Université de Montréal
Rôle de la tolérance centrale et périphérique des
lymphocytes T autoréactifs dans deux nouveaux modèles
murins double transgéniques
par Sylvie Chabot
Département de microbiologie, infectiologie et immunologie Faculté de médecine
Thèse présentée à la Faculté des études supérieures en vue de l’obtention du grade de Ph.D.
en microbiologie et immunologie
Mai 2014
Université de Montréal
Faculté des études supérieures et postdoctorales
Cette thèse intitulée :
Rôle de la tolérance centrale et périphérique des
lymphocytes T autoréactifs dans deux nouveaux modèles
murins double transgéniques
Présentée par : Sylvie Chabot
a été évaluée par un jury composé des personnes suivantes :
Hugo Soudeyns, président-rapporteur Fernando Alvarez, directeur de recherche Idriss Djilali-Saiah, co-directeur de recherche
Mariela Segura, membre du jury Silvia Vidal, examinateur externe
Résumé
Les maladies autoimmunes sont des affections chroniques, le plus souvent invalidantes, qui touchent plus de 5% de la population dans les pays développés. L’autoimmunité résulte de la rupture des mécanismes de tolérance du système immunitaire vis-à-vis des autoantigènes exprimés par les tissus de l’organisme, entraînant la destruction d’un ou de plusieurs organes-cibles par les lymphocytes T et/ou B. L’hépatite autoimmune et le diabète autoimmun se caractérisent par la destruction sélective des hépatocytes et des cellules β pancréatiques, respectivement. De plus en plus d’arguments suggèrent une implication des lymphocytes T CD8+ dans le déclenchement, la progression et la régulation des réponses associées à plusieurs maladies autoimmunes. Dans ce projet, nous avons suivi l’évolution de clones de lymphocytes T CD8+ spécifiques à un antigène particulier dont le site d’expression différait. Pour ce faire, nous avons développé deux nouveaux modèles murins double transgéniques par croisement entre une lignée de souris exprimant un TCR transgénique spécifique à la nucléoprotéine (NP) du virus de la chorioméningite lymphocytaire (LCMV), et une souris exprimant cette NP-LCMV : 1) uniquement dans les hépatocytes (modèle d’hépatite autoimmune), ou 2) simultanément dans le thymus et le pancréas (modèle de diabète autoimmun). L’avidité fonctionnelle des lymphocytes T CD8+ spécifiques à la NP chez les souris TCR transgéniques était inversement proportionnelle au niveau d’expression du TCR. Le répertoire lymphocytaire dans le thymus, la rate, les ganglions et le sang périphérique a été caractérisé pour chacune des lignées de souris double transgéniques, de même que la capacité fonctionnelle et le phénotype (marqueurs d’activation/mémoire) des lymphocytes T CD8+ autoréactifs. Chacun des deux nouveaux modèles présentés dans cette étude ont montré que les lymphocytes T CD8+ spécifiques à la NP sont aptes à briser la tolérance centrale et périphérique et à provoquer une réaction d’autoimmunité spontanée. Dans le modèle d’hépatite autoimmune, où l’expression de l’autoantigène était restreinte au foie, la surexpression du TCR transgénique a entraîné une délétion thymique quasi-totale des lymphocytes T CD8+ spécifiques à la NP prévenant le développement d’une hépatite spontanée. alors qu’un niveau de TCR comparable à celui d’une souris de type sauvage a permis une sélection positive des lymphocytes autoréactifs qui se sont
accumulés dans le foie où ils se sont activés pour provoquer une hépatite autoimmune spontanée. Dans le modèle de diabète autoimmun, où l’autoantigène était exprimé dans le pancréas et le thymus, les souris des deux lignées double transgéniques ont montré une délétion thymique partielle, peu importe le niveau d’expression du TCR. Seuls les mâles adultes développaient un diabète spontané et une partie de leurs lymphocytes T CD8+ exprimaient une combinaison particulière de marqueurs d’activation/mémoire (CD44, CD122, PD-1). Cette population lymphocytaire était absente chez les souris femelles et les mâles sains. L’étude de la tolérance des lymphocytes T CD8+ autoréactifs dans nos deux nouveaux modèles murins double transgéniques a permis d’identifier des mécanismes alternatifs possiblement impliqués dans la tolérance et l’activation, et de mieux comprendre le rôle des lymphocytes T CD8+ autoréactifs dans le processus autoimmun menant à l’hépatite autoimmune et au diabète autoimmun. Ces découvertes seront utiles pour développer de nouvelles approches thérapeutiques ciblant les lymphocytes T CD8+ autoréactifs.
Mots-clés : Souris transgéniques, hépatite autoimmune, diabète autoimmun, lymphocytes T CD8+, tolérance, TCR, site d’expression antigénique, phénotype
Abstract
Autoimmune diseases are chronic and often invalidating disorders affecting more than 5% of the population in developed countries. Autoimmunity results from a breach in tolerance mechanisms towards autoantigens expressed in tissues and organs leading to the destruction of one or more target-organs by T and/or B lymphocytes. Autoimmune hepatitis and autoimmune diabetes are characterised by selective destruction of hepatocytes or pancreatic β cells respectively. Accumulating evidences suggest a direct implication for CD8+ T cells in initiation, progression and regulation processes associated with many autoimmune diseases. To study the development of CD8+ T cell clones towards a particular antigen whose expression site was different, we developed two new double transgenic mice models. Each mice model was obtained after breeding of a mouse expressing a TCR specific for the nucleoprotein (NP) from lymphocytic choriomeningitis virus (LCMV) with a second mouse expressing this LCMV-NP either 1) exclusively in hepatocytes (autoimmune hepatitis model), or 2) broadly in the pancreas and thymus (autoimmune diabetes model). The functional avidity of the NP-specific CD8+ T cells clones in TCR transgenic mice lines was inversely correlated with their respective TCR expression level. The lymphocyte repertoire was characterized in each line of double transgenic mouse, and functional capacities and phenotype (activation/memory markers) of NP-reactive CD8+ T cells were assessed. Both new models of double transgenic mice showed that NP-reactive CD8+ T cells could break central and peripheral tolerance and provoke a spontaneous autoimmune response. In the autoimmune hepatitis model, where autoantigen was expressed only in the liver, overexpression of the transgenic TCR led to massive thymic deletion of the NP-specific CD8+ T cells and no hepatitis developed, whereas TCR expression matching wild-type level allowed positive selection of autoreactive CD8+ T cells which accumulated in the liver, became activated and led to hepatitis development. In the autoimmune diabetes model, where autoantigen was expressed both in the pancreas and thymus, both double transgenic mouse lines showed a partial thymic deletion of NP-specific CD8+ T cells, regardless of the TCR expression level. Only adult male mice developed spontaneous diabetes, and a population of their autoreactive
CD8+ T cells expressed a singular combination of activation/memory markers (CD44, CD122, PD-1) which was not seen in female and healthy male mice. The study of the tolerance of autoreactive CD8 + T cells in our two new double transgenic mouse models led us to identify alternative mechanisms potentially involved in tolerance and activation, and to better understand the role of autoreactive CD8 + T cells in the autoimmune processes leading to autoimmune hepatitis and autoimmune diabetes. These findings will be useful in developing new therapeutic approaches targeting autoreactive CD8+ T cells
Keywords : Transgenic mice, autoimmune hepatitis, autoimmune diabetes, CD8+ T cells, tolerance, TCR, antigen expression site, phenotype
Table des matières
Chapitre 1 Introduction ... 1
1 Le système immunitaire des Vertébrés ... 2
1.1 Les récepteurs antigéniques ... 2
1.2 Les molécules du CMH ... 6
2 La sélection du répertoire des lymphocytes T ... 8
2.1 La sélection des chaînes du récepteur des cellules T (TCR) ... 8
2.1.1 Le cas particulier de la souris avec un TCR transgénique ... 9
2.2 Les mécanismes de tolérance ... 12
2.2.1 L’expression des peptides antigéniques ... 12
2.2.2 La tolérance centrale ... 13
2.2.3 La tolérance périphérique ... 18
2.3 L’activation d’un lymphocyte T ... 30
2.3.1 Affinité et avidité du lymphocyte T ... 30
2.3.2 Voies métaboliques impliquées dans l’activation du lymphocyte T ... 33
3 L’autoimmunité ... 36
3.1 Les maladies autoimmunes ... 36
3.1.1 L’hépatite autoimmune ... 37
3.1.2 Le diabète autoimmun ... 41
4 Problématique ... 43
4.1 Hypothèses ... 45
4.2 Objectifs ... 45
Chapitre 2 Développement d’un nouveau modèle murin double transgénique d’hépatite autoimmune ... 47
Article 1: Mouse liver-specific CD8+ T cells encounter their cognate antigen and acquire capacity to destroy target hepatocytes ... 48
Chapitre 3 Développement d’un nouveau modèle murin double transgénique de diabète autoimmun ... 78
Article 2: New model of double transgenic mouse results in autoimmune diabetes in males ... 79 Chapitre 4 Impact de l’avidité fonctionnelle et du site d’expression antigénique sur la tolérance des lymphocytes T CD8+ autoréactifs ... 94
Article 3: Expression site of antigen and TCR avidity define the selection and fate of CD8+ specific T cell clones ... 95 Chapitre 5 Discussion générale ... 122 1 Évaluation des manifestations pathophysiologiques dans chacun des modèles murins .. 122 1.1 Le modèle d’hépatite autoimmune ... 122 1.2 Le modèle de diabète autoimmun ... 126 2 Sélection du répertoire des lignées de souris transgéniques ... 130 2.1 L’avidité fonctionnelle des lymphocytes T ne correspond pas au niveau d’expression du TCR ... 130 2.2 La surexpression du TCR entraîne la délétion thymique d’un clone de lymphocyte T spécifique à un antigène hépatique ... 132 2.3 Les lymphocytes T autoréactifs des mâles adultes diabétiques présentent un phénotype activé/mémoire particulier ... 136 3 Conclusions et perspectives ... 139
Liste des tableaux
Tableau 1.I Différences et similarités entre les récepteurs antigéniques des cellules B et T (BCR et TCR) ... 5 Tableau 1.II Critères de diagnostic simplifiés pour l’hépatite autoimmune ... 38 Tableau 5.I Comparaison de la tolérance et de la fonctionnalité des LT CD8+ autoréactifs chez les lignées de souris double Tg. ... 141
Supplemental Table 4.I Specific primer sequences used for gene amplification by RT-PCR and PCR ... 121
Liste des figures
Figure 1.1 Structure des récepteurs antigéniques des cellules B et T (BCR et TCR). ... 3
Figure 1.2 Voies de chargement conventionnelles des CMH I et II. ... 7
Figure 1.3 Expression précoce d’un TCR transgénique par rapport à un TCR normal chez la souris. ... 11
Figure 1.4. Interactions entre les thymocytes et les cellules stromales du thymus lors de leur développement en lymphocytes T. ... 15
Figure 1.5. Sélection des thymocytes selon le modèle classique d’affinité. ... 16
Figure 1.6. Déroulement de la réponse immune d’un lymphocyte T. ... 19
Figure 1.7. Mort cellulaire induite par activation dépendante de CD95 (Fas). ... 21
Figure 1.8. Mort cellulaire induite par activation dépendante de HPK1. ... 22
Figure 1.9. Mort autonome de cellule activée. ... 24
Figure 1.10. Distinction entre l’affinité, l’avidité et l’avidité fonctionnelle. ... 32
Figure 1.11. Activation d’un lymphocyte T. ... 34
Figure 2.1 Representative T lymphocyte populations in TNP5 TCR-transgenic mice. ... 70
Figure 2.2 Central and peripheral tolerance in TNP5/TTR-NP double Tg mice. ... 71
Figure 2.3 Functional capacities of Tg TCR CD8+ T lymphocytes. ... 72
Figure 2.4 Autoreactive CD8+ T lymphocytes escape from clonal deletion and accumulate in the liver of TNP5/TTR-NP mice. ... 74
Figure 2.5 Proliferation, activation and acquisition of effector function by liver infiltrating NP-specific CD8+ T cells... 75
Figure 2.6 Capacity of transgenic TCR CD8+ T cells to provoke liver-specific inflammatory process. ... 77
Figure 3.1 Diabetes development in TNP/RIP-NP male mice. ... 91
Figure 3.2 Autoreactive CD8+ T cells from TNP/RIP-NP mice acquire early functional capacities. ... 92
Figure 4.1 Central and peripheral frequencies of NP-specific CD8+ T cells in TCR transgenic
mice. ... 111
Figure 4.2 Expression of transgenic TCR in TNP4 and TNP5 mice. ... 113
Figure 4.3 Functional capacities of Tg TCR CD8+ T cells. ... 114
Figure 4.4 Phenotype of TCR transgenic mice CD8+ T cells. ... 115
Figure 4.5 Functional properties of double transgenic mouse CD8+ T cells. ... 116
Figure 4.6 Phenotype of double transgenic mouse CD8+ T cells. ... 117
Supplemental Figure 4.1 Representative Vα chain transcripts quantification. ... 118
Supplemental Figure 4.2 Central T cell tolerance in TNP4/TTR and TNP5/TTR mice. ... 119
Liste des abréviations
Ac AnticorpsACAD Activated cell autonomous death (mort autonome de cellule activée) ADN Acide désoxyribonucléique
Ag Antigène
AICD Activation-induced cell death (mort cellulaire induite par activation) AID Activation induced cytidine deaminase (désaminase induite par activation) Aire Autoimmune regulator (gène régulateur de l’autoimmunité)
ANA Anti-nuclear antibody (anticorps anti-noyau)
APAF Apoptosis protease activating factor (facteur apoptotique d’activation de la protease) APECED Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy
ARN Acide ribonucléique ARNm ARN messager
ATR Antigène tissulaire restreint
Bak (molécule pro-apoptotique de la famille Bcl-2) Bax (molécule pro-apoptotique de la famille Bcl-2) BCR B cell receptor (récepteur de cellule B)
Bcl-2 (molécule anti-apoptotique de la famille Bcl-2) Bcl-XL (molécule anti-apoptotique de la famille Bcl-2)
Bim (molécule régulatrice de l’apoptose de la famille Bcl-2) BTK Bruton’s tyrosine kinase (tyrosine kinase de Bruton) CD Cellule dendritique
CD40LG CD40 ligand gamma chain (chaîne gamma du ligand de CD40)
cFLIP Cellular FLICE inhibitor protein (protéine cellulaire inhibitrice de FLICE) CMH Complexe majeur d’histocompatibilité
ConA Concanavalline A
CPA Cellule présentatrice d’antigène
CYP : Cytochrome P450
DAI Diabète autoimmun (ou de type I, ou IDDM)
DISC Death-inducing signalling complex (complexe signalétique induisant la mort) DN Thymocyte double négatif
DP Thymocyte double positif
EAE Encéphalite autoimmune expérimentale
ERK Extracellular protein-regulated kinase (ancien nom pour MAPK)
FADD Fas associated protein with death domain : protéine adaptatrice associée à la voie de Fas et contenant un domaine de mort cellulaire.
FLICE FADD-like IL-1β converting enzyme : enzyme se liant à FADD qui prévient la formation du complexe de mort DISC
FoxP3 Forkhead box P3
GAD Glutamic acid decarboxylase (enzyme décarboxylase de l’acide glutamique)
GADA Glutamic acid decarboxylase autoantibody (autoanticorps anti-décarboxylase de l’acide glutamique)
GP Glycoprotéine
GVHD Graft versus host disease (maladie du greffon contre l’hôte) HAI Hépatite autoimmune
HPK1 Hematopoietic progenitor kinase 1 (kinase des progéniteurs hématopoïétiques) IA-2A Insulinoma associated 2 autoantibody (autoanticorps anti-insulinome 2) IAA Insulin autoantibody (autoanticorps anti-insuline)
ICA Islet cell autoantibody (autoanticorps anti-ilot)
IDDM Insulino-dependant diabetes mellitus (diabète insulino-dépendant, aussi appelé diabète autoimmun ou de type 1)
IFN Interféron
Ig Immunoglobuline
IKK I kappaβ kinase (kinase I kappaβ) IL Interleukine
ITAM Immunoreceptor tyrosine-based activation motif (motif d’activation des immunorécepteurs par la tyrosine)
LAP Latency associated peptide (peptide associé à la latence) LB Lymphocyte B (aussi appelé cellule B)
LC1 Liver cytosol type 1 (antigène cytosolique de type 1 du foie)
LCMV Lymphocytic choriomeningitis virus (virus de la chorioméningite lymphocytaire)
LKM Liver kidney microsome (microsome du foie et du rein) LT Lymphocyte T (aussi appelé cellule T)
MAI Maladie autoimmune
MAPK Mitogen-activated protein kinase (protéine kinase activée par la mitogenèse) MBP Myelin basic protein (protéine basique de la myéline)
mTEC Medullary thymic epithelial cell (cellule épithéliale de la médulla du thymus) NHEJ Non-homologous end-joining : système de ligature des extrémités d’un ADN cassé NF-κB Nuclear factor κB (facteur de transcription nucléaire κB)
NOD (mouse) Non obese diabetic (souris diabétique non obèse) NP Nucléoprotéine
OVA Ovalbumine
pCMH Complexe peptide-CMH
PD-1 Programmed death réceptor (récepteur de mort cellulaire programmée)
PD-L1 (or L2) Programmed death réceptor ligand 1 or 2 (ligands du récepteur de mort cellulaire programmée)
PID Primary immunodeficiency disorder (trouble d’immunodéficience primaire) Qa-1 Molécule non-classique du CMH de classe II
RAG Recombination activating gene (gène activateur de recombinaison) RIP Rat insulin promoter (promoteur de l’insuline du rat)
SCID Severe combined immunodeficiency (immunodéficience combinée sévère)
SLA/LP Soluble liver antigen/liver pancreas antigen (antigène soluble du foie/antigène du foie et du pancréas)
SP Thymocyte simple positif
TCR T cell receptor (récepteur de cellule T) Tg Transgénique
TNF Tumor necrosing factor (facteur nécrosant de tumeurs)
TRAF TNF receptor-associated factor (facteur associé au récepteur du TNF) Treg Lymphocyte T régulateur
TTR Transthyrétine
UNG Uracil-DNA glycosylase : glycosylase uracile-ADN VHB Virus de l’hépatite B
VHC Virus de ,hépatite Cl
WAS Waskott-Aldrich syndrome (syndrome de Waskott-Aldrich)
Au bout de la patience, il y a le ciel. Proverbe wolof (Sénégal)
Remerciements
Je voudrais d’abord remercier le Dr Idriss Djilali-Saiah, qui m’a accueillie dans son laboratoire qui en était à ses tous débuts. Un gros merci aussi au Dr Fernando Alvarez, qui a généreusement accepté de prendre le relais pour me superviser durant le dernier droit de mon parcours, c'est-à-dire la rédaction de cette thèse.
Merci à mes collègues qui se sont succédés au laboratoire d’Idriss. Vous avez été des compagnons agréables et aussi des amis(es) sincères : Amin Fakhfakh, Hayet Cherief, Ouiza Zeymour et Thérèse Trinh Huynh, sans oublier mon super-stagiaire Mehdi Alami Hassani que j’ai eu l’immense honneur de superviser durant une session d’été entière.
Je tiens à remercier à Pascal Lapierre et Kathie Béland, au laboratoire du Dr Alvarez, qui se sont toujours montrés ouverts à la discussion lorsque j’avais besoin d’un point de vue supplémentaire sur un résultat qui me laissait perplexe ou pour m’emmener à porter un jugement plus critique sur ma démarche. Merci à Kathie, qui m’a appris de nouvelles techniques que je ne connaissais pas et pour avoir accepté de lire et de me donner ses précieux commentaires sur les manuscrits des articles et sur certaines sections de ma thèse.
Je ne veux surtout pas oublier Sonja Lespérance, technicienne en soins animaliers, qui a bien pris soin de « mes » souris durant toutes ces années et qui s’est toujours montrée patiente et souriante. J’ai toujours pu compter sur elle pour répondre à mes nombreuses questions et demandes de taxi-souris. Merci Sonja, car grâce à toi je suis devenue une vraie pro en gestion…de colonie de souris.
Je veux remercier mes parents Jean-Guy Chabot et Lucille Vallée, ma sœur Nicole, mon frère Éric, et toute ma tribu, la Chabotterie, pour leur soutien indéfectible. Vous m’avez encouragée à persévérer dans mon rêve de devenir une scientifique depuis que je suis toute petite. Un merci spécial à ma petite sœur, Dre Nicole Chabot, pour son écoute et sa solidarité : elle est la mieux placée pour savoir ce que c’est que de poursuivre des études au doctorat, avec
tout le travail, les remises en questions, les joies et le stress que cela implique, car elle est aussi passée par cette épreuve il y a quelques années.
En terminant, je tiens à remercier mon cher époux, Abdou Diop, qui a travaillé très dur durant ces dernières années pour que notre famille ne manque de rien. Il a enduré (presque) sans broncher mes explications passionnées lorsque j’avais obtenus des résultats excitants, même s’il ne comprenait pas grand-chose à mon charabia. Merci aussi à mes trois petits chéris Ahmadou Bamba, Habib et Muhammad, qui ont été mon inspiration et ma force vitale pour aller toujours de l’avant dans les moments de découragement.
Chapitre 1 Introduction
L’autoimmunité résulte d’un dérèglement des mécanismes de tolérance qui ont pour but d’éliminer les lymphocytes réagissant trop fortement vis-à-vis de molécules faisant partie du soi. Dans cette introduction, nous verrons comment le lymphocyte, cellule par excellence du système immunitaire des Vertébrés, a évolué pour devenir une cellule hautement spécialisée grâce à son récepteur unique et spécifique (TCR) qui, de concert avec la molécule du complexe majeur d’histocompatibilité (CMH), permet de discriminer les molécules antigéniques faisant partie du soi et du non-soi.
Nous passerons ensuite en revue la façon dont le répertoire de lymphocytes T est formé, depuis la sélection des chaînes du TCR, en passant par les étapes successives de sélections dans le thymus et les organes périphériques secondaires, et en terminant par les mécanismes d’activation du lymphocyte T lors de sa rencontre avec son antigène correspondant.
Dans la dernière partie, la description de la pathogenèse et les critères de diagnostic des deux maladies autoimmunes faisant l’objet du présent projet, l’hépatite autoimmune et le diabète autoimmun, seront abordés. Les modèles animaux existants pour chacune de ces maladies autoimmunes et sur lesquels les connaissances actuelles ont été obtenues seront brièvement énumérés et commentés.
1 Le système immunitaire des Vertébrés
L’immunité innée est un système d’auto-défense acquis très tôt dans l’évolution, que l’on retrouve chez les organismes unicellulaires primitifs jusqu’aux Vertébrés plus évolués (Cooper and Alder 2006). À la base de ce système de défense de première ligne, des récepteurs constitutifs reconnaissent certains motifs conservés chez différentes classes de pathogènes (bactéries, virus, parasites), permettant l’induction d’une réponse inflammatoire qui limite la progression de l’agent pathogène (Akira, Uematsu et al. 2006). Avec l’émergence des Vertébrés, un système immunitaire adaptatif se développe. On assiste à l’apparition d’un nouveau type de cellule spécialisée, portant un récepteur unique et spécifique à un antigène, capable de donner naissance à une lignée clonale : le lymphocyte.
1.1 Les récepteurs antigéniques
Les récepteurs des lymphocytes B (LB) et des lymphocytes T (LT), respectivement le BCR et le TCR, partagent tous des domaines prototypiques d’immunoglobulines (Ig) (Azumi, De Santis et al. 2003; Cooper and Alder 2006). Leur structure est assez similaire et comprend une région constante C ancrée à la membrane, surmontée par une région variable V impliquée dans la spécificité antigénique (figure 1.1). En raison de la longueur insuffisante des domaines Ig à l’intérieur de la membrane cellulaire, le BCR et le TCR doivent s’adjoindre des complexes invariants qui permettent la transduction du signal lors de la liaison du peptide sur le récepteur. Pour le BCR, il s’agit d’un corécepteur formé des chaînes Igα et Igβ qui contient 2 domaines ITAM (immunoreceptor tyrosine-based activation motif) nécessaires à la transmission du signal. De façon analogue, le TCR requiert la présence du complexe CD3 formé d’une chaîne CD3δ, d’une chaîne CD3γ, de deux chaînes CD3ε et de deux chaînes CD3ζ. Le complexe CD3 contient au total 10 ITAM. Les ITAM sont des domaines hautement conservés chez les mammifères. Leur phosphorylation permet la liaison d’une enzyme (kinase) importante pour initier le signal d’activation du lymphocyte suite à l’engagement du récepteur.
Figure 1.1 Structure des récepteurs antigéniques des cellules B et T (BCR et TCR).
Le BCR est constitué d’un domaine constant (C) et d’un homodimère dont chacun des 2 bras est formé d’une chaîne lourde (H) et légère (L). Ces chaînes comprennent chacune une région constante (C) et une région variable (V). Cette dernière détermine la spécificité antigénique du lymphocyte B. Le corécepteur, formé d’une chaîne Igα et d’une chaîne Igβ, est requis pour le signalement (signalling) grâce à ses 2 motifs ITAMs (rectangle). Le TCR est un hétérodimère formé de 2 chaînes, α et β, comprenant chacune une région constante (C) et une région variable (V) responsable de la spécificité antigénique. L’activation du TCR nécessite la présence du corécepteur CD3, formé des sous-unités εδ, εγ et ζζ, apportant un total de 10 ITAMs pour le signalement. Adapté de (Sadelain, Riviere et al. 2003)
Le système immunitaire adaptatif se caractérise principalement par un répertoire diversifié de lymphocytes arborant chacun un récepteur spécifique à un antigène unique. Pour atteindre un tel niveau de diversité, les Agnathes (Vertébrés sans mâchoire, tel la lamproie) recombinent des segments de gènes riches en leucine, afin d’encoder différents récepteurs de lymphocytes (Cooper and Alder 2006). Suite à l’émergence des Vertébrés à mâchoires, la diversification des récepteurs atteint son apogée avec l’apparition de mécanismes maximisant la variabilité somatique (Azumi, De Santis et al. 2003). La génération de la diversité des récepteurs s’effectue à l’intérieur de la lignée germinale par la recombinaison des segments de variabilité (V), de diversité (D) et de jonction (J) des gènes codant pour les chaînes du récepteur. Ainsi, le BCR est formée d’une chaîne lourde codée par les segments V, D et J et d’une chaîne légère codée par les segments V et J. De même, le TCR est constitué d’une chaîne β (segments V, D et J) et d’une chaîne α (segments V et J). Les gènes activateurs de recombinaisons (RAG1/2, recombination activating genes) sont responsables de l’introduction de recombinaisons aléatoires à la jonction des segments V(D)J des gènes codant pour les chaînes du récepteur. C’est par ce seul mécanisme que la totalité de la diversité des TCR est générée. Le nombre total de combinaisons de TCR possibles a été estimé entre 1012 et 1015 (Davis and Bjorkman 1988). En ce qui concerne le BCR, il existe des mécanismes additionnels qui participent à l’augmentation de la diversité.
La diversité du BCR chez l’humain et la souris peut être augmentée grâce à l’hypermutation somatique et à la commutation isotypique (class-switching), ces deux mécanismes étant sous l’action de l’enzyme AID (activation-induced cytidine deaminase). L’hypermutation somatique se caractérise par l’introduction de mutations ponctuelles dans les régions variables V des chaînes lourde et légère du récepteur. La commutation isotypique permet à une entité V(D)J donnée de s’associer à différentes régions constantes C qui déterminent la classe de l’Ig (isotype). Le mode d’action exact de l’AID reste encore imprécis. Elle pourrait participer à l’édition de l’ARN messager (ARNm), entraînant un bris simple brin qui serait réparé par la machinerie cellulaire, dans le cas de l’hypermutation somatique, ou par le système de réparation NHEJ (non homologous end joining) lors de la commutation
isotypique (Muramatsu, Kinoshita et al. 2000). Plus récemment, il a été proposé que l’AID puisse agir directement sur l’ADN en désaminant une cytidine ce qui résulte en une base uracile (U) qui n’est pas une base habituelle dans l’ADN. Le brin d’ADN erroné est reconnu par l’enzyme UNG (uracile-DNA glycosylase), qui clive l’uracile, laissant ainsi un site abasique qui sera ensuite réparé par d’autres polymérases (Di Noia and Neuberger 2007; Neuberger and Rada 2007).Le brin réparé résultant contient une mutation qui, si elle se situe dans la zone impliquée dans la reconnaissance de l’antigène, donne naissance à un récepteur avec une nouvelle spécificité. Le tableau 1 résume les similarités et les différences entre les récepteurs des cellules B et T.
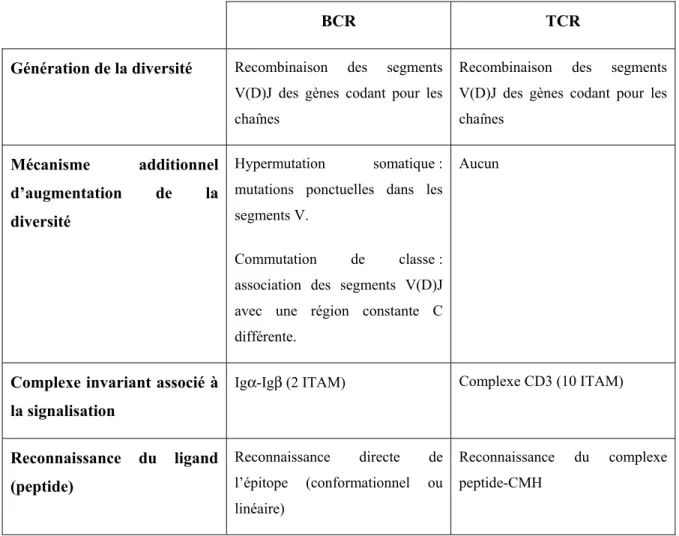
Tableau 1.I Différences et similarités entre les récepteurs antigéniques des cellules B et T (BCR et TCR)
BCR TCR Génération de la diversité Recombinaison des segments
V(D)J des gènes codant pour les chaînes
Recombinaison des segments V(D)J des gènes codant pour les chaînes
Mécanisme additionnel d’augmentation de la diversité
Hypermutation somatique : mutations ponctuelles dans les segments V.
Commutation de classe : association des segments V(D)J avec une région constante C différente.
Aucun
Complexe invariant associé à la signalisation
Igα-Igβ (2 ITAM) Complexe CD3 (10 ITAM)
Reconnaissance du ligand (peptide) Reconnaissance directe de l’épitope (conformationnel ou linéaire) Reconnaissance du complexe peptide-CMH
1.2 Les molécules du CMH
Les molécules du CMH de classe I et de classe II constituent des innovations récentes apparues chez les Vertébrés (Azumi, De Santis et al. 2003). Leur rôle est de présenter des peptides antigéniques aux lymphocytes T (LT), tout en permettant de discriminer le soi du non-soi. La répartition des molécules du CMH dans l’organisme dépend du type de peptide qu’elles doivent présenter (Peters, Neefjes et al. 1991). Toutes les cellules nucléées expriment le CMH de classe I. Cette molécule est formée lors de l’assemblage d’une chaîne lourde de classe I avec la β2 microglobuline (β2m) dans le réticulum endoplasmique (RE). Le CMH de classe I présente aux LT CD8+ des peptides intracellulaires résultant généralement de la dégradation de protéines cytosoliques ou virales par le protéasome ou d’autres protéases. Les fragments (peptides) sont transportés au RE par des protéines transporteuses. C’est à cet endroit que le CMH de classe I et le peptide forment un complexe stable qui est ensuite envoyé à la surface de la cellule (figure 1.2a).
Les molécules du CMH de classe II sont exprimées seulement par un nombre limité de cellules, dont les CPA (macrophages, CD et LB). Le CMH II est aussi présent dans les cellules corticales et médullaires du thymus qui agissent en tant que CPA et jouent un rôle important lors du développement des thymocytes en LT. L’expression du CMH II peut être constitutive ou induite, par exemple par l’IFN-γ chez le macrophage. Les chaînes α et β du CMH II et la chaîne invariante li sont synthétisées et assemblées dans le RE. Le CMH II assemblé transite ensuite via l’appareil de Golgi et le trans-Golgi avant de rejoindre un compartiment endosomal/lysosomal, où il demeure sous une forme inactive en raison de la présence de la chaîne li qui bloque la niche antigénique. Le CMH de classe II présente aux LT CD4+ des peptides provenant de protéines exogènes englouties principalement par endocytose. Ces protéines sont fragmentées dans les endosomes/lysosomes à l’intérieur de la cellule. La chaîne invariante li du CMH II doit être digérée afin de permettre au peptide de s’associer. Le complexe formé est finalement transporté à la membrane cellulaire (figure 1.2b).
Figure 1.2 Voies de chargement conventionnelles des CMH I et II.
(A) Les molécules de CMH de classe I (CMH I) sont chargées avec des peptides dérivés de la dégradation de protéines intracellulaires par le protéasome. (B) Les molécules de CMH de classe II (CMH II) sont chargées avec des peptides provenant de la dégradation de protéines extérieures dans les endosomes/lysosomes. RE, réticulum endoplasmique; TCR, récepteur de lymphocyte T; MIIC, MHC class II compartment (compartiment endosomal tardif); li, chaîne invariante. Adapté de (Klein, Hinterberger et al. 2011)
2 La sélection du répertoire des lymphocytes T
Tous les lymphocytes (B et T) sont issus de progéniteurs communs. Ceux-ci proviennent soit du foie fœtal durant le développement initial du répertoire, soit de la moelle osseuse après la naissance. Pour les LB, le développement des précurseurs continue dans la moelle osseuse. En ce qui concerne les LT, les progéniteurs migrent plutôt vers le thymus afin de poursuivre leur maturation.
2.1 La sélection des chaînes du récepteur des cellules T (TCR)
Les précurseurs des LT qui entrent dans le thymus deviennent des thymocytes. Ils sont dits double négatifs (DN), car ils n’expriment aucun corécepteur CD4 ou CD8. Les chaînes γ et δ du récepteur de cellule T (TCR) sont exprimées à un stade précoce du développement des thymocytes DN immatures. La force du signal du TCR γδ semble jouer un rôle critique dans le choix de la lignée : un signal fort engagerait le thymocyte à poursuivre en tant que γδ, tandis qu’un signal faible amènerait plutôt le thymocyte à s’engager dans la voie αβ (Hayes, Li et al. 2005; Zarin, Wong et al. 2014). Une fraction seulement des précurseurs de cellules T s’engagent dans la lignée γδ. Le lieu de leur maturation est extra-thymique, mais demeure encore inconnu (Bandeira, Itohara et al. 1991). Les LT γδ sont des résidents des tissus épithéliaux, notamment des muqueuses intestinale et pulmonaire. Les LT γδ sont capables de reconnaître un antigène sans que celui-ci n’ait eu besoin d’être traité et présenté par une CPA; toutefois, la majorité des ligands de ces récepteurs demeurent inconnus, de même que les mécanismes de reconnaissance. Le répertoire des LT γδ montre une diversité limitée mais de haute fréquence, ce qui leur permet une réponse rapide sans avoir recours à une expansion clonale préalable (Shin, El-Diwany et al. 2005). Les LT γδ sont impliqués dans la régulation de la réponse immunitaire, mais ne semblent pas jouer de rôle majeur dans la reconnaissance antigénique (Hayday 2000).
La majorité des thymocytes DN s’engagent vers la lignée αβ, ce qui signifie que leur TCR est constitué d’une chaîne α et d’une chaîne β. La maturation du TCR débute avec le réarrangement des segments VDJ des gènes codant pour la chaîne β, jusqu’à l’obtention d’une chaîne fonctionnelle capable de s’assembler avec une chaîne pré-Tα (figure 1.3a). À ce stade, l’exclusion allélique empêche tout nouveau réarrangement de la chaîne β déjà sélectionnée (Uematsu, Ryser et al. 1988). Le réarrangement de la chaîne α, lequel n’est pas soumis à l’exclusion allélique, débute alors. Le réarrangement des segments VJ des gènes de la chaîne α se poursuit jusqu’à ce qu’un TCR fonctionnel, capable de se lier avec un complexe peptide-CMH (ppeptide-CMH) avec une certaine affinité minimale soit assemblé : c’est la sélection positive (Hogquist, Baldwin et al. 2005; Klein, Hinterberger et al. 2009). Lors de la sélection positive qui a lieu dans le cortex du thymus, le thymocyte immature est appelé double positif (DP), puisqu’il exprime à la fois les corécepteurs CD4 et CD8 (Ebert, Ehrlich et al. 2008; Klein, Hinterberger et al. 2009). Les thymocytes qui ne réussissent pas à s’associer peuvent réarranger leur chaîne α jusqu’à ce que le TCR soit capable de se lier efficacement à un complexe pCMH. Un thymocyte qui ne parvient pas à assembler un TCR fonctionnel est éliminé. Selon le type de molécule du CMH avec laquelle le TCR se lie durant la sélection positive, le thymocyte conserve à sa surface l’un des corécepteurs qui le caractérisera, soit le CD4 (avec un CMH de classe II) ou le CD8 (avec un CMH de classe I). Il devient alors un LT CD4+ ou CD8+, respectivement. Les LT CD4+ et CD8+ migrent vers la médulla du thymus où ils subissent une étape de sélection additionnelle, appelée tolérance centrale (Klein, Hinterberger et al. 2009).
2.1.1 Le cas particulier de la souris avec un TCR transgénique
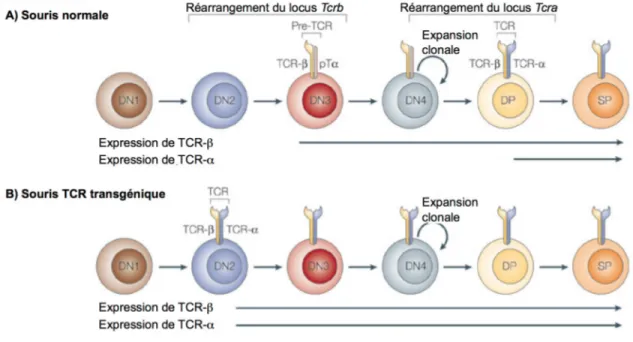
Afin d’étudier les mécanismes de tolérance des LT, plusieurs lignées de souris exprimant un TCR transgénique (Tg) ont été développées (Steinmetz, Bluthmann et al. 1989; Hogquist, Jameson et al. 1994; Barnden, Allison et al. 1998; Mueller, Heath et al. 2002; Bettelli, Pagany et al. 2003). Les gènes codant pour les chaînes α et β du TCR choisi sont introduits dans le pronucléus d’œufs de souris fécondés et implantés chez une souris porteuse.
Les deux chaînes Tg du TCR sont exprimées très tôt durant le développement des LT des embryons, ce qui a pour effet de court-circuiter les étapes de réarrangement et de sélections habituelles (figure 1.3b). Chez ces souris Tg, à cause de l’exclusion allélique, toutes les chaînes β sont Tg (Uematsu, Ryser et al. 1988; Malissen, Trucy et al. 1992). Par contre, l’exclusion allélique ne s’applique pas pour les chaînes α qui peuvent être soit endogènes, soit Tg (Malissen, Trucy et al. 1992). Le répertoire des souris avec un TCR Tg est variable selon la lignée, mais présente une caractéristique particulière. En effet, un TCR Tg restreint par le CMH de classe I favorise un biais (skewing) envers les LT CD8+, i.e. que le répertoire de ces souris Tg montre une plus grande proportion de LT CD8+ comparativement au répertoire d’une souris de type sauvage (Steinmetz, Bluthmann et al. 1989; Hogquist, Jameson et al. 1994; Mueller, Heath et al. 2002). De la même façon, un TCR Tg restreint par le CMH de classe II donnera un répertoire biaisé vers les LT CD4+ (Ho, Cooke et al. 1994; Barnden, Allison et al. 1998; Bettelli, Pagany et al. 2003).
Figure 1.3 Expression précoce d’un TCR transgénique par rapport à un TCR normal chez la souris.
(A) Chez la souris normale, le réarrangement du locus Tcrb codant pour la chaîne β du TCR survient au début du stade DN. Le pré-TCR doit être fonctionnel, i.e. s’associer de façon efficace avec une chaîne pré-Tα (pTα), pour être autorisé à poursuivre avec le réarrangement du locus Tcra, codant pour la chaîne α du TCR, entre les stades DN et DP. (B) Chez la souris avec un TCR transgénique (Tg), les 2 chaînes Tg α et β sont exprimées simultanément dès le début du stade DN et il n’y a pas de réarrangement des locus du TCR. DN, double négatif; DP, double positif; SP, simple positif. Adapté de (Hogquist, Baldwin et al. 2005)
2.2 Les mécanismes de tolérance
Le très grand nombre de combinaisons de chaînes des TCR fait en sorte qu’il y en aura immanquablement quelques uns qui seront spécifiques à des constituants du soi, entraînant ainsi un risque potentiel d’autoimmunité pouvant être à l’origine d’un état pathologique. Afin d’éliminer les clones de LT autoréactifs du répertoire ou de les rendre inopérants, il existe des mécanismes de contrôle. Le premier de ces mécanismes est la tolérance centrale, qui a lieu dans le thymus peu après la génération des lymphocytes T SP. Les clones T autoréactifs ayant survécu à cette sélection et se retrouvant en périphérie seront soumis à la tolérance périphérique, afin d’écarter tout danger potentiel d’autoimmunité.
2.2.1 L’expression des peptides antigéniques
La tolérance centrale implique la présentation de peptides antigéniques aux lymphocytes T nouvellement formés, afin d’éliminer ceux pouvant présenter une réactivité trop forte face aux peptides constituants du soi. Ces derniers représentent un vaste échantillon de protéines pouvant être exprimées simultanément dans plusieurs tissus de l’organisme (antigène ubiquitaire). Par contre, la découverte inattendue dans le thymus de transcrits d’ARN messager codant pour certaines protéines dont l’expression est limitée à seulement quelques tissus isolés a poussé des chercheurs à s’intéresser à la synthèse ectopique de ces protéines (antigènes) tissulaires restreintes à la périphérie.
L’étude de patients atteints d’APECED (autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy), une maladie monogénétique caractérisée par une autoimmunité contre de multiples organes, a permis d’en identifier la cause : une déficience dans l’expression du gène Aire (autoimmune regulator). Des souris chez lesquelles le gène Aire a été artificiellement altéré montrent un défaut de tolérance et développent aussi une réponse autoimmune dirigée contre plusieurs organes : glandes salivaires, follicules ovariens, rétine, etc. (Anderson, Venanzi et al. 2002). Ceci suggère que le gène Aire joue un rôle essentiel dans la délétion clonale des LT spécifiques pour des antigènes tissulaires restreints (ATR) (Liston,
Lesage et al. 2003). De plus, Aire agit comme un facteur de transcription qui contrôle l’expression ectopique de plusieurs ATR à l’intérieur d’une population de cellules épithéliales médullaires du thymus (Derbinski, Schulte et al. 2001; Anderson, Venanzi et al. 2002; Derbinski, Gabler et al. 2005). Ainsi, la tolérance centrale couvre un très large spectre antigénique, au-delà des protéines normalement exprimées dans le thymus, mais englobant aussi des ATR.
2.2.2 La tolérance centrale
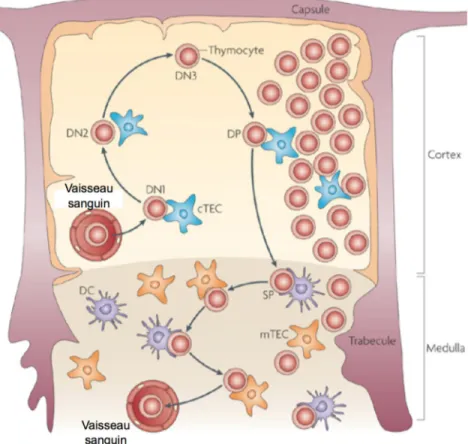
Le thymus est formé de deux régions anatomiques principales. La région externe, appelée cortex, abrite les thymocytes entre les stades DN et DP, tandis que la région interne, la médulla, contient des thymocytes au stade SP (Hogquist, Baldwin et al. 2005; Xing and Hogquist 2012).
La première étape qui façonne le répertoire de LT, la sélection positive, a lieu dans le cortex qui renferme des cellules épithéliales corticales thymiques (cTECs) (figure 1.4). Ces dernières agissent en tant que cellules présentatrices d’antigènes (CPA) et expriment une variété de complexes pCMH pour la sélection positive. Les cellules épithéliales du thymus en général expriment constitutivement un haut niveau de molécules CMH de classe II, mais sont plutôt inefficaces pour présenter des protéines exogènes selon la voie endocytique classique (Kasai, Hirokawa et al. 1996). De plus, des études récentes suggèrent que les cTECs génèrent leurs complexes pCMH en empruntant des voies protéolytiques distinctes des autres CPA présentes dans le thymus ou en périphérie (Klein, Hinterberger et al. 2009). Bien que la gamme complète de pCMH exprimés par les cTECs soit encore indéterminée, il semblerait qu’une diversité limitée de pCMH de haute spécificité soit en mesure de générer un répertoire de LT final très diversifié (Klein, Hinterberger et al. 2009). Les thymocytes DP possédant un TCR capable de former un lien fonctionnel avec un complexe pCMH présenté par une cTEC sont autorisés à poursuivre leur maturation dans la médulla. Par contre, ceux qui échouent meurent par négligence (death by neglect), un phénomène responsable de l’élimination de 80-90% des thymocytes durant le processus de sélection du répertoire (Klein, Hinterberger et al.
2009). Une fraction des thymocytes DP semblent plutôt subir une mort par instruction (death by instruction) : suite à la liaison de la molécule CD8 avec le CMH I en absence d’un engagement productif du TCR, on observe le relâchement de cytochrome c et la fragmentation de l’ADN menant à l’apoptose (Grebe, Clarke et al. 2004).
Les thymocytes positivement sélectionnés s’engagent dans une lignée T en exprimant un seul des marqueurs CD4 ou CD8 avant de passer dans la médulla (figure 1.4). Les thymocytes devenus simple positifs (SP) demeurent dans la médulla pour une période de 4-5 jours. Durant ce séjour, les nombreuses CPA présentes, principalement les cellules épithéliales thymiques de la médulla (mTEC) et exprimant toute une gamme d’antigènes diversifiés sont littéralement «scannées» par les thymocytes en développement. Depuis la dernière décennie, des mécanismes particuliers permettant d’améliorer la présentation d’antigènes très peu exprimés, ou exprimés par un nombre très limité de cellules ont été mis en évidence. Par exemple, lors de la présentation croisée, des ATR exprimés par seulement quelques mTEC, ou encore des molécules pCMH intactes, peuvent ainsi être transférés aux CD voisines qui pourront ensuite les présenter afin d’augmenter la probabilité de rencontre avec un thymocyte spécifique (Klein, Roettinger et al. 2001; Gallegos and Bevan 2004; Millet, Naquet et al. 2008). De plus, des CD migratoires en provenance des organes périphériques seraient aussi capable d’immigrer au thymus afin d’y présenter un antigène non exprimé par les mTEC ou présent en très faible quantité dans la circulation (Bonasio, Scimone et al. 2006; Klein, Hinterberger et al. 2009; Hadeiba, Lahl et al. 2012).
Figure 1.4. Interactions entre les thymocytes et les cellules stromales du thymus lors de leur développement en lymphocytes T.
Les progéniteurs hématopoïétiques provenant de la moelle osseuse arrivent au thymus par la circulation sanguine. Durant son développement dans le cortex du thymus, le thymocyte réarrange d’abord la chaîne β, puis la chaîne α de son TCR. Les contacts périodiques avec les cellules épithéliales du cortex (cTECs) permettent de valider la fonctionnalité des chaînes réarrangées. Une fois que le thymocyte a acquis un TCR fonctionnel au stade double positif (DP), il choisit de s’engager dans la lignée CD4 ou CD8, avant de migrer dans la médulla. Pendant sa résidence de 3-4 jours dans la médulla, le thymocyte scanne les cellules épithéliales médullaires (mTEC) et les cellules dendritiques (CD) présentant toute une panoplie d’antigènes différents qui reflètent l’ensemble des peptides qui composent le soi. Au terme de sa maturation, le thymocyte devient un lymphocyte T CD4+ ou CD8+ et quitte le thymus par la voie sanguine afin de gagner les organes lymphoïdes périphériques (rate, ganglions). Adapté de (Klein, Hinterberger et al. 2009)
Lors de la présentation antigénique, le destin d’un LT dépend du degré d’affinité/avidité de son TCR pour le peptide correspondant. Selon le modèle classique d’affinité (figure 1.5), les LT de faible affinité ne réussissent pas à être positivement sélectionnés et sont éliminés (mort par négligence ou par instruction). Ceux possédant une affinité trop grande sont aussi supprimés (délétion clonale), car ils représentent un danger trop grand d’autoréactivité face au soi. Toutefois, une fraction de ces LT de forte affinité peut être redirigés dans une voie alternative et devenir des cellules T régulatrices (Treg). Seuls les LT avec une affinité intermédiaire sont sélectionnés et peuvent quitter le thymus pour rejoindre les organes lymphoïdes périphériques.
Figure 1.5. Sélection des thymocytes selon le modèle classique d’affinité.
Selon le modèle classique de sélection des thymocytes, l’affinité/avidité du récepteur TCR avec son ligand pCMH détermine le sort de celui-ci. Les thymocytes qui ne réussissent pas à former une association suffisamment forte ou durable avec le complexe pCMH correspondant meurent par négligence. Ceux dont l’affinité avec le pCMH est très forte constituent un risque potentiel d’autoimmunité et sont éliminés par délétion clonale. Toutefois, une fraction des thymocytes de forte affinité peuvent être dérivés dans une voie alternative afin de devenir des cellules T régulatrices (Treg). Adapté de (Xing and Hogquist 2012)
D’autres mécanismes de tolérance centrale plus rares et dont la prépondérance est mal connue peuvent permettre aux thymocytes fortement autoréactifs d’éviter la délétion clonale. Il s’agit de l’édition du récepteur (receptor editing) et de l’induction de l’anergie. L’édition du récepteur consiste en un réarrangement secondaire du locus TCRα entraînant un changement de spécificité du TCR (McGargill, Derbinski et al. 2000). Par ailleurs, l’anergie entraîne un état de non-réponse chez un LT autoréactif, mais ce mécanisme est surtout rencontré en périphérie et fera l’objet d’une section ultérieure (voir section 2.2.3.2).
Les études sur la tolérance centrale des LT ont été réalisées à l’aide de modèles de souris Tg exprimant un TCR et/ou un Ag/superantigène particulier. Selon la nature du TCR, on observe deux catégories de modèles Tg : ceux dont les chaînes du TCR sont exprimées au stade DN, et ceux dont les chaînes du TCR sont exprimées au stade SP (Hogquist, Baldwin et al. 2005). Cependant, dans tous les cas, les chaînes des TCR des souris Tg sont exprimées plus tôt que chez la souris de type sauvage : il y a donc des différences subtiles quant au moment et au lieu de délétion des clones de LT. De manière générale, les modèles de souris Tg exprimant un Ag ubiquitaire (exprimé à la fois dans le cortex et la médulla du thymus) entraînent une délétion clonale précoce dans le cortex. Dans les modèles faisant plutôt appel à un ATR, dont l’expression est contrôlée par Aire chez les mTECs, la délétion clonale se produit plus tardivement dans la médulla (Xing and Hogquist 2012). Étant donné que la chaîne α du TCR chez la souris de type sauvage est exprimée seulement au stade DP, la délétion médullaire (au stade SP) est la plus probable chez les animaux non Tg (Baldwin, Sandau et al. 2005; Xing and Hogquist 2012). C’est aussi au stade SP que les thymocytes des souris normales comptent le plus grand nombre de cellules apoptotiques (Surh and Sprent 1994; Cho, Edmondson et al. 2003).
2.2.3 La tolérance périphérique
Plusieurs mécanismes en périphérie sont mis en place pour réprimer les LT autoréactifs qui ont pu échapper à la tolérance centrale et qui constituent de ce fait un danger potentiel d’autoimmunité. Un LT ayant échappé à la délétion dans le thymus peut facilement être activé en périphérie et provoquer des dommages tissulaires, parce que le seuil critique d’activation serait plus bas en périphérie (Enouz, Carrie et al. 2012). La tolérance périphérique est aussi cruciale pour les LT qui rencontrent leur Ag respectif pour la 1re fois en dehors du thymus comme dans le cas d’un Ag alimentaire ou d’un Ag présent lors d’une infection par un pathogène. Les mécanismes de tolérance périphériques sont dits intrinsèques lorsqu’ils se produisent au sein même du LT concerné, comme par exemple l’apoptose, l’anergie, l’ignorance ou le changement de phénotype. D’autres mécanismes dits extrinsèques impliquent l’intervention de cellules additionnelles, comme les Treg et les CD tolérogènes.
2.2.3.1 L’apoptose
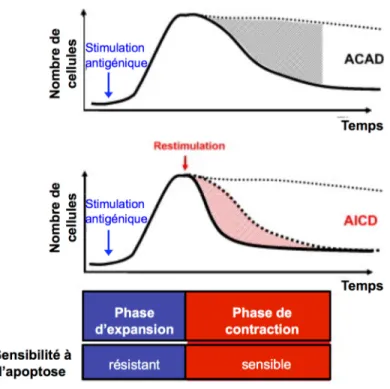
De la même façon que les thymocytes sont sélectionnés dans le thymus, c’est l’avidité du LT qui détermine son sort. L’avidité fonctionnelle se définit comme étant la résultante de l’ensemble des forces de liaison entre le TCR d’un LT avec le complexe pCMH présenté par une CPA. Ce concept englobe l’affinité du TCR pour son ligand, le niveau d’expression du TCR et la quantité du ligand requise pour l’activation du LT, tout en tenant compte également des signaux produits par les autres molécules de costimulation (Ashton-Rickardt and Tonegawa 1994; Hofmann, Radsak et al. 2004; Kroger and Alexander-Miller 2007; Vigano, Utzschneider et al. 2012). En général, un LT mature de haute avidité rencontrant pour la 1re fois son Ag correspondant en périphérie subit une activation initiale, suivie de quelques rondes de division cellulaire; c’est la phase d’expansion durant laquelle les LT sont résistants à l’apoptose (figure 1.6). Par contre, lors de la résolution de la réponse immune (phase de contraction), les LT deviennent sensibles à l’apoptose (Morgan, Kurts et al. 1999; Brenner, Krammer et al. 2008). Il existe deux principaux types de mécanismes apoptotiques. La mort cellulaire induite par activation (AICD, activation-induced cell death) est initiée par des
récepteurs à la surface cellulaire qui se lient à des domaines de mort présents dans le cytoplasme. L’autre mécanisme d’apoptose, la mort autonome de cellule activée (ACAD, activated cell autonomous death) est intracellulaire et indépendante des domaines de mort.
Figure 1.6. Déroulement de la réponse immune d’un lymphocyte T.
Suite à une stimulation antigénique du TCR, les lymphocytes T (LT) subissent une phase d’expansion durant laquelle ils sont résistants à l’apoptose. Au début de la phase de contraction de la réponse immune, les LT deviennent sensibles à l’apoptose. Le type de mort cellulaire qui s’ensuit, autonome (ACAD) ou induite par activation (AICD), dépend respectivement de l’absence ou de la présence de restimulation du TCR par l’antigène. Adapté de (Brenner, Krammer et al. 2008)
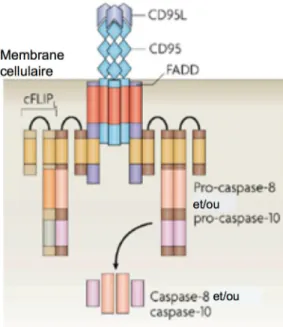
L’activation répétée d’un LT par son Ag spécifique, par exemple lors d’une infection chronique ou d’une maladie autoimmune, peut entraîner l’AICD. L’AICD est une voie apoptotique dépendante du TCR. Cette voie est généralement activée par la molécule CD95 (Fas) qui est exprimée constitutivement sur plusieurs types de cellules dont les LT. L’activation du LT par le lien TCR-pCMH en présence d’IL-2 induit l’expression de CD95L (FasL) par le LT. La liaison de CD95 avec son ligand CD95L entraîne la trimérisation des queues cytoplasmiques de CD95 (figure 1.7). Ensuite, une protéine adaptatrice associée à Fas (CD95) et contenant un domaine de mort (FADD) vient se fixer au site de trimérisation pour former un complexe DISC (death inducing signalling complex) à l’intérieur de la cellule. Le complexe DISC permet aux pro-caspases-8/10 de se fixer pour être activées. Les caspases initiatrices (8/10) activent à leur tour la cascade des caspases effectrices (3 et 7) qui entraînent la mort de la cellule (Krammer, Arnold et al. 2007; Brenner, Krammer et al. 2008).
Figure 1.7. Mort cellulaire induite par activation dépendante de CD95 (Fas).
La mort cellulaire induite par activation (AICD) est souvent dépendante de CD95 (Fas). La liaison de CD95 (Fas) avec son ligand CD95L (FasL) entraîne la trimérisation (regroupement par 3) des parties cytoplasmiques du récepteur CD95 (Fas). Le recrutement de la protéine adaptatrice associée à Fas contenant un domaine de mort (FADD) vient se fixer au site de trimérisation et permet aux pro-caspases initiatrices (8/10) de s’attacher. Le tout forme un complexe signalétique induisant la mort (DISC) qui a la capacité d’activer la cascade de mort constituée par les caspases effectrices (3/7). Adapté de (Krammer, Arnold et al. 2007)
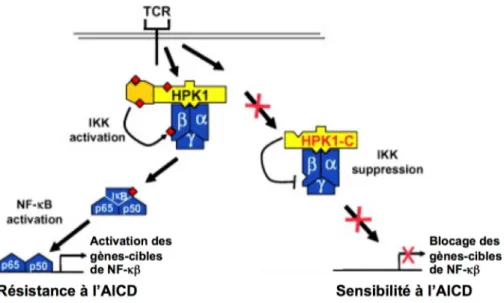
Une autre voie complémentaire, dépendante de HPK1, a aussi été mise en évidence et serait à l’origine de la mort cellulaire en inhibant le complexe IKK, nécessaire à l’activité du facteur de transcription nucléaire NK-κβ (Brenner, Krammer et al. 2008; Israel 2010) (figure 1.8). Les cellules de type Th1 seraient plus susceptibles à la voie induite par CD95L, par contre les Th2 emprunteraient plutôt une autre voie dépendante de la granzyme B (Zhang, Brunner et al. 1997). En effet, des souris déficientes en granzyme B montrent une abolition complète de l’AICD, accompagnée d’une susceptibilité accrue au développement d’une forme d’asthme (Devadas, Das et al. 2006).
Figure 1.8. Mort cellulaire induite par activation dépendante de HPK1.
Suite à l’engagement du TCR, HPK1 (kinase des progéniteurs hématopoïétiques) est clivée en HPK1-N et HPK1-C. La forme HPK1-C rend la cellule sensible à la mort induite par activation (AICD) en bloquant l’activité du complexe IKK (kinase I kappa Β), ce qui prévient l’activation du facteur de transcription nucléaire NK-κβ. Adapté de (Brenner, Krammer et al. 2008)
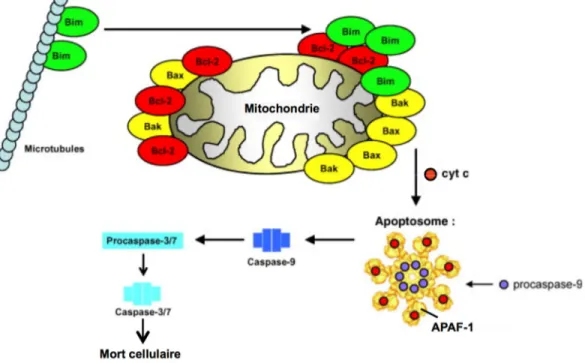
L’apoptose peut aussi être induite de façon indépendante du TCR et des récepteurs de mort cellulaire lors de l’ACAD. En effet, ce mécanisme intracellulaire fait plutôt référence à l’absence de facteurs de survie, ce qui se traduit par la mort de la cellule (Hildeman, Zhu et al. 2002; Brenner, Krammer et al. 2008). Cette voie fait intervenir des molécules de la famille Bcl-2. Cette famille se divise en 3 groupes selon leurs caractéristiques structurales et fonctionnelles (Strasser 2005). Le 1er groupe comprend des molécules anti-apoptotiques telles que Bcl-2 et Bcl-XL. Le 2e groupe englobe des molécules pro-apoptotiques comme Bax et Bak. Le 3e groupe quant à lui est constitué de molécules de régulation, notamment Bad et Bim. Dans la cellule vivante, Bim sous sa forme inactive est fixé sur les microtubules et le niveau de la molécule anti-apoptotique Bcl-2 suffit à contrecarrer les effets pro-apoptotiques de Bax et Bak (figure 1.9). Lors de l’induction de la mort cellulaire, le niveau de Bcl-2 chute et Bim est libéré des microtubules. Celui-ci migre à l’intérieur de la mitochondrie et interagit avec Bax et Bak pour provoquer le relâchement du cytochrome c et l’activation de l’apoptosome. L’apoptosome est constitué du cytochrome c, du facteur apoptotique d’activation de la protéase (APAF-1) et de la pro-caspase 9. Lorsque cette dernière est activée, elle entraîne l’activation des caspases effectrices (3 et 7) qui entraînent la mort de la cellule (Riedl and Salvesen 2007; Brenner, Krammer et al. 2008).
Figure 1.9. Mort autonome de cellule activée.
La molécule régulatrice de l’apoptose, Bim, sous sa forme inactive est liée aux microtubules. Lors de l’induction de la mort autonome de cellule activée (ACAD), Bim devient active et migre vers la mitochondrie, où elle neutralise l’effet anti-apoptotique des molécules Bcl-2 et Bak. Il s’ensuit la libération de cytochrome c (cyt c) qui entraîne l’activation de l’apoptosome, puis la chaîne des caspases initiatrice (9) et effectrices (3 et 7) pour aboutir à la mort de la cellule. APAF-1; apoptotic protease activating factor. Adapté de (Brenner, Krammer et al. 2008)
Le choix de la voie d’élimination des LT lors de la résolution d’une réponse immune dépendrait de la quantité d’antigène présent. Ainsi, à la fin d’une infection aigue, la quantité de l’Ag étranger diminue et l’ACAD contrôlée par Bim sera préférée pour l’élimination des LT (Hildeman, Zhu et al. 2002; Brenner, Krammer et al. 2008). Par contre, lors d’une infection chronique ou d’une réaction auto-immune, les dommages tissulaires libèrent une quantité constante d’Ag, ce qui maintient un niveau relativement élevé de celui-ci. Un niveau élevé d’Ag favorise plutôt l’AICD résultant de l’engagement de CD95/CD95L (Brenner, Krammer et al. 2008).
2.2.3.2 L’anergie
L’activation d’un LT requiert la liaison de son TCR avec le complexe pCMH sur la CPA et la liaison de la molécule CD28 sur le LT avec les molécules CD80 et/ou CD86 (molécules de la famille B7) sur la CPA. L’absence de l’un ou l’autre de ces deux signaux requis pour l’activation du LT provoque un état réfractaire appelé anergie (Harding, McArthur et al. 1992; Schwartz 2003). Le LT anergique se caractérise par la répression du signal du TCR, par l’incapacité à proliférer et par l’absence de production d’IL-2 lors d’un contact avec son Ag spécifique (June, Ledbetter et al. 1990; Linsley and Ledbetter 1993; Lenschow, Walunas et al. 1996; Howland, Ausubel et al. 2000; Xing and Hogquist 2012).
D’autres molécules présentes à la surface cellulaire semblent étroitement associées à l’anergie. Par exemple, la molécule CTLA-4 (cytotoxic T lymphocyte antigen-4) semble être un acteur-clé de l’anergie. En effet, CTLA-4 agit comme un antagoniste de CD28 car il se lie aux molécules de la famille B7 (CD80/CD86) avec une plus forte avidité. Le blocage du lien CD28/B7 provoque la transduction d’un signal négatif empêchant la progression du cycle cellulaire (Tan, Anasetti et al. 1993). Le rôle de CTLA-4 dans l’anergie a été confirmé chez des souris dont l’expression du gène codant pour cette molécule a été aboli (ctla-/-); celles-ci se sont avérées résistantes à l’anergie (Perez, Van Parijs et al. 1997). Par ailleurs, un rôle du récepteur de mort programmée PD-1 dans l’anergie a aussi été proposé. La liaison de PD-1 avec ses ligands PD-L1 et PD-L2 serait en mesure d’inhiber la fonction effectrice des LT
cytotoxiques, soit en freinant la phase initiale d’activation (Probst, McCoy et al. 2005), ou encore en mettant fin au signal du TCR (Fife and Pauken 2011).
2.2.3.3 L’ignorance et le changement de phénotype
L’ignorance peut se produire lorsque l’Ag spécifique à un LT en périphérie n’est pas présent en quantité suffisante pour lui permettre d’atteindre le seuil d’activation. Plus communément, l’ignorance se produit lorsque l’Ag est séquestré dans un lieu immunologique privilégié (cerveau, œil, testicule, utérus); la séparation physique empêche l’Ag d’être présenté dans un organe lymphoïde (ganglion) pour y activer un LT autoréactif spécifique (Miller and Heath 1993). L’ignorance est un mécanisme réversible. Par exemple, lors d’un traumatisme ou d’une infection qui entraînerait le bris de l’étanchéité du compartiment privilégié, l’Ag peut être capturé et entraîné par une CPA dans un ganglion afin d’activer un LT autoréactif.
Par ailleurs, le changement de phénotype consiste en une altération du patron de sécrétion de cytokines des LT auxiliaires Th1 ou Th2, ce qui les rend non pathogéniques (Walker and Abbas 2002). Le changement de phénotype semble être initié, du moins en partie, par le signal des molécules de costimulation lors de l’activation du TCR.
2.2.3.4 Les lymphocytes T régulateurs (Tregs)
Il y plus d’une décennie, des chercheurs ont observé qu’une population de LT CD4+, formés dans le thymus et exprimant la molécule CD25, étaient capables de supprimer l’activité des LT autoréactifs qui avaient échappé à la tolérance centrale (Sakaguchi 2000; Sakaguchi, Sakaguchi et al. 2001). Ces lymphocytes T régulateurs, ou Tregs, comprennent deux populations principales : les Tregs naturels (nTregs) sont formés dans le thymus, alors que les Tregs inductibles (iTregs) se développent en périphérie à partir de LT CD4+ conventionnels (Yuan, Cheng et al. 2014). Ces deux populations de Tregs se caractérisent par l’expression du facteur de transcription Foxp3, qui est indispensable à leur développement et à leur fonction (Hori, Nomura et al. 2003; Fontenot, Rasmussen et al. 2005; Gavin, Rasmussen et al. 2007).
En effet, une diminution du niveau d’expression de Foxp3 mène à la perte de la fonction suppressive des Tregs (Wan and Flavell 2007; Williams and Rudensky 2007).
Durant le processus de sélection des clones de LT dans le thymus, certains LT dont l’affinité est élevée mais qui se situe près du seuil de délétion sont convertis en Tregs au lieu d’être éliminés; c’est ce que l’on appelle la diversion clonale, par opposition à la délétion clonale (Benoist and Mathis 2012). Le signal qui permet la diversion clonale est encore mal connu. Un environnement de cytokines favorable ou encore le fait de posséder un second TCR d’affinité intermédiaire pourrait expliquer ce phénomène (Xing and Hogquist 2012). La génération des Tregs dans le thymus est dépendante de l’engagement du TCR avec le complexe pCMH. La simple présentation de l’Ag par les mTEC semble suffire pour induire la formation de Tregs dans la médulla thymique (Aschenbrenner, D'Cruz et al. 2007). Par ailleurs, la force et/ou la durée du signal TCR pourraient aussi être des facteurs déterminants dans la formation des Tregs. Ainsi, un groupe de chercheurs a montré que l’affaiblissement du signal par le blocage du CMH II sur les mTEC a engendré une formation accrue de Tregs, et une diminution de la délétion clonale (Hinterberger, Aichinger et al. 2010). Enfin, d’autres molécules de costimulation joueraient un rôle prépondérant dans la production des Tregs dans le thymus. La costimulation par CD28 est essentielle et indépendante de l’IL-2 (Tai, Cowan et al. 2005). Par contre, l’IL-2 est nécessaire à la survie des Tregs (Burchill, Yang et al. 2008; Vang, Yang et al. 2008). Pour sa part, l’absence simultanée de TGF-β et d’IL-2 dans le thymus inhibe complètement la formation des Tregs (Liu, Zhang et al. 2008). Les iTregs qui se développent en périphérie possèdent essentiellement les mêmes propriétés suppressives que les nTregs. Ces deux populations de Tregs sont difficiles à distinguer l’une de l’autre, car elles expriment les mêmes marqueurs de surface. Cependant, des études récentes ont identifié des différences spécifiques à chacune de ces populations concernant le nombre de transcrits d’ARNm, l’expression protéique et la stabilité de Foxp3 (Lin, Chen et al. 2013).
En plus des Tregs naturels et inductibles, au moins deux autres populations de LT périphériques possédant des propriétés régulatrices ont été identifiées et désignées sous le nom de lymphocytes Tr1 et Th3. Ces cellules régulatrices n’expriment pas le facteur de
transcription Foxp3 (Lin, Chen et al. 2013). Ce sont des cellules régulatrices secondaires qui exercent leur action par l’entremise de la sécrétion de cytokines suppressives, sans avoir besoin de contact intercellulaire. Les Tr1 ont été décrits pour la première fois chez des souris OVA (avec un TCR Tg spécifique au peptide ovalbumine) qui ont été stimulées de façon répétée avec le peptide OVA et de l’IL-10; les LT naïfs se sont différenciés en LT dont le profil de cytokine différait des Th1 et Th2. Ces Tr1 produisaient majoritairement la cytokine immunosuppressive IL-10, mais peu ou pas de TGF-β, d’IL-2 et d’IL-5 (Groux, O'Garra et al. 1997). Les Tr1 joueraient un rôle dans l’activation des cellules mémoire et dans la suppression des réponses médiées par les Th1 et les Th2 (Groux 2003). Les Th3 ont été identifiés chez des souris suite à l’induction d’une tolérance orale à la protéine basique de myéline (MBP). La majorité des LT CD4+ spécifiques à la MBP sécrétaient du TGF-β. De plus, ces souris étaient résistantes à l’induction d’une encéphalite autoimmune expérimentale (EAE), et cette résistance était annulée par l’injection d’un anticorps anti-TGF-β (Chen, Kuchroo et al. 1994; Fukaura, Kent et al. 1996).
Bien que la majorité des études se soient surtout concentrées sur les Tregs CD4+, de nouvelles observations mentionnent la présence de Tregs CD8+ chez la souris et chez l’homme. Ainsi, des populations de cellules T CD8+Foxp3+, ont montré des propriétés suppressives protégeant contre des maladies autoimmunes ou la maladie du greffon contre l’hôte (GVHD) (Chen, Yan et al. 2009; Kim, Verbinnen et al. 2010; Robb, Lineburg et al. 2012). Des Tregs CD8+Foxp3+ pourraient aussi être la source d’un dysfonctionnement du système immunitaire à l’origine de cancers; en effet, des cellules CD8+Foxp3+ ont été isolées en plus grand nombre dans le sang de patients souffrant de myélomes multiples par rapport à des donneurs sains (Muthu Raja, Kubiczkova et al. 2012). Certaines populations de Tregs CD8+Foxp3+ expriment aussi des chimiokines ou leurs récepteurs, tels que CCL4 ou CCR7 (Joosten, van Meijgaarden et al. 2007; Suzuki, Jagger et al. 2012). Le mécanisme de suppression des CD8+Foxp3+ reste encore mal connu à ce jour, mais une étude récente a suggéré que certains Tregs CD8+Foxp3+ pourraient agir en bloquant les étapes précoces de la signalisation du TCR (Suzuki, Jagger et al. 2012).
D’autre part, les populations de LT CD8+ possédant des capacités régulatrices ou suppressives n’expriment pas toutes le facteur de transcription Foxp3. Ainsi, des cellules CD8+ LAP+(latency associated peptide) converties à partir de LT CD8+ conventionnels en périphérie joueraient un rôle protecteur dans un modèle murin d’EAE (Chen, Yan et al. 2009). Aussi, le rôle de cellules T CD8+ régulatrices restreintes par la molécule non-classique du CMH I Qa-1 a été mis en évidence dans des modèles murins d’EAE et de lupus (Hu, Ikizawa et al. 2004; Kim, Verbinnen et al. 2010). De plus, dans plusieurs modèles murins, différentes populations de cellules T CD8+ suppressives, cette fois avec un phénotype semblable à des cellules mémoires (CD44+CD62L+) ont été identifiées. Ces populations sont caractérisées par l’expression de molécules telles que le CD122 (IL-2Rβ) (Endharti, Rifa et al. 2005; Lee, Ishida et al. 2008; Rifa'i, Shi et al. 2008; Suzuki, Shi et al. 2008), et plus particulièrement PD-1 (Dai, Wan et al. 20PD-10). L’équivalent humain des Tregs CD8+CDPD-122+ n’a pas été formellement identifié, mais une population avec des fonctions très similaires a été isolée; cette dernière se définit comme CD8+CCR3+ (Shi, Okuno et al. 2009).
2.2.3.5 Les cellules dendritiques tolérogènes
Les CD tolérogènes se caractérisent par leur maturation incomplète. Elles sont capables de présenter les Ag aux LT mais sans pouvoir fournir de signal de costimulation adéquat (absence de signal ou signal de coinhibition) (Gallucci, Lolkema et al. 1999). Elles ne proviendraient pas d’une sous-population particulière (Hawiger, Inaba et al. 2001). Étant donné que les LT régulateurs de type Tr1 et Th3 activés produisent de l’IL-10 et du TGF-β, ils sont les principaux suspects impliqués dans la suppression des propriétés stimulatrices des CD, ce qui se traduirait par la formation de CD tolérogènes. La présence de ces dernières favoriserait en retour la différentiation de LT CD4+ naïfs en Tr1 (Jonuleit and Schmitt 2003).