels de2 autres substances : le
fluroxypyr
(8et 14mg/L) dans
l'échantillon
P5 etle clopyralid (63 mg/L)dans
l'échantillon
P8. Lefluroxypyr
est unemoléculedérivée, comme le triclopyr, de l'acide pyridoxyacé¬
tique, le clopyralid étantdérivé de l'acide pyridoxy-2-carboxylique. Le cas n°5 (H, 56 ans) correspondait effectivement à une intoxication modérée par 5 à 6
verres de
GARLON©,
mélangedetriclopyr
etfluroxy¬ pyrà60et20g/L
respectivement.Leprélèvementaété effectué environ à la 7e heure après l'ingestion. Une intoxication modérée par un mélange 2,4-D et MCPP (cas n° 6, H, 42 ans) a cependantmontré des concen¬ trations initiales (T2h)assez importantes etrespective¬ mentégales à 240 et870 mg/L. Maisl'évolution
aété favorablesansdouteenraisondela priseenchargepré¬coce de ce patient en milieu hospitalier spécialisé. Le
cas n° 7 (H, 36 ans) est une intoxication au 2,4-D et 2,4,5-Tavecunesymptomatologiemineureetuneprise encharge àla 3eheure. Le cas n°8 (H, 88 ans) est une
intoxication mortelle par 500 ml de
LONPAR©,
mélange de2,4-D, MCPAetclopyralidrespectivement
à 150, 175 et 35
g/L
(7). Les concentrations plasma¬ tiques, égales à 700mg/L
pour le 2,4-D et 750mg/L
pourleMCPAaumomentdel'admission(T4h), étaient respectivement de 370 et 340 mg/L au moment du décèsà la26e heure.
Enfin le laboratoire utilisant la spectrométrie de masse entandem a signalé laprésencede certains deces phé¬
noxyacidesàl'étatde«traces»(<2mg/L)tantdans les plasmas surchargés, correspondant àdes impuretés des
produits purs, que dans les cas réels pouvant peut-être s'expliquer soit par des impuretés des produits tech¬
niques ingérésparles patientsou paruneexposition de ceux-ci àcesproduits endehors d'uncontexte aigu.
tiondesprotéinesplasmatiques suivied'une
dilution
au1/2 dansl'eau etd'unechromatographieavecdétection
UV
etidentificationspectrale estuneapproche simple, rapide, nécessitant peu d'échantillon plasmatique et largement suffisantepourl'objectif
défini. Dans lescasd'intoxication
légère, le recours à une approche parextraction liquide/liquide, ou peut-être liquide/solide,
d'un
volume plus important de plasma devrait per¬mettred'abaisserla
limite
dedétection. Cette approche devra être envisagéeencas d'expositionprofessionnel¬ le et la spectrométrie de masse en tandem sera le meilleur mode de détection pourmesurer des concen¬ trations faibles dansl'urine
(de quelques pg/L à 0,5 mg/L) de personnes exposées à ces substances lors d'applications agricoles (8-11).Références
Conclusion
Dans cette étude orientée sur le diagnostic analytique
des intoxicationsaiguës parherbicides phénoxyacides, plusieursméthodologies ontété évaluées. La
précipita-1. Fournier J. Identité chimique et activité biologique d'un produit. In :FournierJ., Chimiedespesticides. Lavoisier,
Paris, 1988 : 95-109.
2. Poisindex,Micromedex Healthcareseries, Chicago,2006,
Vol 127.
3. Flanagan
RI,
RuprahM. HPLCmeasurement of chloro-phenoxy herbicides,bromoxynil,andioxynil, in biologicalspecimens to aid diagnosis of acute poisoning. Clin.
Chem. 1989;35 : 1342-7.
4. VanDammeJC,GalouxM. Méthode d'analyse parchro¬
matographieliquidehauteperformancedesformulationsà
base de mélange d'acides phénoxycarboxyliques, de
dicamba, d'ioxynil et de bromoxynil. J. Chromatogr.
1980; 190: 401-10.
5. Prescott LF., ParkI, DarrienI. Treatmentofsevere2,4-D
and mécoprop intoxication with alkaline diuresis. Br. J.
Clin. Pharmacol. 1979 ;7 : 111-6.
6. Turcant A., Premel-Cabic A., Cailleux A., Allain P.
Toxicological screening ofdrugsby microborehigh-per¬
formance liquid chromatography with photodiode-array detection and UV spectral library searches. Clin. Chem.
1991 ;37 : 1210-5.
7. BlanchetIP., Turcant A., Harry P., Durand R., AllainP.
Intoxication mortelle par le LONPAR. Ann. Tox. Anal.
2000; 12(2) 169.
8. Kolmodin-Hedman B.,HôglundS., Âkerblom M. Studies on phenoxy acid herbicides. I. Field study. Occupational
exposure tophenoxy acidherbicidesin agriculture.Arch. Toxicol. 1983 ;54 :257-65.
9. Kolmodin-Hedman B., Hôglund S., Swensson k-., Âker¬
blom M. Studies onphenoxy acid herbicides. II Oral and
dermal uptake and elimination in urine of MCPA in
humans.Arch. Toxicol. 1983 ;54: 267-73.
lO.Shealy D., BoninM., Wooten I, Ashley D.,Needham L. Application ofan improved method for the analysis of
pesticides and their metabolites in the urine of farmer applicators and their families. Environnement International 1996 ; 22: 661-75.
ll.Maroni M., Colosio C, Ferioli A., Fait A. Biological monitoring ofpesticide exposure : areview. Toxicology
2000;143:1-118. Annales deToxicologie Analytique, vol.
XVIII,
n° 2,2006vées : une estérification de ces produits sur la colonne
lors del'étapedelavageau methanol est-elle possible ?
Le pourcentaged'acideformique(2%)danslemethanol est-il suffisantpour éluerdela même façontous lespro¬
duits ? Cependant celaboratoire ayantutilisé le MCPB comme étalon interne etpar ailleurs les gammes d'éta¬ lonnage' ayant étéfaites dans les mêmes conditions que les plasmas tests, ces différences restent difficiles à
expliquer. Un nouvel essai a été effectué par ce même laboratoire,5moisplus tard, sur les mêmeséchantillons, avec une colonne chromatographique neuve et des
gammes d'étalonnage obtenues à partir de nouvelles poudresstandards.Lesrésultatssontpluscohérents avec lesvaleurs attenduesmais 3 concentrations (1 surcharge et 2casréels) sontencoreendehors de± 20%. Laques¬
tionreste doncpourlemomentensuspens.
Deux laboratoires ont mentionnéla présence dans les
casréels de2 autres substances : le
fluroxypyr
(8et 14mg/L) dans
l'échantillon
P5 etle clopyralid (63 mg/L)dans
l'échantillon
P8. Lefluroxypyr
est unemoléculedérivée, comme le triclopyr, de l'acide pyridoxyacé¬
tique, le clopyralid étantdérivé de l'acide pyridoxy-2-carboxylique. Le cas n°5 (H, 56 ans) correspondait effectivement à une intoxication modérée par 5 à 6
verres de
GARLON©,
mélangedetriclopyr
etfluroxy¬ pyrà60et20g/L
respectivement.Leprélèvementaété effectué environ à la 7e heure après l'ingestion. Une intoxication modérée par un mélange 2,4-D et MCPP (cas n° 6, H, 42 ans) a cependantmontré des concen¬ trations initiales (T2h)assez importantes etrespective¬ mentégales à 240 et870 mg/L. Maisl'évolution
aété favorablesansdouteenraisondela priseenchargepré¬coce de ce patient en milieu hospitalier spécialisé. Le
cas n° 7 (H, 36 ans) est une intoxication au 2,4-D et 2,4,5-Tavecunesymptomatologiemineureetuneprise encharge àla 3eheure. Le cas n°8 (H, 88 ans) est une
intoxication mortelle par 500 ml de
LONPAR©,
mélange de2,4-D, MCPAetclopyralidrespectivement
à 150, 175 et 35
g/L
(7). Les concentrations plasma¬ tiques, égales à 700mg/L
pour le 2,4-D et 750mg/L
pourleMCPAaumomentdel'admission(T4h), étaient respectivement de 370 et 340 mg/L au moment du décèsà la26e heure.
Enfin le laboratoire utilisant la spectrométrie de masse entandem a signalé laprésencede certains deces phé¬
noxyacidesàl'étatde«traces»(<2mg/L)tantdans les plasmas surchargés, correspondant àdes impuretés des
produits purs, que dans les cas réels pouvant peut-être s'expliquer soit par des impuretés des produits tech¬
niques ingérésparles patientsou paruneexposition de ceux-ci àcesproduits endehors d'uncontexte aigu.
tiondesprotéinesplasmatiques suivied'une
dilution
au1/2 dansl'eau etd'unechromatographieavecdétection
UV
etidentificationspectrale estuneapproche simple, rapide, nécessitant peu d'échantillon plasmatique et largement suffisantepourl'objectif
défini. Dans lescasd'intoxication
légère, le recours à une approche parextraction liquide/liquide, ou peut-être liquide/solide,
d'un
volume plus important de plasma devrait per¬mettred'abaisserla
limite
dedétection. Cette approche devra être envisagéeencas d'expositionprofessionnel¬ le et la spectrométrie de masse en tandem sera le meilleur mode de détection pourmesurer des concen¬ trations faibles dansl'urine
(de quelques pg/L à 0,5 mg/L) de personnes exposées à ces substances lors d'applications agricoles (8-11).Références
Conclusion
Dans cette étude orientée sur le diagnostic analytique
des intoxicationsaiguës parherbicides phénoxyacides, plusieursméthodologies ontété évaluées. La
précipita-1. Fournier J. Identité chimique et activité biologique d'un produit. In :FournierJ., Chimiedespesticides. Lavoisier,
Paris, 1988 : 95-109.
2. Poisindex,Micromedex Healthcareseries, Chicago,2006,
Vol 127.
3. Flanagan
RI,
RuprahM. HPLCmeasurement of chloro-phenoxy herbicides,bromoxynil,andioxynil, in biologicalspecimens to aid diagnosis of acute poisoning. Clin.
Chem. 1989;35 : 1342-7.
4. VanDammeJC,GalouxM. Méthode d'analyse parchro¬
matographieliquidehauteperformancedesformulationsà
base de mélange d'acides phénoxycarboxyliques, de
dicamba, d'ioxynil et de bromoxynil. J. Chromatogr.
1980; 190: 401-10.
5. Prescott LF., ParkI, DarrienI. Treatmentofsevere2,4-D
and mécoprop intoxication with alkaline diuresis. Br. J.
Clin. Pharmacol. 1979 ;7 : 111-6.
6. Turcant A., Premel-Cabic A., Cailleux A., Allain P.
Toxicological screening ofdrugsby microborehigh-per¬
formance liquid chromatography with photodiode-array detection and UV spectral library searches. Clin. Chem.
1991 ;37 : 1210-5.
7. BlanchetIP., Turcant A., Harry P., Durand R., AllainP.
Intoxication mortelle par le LONPAR. Ann. Tox. Anal.
2000; 12(2) 169.
8. Kolmodin-Hedman B.,HôglundS., Âkerblom M. Studies on phenoxy acid herbicides. I. Field study. Occupational
exposure tophenoxy acidherbicidesin agriculture.Arch. Toxicol. 1983 ;54 :257-65.
9. Kolmodin-Hedman B., Hôglund S., Swensson k-., Âker¬
blom M. Studies onphenoxy acid herbicides. II Oral and
dermal uptake and elimination in urine of MCPA in
humans.Arch. Toxicol. 1983 ;54: 267-73.
lO.Shealy D., BoninM., Wooten I, Ashley D.,Needham L. Application ofan improved method for the analysis of
pesticides and their metabolites in the urine of farmer applicators and their families. Environnement International 1996 ; 22: 661-75.
ll.Maroni M., Colosio C, Ferioli A., Fait A. Biological monitoring ofpesticide exposure : areview. Toxicology