Shared and unique mechanisms of macrophage-like
neutrophils in EAE
Mémoire
Ryder Whittaker Hawkins
Maîtrise en microbiologie-immunologie - avec mémoire
Maître ès sciences (M. Sc.)
Shared and Unique Mechanisms of Macrophage-Like
Neutrophils in EAE
Mémoire
Ryder Whittaker Hawkins
Sous la direction de :
Résumé
L’ensemble des maladies démyélinisantes (e.g. la sclérose en plaques et la neuromyélite optique) représente un fardeau majeur sur la société et sur le bien-être des citoyens affectés. Quoique des progrès aient été fait dans la compréhension des mécanismes biologiques qui y sont sous-jacents, les causes ultimes ne sont pas connues et il y a un besoin de développer des traitements plus pointus. Le symptôme caractéristique de toute maladie démyélinisante est la perte de la myéline qui isole les fibres nerveuses du système nerveux central (SNC). Cette perte est effectuée par l’action néfaste et non-contrôlée de cellules du système immunitaire : les lymphocytes T qui réagissent contre les protéines de la myéline, les cellules B qui sécrètent des autoanticorps, les macrophages qui phagocytent des débris de myéline, et les cellules dendritiques qui orchestrent tout. Cependant, il est clair aujourd’hui d’après le modèle animal de la sclérose en plaques, l’encéphalomyélite autoimmune expérimentale (EAE), que les neutrophiles sont indispensables au développement complet de la maladie. Ainsi, la déplétion des neutrophiles prévient l’apparition des symptômes. Néanmoins, le mécanisme d’action des neutrophiles reste à être élucidée. Le présent mémoire résume d’abord les connaissances actuelles sur les neutrophiles dans l’EAE et la sclérose en plaques et présente ensuite des données originaux permettant de mieux comprendre les fonctions des neutrophiles dans l’EAE. Nous démontrons que les neutrophiles infiltrant le SNC subissent des changements moléculaires qui les activent; que leur transcriptome devient plus similaire à ceux des macrophages et des cellules dendritiques; que les neutrophiles interagissent physiquement avec les lymphocytes T et B in situ dans la moelle épinière enflammée; et que, dans un nouveau modèle de la sclérose en plaques dépendant de cellules B, les neutrophiles utilisent la protéase ASPRV1 pour prolonger l’inflammation à long terme. Ces observations améliorent notre compréhension des maladies démyélinisantes et servent de base pour de prochaines expériences sur le rôle des neutrophiles en général.
Abstract
Autoimmune demyelinating diseases (ADDs) are a leading cause of neurological disability in youth and adults, especially in Canada; the best-known of which is multiple sclerosis (MS); others include neuromyelitis optica spectrum disorder. These diseases are characterized by destruction of myelin and loss of nerve conductivity leading to motor deficits and deteriorating quality of life. ADDs have been studied for nearly two centuries and disease-mitigating therapies are now available for patients; however, a cure has not yet been found. Demyelination proceeds largely via the reaction of autospecific T cells with endogenous myelin proteins, in cooperation with B cells, macrophages and dendritic cells; the root cause of this autoreactivity is still unknown. Yet it is becoming increasingly clear from the study of animal models of MS that depletion of neutrophils, an abundant innate leukocyte population, has the potential to block the development of disease symptoms. We therefore aim to comprehend the molecular reasons behind this phenomenon in mice and translate our findings to the human case. This work aims, firstly, to summarize the facts known about neutrophils in MS, neuromyelitis optica, and other ADDs, as well as in mouse models of demyelination; secondly, to present the results of experiments on neutrophils with the model system experimental autoimmune encephalomyelitis (EAE). We have found that neutrophils in EAE that migrate to the central nervous system undergo transcriptional and proteomic changes that leave them in a putatively activated state. These activated neutrophils physically interact with T and B lymphocytes in the inflamed spinal cord. Furthermore, we use an improved model of EAE, that better describes MS, to show that neutrophils act through the novel gene Asprv1 to prolong and worsen inflammation. This study sheds light on the subtleties of neutrophils in a societally relevant context and provides data for the continued investigation into neutrophil biochemistry and systems biology.
Table of Contents
Résumé ... iii
Abstract ...iv
Table of Contents ... v
List of Figures ... vii
List of Tables ... viii
Acknowledgments ...xi
Foreword ... xii
Introduction... 1
The Development of Autoimmunity ... 2
Current State of Treatment ... 3
Studying Demyelination in the Laboratory ... 5
Neutrophils are Essential for Demyelination... 7
Chapter 1: Neutrophils in ADDs and EAE: Current State of Knowledge ... 9
The mechanism of neutrophil recruitment in EAE ... 10
Expansion and mobilization ... 13
Rolling, adhesion and crawling ... 14
Extravasation ... 15
Are Neutrophils Essential for EAE? ... 16
BBB disruption ... 16
Immunomodulation ... 18
Antigen presentation ... 18
Myelin degradation and phagocytosis ... 19
How do neutrophils contribute to human ADDs?... 19
Clinical Potential for Neutrophil-Related Biomarkers... 21
Next Steps ... 22
Chapter 2: ICAM1 Identifies Transcriptionally Unique Macrophage-Like Neutrophils in EAE ... 25
ICAM1 distinguishes extra- from intravascular neutrophils in the CNS of EAE mice ... 26
ICAM1+ neutrophils have a distinctive transcriptional profile revealing a potential for antigen presentation and immunostimulation ... 31
ICAM1+ neutrophils form immunological synapses with T and B cells ... 37
Chapter 3: The Protease ASPRV1 is a Neutrophil Gene and Worsens EAE via B Cells ... 43
ASPRV1 is specific to neutrophils and increases in the CNS during EAE and severe forms of MS ... 46
ASPRV1 is required for the chronic phase of a B cell-dependent EAE model ... 47
Methods ... 50
Chapter 4: Discussion and General Conclusions ... 53
Catchup ... 54 ICAM1 ... 54 Transcriptomes ... 56 MHCII... 57 bMOG ... 59 ASPRV1... 59 Future Directions ... 60 References ... 62
List of Figures
Introduction
Fig. 0.1: Appearance of multiple sclerosis lesion ... 1
Fig. 0.2: Transfer of neuromyelitis optica ... 2
Fig. 0.3: Macrophage APCs infiltrate the spinal cord in EAE... 6
Fig. 0.4: Neutrophils infiltrate the spinal cord in EAE ... 7
Fig. 0.5: Depletion of neutrophils reduces EAE severity... 8
Chapter 1: Neutrophils in ADDs and EAE: Current State of Knowledge Fig. 1.1: Neutrophils expand during EAE and depend on Th17 cells and Cxcr2 ... 11
Fig. 1.2: Recruitment and functions of neutrophils in EAE: a working model ... 12
Fig. 1.3: The effects of G-CSF in EAE ... 13
Fig. 1.4: BBB destruction in EAE and its rescue by depleting neutrophils... 17
Chapter 2: ICAM1 Identifies Transcriptionally Unique Macrophage-Like Neutrophils Fig. 2.1: The spinal cord of EAE mice contains two subsets of neutrophils ... 27
Fig. 2.S1: Generation of Catchup × Ai6 mice ... 27
Fig. 2.2: ICAM1+ and ICAM1− neutrophils are differently distributed... 28
Fig. 2.S2: Validation of the Catchup × Ai6 system... 29
Fig. 2.S3: Sort scheme ... 32
Fig. 2.3: ICAM1+ neutrophils have a distinct transcriptional profile ... 33
Fig. 2.4: Protein-protein interaction networks ... 34
Fig. 2.5: Co-stimulatory molecule expression ... 35
Fig. 2.6: Neutrophils form immune synapses with lymphocytes ... 37
Fig. 2.S4: Excision of H2-Ab1 ... 39
Chapter 3: The Protease ASPRV1 is a Neutrophil Gene and Worsens EAE via B Cells Fig. 3.1: ASPRV1 is a neutrophil-specific marker in mouse and human ... 44
Fig. 3.2: Molecular dynamics simulation of murine ASPRV1... 46
Fig. 3.S1: B cells are required for bMOG EAE ... 47
Fig. 3.3: ASPRV1 is required for the chronic phase of EAE ... 48
Fig. 3.4: Stereological investigation of macrophages in Asprv1 mice... 49
Chapter 4: Discusssion & General Conclusions Fig. 4.1: Neutrophil transcriptomes in other diseases ... 57
List of Tables
Introduction
Table 0.1: Comparison of demyelinating autoimmune diseases ... 4 Chapter 1: Neutrophils in ADDs and EAE: Current State of Knowledge
Acknowledgments
First of all, I thank my supervisor, Dr. Luc Vallières, for his supervision and direction. Dr. Vallières conceived of the project idea and supported me all the way, from applying for the scholarship that helped fund the project, to the interpretation and presentation of results. My thanks go equally to Drs. Alexandre Patenaude and Aline Dumas, my coauthors on this work. Drs. Dumas and Patenaude’s expertise not only in technical matters, but in laboratory best practices, data analysis, planning of experiments, and long hours in the laboratory were invaluable to the success of this project. I really couldn’t have done it without them. As well, this work was the fruit of international collaboration. Thanks to Dr. Matthias Gunzer for consultation on the manuscript, on our healthy collaboration and on providing the mice that drove this paper forward. Although we never met, thanks to Dr. Takeshi Matsui for his insights into the ASPRV1 side of the work. Our work was also helped by a collaboration with some fellow MS researchers at Western University, the team of Dr. Steven Kerfoot and his two wonderful students, Rajiv and Yodit. I thank all of them for their experimental contributions to the work. I thank as well Drs. Poubelle, Larochelle and Pelletier, for their local collaboration. For the review chapter, thanks to my coauthors Drs. Courtney Casserly and Julia Nantes, who wrote the review with myself and Dr. Vallières. This thesis was supported by an endMS studentship from the Multiple Sclerosis Society of Canada, who also sponsored the review paper as part of their SPRINT program. Finally, I thank the Université Laval for the supportive, well-connected, and encouraging environment, which supported my scientific career through conferences, seminars, courses and close networking between labs.
Foreword
The Introduction is an original work by the student. Chapter 1 is taken from the published review article in Autoimmunity Reviews, on which the student was third author, entitled “Neutrophil perversion in autoimmune demyelinating diseases: mechanisms to medicine” (1). The student worked in close collaboration in the researching and writing of this review paper on neutrophils. Only the passages and figures that were directly relevant to the content of the thesis were included. As well, figures were added by the student during the writing of the thesis in order to improve legibility and to better summarize the progress in the field.
Chapters 2 and 3 are taken from the student’s first-author published article in JCI Insight (2). The student contributed both to the experimental and writing aspects of this paper. The introductions and conclusions of the article were shortened for continuity. To better introduce each subject of the article, the paper was divided into two logically coherent sections. As well, new original figures have been added to give a better idea of the effort that went into the project, even those parts that were considered not pertinent for formal publication. All supplementary information to the published article can be found on the journal website.
Chapter 4 is original and situates the conclusions and results of the two included articles in the greater scientific context. This chapter extends and replaces much of the discussion sections of the two published articles.
A full list of references is included at the end of the thesis.
Introduction
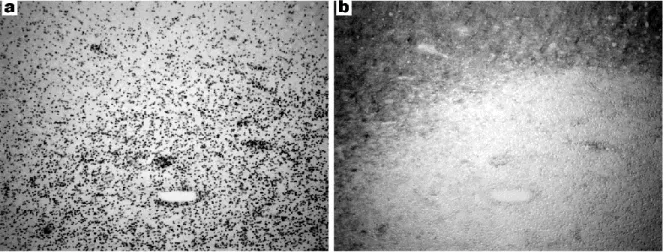
Multiple sclerosis (MS) is a leading cause of neurological disability that strikes spontaneously in early age and persists throughout life. The disease is common, with a Canadian incidence of around 200 per hundred thousand for a total of around 100,000 in the nation with a population near 35 million. Patients can expect a chronic worsening of symptoms, being dependent on a cane or wheelchair after 15 years, and a reduced life expectancy (3). The malady is defined by cm-sized lesions (Fig. 0.1) on the nerve fibres of the central nervous system (CNS), which must be disseminated across anatomical space and evolve throughout time (4). The lesions lack myelin* as visualized by a Luxol fast blue stain (5). Often the brainstem and spinal cord are affected, and the location of lesions will dictate the symptoms experienced: ocular disorders, unsteadiness or ataxia, or paresthesia or loss of sensation (4).
Fig. 0.1 Appearance of multiple sclerosis lesion in human white matter. a, Perivascular
infiltration of immune cells; b, Loss of myelin stained dark by Luxol Fast Blue. The plaque appears as a white area surrounding the blood vessel. From the author’s own unpublished work.
Lymphocytes infiltrate in perivascular cuffs in man, where a majority of said cells are CD8+ and a variable number are CD4+. MHC class II-positive macrophages and some few B cells are present in lesions themselves (6). Within these lesions, the myelin sheath surrounding axons is extensively eroded, which results in the observed neurological symptoms (3,4). In chronic cases, plaques become “inactive”, with most of the cellular infiltrate dispersing and leaving behind a totally demyelinated area; the cycle is then repeated at other sites in the CNS. In addition, some re-myelination can occur, and has the appearance of light myelin staining in an obviously damaged area (3). Within lesions, concomitant with demyelination, the oligodendrocytes are destroyed and microglia and astrocytes proliferate and adopt ‘reactive’ character (7-11).
MS belongs to the broader family of autoimmune demyelinating diseases (ADDs), which differ in their specific histological details, but which all share the characteristic of loss of myelin and an autoimmune component. Notable other ADDs are NMOSD (neuromyelitis optica spectrum disorder), a relapsing disease with prominent inflammation of the optic nerve and large lesions in the spinal cord (3), and ADEM (acute disseminated encephalomyelitis), a serious acute, monophasic disorder that often complicates childhood measles, chickenpox, or smallpox or vaccinations (Table 0.1)(3,4). The NMOSD autoantigen is known: the aquaporin (AQP)-4 water channel protein, a part of the dystroglycan complex (12,3). Autoantibodies targeting AQP-4, which is expressed on astrocytic end-foot processes, localize to the cerebral capillary endothelium and cause the degradation of the blood-brain barrier (BBB) (3). Importantly, antibodies collected from NMOSD-patient cerebrospinal fluid are capable of transmitting demyelination to rats (13,14), a functional proof reminiscent of Koch’s postulates (15), which furthermore seems to depend on neutrophils (16)(Fig. 0.2). Cellular infiltrates in NMOSD comprise macrophages, neutrophils and eosinophils (17). On the contrary, ADEM plaques are mainly perivascular and contain macrophages, T and B cells, and occasional plasma cells and granulocytes (3). Additionally, ADEM patients are sometimes seropositive for IgG autoantibodies towards myelin basic protein (MBP), proteolipid protein (PLP), MOG and crystallin epitopes (18). It should be noted that oligoclonal IgG in the CSF is a hallmark of MS, but the targeted antigen is not known. These two examples, along with the case of MS above, serve to highlight the diversity of clinical pictures associated with a similar demyelinating process.
Fig. 0.2 Neuromyelitis optica can be transferred to animals by injection of NMO-patient serum. a-e, disease is worsened by the injection of anti-AQP4 NMO patient serum, but not by serum from
NMO patients without anti-AQP4. Statistics tabulated at right. From (13).
The Development of Autoimmunity
For autoimmunity to develop, several conditions must be met. First, mutations in susceptibility genes are often noted in patients with familial autoimmune diseases, including MS and arthritis, and the most salient of these such mutations lie in the major histocompatibility complex* (or HLA genes) (19,20). These mutations can lead to the loss of self-tolerance, whether central (by the elimination of autoreactive T cells) or peripheral (by the action of the regulatory cells—Tregs and Bregs). Once tolerance fails, autospecific T cells can persist in the body. Upon triggering by an infection, environmental shift, injury or other immunologically pertinent change,
the autoantigen can be brought into contact with the self-reactive T cells. Due to non-functional mechanisms of tolerance, the self-antigen activates the autospecific cell. One theory, the shared epitope hypothesis, holds that the nature of the mutation is to produce an MHC that inappropriately binds self-antigen. The structure of the major MS-predisposing MHCII allele, HLA-DRB1*1501, with its antigen was solved by Wucherpfennig and colleagues in 1998 (21); yet, we are no closer to an understanding of the precise effects of genetics on MS. Mutations in virtually any step of the tolerance machinery, such as a reduction in the T-cell activation threshold or a failure in apoptosis of autoreactive cells, as well as molecular mimicry by a pathogen, could lead to autoimmunity. The final result is an immune response directed against self-proteins (22). A provocative hypothesis is that MS is caused by a lack of neonatal exposure to Epstein-Barr virus (EbV) (which would be asymptomatic and protective), but instead an infection by EbV in adolescence, leading to mononucleosis and, later on, MS; this is supported only on the basis of statistics (23). This would explain partly why the incidence of MS is inversely correlated with prevalence of infectious disease and with GDP (24,25). Bearing the above points in mind, it is important to further investigate any and all genetic effects on ADDs, using animal models if necessary, in order to chart the molecular steps that lead to disease.
Current State of Treatment
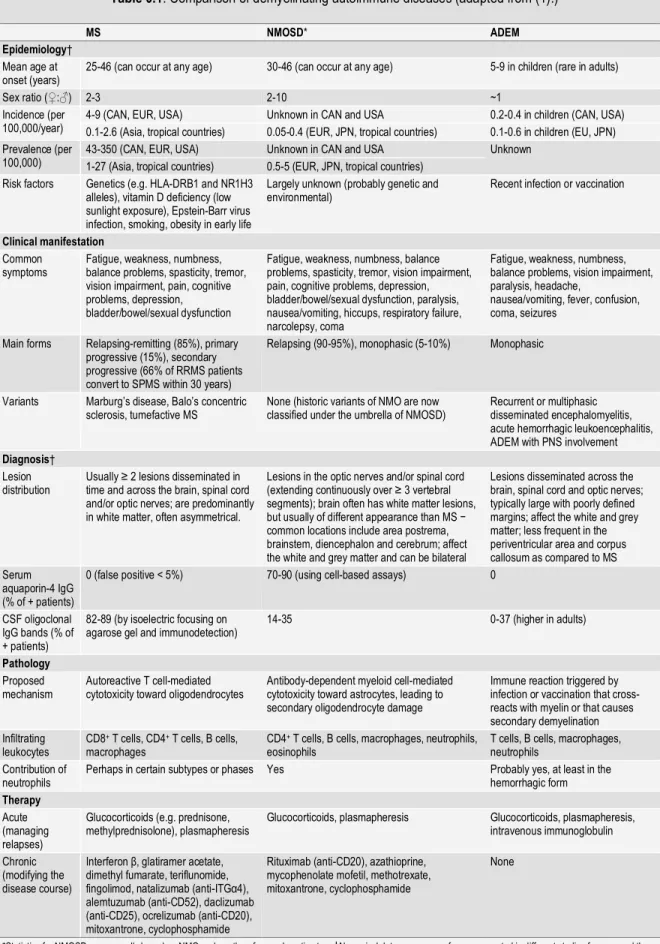
While disease-mitigating therapies are available for patients with MS and other ADDs (26-37)(Table 0.1), there is currently no cure. Desensitization with myelin basic protein was tried and yielded poor results (38,39). Some landmark therapies were the development of glatiramer (copolymer-1) in the 1990s, and more recently, the application of immunomodulatory biologics such as rituximab and ocrelizumab. In Chapter 1 we will discuss possible neutrophil-focused therapies in more detail.
Table 0.1. Comparison of demyelinating autoimmune diseases (adapted from (1).)
MS NMOSD* ADEM
Epidemiology†
Mean age at
onset (years) 25-46 (can occur at any age) 30-46 (can occur at any age) 5-9 in children (rare in adults)
Sex ratio (♀:♂) 2-3 2-10 ~1
Incidence (per
100,000/year) 4-9 (CAN, EUR, USA) 0.1-2.6 (Asia, tropical countries) Unknown in CAN and USA 0.05-0.4 (EUR, JPN, tropical countries) 0.2-0.4 in children (CAN, USA) 0.1-0.6 in children (EU, JPN) Prevalence (per
100,000) 43-350 (CAN, EUR, USA) 1-27 (Asia, tropical countries) Unknown in CAN and USA 0.5-5 (EUR, JPN, tropical countries) Unknown Risk factors Genetics (e.g. HLA-DRB1 and NR1H3
alleles), vitamin D deficiency (low sunlight exposure), Epstein-Barr virus infection, smoking, obesity in early life
Largely unknown (probably genetic and
environmental) Recent infection or vaccination
Clinical manifestation
Common
symptoms Fatigue, weakness, numbness, balance problems, spasticity, tremor, vision impairment, pain, cognitive problems, depression, bladder/bowel/sexual dysfunction
Fatigue, weakness, numbness, balance problems, spasticity, tremor, vision impairment, pain, cognitive problems, depression, bladder/bowel/sexual dysfunction, paralysis, nausea/vomiting, hiccups, respiratory failure, narcolepsy, coma
Fatigue, weakness, numbness, balance problems, vision impairment, paralysis, headache,
nausea/vomiting, fever, confusion, coma, seizures
Main forms Relapsing-remitting (85%), primary progressive (15%), secondary progressive (66% of RRMS patients convert to SPMS within 30 years)
Relapsing (90-95%), monophasic (5-10%) Monophasic
Variants Marburg’s disease, Balo’s concentric
sclerosis, tumefactive MS None (historic variants of NMO are now classified under the umbrella of NMOSD) Recurrent or multiphasic disseminated encephalomyelitis, acute hemorrhagic leukoencephalitis, ADEM with PNS involvement
Diagnosis†
Lesion
distribution Usually ≥ 2 lesions disseminated in time and across the brain, spinal cord and/or optic nerves; are predominantly in white matter, often asymmetrical.
Lesions in the optic nerves and/or spinal cord (extending continuously over ≥ 3 vertebral segments); brain often has white matter lesions, but usually of different appearance than MS − common locations include area postrema, brainstem, diencephalon and cerebrum; affect the white and grey matter and can be bilateral
Lesions disseminated across the brain, spinal cord and optic nerves; typically large with poorly defined margins; affect the white and grey matter; less frequent in the periventricular area and corpus callosum as compared to MS Serum
aquaporin-4 IgG (% of + patients)
0 (false positive < 5%) 70-90 (using cell-based assays) 0
CSF oligoclonal IgG bands (% of + patients)
82-89 (by isoelectric focusing on
agarose gel and immunodetection) 14-35 0-37 (higher in adults)
Pathology
Proposed
mechanism Autoreactive T cell-mediated cytotoxicity toward oligodendrocytes Antibody-dependent myeloid cell-mediated cytotoxicity toward astrocytes, leading to secondary oligodendrocyte damage
Immune reaction triggered by infection or vaccination that cross-reacts with myelin or that causes secondary demyelination Infiltrating
leukocytes CD8
+ T cells, CD4+ T cells, B cells,
macrophages CD4
+ T cells, B cells, macrophages, neutrophils,
eosinophils T cells, B cells, macrophages, neutrophils Contribution of
neutrophils Perhaps in certain subtypes or phases Yes Probably yes, at least in the hemorrhagic form
Therapy
Acute (managing relapses)
Glucocorticoids (e.g. prednisone,
methylprednisolone), plasmapheresis Glucocorticoids, plasmapheresis Glucocorticoids, plasmapheresis, intravenous immunoglobulin Chronic
(modifying the disease course)
Interferon β, glatiramer acetate, dimethyl fumarate, teriflunomide, fingolimod, natalizumab (anti-ITGα4), alemtuzumab (anti-CD52), daclizumab (anti-CD25), ocrelizumab (anti-CD20), mitoxantrone, cyclophosphamide
Rituximab (anti-CD20), azathioprine, mycophenolate mofetil, methotrexate, mitoxantrone, cyclophosphamide
None
*Statistics for NMOSD are generally based on NMO and are therefore underestimates. †Numerical data are ranges of means reported in different studies from around the world. Abbreviations: CAN, Canada; EUR, Europe; IgG, immunoglobulin G; JPN, Japan; PNS, peripheral nervous system; USA, United States of America.
Studying Demyelination in the Laboratory
Much of our knowledge about ADDs comes from the animal model experimental autoimmune encephalomyelitis (EAE), in which animals (chiefly rodents) are immunized with a myelin antigen or brain extract in order to reproduce the aforementioned symptoms of immune-driven demyelination and neurological deficits (40,41). This paradigm can be manipulated in three distinct strategies, depending on the mechanism step studied:
1) active EAE, induced by direct injection of myelin peptide or protein (such as MOG35-55*) with
an immunogenic adjuvant – to study antigen-mediated effects (42,43);
2) passive EAE, caused by the transfer of autospecific T cells from an active-EAE animal to an unimmunized animal – to study the encephalitogenic potential of T-cell subsets (44,42); and
3) transgenic EAE models, in which EAE is induced spontaneously by virtue of a hard-coded myelin-specific receptor – to study T-cell specificities, single-epitope immune responses, or epitope spreading, or for combining EAE to other mouse models. For example, in the 2D2 mouse, the TCR † from a MOG35-55-reactive T-cell clone was cloned and knocked in to
generate a strain with monospecific autoreactive T-cells (45); in another case, an immunoglobulin heavy chain knock-in mouse, IgHMOG, produces a B-cell repertoire with a
high-frequency of anti-MOG antibody-secreting cells and is hypersensitive to injection of MOG (46).
The pathological profile of EAE mirrors that of human ADDs, particularly ADEM (47): perivascular infiltration of immune cells including neutrophils and lymphocytes; the symptoms comprise ascending flaccid paralysis of the limbs and occasional motor incoordination (48). Since the causative agent in EAE is defined, while that of ADDs is unknown, EAE does not perfectly mimic human disease. Rather, by attempting to recreate the symptoms instead of the cause, EAE provides a reproducible test-bed in which to pick apart the immune cascades responsible for demyelination.
Fig. 0.3 Macrophages, which can serve as APCs, infiltrate the ventral median fissure of the spinal
cord in EAE. Immunohistochemistry for F4/80 antigen.
EAE in the C57BL/6 mouse is induced by injection of MOG35-55 using pertussis toxin as an adjuvant,
and since genetic manipulation in C57BL/6 mice is commonplace, this model can be used to understand the effects of gene knockouts on EAE and hence autoimmunity. As outlined above, autoimmunity is the (mis)direction of normally homeostatic immune processes towards destroying the structures of the self. As such, the processes of immunity and autoimmunity are largely conserved. Self-peptide presentation to naïve CD4+ T cells bearing cognate TCRs stimulates T-cell proliferation and, depending on the local cytokine environment, the commitment to one of two lineages, Th1 or Th17 (1). The MOG35-55 peptide is presented by MHC class II*, and
so the resulting T-cell response is (at least initially) CD4+ and class II-restricted. Th1 and Th17 cells accumulate
in the meninges and perivascular spaces of the CNS, where they re-encounter APCs† presenting the myelin antigen for which they are specific. Upon restimulating the T cell, the APC releases the pro-inflammatory factors interleukin (IL)-12 and IL-23 that engender the secretion of growth factors and chemokines and further proliferation of the autospecific T cell (49). These secreted factors mobilize classical, Ly-6Chi, monocytes from
the bone marrow to the CNS and give rise to macrophages (Fig. 0.3)(shown originally by the fact that bone-marrow chimeras bear MHC complexes of the haplotype of the donor on meningeal monocytes and macrophages in (50)).
These macrophages can initiate demyelination through phagocytosis (51) and seem to be required for EAE, since disease is blocked in Ccr2 mice that are unable to recruit macrophages to the spinal cord (52). Monocytes also give rise to CD11c+ DCs that amplify and perpetuate the immune response. Microglia, in contrast, are not replaced by hematopoietic cells, but also contribute in their own way in EAE (50). B cells aid in
* class of MHC proteins responsible for presentation of infectious and autoimmune epitopes, borne only on immune cells. † antigen-presenting cells: canonically, dendritic cells (DCs), B lymphocytes or macrophages, bearing MHC class II complexes.
the generation of autospecific Th cells and promote their migration into the CNS; however, B cells also suppress
the early stages of EAE by secreting anti-inflammatory interleukin-10 (53).
Fig. 0.4 Neutrophils infiltrate the spinal cord and brain in EAE. a, b, neutrophils stained for Ly6G
or marked with fluorescent reporter infiltrate the median fissure of the spinal cord (a) and brain ventricles (b) during EAE. c, neutrophils marked for Ly6G infiltrate the spinal cord beginning at day 7 post-immunization (d.p.i.) and are abundant by day 14. d, e, early observations of neutrophils in EAE; anti-Gr-1. f, g, neutrophils are abundant in the onset and peak phases of EAE and decline afterwards. From (54), (55), (56), and author’s own observations.
Neutrophils are Essential for Demyelination
Notably, neutrophils are abundantly present in CNS lesions in EAE (Fig. 0.4). These short-lived innate immune effector cells originate from the myeloid lineage and are stockpiled in great numbers in the bone marrow in preparation for infection or injury (57,58). Neutrophils are the first cellular line of defense towards damage to the body, and are mobilized both to destroy pathogens and to repair organs after sterile insult (59). Neutrophils’ presence in EAE has been known from the investigations of Hoenig (60) with dyestuffs, of Määttä (55) with the anti-Gr-1 antibody RB8-6A5, and of Segal (61) using the more specific anti-Ly6G 1A8 antibody. More details on what is known of neutrophils in EAE are given in Chapter 1.
An important study showed that anticipatory depletion of neutrophils using anti-Ly6G could suppress the symptoms of EAE modestly to almost totally, depending on the dose (62) (Fig. 0.5). They found that neither myeloperoxidase nor elastase were responsible for the observed function, but hypothesized neutrophils influenced the local environment to stimulate the expression of MHCII and co-stimulatory molecules on macrophages and microglia. This finding has been reproduced independently (63,64,54). Work from Segal and colleagues has shown correlative evidence for neutrophil-derived and -activating factors in the blood and cerebrospinal fluid (CSF) of MS patients (65); yet the relevance of these observations on human disease is not
Fig. 0.5 Depletion of neutrophils reduces EAE severity. a, depletion with anti-Gr-1; bcdef,
depletion with anti-Ly6G; g, h, inhibition of CXCR2 has an effect reminiscent of neutrophil depletion. From (63), (62), (54), (64), and (61).
To address this lack of knowledge into neutrophils’ exact functions, we performed a study on the cell-surface characteristics, transcriptional landscape, anatomical distribution, and cellular interactions of neutrophils in EAE (Chapter 2). We found that neutrophils that enter the CNS parenchyma during active MOG35-55-induced
EAE are distinguished by their high surface expression of ICAM1*. Transcriptomic analysis suggested that neutrophils expressing ICAM1 are substantially different from those that do not, with ICAM1+ neutrophils more resembling macrophages and DCs. Various aspects of the transcriptomic data were tested in vitro and in vivo using knockout mice. Finally, a novel model for EAE that more accurately mimics MS was developed: the bMOG model, a collaboration with Dr. Steven Kerfoot and colleagues at the University of Western Ontario (Chapter 3). Study of this model revealed the protease ASPRV1 as a neutrophil effector molecule, previously uncharacterized in the immune system, that contributes to the development of chronic inflammation in EAE, and so possibly in human ADDs.
Chapter 1:
Neutrophils in ADDs and EAE:
Current State of Knowledge
Adapted in part from ‘Neutrophil perversion in demyelinating autoimmune diseases: mechanisms to medicine’ (1)
Résumé : Les neutrophiles sont essentiels pour la santé et l’unité du corps, mais constituent un danger si mal contrôlé. La perversion de neutrophiles à une fonction autre que celle de l’homéostasie est bien documenté dans plusieurs maladies autoimmunes, telles le lupus, le psoriasis et l’arthrite, mais peu connu dans le cadre de la démyélinisation. Grâce au modèle animal encéphalomyélite autoimmune expérimentale (EAE), certaines molécules qui déterminent l’invasion de neutrophiles dans le système nerveux central ont été identifiées. Des mécanismes par lesquels les neutrophiles pourraient empirer la démyélinisation ont également été proposés. Chez l’humain, les neutrophiles sont abondantes dans le système nerveux dans des cas de neuromyélite optique et d’autres maladies démyélinisantes sévères; cependant, l’évidence concrète pour la présence de neutrophiles dans la sclérose en plaques manque. En dépit de ce fait, des molécules de recrutement et d’activation de neutrophiles (e.g. CXCL1, CXCL5, élastase) restent des indicateurs de sévérité pour la sclérose en plaques. L’effet des traitements courants sur les neutrophiles est discuté.
Neutrophils are essential to a healthy life, yet pose a threat if improperly controlled. Neutrophil perversion is well documented in a variety of inflammatory disorders (e.g. arthritis, lupus, psoriasis), but is only beginning to be demystified in autoimmune demyelination, the most common cause of neurological disability in young adults. Using the animal model experimental autoimmune encephalomyelitis (EAE), several molecules that help neutrophils invade the central nervous system (CNS) have been identified. Mechanisms by which neutrophils may contribute to demyelination have also been proposed (e.g. secretion of endothelial/leukocytic modulators, antigen presentation to T cells, myelin degradation and phagocytosis). In human, neutrophils are seen in the CNS of people with neuromyelitis optica spectrum disorder and other severe variants of autoimmune demyelinating diseases. At the time of autopsy for multiple sclerosis (MS) — often many years after its onset — neutrophils appear to have escaped the scene of the crime. However, new clues implicate neutrophils in MS relapses and progression. This warrants further investigating 1) the differential importance of neutrophils among demyelinating diseases, 2) the largely unknown effects of current MS therapies on neutrophils, and 3) the potential of neutrophil proteins as clinical biomarkers or therapeutic targets.
Studies on injury and infection tell us that neutrophils quickly invade the affected tissues to execute different functions (e.g. phagocytosis, degranulation, production of reactive oxygen species, extracellular trap formation, antigen presentation) (58). Is the same true in the context of ADDs? In this chapter, we take a comprehensive look at how neutrophils gain access to the CNS and contribute to demyelination in EAE. We highlight the importance of neutrophils in human diseases, such as MS and NMOSD, and propose clinical directions. Finally, we outline future challenges in this emerging area of research.
The mechanism of neutrophil recruitment in EAE
Under normal physiological conditions, neutrophils, like most other leukocytes, are excluded from the CNS parenchyma* by the blood-brain barrier (BBB) (reviewed in (66)), a functional obstacle of specialized endothelial cells interconnected by tight junctions (67). Despite, the CNS vasculature is constantly patrolled by leukocytes that crawl on its luminal surface. These sentinel cells were discovered in 2003 (68) by transplantation of GFP-tracked blood cells into lethally irradiated mice and were later found to develop into perivascular macrophages under inflammatory stress (69). These cells comprise 25% neutrophils (CD11b+Ly6G+) and 75% monocytes (CD11b+Ly6G−) (70), expand under the influence of TNF-α, interleukin-1β, and angiopoietin-2, and are rod-shaped when patrolling through capillaries. Indeed, their movement on the endothelium is driven by a leading edge (where actin polymerizes to push the cell front forward) and by a uropod (where microtubules reorganize to allow retraction of the rear edge). While some rod-cells cross the BBB to replenish perivascular macrophage pools, neutrophils do not reside in the healthy CNS.
In EAE mice (which, the reader will recall, are mice injected with myelin antigen and adjuvants), or in mice treated with pertussis toxin (PTX), rod-shaped neutrophils expand at least four-fold in the brain circulation (70). In contrast, the infiltration of neutrophils into the CNS parenchyma is a more specific event that occurs in EAE, but not in response to toxin alone (as will be clear from the experiments in Chapter 2), nor to an irrelevant antigen, nucleocapsid (61) (and see also (56,55)). These neutrophils appear in meningeal and perivascular inflammatory foci shortly before the onset of clinical symptoms, and at the peak of disease are observed in the CNS parenchyma within areas of demyelination and axonal damage (Fig. 0.4, Fig. 1.1a). CNS infiltration of neutrophils (and expression of neutrophil chemokines) is also observed in passive EAE, but only when transferring Th17 cells and not Th1 cells (61) (71)(Fig. 1.1bd). In other words, neutrophils can be engaged during
certain forms of EAE depending on the nature of the effector T cells (and thus the overall character of the response).
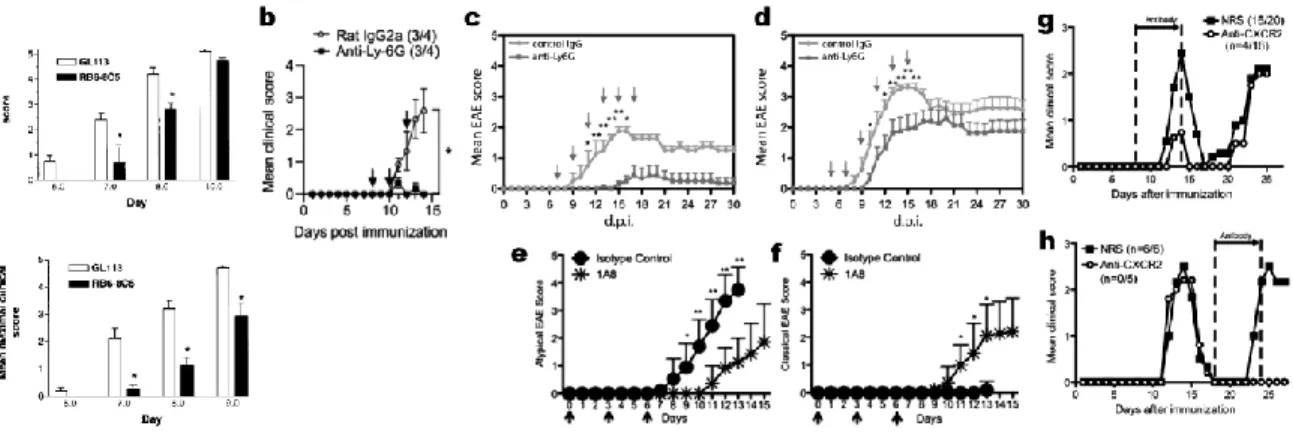
Fig. 1.1 (next page) Neutrophils expand during EAE and their recruitment depends on Th17 cells and Cxcr2. a, Ly6G-expressing neutrophils expand during EAE; b, Passive transfer of Th17
cells induces a neutrophil-centric EAE; c, EAE is absent in Cxcr2 mice and is restored by transfer of
Cxcr2+ neutrophils; d, Passive transfer of Th17 cells results in upregulation of the neutrophil chemokines CXCL1 and CXCL2; e, the blood-brain barrier is permeabilized in EAE, but not when neutrophils are pre-emptively depleted using anti-Ly6G. From (61) and (71).
* The grey and white matter substance of the CNS, as opposed to the connective-tissue meninges, which overlie it. The parenchyma is on the other side of the BBB.
While the mechanism of neutrophil recruitment has been more extensively studied in peripheral tissues and ex vivo models, it appears to be similar in the CNS. As illustrated in Figure 1.2 and reviewed elsewhere (72), neutrophil recruitment includes the following general steps: 1) under the influence of pro-inflammatory cytokines, neutrophils emigrate from the bone marrow, while endothelial cells up-regulate the expression of chemokines and adhesion molecules; 2) these chemokines support the conversion of integrins to a high-affinity state allowing the anchorage of neutrophils to the endothelium; 3) these adherent neutrophils adopt a polarized shape and monitor the extracellular milieu as they crawl along the luminal wall; and 4) specific guiding cues signal neutrophils to cross the endothelium towards the parenchyma. Molecules in each of these important steps have been identified in the context of CNS inflammation, and are discussed below.
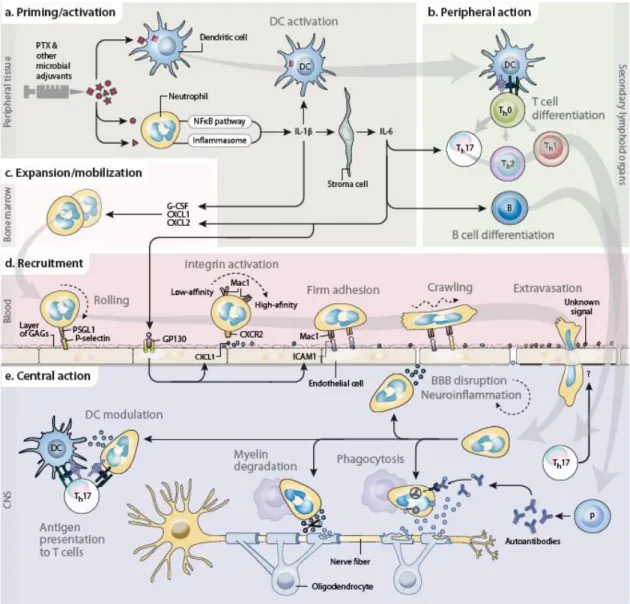
Fig. 1.2 Recruitment and functions of neutrophils in EAE: a working model. a, Neutrophils
sense PTX — and the other bacterial adjuvants used for immunization — through pattern-recognition receptors. This leads to the production of IL-1β, which triggers a systemic inflammatory cascade involving stromal cell-derived IL-6. At the same time, DCs take up myelin peptides and are activated by the adjuvants and neutrophil-derived IL-1β. b, Myelin-antigen-exposed DCs migrate to secondary lymphoid organs to activate T cells; the direct and indirect production of IL-1β and IL-6 by neutrophils favors the differentiation of Th0 cells into Th17 and Tfh cells. IL-6 also induces the differentiation of B cells. c, G-CSF and ELR+ chemokines (e.g. CXCL1 and CXCL2) — generated by the inflammatory cascade downstream of adjuvant-activated neutrophils and differentiated T cells — stimulate the expansion and mobilization of pools of neutrophils harbored in the bone marrow. d, Neutrophils roll on the cerebral vascular luminal wall by selectin-selectin receptor interaction. IL-6-activated endothelium allows neutrophil firm adhesion through ELR+ chemokines and adhesion molecules (e.g. ICAM1, Mac1). Thereafter, neutrophils crawl and can leave the blood through diapedesis in response to an unknown (Th17-derived) signal. e, Under the control of T cells and together with other myeloid cells (macrophages, DCs), infiltrating neutrophils participate in inflammation and demyelination by exerting immunoregulatory and effector functions (e.g. secretion of molecules disrupting the BBB and modulating other leukocytes, presentation of myelin antigens to T cells, and myelin degradation and phagocytosis via antibody-dependent and -independent mechanisms). Abbreviations: B, B cells; FcγR, Fc-gamma receptor; GAGs, glycosaminoglycans; Mac, macrophage; P, plasma cells; SELPLG, selectin P ligand. From (1).
Expansion and mobilization
Born from stem cells in the red bone marrow, neutrophils are retained there through the CXCL12−CXCR4 signaling pathway. They egress into the bloodstream when stimulated by CXCL1 and CXCL2, the principal ELR motif* chemokines (73,74), acting through CXCR2. Upon inflammation, the production of neutrophils must be scaled up to meet the higher demand for these short-lived cells, a response referred to as emergency granulopoiesis (75). The granulocyte colony-stimulating factor (G-CSF or CSF3) plays a central role in this process by downregulating CXCL12 and CXCR4 while upregulating CXCL1, CXCL2 and CXCR2 (76,77). G-CSF also augments granulopoiesis by stimulating the proliferation and differentiation of neutrophil precursors (75). G-CSF is produced by brain endothelium in response to IL-1β (78).
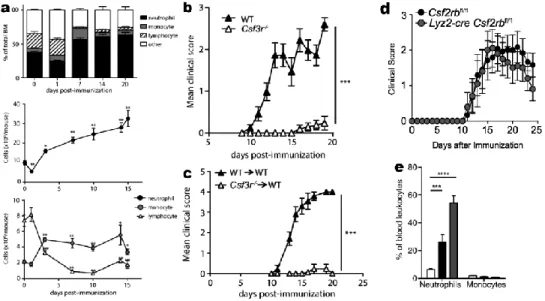
In agreement, neutrophils expand in the bone marrow and accumulate in peripheral hematopoietic compartments (blood, spleen) during the presymptomatic phase of active EAE (65)(Figs. 1.1a, 1.3a). This response is induced via the release of G-CSF and CXCL1 into the blood, since it is blocked in Csf3r mice (Fig. 1.3bc) and by anti-CXCR2 (Fig. 1.1ce). The related cytokine GM-CSF is not involved since even neutrophil-specific Csf2rb knockout does not alter EAE (79)(Fig. 1.3d). Furthermore, adoptively transferred Th17 cells
increase the number of neutrophils in the blood (65)(Fig. 1.3e), providing convincing evidence that neutrophils are mobilized not only in response to adjuvants, but secondary to the presence of autoreactive T cells.
Fig. 1.3 The effects of G-CSF in EAE—literature review. a, The bone marrow produces neutrophils
in EAE; b, c, EAE is absent in Csf3r mice or Csf3r-to-wild-type bone marrow chimeras; d, EAE does not depend on the GM-CSF receptor, since EAE develops normally in Csf2rb mice; e, Neutrophils are increased in the blood when EAE is induced by passive transfer of Th17 cells (grey), but less so when induced by transfer of Th1 cells (black); compared with no injection (white). From (65) and (79).
As mentioned above, G-CSF is produced by brain endothelium upon exposure to IL-1β in culture; it is also detected in the meninges ten days after MOG or adjuvant injection (80). Since G-CSF production is downstream of T-cell activation, the T-cell factors that regulate G-CSF are also important to study. The Th17
cytokine IL-17 can mobilize neutrophils in EAE (71) and stimulates tissue cells to produce neutrophil-active chemokines (57). It promotes neutrophil mobilization via CSF and CXCR2 ligands in other contexts. The G-CSFR is expressed on myeloid progenitor cells, neutrophils, platelets, monocytes, and some nonhematopoietic cell types, including endothelium (81). EAE depends on G-CSFR in hematopoietic cells, since WT mice with Csf3r bone marrow do not develop EAE (65)(Fig. 1.3c). Since the opposite chimeras were not tested, nonhematopoietic cells (e.g. microglia) may also respond to G-CSF. It remains to be seen whether neutrophil mobilization in EAE is modulated by other cytokines such IL-6, which is expressed in EAE and can synergize with G-CSF in other circumstances (69,70,82).
Rolling, adhesion and crawling
To access sites of inflammation, circulating neutrophils must first attach to the vasculature. This is normally prevented by the glycocalyx, a layer of glycoproteins and polysaccharides that coats the luminal endothelial surface and masks potential binding sites. Under the action of inflammatory mediators (e.g. IL-1β, TNF, IL-6, histamine), this protective layer is partially shed by proteases (e.g. heparanase), while endothelial cells produce transmembrane adhesion proteins (e.g. E-selectin, ICAM1, VCAM1) and chemoattractants (e.g. CXCL1, CXCL2). The latter can be released into the circulation or retained on the thinned glycocalyx by binding to heparan sulphate proteoglycans (72). Consequently, circulating neutrophils roll on the endothelium in a stop-and-go manner through the glycoprotein selectin P ligand (SELPLG), which binds transiently and with low affinity to endothelial selectins. This rolling motion slows down the flow of neutrophils, giving them more time to interact with glycocalyx-bound chemokines via CXCR2 accumulated at the leading edge (83). The CXCR2 signaling pathway, among other neutrophil-specific signals, then stimulates heterodimeric integrins (e.g. Mac-1, LFA-1) to adopt an active conformational state that has high affinity for the corresponding ligands (e.g. ICAM1, VCAM1). This allows neutrophils to firmly adhere to the vasculature, and subsequently spread and crawl along the chemokine gradient (84). This process is promoted by collisions of platelets with the neutrophil’s uropod (83).
Evidence to date suggests that the recruitment of neutrophils follows the same general principles in the CNS as in the periphery, although few studies have directly demonstrated the specific nature and dynamics of the molecules involved in EAE. The increased number of crawling neutrophils in the CNS of EAE mice can be attributed to two causes: the microbial adjuvants used for immunization (Freund’s adjuvant, PTX) and the antigen-specific T cell response within the CNS (Fig. 1.2). In the periphery, the adjuvants are detected by immune and non-immune cells through membrane-bound and cytoplasmic pathogen recognition molecules (e.g. Toll-like and Nod-like receptors). The signaling pathways of these receptors converge to induce the transcription
factor NF-κB, which drives the expression of inflammatory mediators, notably IL-1β. The latter is a master regulator that triggers a systemic molecular cascade, leading to increased neutrophil patrolling. This recruitment can be further enhanced by IL-1β produced locally in the CNS by different cells after reactivation of myelin-specific T cells (78).
The bacterial toxin PTX from B. pertussis is commonly used as an immunogenic adjuvant to potentiate EAE and can be considered a simplified system for studying neutrophil activation and recruitment. PTX’s ADP-ribosyltransferase activity modifies α subunits of G proteins, leading to recognition via TLR4. Myeloid-cell activation of TLR4 occasions the synthesis of IL-1β under an inactive form (85). In parallel, PTX activates, through GTPase modifications, the inflammasome sensor pyrin, which in turn induces the activation of caspase-1 and the cleavage of pro-IL-caspase-1β (85,86). Now in its active form, IL-caspase-1β stimulates nearby stromal cells to release IL-6, which circulates to induce expression of ICAM1 and CXCL1 on the cerebral endothelium (70,82). Neutrophils can then roll on the endothelium via P-selectin and adhere via CXCR2 and Mac-1 (discussed above). In sum, upon activation in peripheral tissues, the innate immune system can induce neutrophil crawling at distant sites, including the CNS vasculature, via a pyrin−IL-1β−IL-6 cascade (Fig. 1.2).
Alternative mechanisms have also been found to induce the recruitment of crawling neutrophils in the CNS vasculature. In the presence of bacterial lipopolysaccharide (LPS), neutrophil adhesion is not mediated by IL-6, but rather involves a direct action of IL-1β and TNF on the endothelium (69). LPS also induces the synthesis of angiopoietin-2 (69), which potentiates the action of IL-1β and TNF on the endothelium. Angiopoietin-2 acts by antagonizing the inhibitory effect of its receptor, Tie2, on the NF-κB pathway.
Other alternative mechanisms of neutrophil recruitment include as a feature the engagement of diverse pathways leading to IL-1β processing. Besides pyrin, four other sensors have been established so far to form functional inflammasomes, each responding to a variety of stimuli (87) (88). The Freund’s adjuvant used to induce EAE is a mixture of bacterial components comprising such stimuli. IL-1β can also be processed by inflammasome-independent mechanisms; it is therefore not surprising that EAE is not attenuated (or only partially (89)) in mice lacking inflammasome sensors. Further research is required to elucidate the complex mechanism of neutrophil recruitment in EAE. Special emphasis should perhaps be placed on convergence points among the various inflammatory pathways, which may represent more optimal therapeutic targets.
Extravasation
During intraluminal crawling, neutrophils screen the vascular surface for signs of inflammation. From there, they can leave the circulation by a process termed extravasation. Two routes have been described: paracellular (between endothelial cells) and transcellular (through an endothelial cell). Extravasation requires an
cadherin, Mac1, VLA4) and cytoplasmic proteins (e.g. LSP1, cortactin, GTPases, kinases) (90,91). Very little research has investigated this process for neutrophil entrance into the CNS. Ultrastructural studies on cultured cerebral endothelial cells support the notion that neutrophils primarily take a transcellular route to penetrate the BBB (92,93). Future therapies should aim to address the following two questions:
1) does CNS extravasation involve BBB-specific molecules; and
2) what signals inform neutrophils where to extravasate during EAE?
One could expect that these signals include CXCR2 ligands; however, these ligands probably influence extravasation indirectly by acting on earlier steps (mobilization, adhesion, crawling). This is supported by the observation that CXCR2 ligands are abundantly produced by cerebral endothelial cells under other types of inflammatory conditions (e.g. endotoxemia) in which neutrophils do not infiltrate the brain (82). Alternatively, it is possible that neutrophils mainly extravasate in BBB-devoid regions (e.g. the meninges), from where they could cross the parenchyma-limiting membrane (glia limitans). This would be consistent with the fact that neutrophils are mainly found in submeningeal inflammatory foci at the peak of EAE (56,80).
Are Neutrophils Essential for EAE?
Are neutrophils a necessary condition for EAE? The literature so far indicates “yes”. Neutrophils have long been overlooked in the field of autoimmunity, possibly since functional studies have been hampered by the paucity of tools for neutrophil-specific identification and manipulation. However, recent observations demonstrate the critical importance of neutrophils in active EAE: administration of anti-Ly6G or anti-Gr1 antibody impedes EAE development (Fig. 0.5). This effect is due to the elimination of neutrophils by antibody-dependent phagocytosis in macrophages (94,95) and not to a loss of function of Ly6G, since Ly6G-deficient mice normally develop EAE (96). Interfering with the mechanism of neutrophil recruitment at any level blocks or attenuates EAE. This is seen with inhibition of CXCR2 (61,65,64,97), CXCL1 (82,65), G-CSF and G-CSFR (65), IL-1R (98), pyrin (85) and ceramide synthase 2 (99), and the resistance of Cxcr2 mice to EAE can be overcome by transfer of Cxcr2+ neutrophils (Fig. 1.1c). Furthermore, EAE is exacerbated by factors that positively influence neutrophil
recruitment and action, such as a deficiency in ceramide synthase 6 (100) and administration of G-CSF between immunization and disease onset (101). Overall, these studies demonstrate the harmful nature of neutrophils in EAE. Although the underlying mechanism is still obscure, four non-mutually exclusive hypotheses are described below.
BBB disruption
The BBB is temporarily altered in EAE, allowing the intrusion of leukocytes and blood-borne molecules (e.g. immunoglobulin, fibrinogen, and test materials such as fluorescent tracers, Evans blue dye or gadolinium)
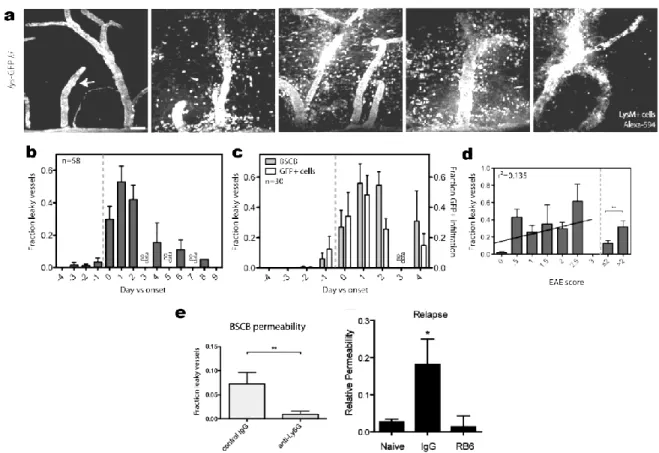
into the CNS (Fig. 1.4) (102). This likely results from the breakdown of endothelial tight junctions and substrate-specific transporters as well as the degradation of the vascular wall matrix and basement membrane (103). While several mechanistic explanations have been proposed, recent studies suggest that neutrophils are the main culprit. As one of the first responders in EAE, neutrophils appear in the CNS shortly before the emergence of clinical symptoms and concomitantly with BBB disruption (61,98) (Figs. 0.4, 1.1, 1.4). Further, depleting neutrophils in circulation mitigates both BBB permeability and clinical severity of EAE (61,98) (Figs. 0.5 and 1.4). In theory, neutrophils could contribute to BBB disruption via contact-dependent mechanisms and by releasing a variety of molecules, including cytokines (IL-1β, TNF, IL-6, IL-23), free radicals (nitric oxide, reactive oxygen species) and proteases (e.g. MMP8). However, while neutrophils are known to express these molecules in EAE, a functional experiment has yet not been performed. As BBB disruption can also occur in EAE models without neutrophil infiltration, it seems that neutrophils are not the only ones with the key to unlock the BBB.
Fig. 1.4 BBB destruction in EAE and its rescue by depleting neutrophils. a, The BBB is
permeabilized (as judged by release of blood Alexa-594 into parenchyma) starting ~1 day before onset (panel 2) and concomitantly with LysM+ neutrophil recruitment. Panels: 2 days before disease onset, 1 day before onset, day of onset, 2 days after onset and 4 days after onset. b-d, Quantification of leakiness of the BBB to Alexa-594 (b); to Alexa-594 and to cells (c); and its correlation with EAE score (d). e, BBB permeability is inhibited by depletion of neutrophils with Ly6G (left) or with anti-Gr-1 (right). From (98) and (61).
Immunomodulation
To mount coordinated attacks, immune cells must communicate with each other through contact-dependent mechanisms and extracellular chemical messengers (e.g. interleukins, chemokines, growth factors, lipid mediators, alarmins). Accordingly, neutrophils have been shown to interact with DCs, T cells and B cells in different experimental settings (104-107). In EAE, a recent study presents neutrophils as stimulators of DCs (62), which is in agreement with their overall deleterious effect. The authors found that neutrophil depletion does not affect the peripheral priming and CNS infiltration of T cells, but reduces the number and activation of CD11c+ cells (62). Providing more precise support that neutrophils promote APC maturation, these authors found that neutrophils isolated from the CNS of EAE mice could stimulate the expression of MHCII and co-stimulatory molecules (CD80, CD86) on in vitro-differentiated bone marrow-derived DCs. Using a transwell assay, it was demonstrated that this effect is mediated by a yet unknown soluble mediator that is not IL-6, IL-12, IFNγ or a reactive oxygen species (ROS) (62).
The deleterious interaction of neutrophils with DCs does not exclude the possibility that neutrophils interact with other immune cells, nor that they have immunosuppressive effects. For example, neutrophils, upon stimulation with G-CSF, release TNF ligand superfamily member 13b (BAFF), a molecule implicated in B cell proliferation and maturation (108,109). As B cells contribute to MS and NMOSD (110), it would be relevant to investigate the possible interaction of neutrophils with B cells using appropriate EAE models, in which B cells are pathogenic. Furthermore, neutrophils, under the influence of IFNγ, suppress myelin-reactive T cell proliferation via the production of nitric oxide (NO) (111). Although this effect may seem contradictory to the deleterious nature of neutrophils in EAE, it may be part of a negative feedback loop that might coordinate the actions of T cells and neutrophils. This would explain why EAE is more severe in mice lacking IFNγ, IFNγR or NO synthase (NOS2 or iNOS) (111). Another study has shown that adoptive transfer of spleen Ly6G+ neutrophils (referred to as granulocytic myeloid-derived suppressor cells) attenuates EAE by inhibiting T cell priming in lymph nodes via PD-L1 (112). However, the question of why exogenous splenic neutrophils behave differently than endogenous CNS-infiltrating neutrophils remains unanswered. It is possible that splenic neutrophils have an immature phenotype or that the act of transferring them modifies their fate. In sum, there is compelling evidence that neutrophils exert immunomodulatory functions in EAE. This provides hope that manipulating these functions with specificity may influence the course of human demyelinating diseases.
Antigen presentation
Presentation of antigens to Th cells by APCs via MHCII molecules is a critical step in adaptive immunity.
It is therefore not surprising that MHCII alleles are the strongest genetic risk factors for MS (113). First, the process involves the uptake of extracellular proteins by APCs and their processing into short peptides (14-20 amino acids), which are then loaded onto MHCII molecules. These steps are mediated by proteases (e.g.
cathepsins, legumain, IFI30) and chaperones (e.g. CD74 and H2-DM) in a series of endosomal compartments (57). Second, the APCs must establish physical contacts with Th cells to form immunological synapses. At these
structures, cell-surface receptors and signaling components gather to generate intracellular signals that dictate cytoskeletal and transcriptional changes, leading to T cell activation. Monocyte-derived CD11c+ DCs are considered the main APCs in EAE (discussed in Introduction). However, a recent study challenged this view by showing that depletion of DCs interferes with immune tolerance and aggravates EAE (114), and DCs are not always required for the development of Th responses and can be replaced by other APCs.
An interesting possibility is that neutrophils serve as alternative APCs in EAE or contribute to epitope spreading, whereby distinct epitopes become new targets in an ongoing immune response (115). Many studies suggest that neutrophils can present antigens to T cells (116). While not equipped for this in steady state conditions, neutrophils can express MHCII and co-stimulatory molecules during inflammation (62,117-120) or when cultured with cytokines such as GM-CSF or IFNγ (117,118,120-129). Functional studies have shown that neutrophils can process and present antigens to prime Th cells (119,127,130,131). Others have shown that
neutrophils can also cross-present exogenous antigens to CD8+ cytotoxic T cells (120,132,133). Chapter 2 will deal more explicitly with a characterization, and an exciting finding, of MHC class II on neutrophils. The acquisition of antigen-presenting capacity by neutrophils may well be a missing link between innate and adaptive immune responses that causes or influences autoimmune disorders.
Myelin degradation and phagocytosis
A widely held concept is that demyelination in EAE results from the action of myeloid cells, such as macrophages under the command of Th cells. The mechanistic details are unknown, but the final stage is
ostensibly phagocytosis of oligodendrocyte remains. A recent study has sparked renewed interest in this process by reporting that monocyte-derived macrophages, in the CNS of EAE mice, exhibit an inflammatory phenotype and initiate demyelination, often at nodes of Ranvier (51). This conclusion is based, in part, on 3D ultrastructural images showing macrophages engulfing myelin debris. Interestingly, this study has also provided a few images of neutrophils phagocytosing myelin. Another study has reported that EAE develops in Ccr2 mice, even though CNS-infiltrating macrophages are largely replaced by neutrophils (134,135). It seems possible that neutrophils and macrophages act similarly to degrade myelin in an antibody-dependent or independent manner. Phagocytosis is an established function of neutrophils that deserves to be scrutinized in EAE in terms of molecular mechanism and pathophysiological significance.
How do neutrophils contribute to human ADDs?
been directly observed in the CSF (136,137) and CNS lesions (136,17,138,16). Furthermore, injecting autoantibodies extracted from NMOSD patients into the CNS of rodents causes neutrophil infiltration alongside demyelination (Fig. 0.1). It has been proposed that neutrophils contribute to NMOSD through antibody-dependent cell-mediated cytotoxicity (ADCC) (139), but this remains to be proven experimentally and does not exclude other mechanisms. For MS and ADEM, direct evidence of CNS neutrophil infiltration has been limited to rare variants of these illnesses that are severe and often fatal (i.e. Marburg's disease and acute hemorrhagic leukoencephalitis (17)). However, recent studies suggest that neutrophils play a role even in the more prevalent forms of these diseases.
As explained in the Introduction, MS is characterized by acute inflammatory brain lesions, and often first presents with a relapsing-remitting course (RRMS), such that lesions coincide with focal neurological symptoms that subsequently improve. There are also the primary and secondary progressive forms of MS (PPMS and SPMS, respectively), in which irreversible neurodegeneration is thought to be the driving force behind clinical disability. In these main forms, direct evidence for CNS neutrophil infiltration has generally not been found (17), but this does not necessarily exclude a role for neutrophils in MS. It may be that available tissue samples from autopsies of people with this chronic, multiphase disease do not capture the transient role neutrophils could have had in the initial formation of CNS lesions. Another possibility is that neutrophils have been missed due to their plastic nature, allowing them to adopt macrophage and DC characteristics under inflammatory conditions (116). This may make neutrophils difficult to distinguish from other myeloid cells using conventional histological methods, especially given that there is no known neutrophil-specific marker for humans. A further possibility is that, in MS, neutrophils have their effect mainly in the periphery, rather than directly within the CNS. For example, by producing inflammatory mediators in response to innate immune stimuli, they may reduce the activation threshold for autoreactive T and B cells in lymphoid organs (140). All things considered, it is important to look beyond histopathology when studying the possible contribution of neutrophils in MS.
Several lines of indirect evidence suggest that neutrophils play a role in human multiple sclerosis. First, the levels of many neutrophil-related molecules (G-CSF, CXCL1, CXCL8, CXCL5, neutrophil elastase) increase in the brain (141), CSF (142) and blood (65,143,144,145) of MS patients, although probably less than in NMOSD (146,147) and ADEM patients (148). At least some of these markers (CXCL1, CXCL5, neutrophil elastase) positively correlate with neurological disability and radiological measures of lesion burden and activity (65). One study even reports higher blood levels of CXCL1 and neutrophil elastase over the course of SPMS compared to that of RRMS (145). Second, neutrophils extracted from the blood of MS patients are more primed (i.e. potentiated and ready to fully respond to further stimulation). This was evidenced by increases in their expression of inflammatory markers, resistance to apoptosis, and propensity for degranulation, oxidative burst and extracellular trap formation (149,150,151). Third, a recent study found that neutrophils are present in the CSF of
MS patients during relapse at an early stage of the disease, and correlate with the levels of IL-17A (152), a cytokine that supports neutrophil activation and recruitment. This is in agreement with other studies that also report evidence of increased neutrophil activity during relapse relative to the stable remission phases of MS. Fourth, gain-of-function mutations in Mefv, the gene encoding pyrin, an inflammasome predominantly expressed in neutrophils and mediating the effect of PTX on EAE (85), are associated with a higher susceptibility of developing a more progressive or severe form of MS (153,154,155,156). Finally, and not least importantly, administration of the neutrophil growth factor G-CSF exacerbates RRMS (157,158,159,160), as has also been observed in NMOSD (159). Collectively, these observations could be unified by the hypothesis that neutrophil activity is a factor that influences MS development and severity.
Clinical Potential for Neutrophil-Related Biomarkers
The human-based literature summarized above brings hope that neutrophils and associated proteins may serve as clinically useful biomarkers. At present, some paraclinical tools are used to aid differential diagnosis and management of demyelinating autoimmune diseases (Table 0.1). For example, magnetic resonance imaging (MRI) is the gold standard to determine the spatiotemporal pattern of demyelination (161,162), while serological testing for the anti-aquaporin-4 antibody is important to definitively distinguish NMOSD from MS (162,163). These tools are nevertheless far from perfect. For example, the specificity and sensitivity vary greatly according to the serological test used (163), which could lead to a delay in diagnosis or misdiagnosis. Furthermore, there is no reliable biomarker to identify MS subtypes and predict clinical course (e.g. relapses, conversion of clinically isolated syndrome to RRMS or of RRMS to SPMS). We have also limited means to predict whether a patient will respond to a given pharmacological therapy or have an adverse reaction to the drug.
While there is a need for additional biomarkers, we are currently several steps away from translating emerging theory on neutrophils into clinically useful tools. To assess the potential of neutrophil-related markers, larger-scale studies are required to confirm the previously observed differences among MS subtypes and the other autoimmune demyelinating diseases. If the presence of neutrophils in the CNS is only confirmed in certain subsets of MS patients, this may provide an opportunity to classify patients in ways that have important implications for clinical care. Furthermore, it might be important to assess the impact of factors such as age, sex, disease duration and medications on the prospective biomarkers, as these factors have not always been accounted for in prior investigations. Controlled interventional studies are also needed to determine if neutrophil markers could be useful to predict individual treatment response. We also must assess the added value of such biomarkers compared to those currently available or in the development pipeline.
Next Steps
The sum of the research over the last decade demonstrates the importance of neutrophils not only in the animal model EAE, but also in human ADDs. Direct evidence of neutrophil perversion has primarily been found for severe forms of diseases, including NMOSD and rare variants of MS. Indirect evidence supports the hypothesis that neutrophils participate, or at least exert an influence, in common forms of MS. The involvement of neutrophils in ADEM also seems likely, but remains to be firmly established.
Neutrophil neutralization thus emerges as a logical and potentially feasible goal, at least for NMOSD (Box 1.1), and should be achieved preferably using disease-modifying therapies (DMTs) that would modulate neutrophil activity with specificity, since wholesale depletion of these key players of innate immunity is too hazardous. The little we know of the currently available DMTs for MS and NMOSD suggests that they affect neutrophils in a partial and/or non-specific manner. Neutrophils should be more closely examined in future clinical trials to better understand their relevance to treatment effect. Moreover, neutrophil-specific drugs, like sivelestat, should be developed and tested.
We anticipate that neutrophil-related molecules will not offer a significant advantage over the established biomarker anti-aquaporin-4 antibody in distinguishing NMOSD from MS. Rather, neutrophils could prove useful to identify variants or stages of MS that may require a different therapeutic approach. In such a scenario, neutrophils could be exploited to guide the choice of treatment and predict the patient’s response to therapy.
New neutrophil-based targets and biomarkers could flow from the study of EAE. This model has already taught us much about the mechanisms of neutrophil recruitment and action in the CNS (Fig. 1.2). It appears so far that these mechanisms are comparable to those observed in other tissues, although differences exist (e.g. in EAE, neutrophils do not appear to form extracellular traps like those seen in other autoimmune diseases such as lupus). Although EAE is an imperfect representation of its human counterparts, it remains a valuable tool to
Box 1.1. Key questions to answer to understand and exploit neutrophils in demyelinating
autoimmune diseases.
• What molecule(s) indicate neutrophils when and where to infiltrate the CNS in EAE? While ELR+ chemokines are
involved in earlier steps of recruitment, the molecule(s) triggering extravasation remain unknown. • Are BBB-specific molecules involved in neutrophil recruitment?
• By which mechanisms do neutrophils contribute to demyelination in EAE?
• Could new tools for neutrophils-specific gene deletion (e.g. Catchup mouse) be exploited to study the biology of neutrophils in EAE?
• Do neutrophils influence MS and how? For example, considering that B cell activation and CSF oligoclonal bands are hallmarks of MS, can neutrophils influence MS indirectly via B cells as they do in other contexts?
• Could neutrophils or related molecules be exploited as biomarkers to 1) identify variants or stages of MS, 2) guide treatment, and 3) predict patient responses?
• Could new drugs be designed to specifically neutralize neutrophil functions and mitigate demyelinating diseases without causing neutropenia?
• Do neutrophil-specific proteins exist in human? These could facilitate the identification of neutrophils from other myeloid cells in histopathological samples.