Habilitation à diriger des recherches
Discipline : Chimie théorique, physique, analytique
Développement de matériaux à porosité hiérarchisée
pour la catalyse hétérogène
Jean-Philippe DACQUIN
Docteur de l’université de Lille 1, Sciences et Technologies
Ecole doctorale, Sciences de la Matière, du Rayonnement et de l’Environnement
HDR préparée et soutenue publiquement le 25 Juin 2019
Rapporteurs :
Gérard Delahay, Directeur de Recherche CNRS, ICGM, Montpellier. Bénédicte Lebeau, Directeur de Recherche CNRS, IS2M, Mulhouse. Philippe Vernoux, Directeur de Recherche CNRS, IRCELYON, Lyon. Examinateur:
Sébastien Royer, Professeur des Universités, Université de Lille. Président du jury et examinateur:
Rose Noëlle Vannier, Professeur des Universités, Directrice de l’ENSCL, ENSCL. Garant et examinateur :
RESUME :
Les années 90 ont été un tournant dans le développement de matériaux à porosité contrôlée grâce aux découvertes successives de la mésostructuration et de la macrostructuration par voie douce. Les objets poreux à structure hiérarchisée qui en découlèrent ont ainsi permis de pouvoir coupler des propriétés liées à des dimensions différentes. En effet, alors que la transformation catalytique dépend de l'échelle la plus petite, les propriétés de transport des espèces dépendent des échelles de porosité supérieures (ex : macroporosité). Le dimensionnement et les propriétés morphologiques des réseaux poreux à l’échelle nano- et macro- du support doivent par conséquent être bien ajustés afin d’optimiser le transport des réactifs et produits au sein du grain de catalyseur où sont localisés les sites actifs. Au regard de ce constat, j’ai conduis mes travaux sur différentes approches et stratégies de structuration de supports type oxyde avec l’aide d’agents structurants (microsphère de différents polymères, micelle de molécules de tensioactifs…) dont la taille peut être modulée de l’échelle du nanomètre à plusieurs centaines de micromètres. Durant ces dix dernières années, je me suis employé à optimiser les propriétés poreuses et de surface de différents matériaux pour améliorer leurs performances catalytiques en essayant de prendre en compte les phénomènes intervenant dans leur efficacité globale. De nombreuses applications peuvent tirer profit de l’aspect multi-échelles de ces matériaux et de leurs architectures complexes.
MOTS CLEFS: Matériaux poreux, Texturation contrôlée, Catalyse environnementale, Oxydes, Pérovskites
ABSTRACT :
The nineties constitute a turning point in the development of the materials with controlled porosity thanks to the successive discoveries of mesostructuration and macrostructuration routes. Derivated hierarchical porous objects are able to cumulate properties related to the different dimensions afforded. Whereas catalytic transformation is strictly depending on the smallest scale, transport properties of species are depending of higher porosity scales (i.e. macroporosity). Dimension, morpholigical properties of porous networks at the nano- and macro- scales of the support must be well balanced in order to maximise reactant transport within the catalyst grain. At the light of these considerations, I dedicated my work to the application of different approaches and strategies to design structured oxides with the help of structuring agents (microspheres of different polymers, micelle made from surfactant molecules…) of which the size can be modified from the nanometer scale to the micrometer scale. Hence, for the last ten years, I have developed and optimized porous and surface properties of different classes of materials in order to improve their catalytic performances, while trying to take into account phenomena involved in their global efficiency. Numerous applications can be valorized from the use of these multiporous objects exhibiting complex architectures.
1
SOMMAIRE
INTRODUCTION ... 3
Chapitre I : Travaux de thèse et stage post-doctoral ... 6
I. Travaux de Thèse : ... 6
I.1. Contexte et objectifs : ... 6
I.2. Etude de la réaction de décomposition simultanée de N2O(g) et NO(g). ... 8
I.1.1. Choix du support LaCoO3 ... 8
I.1.2. Etude des phénomènes de reconstruction de surface du système Pd/LaCoO3. ... 10
I.3. Etude de la réaction de réduction catalytique simultanée de N2O(g) et NO(g) en présence d’hydrogène. ... 13
I.3.1. Choix du système catalytique Pt/LaFeO3... 13
I.3.2. Etude des phénomènes de reconstruction de surface du système Pt/LaFeO3. ... 14
I.4. Influence de la méthode de préparation : ... 17
II. Travaux de stage post-doctoral ... 19
II.1 Développement expérimental de catalyseurs à porosité contrôlée et hiérarchisée : application pour la synthèse de biodiesel. ... 19
II.1.1 Catalyseurs solides acides pour la synthèse de biodiesel ... 20
II.2. Synthèse de l’alumine macroporeuse mésoporeuse organisée ... 25
CHAPITRE II : Thèmes de recherche développés à l’Université de Lille ... 27
I. Matériaux pour la catalyse environnementale (équipe REMCAT) ... 27
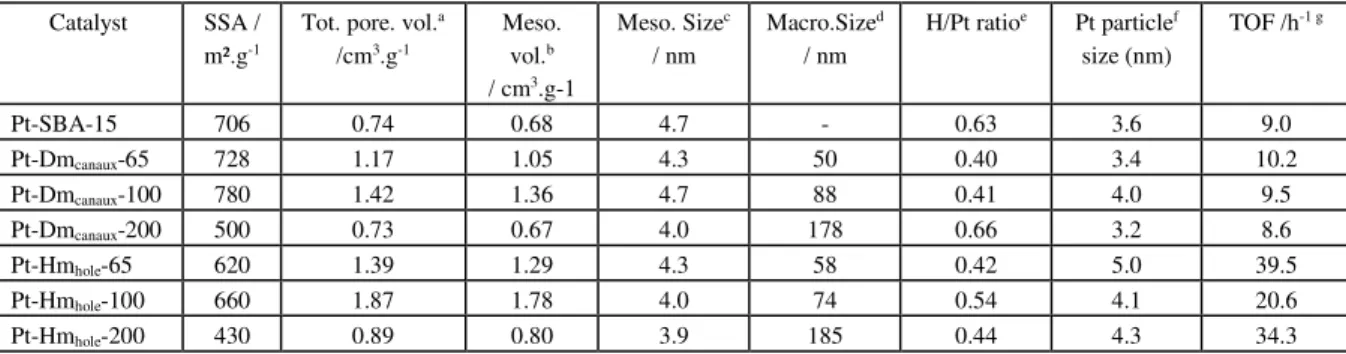
I.1. Contrôle des propriétés poreuses d’oxydes et d’oxydes mixtes pour l’hydrogénation catalytique des nitrates en phase aqueuse ... 27
I.1.1 Impact des propriétés poreuses sur les propriétés catalytiques ... 28
I.1.2 Effet de la mésotructuration sur les propriétés catalytiques de catalyseurs au Pd supporté sur cérine-zircone ... 36
I.2. Oxydes mixtes pour le traitement de polluants atmosphériques ... 38
I.2.1. L’orthovanadate de cérium ... 38
I.2.2. Les pérovskites ... 41
I.2.2.1. Influence de la composition ... 41
I.2.2.2. Influence de la macroporosité... 46
II. Matériaux pour la catalyse hétérogène (Equipe MATCAT) ... 51
II.1. Oxydes métalliques ... 51
II.1.1. Oxydes de manganèse ... 51
II.1.1. Oxydes de cobalt ... 54
2
CHAPITRE III : Conclusion et Projets de Recherche ... 59
III.1. Conclusion ... 59
III.1. Projets de recherches ... 60
III.1.1.Utilisation de bioporogènes pour la préparation d’oxydes simples ou mixtes à porosité hiérarchisée ... 60
III.2. Mise en forme de matériaux microporeux et macro-microporeux ... 62
III.3. Vers la structuration d’oxydes métalliques induite par plasma froid ... 63
Références bibliographiques ... 64
ACTIVITES D'ENSEIGNEMENT……..………69
CURRICULUM VITAE……….………71
3
INTRODUCTION
En 2008, après ma thèse à l’Unité de Catalyse et de Chimie du Solide consacrée au développement de matériaux de structure type « pérovskite » pour le traitement de gaz à effet de serre, je changeai d’environnement et de sujet pour aller en stage post-doctoral à l’Université de York, puis à l’Institut de Catalyse de Cardiff, où j’ai travaillé sur la préparation de matériaux à porosité hiérarchisée pour la synthèse de carburant alternatif de type biodiesel. J’ai été ensuite recruté en 2010 à l’UCCS au sein de l’équipe Environnement dirigée par Pascal Granger sur un profil ayant pour thème : « matériaux mésostructurés pour la catalyse ». A partir de cette période, mon temps de recherche a été dédié à l’application de catalyseurs de type oxydes (silice, alumine) et oxydes mixtes (pérovskite, cérine-zircone et orthovanadate de cérium) pour le traitement de polluants azotés en phase liquide et gazeuse. Le bon déroulement de ces activités de recherche n’aurait pas pu se faire sans le soutien de l’équipe d’accueil d’un point de vue scientifique mais aussi pour l’obtention de différents équipements et contrats. En 2016, j’ai souhaité être redéployé dans une nouvelle équipe de recherche de l’UCCS appelée «Matériaux Catalytiques » et constituée par Sébastien Royer. L’activité de l’équipe est centrée sur la conception, l’activation et la caractérisation de matériaux catalytiques, domaines dans lesquels j’ai pu intégrer mes compétences.
L’ensemble de mes travaux s’articule ainsi autour de 3 thématiques de recherche : • La préparation de catalyseurs à porosité hiérarchisée:
Les années 90 ont été un tournant dans le développement de matériaux à porosité contrôlée grâce aux découvertes successives de la mésostructuration et de la macrostructuration par voie douce. Les objets poreux à structure hiérarchisée qui en découlèrent ont ainsi permis de pouvoir coupler des propriétés liées à des dimensions différentes. En effet, alors que la transformation catalytique dépend de l'échelle la plus petite, les propriétés de transport des espèces dépendent des échelles de porosité supérieures (ex : macroporosité). Le dimensionnement et les propriétés morphologiques des réseaux poreux à l’échelle nano- et macro- du support doivent par conséquent être bien ajustés afin d’optimiser le transport des réactifs et produits au sein du grain de catalyseur où sont localisés les sites actifs. Au regard de ce constat, j’ai conduis mes travaux sur différentes approches et stratégies de structuration de supports type oxyde (alumine, silice) avec l’aide d’agents structurants (microsphère de différents polymères, micelle de molécules de tensioactifs…) dont la taille peut être modulée de
4 l’échelle du nanomètre à plusieurs centaines de micromètres. Le bon déroulement de l’assemblage (hydro)oxyde-agent est crucial car il va définir les propriétés finales du support poreux (taille, volume poreux, morphologie, tortuosité, connectivité intra- ou inter-réseau). Par ailleurs, la structuration de céramiques denses (ex : pérovskite, cérine-zircone) est également un objet de recherche d’intérêt particulier dans les domaines de l’environnement et de l’énergie. Leurs sels métalliques étant très réactifs envers la précipitation en phase aqueuse, la mise en œuvre de méthodes de structuration se révèle plus sophistiquée en comparaison avec la silice. En outre, l’identification et l’ajustement de nombreux paramètres expérimentaux tels que la composition du mélange de solvants, la nature de l’agent complexant et des sels métalliques, la température, le pH, le taux d’humidité ainsi que l’étape de libération de la porosité doivent être privilégiés dans l’élaboration du matériau.
• La préparation de catalyseurs à structure type « pérovskite » : optimisation de la composition et des propriétés de surface :
Une partie de mes activités est également dédiée à la recherche de la composition « idéale » de la pérovskite associée étroitement aux conditions d’activation de celle-ci en fonction du type de réaction catalytique mis en jeu (ex : catalyse 3-voies). Initialement, les pérovskites sont surtout connues pour leurs propriétés d’oxydation. Néanmoins, une des singularités de la structure pérovskite est d’être en capacité d’accepter un grand nombre d’éléments dans sa structure de type ABO3, sous la condition de
respecter un facteur de tolérance t compris entre 0,75 et 1,13. Ce facteur de tolérance, défini en fonction des rayons ioniques et égal à 1 lorsque la structure idéale est atteinte, permet de prédire la formation ou non d’un réseau cristallin appartenant à la structure pérovskite. En fonction de la nature du cation ajouté (taille, degré d’oxydation, électronégativité), une modification de la stabilité et des propriétés redox du solide est observée. Les phénomènes de redispersion de différents métaux nobles à la surface de la pérovskite peuvent également être bénéfiques à différentes applications environnementales et suscitent la recherche des conditions optimales d’activation du catalyseur supporté par le biais de différents types de traitement thermiques. Néanmoins, l’application de ces solides dans l’industrie reste peu fréquente. En effet, la faible activité massique observée pour ces solides a pour origine leur très faible surface spécifique (<10 m².g-1). Ainsi la réduction de la taille des
cristallites d’oxydes mixtes par leur dispersion dans une matrice poreuse de haute surface spécifique représente, avec la structuration poreuse mentionnée précédemment, une des voies étudiées en vue de leur application en catalyse.
5 • Applications de solides originaux pour la catalyse hétérogène :
A partir des catalyseurs élaborés dans les deux précédents axes, plusieurs réactions catalytiques d’intérêt environnemental ont été ciblées :
- Elimination des composés azotés provenant de sources fixes et mobiles.
- Hydrogénation catalytique des nitrates en phase aqueuse en présence d’hydrogène. - Synthèse de carburant alternatif de type biodiesel.
- Hydrogénation du CO2 en phase gaz.
En pratique, j’ai surtout cherché à montrer l’intérêt de (i) structurer la porosité des matériaux pour améliorer l’efficacité de la réaction catalytique (ii) optimiser la composition de bulk et de surface d’oxydes mixtes par dopage, substitution partielle et modification des conditions de préparation du solide. Ceci n’est possible que par l’utilisation d’outils de caractérisation des propriétés critiques des solides (texture – structure – fonctions) à partir de techniques (i) massiques (Porosimétrie d’adsorption azote, TPR-H2..), (ii) spectroscopiques (DRX, RAMAN, XAS, XPS, FTIR) et (iii) d’imagerie électronique
(MEB, MET, Tomographie électronique) accessibles au laboratoire mais également à travers différentes collaborations internes et externes.
J’ai organisé ce document en mentionnant d’abord les points-clés de ma propre thèse et de mon stage post-doctoral, puis la première thèse que j’ai co-encadrée (Abdelali Zaki, thèse soutenue en 2014), intitulée « Développement de matériaux à porosité hiérarchisée pour le traitement catalytique de composés azotés dans l’eau». Je mentionne ensuite les travaux réalisés sur l’application en catalyse de l’alumine et de la cérine-zircone dopés par des métaux nobles suite aux suivis scientifiques de deux doctorants issus de collaboration avec le NCL Pune (Sunil Sekhar) et l’université de Bucarest (Simona
Troncéa). Je reviens ensuite sur la deuxième thèse que j’ai co-encadrée et qui s’inscrit dans la poursuite
de mes travaux initiés pendant mon doctorat sur le développement de systèmes pérovskites à base de fer et l’influence de la méthode de préparation sur les performances catalytiques en catalyse 3-voies (Anke Schön, thèse soutenue en 2015). Enfin, suite à mon redéploiement dans l’équipe MATCAT en 2016, j’ai eu l’opportunité d’amorcer de nouvelles thématiques scientifiques et de nouveaux co-encadrements de thèse dont l’activité principale s’oriente sur la préparation de solides à porosité hiérarchisée et leur mise en forme (Yin Xu et Bakytzhan Yeskendir). La mise en place progressive de ces nouvelles activités constituera une partie de mon projet de recherche qui sera présenté à la suite de mes travaux.
6
Chapitre I : Travaux de thèse et stage post-doctoral
I. Travaux de Thèse :
I.1. Contexte et objectifs :
Titulaire d’une bourse ADEME-Région dans l’équipe Environnement de l’unité de Catalyse et de Chimie du Solide entre 2005 et 2008, mes travaux de thèse ont contribué à l’avancement d’un projet européen « Eureka » (StatioNOCat) coordonné par Gaz de France, et dont le sujet portait sur l’application d’un procédé catalytique basse température pour l’élimination simultanée du N2O(g) et du NO(g) provenant
des gaz de queues d’ateliers de production d’acide nitrique (figure 1, stratégie n°3). A l’époque, le contexte environnemental et législatif européen avait été marqué par l’entrée en vigueur du protocole de Kyoto en 2005 et la mise en place consécutive d’un système de permis d’émissions de gaz à effet de serre négociables entre les autorités publiques et les industries concernées. Les collaborations créées autour de ce projet regroupaient des industriels et universitaires français et polonais, celles-ci devant aboutir à la formulation d’un catalyseur efficace pour l’élimination de N2O(g) et NO(g) entre
200°C et 400°C, opérant dans un environnement de composition complexe en présence d’eau et d’un large excès d’oxygène.
7 Initialement, le traitement de telles émissions provenant de sources stationnaires ou mobiles nécessitent généralement la présence de métaux nobles.1 Les catalyseurs préparés par addition de ces
métaux sur les supports conventionnels utilisés (alumine, silice) sont généralement sujets aux effets inhibiteurs liés aux fortes adsorptions de NO(g) et O2(g), accentuées ici par la faible température de
fonctionnement (<400°C). La forte adsorption de l’oxygène pourrait être atténuée sur des oxydes mixtes de type pérovskite (dopés ou non par des métaux nobles) présentant une mobilité des espèces oxygènes plus importante.2 Cette mobilité pourrait favoriser la formation de lacunes de surface,
facilitant la désorption de l’oxygène issu de la décomposition de N2O(g) ou NO(g), étape déterminante
de la vitesse de réaction en décomposition de ces oxydes d’azote.3 En 1997, Guilhaume et coll. ont
démontré la conservation des propriétés catalytiques de LaMn0,976Rh0,024O3 dans des conditions
opératoires oscillant entre des compositions réductrices et oxydantes suggérant qu’il existait, au même titre que le catalyseur de référence Pt-Rh/CeO2-Al2O3, une capacité de stockage de l’oxygène.4
Cependant, contrairement à Pt-Rh/CeO2-Al2O3, il a été démontré sous ces conditions cycliques que
l’incorporation directe de rhodium sous la forme Rh3+ dans la structure pérovskite évitait
l’agglomération ou la volatilisation des particules de métal noble grâce à la forte interaction métal/support. Suite à cet intéressant résultat, une équipe japonaise a étudié le comportement considéré « intelligent » d’un catalyseur à base de palladium inséré également dans la structure pérovskite (LaFe0,95Pd0,05O3).5 Selon leurs différents travaux,6 le matériau se caractérise par sa capacité
à adapter sa structure en fonction des changements environnementaux, de manière à augmenter considérablement l’efficacité et la durée de vie du catalyseur. Lorsque le catalyseur est soumis à des cycles successifs oxydo-réducteurs, le palladium migre de façon réversible à l’intérieur et à l’extérieur du réseau cristallin de la structure pérovskite (Figure 2). Ce comportement dynamique tend à limiter les phénomènes de frittage des particules de palladium métallique observés de manière notoire sur Pd/Al2O3, et ainsi, améliore la durée de vie du catalyseur et son efficacité dans les réactions DéNOx.
Figure 2 : Schéma comparatif montrant la fonction de régénération du catalyseur intelligent et la détérioration du catalyseur conventionnel lors de cycles successifs oxydo-réducteurs présents dans les gaz d’échappement de sources mobiles.
8 Au regard des résultats obtenus par Guilhaume et coll. et Tanaka et coll., l'effet inhibiteur de l’oxygène sur la vitesse de décomposition de N2O(g) pourrait être atténué après déposition d’un métal noble en
interaction avec un support réductible comme la pérovskite. Il était également intéressant de remarquer pour ces catalyseurs que le métal noble était inséré directement dans le site B de la structure pérovskite. Par conséquent, il semblait peu probable que l’ensemble des particules de métal noble participe à la réaction catalytique, puisqu’une partie serait partiellement noyée dans la structure du support. C’est d’ailleurs une raison pour laquelle Tanaka et coll. ont utilisé une quantité massique de palladium plus importante dans leurs catalyseurs (≈ 3% en masse de catalyseur) afin d’obtenir assez de sites actifs disponibles pour la conversion catalytique.
A partir de ces différents constats, nous avons axé nos recherches sur le dopage post-synthèse par des métaux nobles et l’optimisation du traitement d’activation des catalyseurs pour tenter d’améliorer l’élimination de N2O(g) et NO(g) à basse température.
I.2. Etude de la réaction de décomposition simultanée de N
2O
(g)et NO
(g).
I.1.1. Choix du support LaCoO3
Pour une utilisation comme support de catalyseur hétérogène, l’alumine sous la forme γ-Al2O3
présente des propriétés physiques (surface spécifique et porosité texturale) et de surface (densité importante de groupements de type OH) avantageuses pour disperser une phase active métallique. Nous avons ainsi sélectionné un support commercial comme référence (SBET = 100 m².g-1) pour
disperser le palladium comme phase active.
Dans la pérovskite de formule ABO3, il est reconnu que le cation en position A dans la structure
influence la stabilité thermique de l’oxyde, tandis que le cation B est responsable de l’activité catalytique.7 En jouant sur différentes substitutions cationiques de la structure pérovskite en site B,
deux équipes de recherche ont montré que la force de la liaison métal-oxygène jouait un rôle important dans l’ordre d’activité des solides LaBO3 en décomposition catalytique de N2O(g)
(Co>Ni,Cu>Fe>Mn>Cr).8 D’autres travaux ont montré que la stabilité sous atmosphère oxydante de la
pérovskite croît selon la séquence suivante : GdCoO3 < SmCoO3 < NdCoO3 < PrCoO3 < LaCoO3.9 A partir
de cet état de l’art, nous nous sommes orientés sur l’utilisation de la structure pérovskite LaCoO3
9 Etude comparative des systèmes 1 wt.% Pd/γ-Al2O3 et 1 wt.% Pd/LaCoO3.
Tout d’abord, nous avons adopté une stratégie alternative aux travaux de Tanaka et coll. pour introduire et disperser les métaux dans des proportions beaucoup plus faibles (1% en masse de catalyseur), le but étant de retrouver le maximum d’atomes de palladium en surface accessibles aux réactifs. La procédure consiste à imprégner le support par le précurseur au palladium (Pd(NO3)2,2H2O)
avec un léger excès d’eau et d’évaporer de manière contrôlée la solution à 40°C pendant 4 à 6 heures. Le solide résultant est séché pendant 24h à 80°C et un traitement sous air est opéré à 400°C.10 Les
catalyseurs frais Pd/γAl2O3 et Pd/LaCoO3 ont été ensuite comparés par plusieurs tests réalisés en
montée de température programmée entre la température ambiante et 500°C en présence de N2O(g)
(1000 ppm) et NO(g) (1000ppm) dilués dans l’hélium. Avant le démarrage de la réaction catalytique,
plusieurs activations simples (tableau 1) ont été réalisées sur les échantillons placés dans le réacteur, de manière à observer un éventuel changement dans le comportement catalytique des solides. Tableau 1 : Propriétés catalytiques obtenues à partir des réactions en température programmée réalisées sur les systèmes Pd/γAl2O3 et Pd/LaCoO310
(a) Etat d’oxydation du palladium déterminé par mesures XPS ex situ. (b) Calculé à 460°C Catalyseur Prétraitement “in-situ” avant test Conversion de N2O(b) (%) Conversion de NO (%)(b) Vitesse spécifique (b) (mol.h-1.g-1) Energie d’activation apparente (kJ.mol-1)
Pd°/Al2O3(a) Réduction sous
H2(g) à 250°C 4 0 3 x 10
-5 89±5
PdO/Al2O3(a) Traité sous air à
400°C 15 0 1 x 10-4 92±5
Pd°/LaCoO3(a)
Réduction sous H2(g) à 250°C
59 5 / /
PdO/LaCoO3(a) Traité sous air à
400°C
32
0 3 x 10-4 55±3
Les conversions et les valeurs d’énergies d’activation rapportées dans le tableau 1 montrent clairement une activité catalytique dédiée exclusivement à la conversion de N2O dans la gamme de température
étudiée. Il est intéressant d’observer que la réduction préalable des particules de palladium sur alumine ne modifie pas l’énergie d’activation apparente, suggérant que les états de surface impliqués sous milieu réactionnel sont identiques. Dans le cas du solide PdO/LaCoO3, celui-ci développe une
activité catalytique plus importante que PdO/Al2O3 malgré sa plus faible dispersion métallique. Cette
observation peut être expliquée par la participation de LaCoO3 à la décomposition catalytique de
10 singulier associé à une superposition de la réaction catalytique avec la réoxydation du support (pas de production d’oxygène observée pendant la réaction en température programmée). Par conséquent, la définition des sites actifs sur Pd/LaCoO3 semblent plus compliqués à identifier que sur Pd/Al2O3 au
regard de la réductibilité et de l’activité intrinsèque du support pérovskite.
I.1.2. Etude des phénomènes de reconstruction de surface du système Pd/LaCoO3.
Sur la base de ces premiers résultats, nous nous sommes intéressés à l’activation du solide en réalisant une étape de réduction sous hydrogène suivi d’un vieillissement sous milieu réactionnel en atmosphère sèche puis humide (figure 3). Ce vieillissement permettra entre autres la réoxydation massique et de surface complète du système Pd-LaCoO3 et ainsi d’étudier réellement la réaction
catalytique de décomposition de N2O(g) et NO(g) sans interférences avec d’autres processus.
Figure 3 : Représentation schématique des étapes de traitements thermiques appliqués aux différents solides Pd/LaCoO3 étudiés.
Ainsi, après avoir démontré l’avantage du système PdO/LaCoO3 par rapport à la référence PdO/Al2O3,
nous avons comparé les performances catalytiques des échantillons LaCoO3 et Pd/LaCoO3 ayant subis
différents traitements d’activation comme illustrés sur la figure 4. Tout d’abord, une sensibilité particulière de la conversion de N2O en fonction de la température de réduction initiale du solide est
remarquée. En effet, un traitement sous hydrogène à 250°C suivi d’une réoxydation sous milieu réactionnel à 500°C (Pd/LaCo(R250V500) permet d’atteindre des performances catalytiques nettement supérieures à la référence fraîchement calciné Pd/LaCo(C400). Lorsque la température de réduction du solide est augmentée à 500°C (Pd/LaCo(R500V400), l’activité catalytique devient
T em pé ra tu re ( °C ) Temps (h) 12h 24h 0 RXXX VXXX Réaction N2O/NO/He
Traitements successifs sous environnements réducteur et réactionnel
XXX: 250°C/400°C/ 500°C
Réduction
11 comparable à celle obtenue sans traitement réducteur Pd/LaCo(C400). En effet, contrairement à un traitement réducteur à 250°C, une réduction sévère sous hydrogène pur à 500°C ne mène pas à la restauration de la structure pérovskite après vieillissement sous mélange réactionnel mais essentiellement à la ségrégation d’oxydes simples La2O3 et Co3O4 (Figure 4B).
Figure 4 : (A) Courbes de conversion de N2O (a) et de production de O2 (b) pour les catalyseurs LaCoO3 (LaCo-) et Pd/LaCoO3 (Pd/LaCo-) avec ou sans traitement d’activation (R : Réduction et V : Vieillissement) ; (LaCo(R250V500) (■),Pd/LaCo(C400) (▲), PdLaCo(R250V250) (○), ; PdLaCo(R250V500) (♦) ; PdLaCo(R500V400) (□) ; (B) Diagrammes de diffraction X obtenus sur les catalyseurs avant test catalytique (a) Pd/LaCo(C400); (b) Pd/LaCo(R250V250); (c) Pd/LaCo(R250V500);(d) Pd/LaCo(R500V500)11
Ensuite, différents états de surface peuvent expliquer les déviances observées dans les activités catalytiques (tableau 2). La comparaison entre les rapports atomiques Pd/La suggère une plus faible accessibilité des particules de palladium en surface pour l’ensemble des catalyseurs en comparaison au solide Pd/LaCo(R250V500). L’analyse de l’énergie d’activation apparente et des énergies de liaison des photopics du Pd 3d calculée pour Pd/LaCo(R250V500) est particulièrement intéressante. Une corrélation est obtenue entre les données cinétiques et spectroscopiques, confirmant l’obtention d’une plus faible énergie d’activation apparente (42 kJ.mol-1 contre 55 kJ.mol-1 pour Pd/LaCo(C400))
avec une énergie de liaison plus élevée pour le palladium glissant de 336,8 à 337,2 eV. Par conséquent, la meilleure activité de Pd/LaCo(R250V500) est légitimement corrélée à une redispersion des espèces Pd stabilisées dans un état d’oxydation singulier en comparaison aux valeurs d’énergie de liaison couramment rapportées pour les référence PdO et PdO2 supportés sur des supports de type silice ou
12 Tableau 2 : Evolution de la composition de surface de Pd/LaCoO3 sous atmosphères réductrice et oxydante.
Par conséquent, une pré-réduction à 500°C entraîne un effet néfaste sur les propriétés structurales et catalytiques probablement dû à un phénomène de frittage qui pourrait inhiber la reconstruction de la pérovskite et la redispersion des espèces Pd stabilisées par la structure. Dans ces conditions, l’effet inhibiteur de l’oxygène affecterait plus sévèrement la vitesse de décomposition de N2O(g) en
s’adsorbant préférentiellement sur les particules PdOx plus faiblement dispersés en surface. La
reconstruction de la structure pérovskite pendant le vieillissement thermique pourrait être le paramètre clé du phénomène de redispersion du Pd, prenant également en compte l’incorporation partielle du métal noble dans le réseau cristallin de la structure. Une telle configuration pourrait diminuer la force de la liaison Co-O avec la formation relative de lacunes anioniques favorable à la dissociation de N2O(g) et expliquer ainsi l’augmentation de la vitesse de conversion de N2O(g).
Néanmoins, en ajoutant la présence d’eau dans le milieu réactionnel (0,5 vol.%), le phénomène de frittage est favorisée même sur le meilleur catalyseur (PdLaCo(R250V500H2O)) comme indiqué par les
plus faibles vitesses de décomposition de N2O(g) obtenues.10 En observant les états de surface à
posteriori, la formation de particules PdO à 336,9 ±0.2 eV, similaires à celles dispersés sur un Pd/LaCo(C400), est à nouveau stabilisée sur le support, ce qui expliquerait ainsi l’accroissement de l’effet d’inhibition par O2(g) et H2O(g). Finalement, la technique XPS a largement contribué à établir une
certaine compréhension des phénomènes de reconstruction de surface de la pérovskite en présence d’un métal noble. Néanmoins, l’analyse sur une profondeur d’environ 5 à 10 nm ne peut fournir qu’une tendance moyennée concernant ces processus d’agglomération ou de redispersion des espèces métalliques.
13
I.3. Etude de la réaction de réduction catalytique simultanée de N
2O
(g)et NO
(g)en
présence d’hydrogène.
I.3.1. Choix du système catalytique Pt/LaFeO3
Afin d’exalter les propriétés catalytiques à basse température, l’ajout de platine a été étudié sur des supports de type pérovskite LaFeO3. Malgré leur plus faible réductibilité sous atmosphère réductrice
vis-à-vis du palladium et de LaCoO3, il est possible néanmoins d’augmenter la fenêtre de température
de réduction lors du traitement d’activation. Ces deux caractéristiques associées respectivement au métal noble et au support sont supposées être des éléments déterminants dans le mécanisme de reconstruction de la structure pérovskite et de redispersion du métal lorsque le solide est soumis à différentes atmosphères oxydo-réductrices. Nous avons montré précédemment que ces processus avaient un effet bénéfique sur les propriétés catalytiques en décomposition de N2O(g) pour le système
Pd/LaCoO3. Cependant, une température de pré-réduction à 500°C provoque une chute des
performances liée à la non-reconstruction du support sous milieu réactionnel. Ainsi, nous avons voulu vérifier si de tels processus pouvaient avoir les mêmes conséquences sur le système Pt/LaFeO3 dans la
réduction de N2O(g) et NO(g) par H2(g). Comme pour LaCoO3, la procédure de préparation de LaFeO3 a
été établie à partir d’une méthode par complexation à l’acide citrique.12 Les catalyseurs au platine
supporté sur LaFeO3 ont été préparés selon une méthode classique d’imprégnation avec un léger excès
d’eau en utilisant une solution d’acide hexachloroplatinique. Les échantillons imprégnés ont été séchés à 80°C puis traités sous air à 400°C. Les catalyseurs calcinés seront nommés respectivement LaFe(C400), Pt/LaFe(C400) et ceux réduits Pt/LaFe(R300) et Pt/LaFe(R500). Les catalyseurs vieillis sont décrits figure 5 et seront nommés Pt/LaFe(R300V500R300) et Pt/LaFe(R500/V500/R500).
Figure 5: Schéma présentant les étapes successives du traitement d’activation réalisé sur les catalyseurs Pt/LaFeO3 avant la réaction de montée en température programmée; R300 et R500: traitement réducteur sous hydrogène pur respectivement à 300°C et à 500°C, V500 : traitement thermique effectué à 500°C en présence de 1000ppm N2O, 1000ppm NO, 3vol.% O2, 0,5vol.% H2O et 0,5vol.%H2.
T emp ér at ur e (° C ) Temps (h) 12h 24h 36h 500 400 300 200 100 0 R300 V500 R300 Réaction N2O/NO/O2/H2O+H2
14
I.3.2. Etude des phénomènes de reconstruction de surface du système Pt/LaFeO3.
Etude comparative des systèmes (Pt/γ-Al2O3 et Pt/LaFeO3)
Le choix de la structure pérovskite LaFeO3 s’est avéré judicieux dans la mesure où elle possède une
stabilité thermique sous atmosphère réductrice suffisante pour activer la réduction du platine tout en préservant ses propriétés structurales au même titre que γ-Al2O3. Les modifications structurales et de
surface ont été suivies ex-situ par DRX, H2-TPR, XPS, et HRTEM. Les résultats ont été corrélés avec les
performances catalytiques en termes d’activité et de sélectivité. Sans vieillissement préalable, les résultats catalytiques en réduction simultanée de N2O(g) et NO(g) demeurent meilleurs lorsque le platine
est activé sous la forme métallique et dispersé sur alumine. Ce comportement catalytique initial peut s’expliquer par une meilleure dispersion des espèces Pt sur Al2O3 caractérisé par une aire spécifique
quatre fois plus importante par rapport à LaFeO3 (100m².g-1 contre 25m².g-1). Cependant, après
traitements thermiques oxydo-réducteur (figure 5), des changements morphologiques importants interviennent, conduisant à un frittage significatif des particules de platine sur les systèmes 4 wt.% Pt/γ-Al2O3 ou 1wt.%Pt/γ-Al2O3 (figure 6).12 Parallèlement, une forte diminution de la conversion
simultanée en N2O(g) et NO(g) est observée plus particulièrement à haute température. Ces
observations sont également obtenues par les travaux de Yang et coll. sur l’effet de la température en réduction des NOx(g) par l’hydrogène sur les catalyseurs Pt/Al2O3.13 Dans leurs conditions (0,2%NO, 1%
H2, 5%O2 dilué dans l’hélium), la réduction par l’hydrogène interviendrait à basse température (<150°C)
sur les sites Pt partiellement oxydés conduisant à une formation prépondérante de N2O(g). Concernant
les systèmes 4wt.% Pt/LaFeO3 ou 1wt.%Pt/LaFeO3, la stabilisation initiale des particules de platine sous
la forme PtO2 à la surface de la pérovskite (XPS) est suggéré responsable de la plus grande résistance
de ces catalyseurs vis à vis des phénomènes de frittage (figure 6).
Figure 6: Histogrammes de distribution des particules Pt en taille sur les catalyseurs (a) 1wt.%Pt/Al2O3 et (b) 1wt.%Pt/LaFeO3 traités successivement sous différentes atmosphères réactionnelles.
15 Ce comportement singulier peut être expliqué par une interaction métal/support exacerbée après traitement réducteur à 300°C et/ou vieillissement sous atmosphère réactionnelle à 500°C. Afin de mieux visualiser le phénomène, une étude complémentaire a été réalisée par microscopie électronique à transmission haute résolution (HRTEM) en collaboration avec le groupe de Claude Henry du centre des nanosciences de l’université de Marseille. Sur l’ensemble des clichés réalisés, l’identification des distances interréticulaires appartenant aux plans cristallins (002), (111), (200), et (202) de la phase orthorhombique de LaFeO3 confirme que la structure est bien conservée après
réduction sous hydrogène à 300°C et 500°C. Les particules de platine sont également observées avec des distances correspondant majoritairement aux plans (111) du platine métallique. De manière surprenante, parmi les particules observées par HRTEM sur 4%Pt/LaFe(R300), on visualise aisément une orientation préférentielle des plans cristallins (111) des particules Pt avec les plans caractéristiques de LaFeO3 (200), (111), (002), et (202). Des observations similaires sont rapportées pour les catalyseurs
4%Pt/LaFe(R300A500R300) (Figure 7), ce qui révèle une forte interaction du métal avec le support pérovskite même après un traitement de vieillissement en présence d’eau et d’oxygène.12,14
Ainsi, sur les catalyseurs Pt/Al2O3 pré-réduits, la diminution de la teneur en platine de 4% à 1% entraîne
une dégradation de l’activité à basse température sans changement significatif de la sélectivité envers N2(g).12 Par contre, des tendances différentes sont obtenues sur les catalyseurs Pt/LaFeO3 pré-réduits
où la diminution de la teneur en platine d’un facteur 8 sur le support LaFeO3 n’altère pas la conversion
de NO(g) à basse température, en particulier sur le catalyseur 0,5%Pt/LaFe(R300V500R300) (Figure 7).
Malgré une meilleure dispersion conduisant généralement à une plus forte exposition des particules de platine au phénomène de frittage, 0,5%Pt/LaFe(R300V500R300) offre la meilleure sélectivité envers N2(g) parmi les catalyseurs étudiés et une résistance remarquable à la croissance des particules
métalliques. Dans ces conditions, l’orientation préférentielle des plans du métal noble vis-à-vis des plans de la pérovskite pourrait être un des paramètres permettant d’inhiber la réaction de frittage.
Figure 7 : Activité catalytique des solides Pt/LaFeO3 en conversion de NO sous milieu réactionnel 1000ppm NO/1000ppm N2O/3 vol.%O2/0.5 vol.%H2O et cliché HRTEM de 4%Pt/LaFe(R300V500R300)12
16 Influence de la température de réduction sur l’interaction métal/support du système 1%Pt/LaFeO3
Figure 8 : Clichés HRTEM du catalyseur (a) 1wt.%Pt/LaFe(R500) et (b) 1wt.%Pt/LaFe(R500V500R500)
La sensibilité des catalyseurs à un traitement réducteur plus sévère a été également étudiée par DRX, TPR-H2, XPS, HRTEM et réactions en montée de température programmée.14 Malgré une faible
réductibilité identifiée pour le support et le métal noble, la nature de l’interaction entre Pt et LaFeO3
a étroitement dépendu de la température de réduction du traitement d’activation. Une pré-réduction à 500°C mène à une suppression de l’interaction métal/support correspondant à la stabilisation de particules de platine avec un caractère métallique plus marqué (XPS). Ce nouvel état de surface est caractérisé par des orientations aléatoires des plans du métal noble avec LaFeO3 sur les
différents clichés obtenus sur les catalyseurs Pt/LaFe(R500) et Pt/LaFe(R500V500R500) (figure 8). Il apparaît clairement qu’une réduction sous hydrogène à 500°C supprime l’interaction observée précédemment pour les catalyseurs réduits à une température plus douce (Pt/LaFeR300 et Pt/LaFeR300V500R300) (Figure 9).
Figure 9 : Clichés HRTEM du catalyseur (a) 1wt.%Pt/LaFe(R300) et (b) 1wt.%Pt/LaFe(R300V500R300)
En ajustant une température de pré-réduction du catalyseur Pt/LaFeO3 à 500°C, l’analyse des
performances catalytiques a révélé une conversion de NO(g) à basse température (<200°C) similaire au
17 température de l’étude. Néanmoins, un vieillissement sous milieu réactionnel et une réduction à 500°C supplémentaires implique une chute globale de l’activité catalytique du catalyseur Pt/LaFeR500V500R500 que nous supposons lié à la suppression de l’interaction métal-support (figure 8) probablement provoquée par le caractère fortement métallique du platine (XPS). D’autre part, les performances obtenues sur les catalyseurs pré-réduits à 300°C ne peuvent pas être strictement reliées à une phase active composée uniquement de platine mais plus probablement à la stabilisation de nouveaux sites actifs à l’interface métal-support comme rapportés dans plusieurs exemples de la littérature.15
Les performances catalytiques obtenues sur les systèmes Pd/LaCoO3 (réaction de décomposition) et
Pt/LaFeO3 (réduction en présence d’hydrogène) sont intéressantes d’un point de vue du
comportement du matériau sous conditions réactionnelles mais malheureusement leurs activités limitées en catalyse impliquent la recherche de solutions alternatives pour un procédé à basse température permettant de convertir simultanément N2O(g) et NO(g) à partir de 200°C. Néanmoins, les
résultats ont montré qu’une conversion sélective de N2O(g) est possible jusqu’à 600°C sans production
significative de NO(g), ouvrant la voie à une application potentielle de ces systèmes catalytiques pour
un procédé de dépollution à haute température (Figure 1, stratégie n°2).
I.4. Influence de la méthode de préparation :
Parallèlement à l’étude menée sur l’activation des catalyseurs mentionnée précédemment, j’ai évalué différentes méthodes de préparation de la pérovskite LaCoO3 dans l’objectif d’abaisser la température
d’activation de la décompostion de N2O(g). La première méthode, nommée broyage réactif, consiste à
réduire la taille des cristaux de pérovskite formés à la synthèse en remplaçant l’énergie thermique par l’énergie mécanique. Cette stratégie est issue des travaux du Professeur Kaliaguine16 de l’Université
Laval (Québec, Canada) qui m’avait accueilli dans son laboratoire en octobre 2006 pour un séjour d’un mois. La seconde méthode est la technique des opales inverses permettant d’ouvrir la porosité du solide par l’utilisation de billes de polymère auto-organisées. La préparation de ces solides poreux a également eu lieu au département de Génie Chimique de l’Université Laval avec l’aide du docteur Sébastien Vaudreuil. A l’époque, nous avions adapté un protocole expérimental mis au point par Ueda et coll.17 dans lequel l’imprégnation était effectuée par infiltration d’une solution (mélange
d’éthylène-glycol/méthanol (40vol.%) contenant les précurseurs métalliques) dans le réseau interstitiel de billes de polystyrène pré-arrangés dans un entonnoir Büchner. Les billes « sacrificielles » étaient ensuite décomposées par traitement thermique sous air (1°C/min, 600°C, 4 heures) afin d’obtenir la structure type pérovskite LaCoO3.18 Ainsi, nous avons pu mener une étude sur l’influence de la méthode de
18 physico-chimiques des divers solides ont été examinées par DRX, H2-TPR, TEM, BET, XPS et TPD-O2.18
Les résultats ont montré que les changements morphologiques et de surface induisent des variations significatives dans l’activité catalytique de LaCoO3 en décomposition de N2O(g), et cela malgré la
présence d’une structure cristalline rhomboédrique pour tous les solides. Une augmentation significative de la conversion de N2O(g) fut observée sur les échantillons préparés par broyage réactif
développant une surface spécifique importante associée à une plus faible taille des cristallites (d = 13,5 nm contre d = 20 nm pour la voie citrates). Cependant, l’amélioration des vitesses de conversion de ce solide n’est pas uniquement reliée à l’augmentation de la densité de sites mais est aussi fortement influencée par la réactivité plus sensible des espèces oxygène de surface de la pérovskite (TPD-O2).18
Les mesures XPS ont par ailleurs montré une plus faible stabilisation d’espèces carbonates sur ce solide, pouvant raisonnablement être reliée à un plus faible enrichissement de la surface en lanthane comparativement aux autres méthodes (tableau 3). L’agrégation des différentes caractéristiques présentées par LaCoO3 via un broyage réactif favoriserait par conséquent l’activation de la réaction de
décomposition de N2O(g) à plus basse température comparativement aux autres méthodes de
préparation.
Tableau 3 : Comparaison des surfaces spécifiques, des compositions relatives de surface, et des activités en décomposition de N2O développées par les différents solides LaCoO3.
a :Température atteinte pour une conversion de N2O(g) égale à 50%. Catalyseur Préparation T50 (°C)a Conversion de
N2O à 468°C (%) Surface Spécifique (m².g-1) Co/La (XPS) LaCoO3 Céramique / / 0,5 m².g-1 / LaCoO3 Opales inverses 592 4 12 m².g -1 0.40 LaCoO3 Citrates 532 7 20 m².g-1 0.62
19
II. Travaux de stage post-doctoral
Ce stage post-doctoral a été effectué au département de Chimie dans l’équipe de Karen Wilson et Adam F. Lee située à l’Université de York puis à l’Institut de Catalyse de Cardiff. Ces travaux sont focalisés sur la préparation de différentes classes de matériaux à porosité mono- et bimodale pour la synthèse de biodiesel à partir d’huile de Jatropha. Ce sujet de recherche s’inscrivait dans un projet EPSRC impliquant l’université de Manchester (Dr. Jhuma Sadhukhan, modèles de diffusion), l’université de York (matériaux catalytiques), et l’université de Newcastle (Dr. Adam Harvey, ingénierie des réacteurs)
II.1 Développement expérimental de catalyseurs à porosité contrôlée et hiérarchisée : application pour la synthèse de biodiesel.
Le biodiesel est considéré comme une future source de carburant propre et une alternative au diesel pétrosourcé utilisé dans le domaine des transports. Le biodiesel est constitué d’esters d’acides gras dérivés d’huiles végétales ou de graisses animales contenant initialement des triglycérides et des acides gras. L’utilisation du biodiesel est attractive car la présence de fonctions oxygénées et l’absence de soufre améliorent nettement la combustion, menant notamment à une réduction importante de la teneur en particules de suies émises par les gaz d’échappement. Parmi les technologies disponibles, les triglycérides et acides gras contenus dans les sources d’huiles végétales de première ou seconde génération peuvent être classiquement convertis par catalyse homogène acide ou basique en présence d’un alcool à chaîne courte (figure 10).
O O O O O O R R R OH O O OMe R O OMe O H OH OH O OMe O OMe R O OMe O H OH OH O O O O O O R R R MeOH/H+ MeOH/NaOMe 3 MeOH/H2 SO4 3
Figure 10: Synthèse du biodiesel par voie catalytique homogène acide et/ou basique
La production industrielle de biodiesel nécessite la présence d’acide sulfurique et/ou la présence de solutions basiques à base d’alcoolates pour permettre la transestérification des triglycérides en leurs méthylesters d’acide gras respectifs. Le glycérol est obtenu comme produit secondaire de la réaction de transestérification (figure 10). Cependant, en milieu basique, il faut préalablement estérifier les acides gras par une solution acide afin de limiter la réaction de saponification, celle-ci augmentant les coûts de séparation des produits finaux de manière significative. Par conséquent, la limitation de la teneur en acide gras <0,5% demeure une contrainte préalable avant une éventuelle application du procédé biodiesel. De plus, il faut s’assurer de l’absence d’eau dans le milieu réactionnel pouvant
20 mener à l’hydrolyse des esters en acides gras et indirectement à la réaction de saponification. Ces différentes routes impliquent tout un processus d’extraction/séparation afin d’éviter les problèmes de corrosion dans les réacteurs ou dans les différents organes du moteur pendant son fonctionnement.
19 L’emploi alternatif de catalyseurs hétérogènes non corrosifs et plus facilement recyclables est une
voie d’amélioration du procédé biodiesel tant au niveau environnemental qu’économique. Dans cette optique, la préparation et la fonctionnalisation de supports à porosité simple ou multiple ont été étudiées puis valorisées pour la transformation de molécules modèles d’acides gras (acide palmitique) et de triglycérides (tricapryline(C8)-trioléine(C18)) par estérification/transestérification en conditions douces (<100°C, 1bar).
II.1.1 Catalyseurs solides acides pour la synthèse de biodiesel
Silice macroporeuse mésoporeuse sulfonée
Divers catalyseurs solides acides et basiques incluant les zéolites, les résines, les oxydes de terres rares, les hydrotalcites, et autres oxydes mixtes ont été évalués pour la réaction de transestérification de triglycérides.20 Cependant, les systèmes catalytiques micro- et mésoporeux étudiés jusqu’alors
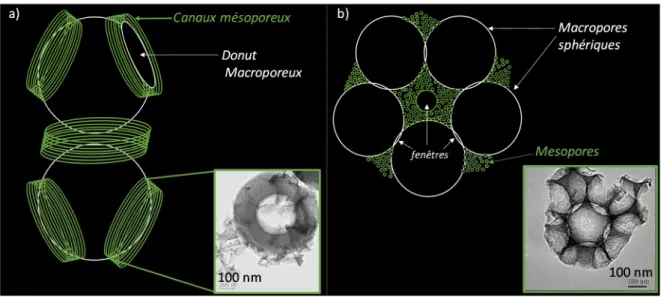
n’étaient pas optimisés pour la transformation de molécules triglycérides d’huiles végétales visqueuses comprenant principalement des chaînes alkyles C16 et C18 (Figure 11(A)). L’élargissement du diamètre d’ouverture des pores était par conséquent un aspect intéressant à étudier pour cette réaction particulièrement assujettie aux restrictions diffusionnelles internes. Si la mésoporosité de la silice pouvait être calibrée pour former des pores plus larges (4 nm< Dp <15 nm), les matériaux à porosité sulfonés dérivés de la SBA-15 montrèrent une activité limitée par la présence de canaux mésoporeux de grande longueur (>100nm) et souvent isolés les uns des autres, impliquant une diffusion intra-granulaire très contrainte pour les espèces et l’obtention de faibles vitesses de réaction. 21
Figure 11: (A) Etapes mises en jeu pendant la synthèse de biodiesel par transestérification et estérification catalytiques et (B) Principe de la double structuration utilisée pour incorporer de la macroporosité dans une silice mésostructurée 19
21 L’alternative choisie a été d’insérer au sein de la mésoporosité une porosité encore plus large correspondant à des diamètres de pores de quelques centaines de nanomètres (figure 11B). De tels réseaux macro- et mésoporeux possédant chacun une dimension bien définie seraient particulièrement intéressants pour améliorer l’efficacité du transport des espèces dans une réaction en phase organique. Pour préparer ce type de matériaux, des agents de structuration supramoléculaires peuvent diriger la formation du réseau mésoporeux alors que des objets ou substances de grande taille (cristaux colloïdaux, mousse de polymère, cellulose, émulsion) peuvent être utilisés pour former le squelette macroporeux.22 Ayant déjà travaillé sur la préparation de billes
de polystyrène (PS) lors de mon séjour au Canada, la porosité bimodale au sein du squelette silicique a été élaborée par une double structuration basée sur le procédé sol-gel couplé avec la technique de sédimentation-agrégation et l’auto-assemblage coopératif.23 L’incorporation des macropores est
obtenue à partir de sphères de polymère (dmoy = 320 nm) et l’addition de copolymères amphiphiles
triblocs non ionique (P123) mène à une porosité calibrée entre 4 et 6 nm.24 Par cette voie originale,
une série de matériaux macroporeux mésoporeux dérivée de la SBA-15 a été préparée en faisant varier le rapport massique PS/TEOS (MM-15-X où X correspond au rapport massique PS/TEOS). La SBA-15 de référence et les matériaux dérivés ont maintenu des surfaces spécifiques élevées (>900 m².g-1)
accompagnées d’une mésoporosité uniforme (entre 4 et 6 nm) après élimination de la partie organique par une lente calcination sous air à 550°C pendant 6 heures (0,5°C/min). L’analyse par microscopie électronique des différents solides a montré que l’augmentation du rapport PS/TEOS entraîne la formation progressive d’un squelette macroporeux dont les murs sont constitués majoritairement de canaux mésoporeux circulaires. (figure 12A). De plus, le caractère macroporeux du solide entraîne indubitablement une diminution en taille des domaines mésoporeux en comparaison avec la SBA-15.
Figure 12 : (A) Clichés TEM du solide MM-SBA-15-4 (B) a-Synthèse et b-fonctionnalisation acide d’une silice à porosité hiérarchisée MM-SBA-15-4 ; Valeurs de TOF des solides MM-SBA-15-X (basées sur le nombre moyen de sites SO3H obtenu par ATG et Fluorescence X) pour la réaction d’estérification de l’acide palmitique (rouge) et la réaction de transestérification de la tricapryline (bleu).
22 Suite à la fonctionnalisation de la surface des supports par greffage de sites -SO3H (Figure 12B),25 les
solides sulfonés ont été évalués pour les réactions d’estérification de l’acide palmitique (C16) et de transestérification de la tricapryline (C8) reconnues comme étant des molécules modèles pour ces deux réactions (figure 12B). Comme illustré, l’insertion progressive de macroporosité est corrélée avec l’élévation des valeurs de TOF pour la réaction d’estérification de l’acide palmitique, et dans une moindre mesure pour la réaction de transestérification de la tricapryline par rapport au catalyseur mésoporeux sulfoné de référence SBA-15-SO3H. Au regard des résultats catalytiques et de la
caractérisation des solides, l’amélioration de l’efficacité catalytique observée sur les catalyseurs à porosité hiérarchisée (MM-SBA-15-SO3H-X) résulte raisonnablement d’une diffusion intra-granulaire
facilitée par la présence de macropores ouverts jouant le rôle d’accès privilégié vers le bulk mésoporeux du solide. La réduction du domaine mésoporeux (100nm pour la SBA-15 contre 35 nm pour MM-SBA15-4) est aussi un élément à prendre en compte puisqu’elle induit une exposition plus importante des sites SO3H à la surface. Cette étude préliminaire montre ainsi l’importance d’ajuster
les propriétés poreuses du solide en fonction de l’application catalytique visée.
Silices mésoporeuses sulfonées : influence de la taille des pores et de la connectivité
Des travaux considérables ont été dédiés à la compréhension de la formation du matériau SBA-15 afin de pouvoir rationaliser les propriétés physico-chimiques en vue d’une application dédiée. Ces différents travaux ont ainsi montré que les propriétés telles que la morphologie, la porosité, la surface spécifique ou encore la chimie de surface dépendent étroitement des paramètres de synthèse.26 Par
exemple, la dimension des mésopores de la SBA-15 est strictement limitée par la taille physique du tensioactif non ionique et/ou les conditions du traitement hydrothermal. Dans notre étude, nous avons évalué l’effet de l’addition d’agents de gonflement (1,3,5 triméthylbenzène :TMB) sur le diamètre de pore final du support silicique dérivé de la SBA-15.27 Pour cela, une série de solides
SBA-15-X (X : diamètre de mésopore) a été préparée en ajoutant initialement du TMB dans la solution réactionnelle en présence du copolymère amphiphile triblocs P123. Au regard de cette association, l’ajout de TMB agit comme agent de gonflement des micelles sphériques du pluronic et génère des gouttelettes homogènes P123/TMB qui peuvent permettre un élargissement de la taille des mésopores jusqu’à environ 15 nm tout en conservant une structure poreuse hexagonale. 28 Les
limitations de cet élargissement sont directement liées à la faible solubilisation du TMB au sein de ces micelles sphériques. Une introduction trop importante de TMB entraîne une déstructuration du système hexagonal de la SBA-15, se traduisant généralement par une transition de phase vers une structure type mousse mésocellulaire. Prenant en compte les limitations de cette stratégie, nous avons
23
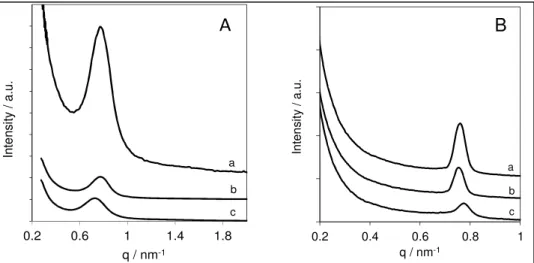
Figure 13 : (A) Diagrammes de diffraction aux bas angles des solides SBA-15-6, SBA-15-8 et SBA-15-14 ; (B) Propriétés physiques de la SBA-15 parent et des matériaux dérivés à porosité élargie (les données pour les solides greffés sont montrées en italique)
pu élargir le diamètre des mésopores de la SBA-15 jusqu’à 14 nm tout en maintenant raisonnablement les propriétés physiques de la SBA-15 parent (figure 13 et 14 ).28Après greffage de groupements -SO
3H
et vérification de la robustesse de la structure poreuse, les catalyseurs solides acides ont tous montré une activité catalytique supérieure à la résine commerciale Amberlyst dans les réactions d’estérification de l’acide palmitique et de transestérification de la tricapryline (C8) et de la trioléine (C18). Dans notre cas, l’élargissement plus sensible de la porosité semble atténuer les limitations diffusionnelles par une meilleure accessibilité des sites actifs. Ces solides à mésoporosité large
Figure 14 : (A) Distribution poreuse des catalyseurs sulfonés obtenue par porosimétrie azote, (a) SBA-15-6, (b) SBA-15-8, (c) SBA-15-14. (B) TOF obtenus pour les réactions d’estérification de l’acide palmitique et de transestérification de la trioléine et de la tricapryline.
24 surclassent également les catalyseurs solides macro-mésoporeux sulfonés cités précédemment (MM-SBA-15-X), suggérant que la taille des mésopores rapportée dans les solides à porosité hiérarchisée MM-SBA-15-X (entre 4 nm et 6 nm) est trop proche des dimensions moléculaires des triglycérides (~5nm) pour permettre un passage efficace des réactifs au sein des canaux mésoporeux, limitant les réactions d’estérification/transestérification aux groupements sulfoniques greffés à l’entrée des macropores. Par conséquent, la sensibilité du catalyseur pourrait être encore améliorée en élargissant les mésopores eux-mêmes ou alors en modifiant les propriétés morphologiques des réseaux poreux. Concernant ce dernier point, les canaux de longueur micrométrique de la structure poreuse de la SBA-15 limitent la vitesse d’échange moléculaire avec le milieu réactionnel visqueux, c’est pourquoi des canaux tridimensionnels interconnectés entre eux pourraient offrir une des solutions alternatives pour améliorer l’accessibilité des sites sulfoniques des catalyseurs siliciques. Dans la littérature, une efficacité accrue du transport moléculaire pour l’immobilisation de biomolécules avait déjà été observée dans la structure poreuse de la KIT-6 en comparaison à la SBA-15.29
Nous avons donc rapporté une autre voie d’application de solides mésostructurés en prenant avantage de l’architecture poreuse de la KIT-6 et en ajustant différentes tailles de pores (5, 6 et 7 nm). Pour ce dernier point, nous avons fait varier la température de l’étape de vieillissement hydrothermal entre 80°C et 120°C pendant 24h. Il est à noter que l’ajout de TMB entraîne la formation de vésicules et la perte de l’architecture poreuse de la KIT-6 parent.30 La surface des matrices siliciques a ensuite été
fonctionnalisée par des groupements propylsulfoniques via un protocole similaire aux études précédentes. Les solides PrSO3H-KIT-6-X (où X correspond à la température de vieillissement), ont été
évalués pour différentes réactions d’estérification d’acides gras en présence de méthanol en conditions douces (figure 15). Tous les catalyseurs à base de KIT-6 ont montré une amélioration entre 40% et 70% de la fréquence de turnover (TOF) respectivement pour l’estérification de l’acide propanoique et hexanoique, par rapport au catalyseur Pr-SO3H-SBA-15 (Dp = 5 nm). Cependant, les
mesures différenciées des activités entre les catalyseurs PrSO3H-KIT-6-X peuvent être reliées à la
longueur de la chaîne alkyle de l’acide gras utilisée pour la réaction. En effet, l’accessibilité des sites pour le solide PrSO3H-KIT-6-80 (dp = 5nm) est limitée par l’introduction d’acide palmitique ou laurique
dans le mélange réactionnel. L’élargissement sensible de la taille des mésopores à 7 nm par l’application d’un traitement hydrothermal plus sévère permet néanmoins de doubler la fréquence de turnover du catalyseur PrSO3H-KIT-6-120 (dp = 7 nm) pour l’estérification de l’acide palmitique et
25 Figure 15: Comparaison des performances catalytiques de différentes familles de catalyseurs solides acides (Amberlyst, SBA-15 sulfonée et KIT-6 sulfonées) pour la synthèse de biodiesel.
II.2. Synthèse de l’alumine macroporeuse mésoporeuse organisée
Le développement parallèle des voies de macrostructuration et de mésostructuration a rapidement mené aux silices macro-mésostructurées organisées.31 Concernant l’alumine, outre la possibilité
d’obtenir une alumine amorphe mésostructurée par le procédé d’auto-assemblage induite par évaporation (EISA),32 il est également possible d’obtenir ces solides avec une porosité hiérarchisée.
Plusieurs études ont démontré la faisabilité de former de tels solides complexes, cependant les réseaux bimodaux se présentent dans la majorité des cas sous la forme de réseaux poreux désorganisés.33 A
notre connaissance, seuls Li et coll. rapportent la production de monolithes macroporeux à mésoporosité organisée.34 En collaboration avec Sébastien Royer et Daniel Duprez de l’université de
Poitiers, travaillant à l’époque sur la modulation de la structure poreuse hexagonale de l’alumine amorphe, l’étude d’une voie permettant d’obtenir un réseau régulier aux deux échelles méso- et macro- a été initiée. Nous avons sélectionné le procédé EISA en présence d’un copolymère triblocs de type pluronic P123 pour former la mésostructure, alors que la formation du réseau macroporeux est obtenue par l’utilisation de billes de polystyrène calibrées en taille. La phase hybride macro-mésoporeuse se forme lors de l’étape de condensation par évaporation lente du solvant, après une étape préalable de sédimentation des billes de polystyrène en un réseau organisé compact (hexagonal et/ou cubique centré). La formation de la macrostructure est directement visualisée par MEB (figure 16.A) et par TEM à faible grossissement (Figure 16B). Le réseau mésoporeux est également observable à plus fort grossissement par TEM (Figure 16C). La formation d’une structure hiérarchisée organisée
26 est confirmée par la présence de réflexions aux bas angles caractéristiques de la formation d’un réseau mésoporeux organisé.35 Cette approche par double structuration (EISA/Sédimentation-Agrégation)
permet également une flexibilité de la taille des macropores, en modifiant initialement la taille des billes de polystyrène utilisée. La combinaison de deux réseaux poreux organisés, présentant des pores de taille définies et homogènes aux deux échelles (macro- et méso-), font de ces matériaux des supports modèles ayant des propriétés diffusionnelles prédictibles, adéquats pour une utilisation dans des réactions impliquant des restrictions diffusionnelles internes.
27
CHAPITRE II : Thèmes de recherche développés à l’Université de Lille
I. Matériaux pour la catalyse environnementale (équipe REMCAT)
Suite à mon recrutement sur le profil de poste « Matériaux mésostructurés pour la catalyse » en septembre 2010, j’ai été rattaché à l’Unité de Catalyse et de Chimie du Solide, dans l’équipe de recherche « Environnement » dirigé par Pascal Granger. La thématique principale de l’équipe porte sur le post-traitement de polluants atmosphériques, principalement les oxydes d’azote (NOx et N2O) et les
composés organiques volatils (toluène, HCHO). Dès mon arrivée, mes travaux de recherche ont été focalisés sur le contrôle des propriétés physiques de catalyseurs à porosité mono- et bimodale et la mise en place d’un procédé catalytique pour le traitement de polluants (nitrates et nitrites) en phase liquide. J’ai également eu l’opportunité de poursuivre mes recherches avec les membres de l’équipe sur l’optimisation des propriétés catalytiques de différents oxydes et oxydes mixtes (pérovskite, cérine-zircone, orthovanadate de cérium, oxydes de manganèse…), en vue de leur application en phase gazeuse (catalyse 3-voies, catalyse 4-voies, oxydation du méthane, oxydation du toluène…).
I.1. Contrôle des propriétés poreuses d’oxydes et d’oxydes mixtes pour l’hydrogénation catalytique des nitrates en phase aqueuse
L’élimination catalytique des nitrates dans l’eau potable a constitué un nouvel axe de recherche au laboratoire. Ce dernier a pu se formaliser par le biais d’un contrat doctoral (Bourse MRT, A. Zaki,
2011-2014), et de collaborations avec le National Chemical Laboratory de Pune (Bourse LIA, S. Sekhar, 2013-2014) et l’université de Bucarest (Bourse ambassade de France, S. Troncea, 2013-2015). Cette
thématique environnementale est associée à l’augmentation continuelle de la concentration des nitrates dans les nappes phréatiques, concentration dont le seuil a été limité par la commission européenne à 50 mg.L- (10mg.L-1 aux Etats Unis) pour des raisons de santé.36 La mise en œuvre de
procédés d’élimination des nitrates dans l’eau potable à température ambiante n’est pas triviale en raison de la stabilité de ces espèces ioniques. Par rapport aux procédés biologiques et physico-chimiques déjà existants, mais possédant des limitations associées au fonctionnement (température, pH) et/ou à la transformation directe du polluant en azote gazeux, la solution catalytique peut se révéler avantageuse. C’est un procédé basé sur l’hydrogénation des nitrates en nitrites, puis l’élimination des nitrites en azote gazeux (Figure 17).37 Alors que la première étape est considérée
28 nitrites est sensible à la structure du catalyseur, influençant la sélectivité (i) vers le produit ciblé (azote gazeux) ou (ii) vers les produits secondaires non désirées (ammoniaque).
Figure 17 : Schéma simplifiée de la réaction d’hydrogénation des nitrates sur des catalyseurs à base de métaux nobles.
I.1.1 Impact des propriétés poreuses sur les propriétés catalytiques
L’élimination de la source de nitrates en azote nécessite souvent la présence d’un agent réducteur (H2(g)). A cet égard, la réaction s’effectue en milieu triphasique (gaz/liquide/catalyseur solide) et
engendre des contraintes diffusionnelles favorisant une plus faible accessibilité des sites actifs. L’efficacité catalytique peut donc être altérée par d’importantes modifications des propriétés de la particule métallique pendant la réaction mais également par un transport contraint des réactifs et des produits dans la porosité du grain de catalyseur. Pour répondre à cette dernière problématique, nous avons évalué le potentiel de plusieurs systèmes catalytiques comprenant des nanoparticules métalliques dispersés dans la porosité du support hôte. Dans un premier temps, le choix de la nature des supports s’est préalablement basé sur des oxydes simples de type silice et alumine amorphes, dont le contrôle des propriétés poreuses avait déjà été initié pendant le stage post-doctoral.
Etude du système catalytique Pt/SiO2
Ce projet de recherche reprend l’étude de la synthèse des silices à double porosité initiée lors du stage post-doctoral. L’étude m’a notamment permis d’ouvrir de nouvelles collaborations en interne avec Christine Lancelot (UCCS, TEM), Grégory Stoclet (SAXS, Unité Matériaux et Transformations (UMET)), Alexandre Mussi (Tomographie électronique, UMET) et Jean-François Dhenin (Tomographie électronique, UMET) et en externe avec Sandra Casale de l’Université Pierre et Marie Curie (MEB, TEM-EDS, HRTEM).
Parmi les différentes voies de synthèse utilisées dans la littérature, l’approche par double structuration, employant un tensioactif non ionique directeur de la mésostructure et des billes de polymère pré-arrangées sous la forme de cristaux colloïdaux, demeure jusqu’à maintenant la technique la plus adaptée lorsqu’on recherche un degré de contrôle élevé des propriétés poreuses du matériau. Un contrôle fin sur les propriétés des différents réseaux poreux peut donner lieu à des supports qui possèdent des propriétés de diffusion prédictibles et idéales pour optimiser mais aussi