Pour l’obtention du Grade de
Docteur de l’Université de Poitiers
Faculté des Sciences Fondamentales et Appliquées(Diplôme National – Arrêté du 7 Aout 2006)
Ecole Doctorale BioSanté
Secteur de Recherche Aspects Moléculaires et Cellulaires de la Biologie
Présentée par
Stéphanie COCHAUD
Implication du système VIP-récepteurs dans la migration et
l’invasion in vitro de cellules de glioblastome humain
Directeur de thèse : Professeur Jean-Marc MULLER Co-directeur de thèse : Docteur Corinne CHADENEAU
Soutenue publiquement le 17 Décembre 2009 Devant la commission d’examen
JURY
Dr. Didier PELAPRAT – Rapporteur Directeur de Recherche, Inserm U773, Faculté de Médecine Bichat, Paris Dr. David VAUDRY – Rapporteur Chargé de Recherche, Inserm U413,
Université de Rouen
Dr. Corinne CHADENEAU – Examinatrice équipe PCDC, IPBC, UMR 6187 CNRS, Université de Poitiers
Dr. Jean MAZELLA – Examinateur Directeur de Recherche, IPMC, UMR 6097 CNRS Université de Nice-Sophia Antipolis
Pr. Jean-Marc MULLER – Examinateur équipe PCDC, IPBC, UMR 6187 CNRS, Université de Poitiers
Remerciements
Je remercie le Professeur Jean-Marc Muller pour m’avoir accueillie au sein de son équipe, pour son aide pour la rédaction de ce mémoire et pour m’avoir transmis ses connaissances de la pharmacologie des récepteurs.
Je tiens à remercier tout particulièrement le Docteur Corinne Chadéneau pour m’avoir encadrée pendant ce stage et pour m’avoir fait partager ses connaissances et sa rigueur. Merci de votre disponibilité et de votre écoute.
Je remercie les personnes qui m’ont fait l’honneur d’accepter de juger ce travail, de croire en l’expression de ma reconnaissance respectueuse.
Je remercie Annie-Claire Balandre et Gaël Epistolin, pour leur aide technique, leurs conseils tant au niveau professionnel que personnel, et leur bonne humeur.
Je remercie le Cancéropôle Grand-Ouest, à l’inititative de ce projet de thèse et pour leur soutien financier, ainsi que la Ligue Contre le Cancer.
Je remercie toutes les personnes du laboratoire pour les conseils techniques, leur soutien et leur bonne humeur. Merci de mettre une bonne ambiance dans le laboratoire.
Merci également à tous mes amis de près ou de loin, qui m’ont soutenue depuis le Master et qui ont cru en moi. Je pense à toi également Jérôme, merci d’avoir supporté mes hauts et mes bas et de m’avoir permis de vivre au mieux le passage critique de la rédaction de la thèse. Merci à vous tous.
Liste des abréviations
……….………..1
1
èrepartie : Etude bibliographique
………..…4
Chapitre 1 : Le glioblastome multiforme
………..………..5
1. Les données cliniques ... 5
A. Généralités ... 5
B. Incidence et facteurs de risque ... 6
1. Incidence ... 6
2. Facteurs de risque ... 6
a. Les rayonnements ionisants……….6
b. La prédisposition génétique………....7
C. Origine ... 8
1. Théorie de la dédifférenciation des astrocytes ... 8
2. Théorie des cellules souches ... 9
D. Anatomie du GBM ... 11
E. Diagnostic ... 12
2. Les données biologiques ... 13
A. Amplification du récepteur de l’EGF ... 13
1. Généralités ... 13
2. Implication de l’EGFR dans le GBM ... 14
B. Les altérations de INK4A/ARF ... 15
1. Généralités ... 15
2. Délétion du locus INK4A/ARF dans le GBM ... 16
C. Les altérations de p53 ... 17
D. Les altérations de PTEN ... 18
E. La perte d’hétérozygotie du chromosome 10 (LOH 10) ... 20
4. Les traitements ... 21
A. La chirurgie ... 21
B. La radiothérapie ... 22
C. La chimiothérapie ... 23
Chapitre 2 : Les neuropeptides VIP, PACAP et leurs récepteurs…………25
1. Les neuropeptides de la famille du VIP ... 25
A. Généralités ... 25
B. Découverte des neuropeptides VIP et PACAP ... 25
1. Mise en évidence et caractérisation du VIP ... 25
2. Mise en évidence et caractérisation du PACAP ... 26
C. Biosynthèse des neuropeptides ... 26
1. Biosynthèse du VIP ... 26
2. Biosynthèse du PACAP ... 28
2. Les récepteurs du VIP et du PACAP ... 28
A. Des récepteurs couplés aux protéines G ... 28
B. Nomenclature des récepteurs du VIP et du PACAP ... 30
C. Les récepteurs VPAC1 et VPAC2 ... 31
1. Les récepteurs VPAC1 et VPAC2 ... 31
2. Les isoformes des récepteurs VPAC1 et VPAC2 ... 31
D. Le récepteur PAC1 ... 32
1. Le récepteur PAC1 ... 32
2. Les isoformes du récepteur PAC1 ... 32
E. Agonistes et antagonistes des récepteurs ... 33
1. Les agonistes ... 34
2. Les antagonistes ... 35
F. Principales voies de signalisation contrôlées par le VIP et le PACAP ... 35
3. Fonctions des neuropeptides VIP et PACAP ... 36
A. Principales fonctions du VIP et du PACAP ... 36
4. Pathologies liées au VIP, au PACAP et à leurs récepteurs ... 40
A. Pathologies non cancéreuses ... 40
1. Hyperproduction de VIP ... 40
2. Pathologies liées à une déficience de l’innervation VIPergique ou PACAPergique .... 40
3. Autres pathologies ... 41
B. Les pathologies cancéreuses ... 42
1. Les VIPomes ... 42
2. Expression des récepteurs du VIP et du PACAP dans les cellules cancéreuses ... 42
3. La prolifération ... 42
4. La différenciation ... 43
5. L'angiogénèse……….44
6. La migration cellulaire………...45
Chapitre 3 : Migration et invasion des cellules tumorales, régulation par le
système VIP-récepteurs……….46
1. Mécanismes régulant la migration et l’invasion des cellules tumorales .. 46
A. Généralités ... ….46
B. Réorganisation du cytosquelette d’actine ... 47
1. Remodelage du cytosquelette d’actine au cours de la migration ... 47
2. Régulation du cytosquelette d’actine par les petites GTPases de la famille Rho ... 48
3. Régulation du cytosquelette d’actine par les signaux externes : rôle des protéines d’adhérence focale ... 50
C. Dégradation de la matrice extracellulaire ... 51
2. Régulation de la migration et de l’invasion par les neuropeptides VIP et
PACAP ... 52
A. Au cours du développement ... 52
B. Dans la réparation de l’épithélium bronchique ... 53
C. Dans la réponse immunitaire ... 53
Chapitre 4 : Présentation du projet de recherche………..55
2
èmepartie : Présentation des travaux……….58
Chapitre 5 : Présentation de l'article n°1………...….59
Chapitre 6 : Annexe 1 de l'article n°1………..90
Chapitre 7 : Annexe 2 de l'article n°1………..94
Chapitre 8 : Présentation de l'article n°2………98
3
èmepartie : Discussion générale et perspectives
...
121
1. Hétérogénéité inter- et intratumorale du GBM ... 122
2. Les neuropeptides VIP et PACAP : des régulateurs potentiels de la
migration et de l’invasion des cellules de GBM ... 123
A. Régulation de la migration et de l’invasion par le VIP et les peptides apparentés 123 B. Mécanismes moléculaires impliqués dans la régulation de la migration et de l’invasion par le VIP et les peptides apparentés ... 125
1. Le récepteur VPAC1 ... 125
2. Présence de sites de très haute affinité ... 126
3. Implication de la protéine kinase A ... 127
4. Remodelage du cytosquelette d’actine ... 128
3. Les neuropeptides VIP et PACAP et les cellules souches tumorales... 130
4
èmepartie : Annexe……….132
Annexe 1 : Vasoactive intestinal peptide decreases MYCN expression and synergizes with retinoïc acid in a human MYCN-amplified neuroblastoma cell line………...133
Annexe 2 : Hedgehog, Notch and Wnt developmental pathways as target for anti-cancer drugs………...143
~ 1 ~
Les termes suivis d’un astérisque sont en anglais
AC Adenylyl Cyclase*
ACNU autre nom de la Nimustine
AIC 5-AminoImidazole-4-Carboxamide ARF ADP Ribosylation Factor*
BCNU 1,3-Bis(2-Chloroéthyl)-1-NitrosoUrée,autre nom de la Carmustine
Β-PIX Pack-Interacting eXchange factor-beta*
CCNU autre nom de la Lomustine CNF Ciliary Neurotrophic Factor*
CREB cAMP Response Element Binding*
DNA-PK DNA-Protein Kinase*
E jour de vie embryonnaire
EC boucle extracellulaire EGF Epidermal Growth Factor*
EGFR Epidermal Growth Factor Receptor* ELMO1 EnguLfment and MOtility 1*
ENU EthylNitrosoUrée*
ERK1/2 Extracellular-Regulated Kinase ½* FAK Focal Adhesion Kinase*
GAP GTPase-Activating Protein* GBM GlioBlastome Multiforme
GDI Guanine nucleotide Dissociation Inhibitor* GEF Guanine nucleotide exchange Factor* GFAP Glial Fibrillary Acidic Protein* GHF-1 Growth Hormone Factor-1*
GRF Growth hormone Releasing Factor*
GTP Guanosine TriPhosphate*
IC boucle intracellulaire
IL InterLeukine
IP3 Inositol TriPhosphate
IRM Imagerie par Résonance Magnétique
~ 2 ~
KO Knock Out*
LIF Leukemia Inhibitory Factor* LLA Leucémie Lymphoblastique Aigüe LMC Leucémie Myéloïde Chronique
LOH Loss Of Heterozygoty*
MAPK Mitogen-Activated Protein Kinase* MLC Chaine Légère de Myosine
MMP Matrix MetalloProteinase*
Mr Masse relative
MTIC 5-(3-diMéthyle-1-Triazényle)Imidazole-4-Carboxamide NF-κB Facteur Nucléaire κB
NSCLC cancer du poumon non à petites cellules N-WASP Neural Wiskott-Aldrich Syndrom Protein* OMS Organisation Mondiale de la Santé
NRSF Neuron-Restrictive Silencer Factor*
P jour post-natal
PAC1 récepteur spécifique du PACAP
PACAP Pituitary Adenylate Cyclase-Activating Polypeptide* PDGF Plateled Derived Growth Factor*
PHI Peptide Having Carboxyterminal Isoleucine* PHM Peptide Having Carboxyterminal Methionine* PHV Peptide Histidine Valine
PI3K Phospho-Inositide 3 Kinase*
PIP2 PhosphatidylInositol-4,5-bisPhosphate PIP3 PhosphatidylInositol-3,4,5-triPhosphate PKA Protéine Kinase A
PKC Protéine Kinase C PKL Paxillin-Kinase Linker*
PLC PhosphoLipase C
PMA Phorbol-12-Myristate-13-Acetate (ou Acétate de Phorbol Myristate) PrP PACAP related Peptide*
PTEN Phosphatase and TENsin homologue deleted on chromosome 10* RCMI Radiothérapie Conformationnelle avec Modulation d’Intensité RCPG Récepteur Couplé aux Protéines G
~ 3 ~
RIP1 Receptor Interacting Protein 1*
ROCK Rho-associated Coiled-coil forming Kinase* RTK Récepteur Tyrosine Kinase
SCID Severe Combined ImmunoDeficient*
SH2 Src Homology 2*
SNAP-25 Synaptosomal-Associated Protein of 25 kDa* SNC Système Nerveux Central
SVZ Zone Sous Ventriculaire
TGFα Transforming Growth Factor α*
TM domaine transmembranaire
TMZ TéMoZolomide
TPA 12-O-Tétra-décanoylPhorbol 13-Acétate*
TRE TPA/PMA-Responsive Element*
VEGF Vascular Endothelial Growth Factor* VIH Virus de l’Immunodéficience Humaine VIP Vasoactive Intestinal Peptide*
~
4~
lio sa rc o m e lio b la st o m e à ce llu le s g éa n te s lio b la st o m e st ro cy to m e an ap la si q u e an th o as tr o cy to m e p lé o m o rp h iq u e st ro cy to m e d if fu s st ro cy to m e p ilo m y x o ïd e st ro cy to m e p ilo cy ta ir e st ro cy to m e su b -é p en d y m ai re à el lu le s g éa n te s u m eu rs a st ro cy ta ir es G ra d e • • I • • • II • III • • • IV E p en d y m o m e an ap la si q u e E p en d y m o m e m ix o p ap ill ai re E p en d y m o m e m ix o p ap ill ai re S u b é p en d y m o m e T u m eu rs é p en d y m a ir es O lig o as tr o cy to m e an ap la si q u e O lig o as tr o cy to m e T u m eu rs o lig o a st ro cy ta ir es O lig o d en d ro g lio m e an a p la si q u e O lig o d en d ro g lio m e T u m eu rs o lig o d en d ro g lia le s G ra d e • • I • • • II • • • II I IV le a u I : C la ss if ic a tio n d e s g lio m es , O rg a n is a tio n M o n d ia le d e la S a n té 2 0 0 7 . S im p lif ié e d ’a p rè s L o u is e t a l., 2 0 0 7 .
1ère Partie – Etude bibliographique
Chapitre 1: Le glioblastome multiforme
~ 5 ~
Chapitre 1
Le glioblastome multiforme
1. Les données cliniques
A. Généralités
Les tumeurs gliales ou gliomes, originaires du tissu glial, comptent pour 80% des tumeurs primaires du système nerveux central (SNC) chez l’adulte. L’organisation Mondiale de la Santé (OMS) a classé ces tumeurs selon leur origine cellulaire : il s’agit des tumeurs astrocytaires, les oligodendrogliomes, les épendymomes et les gliomes mixtes (mélange d’éléments d’oligodendrogliome et d’astrocytome). Ces gliomes sont également sous-divisés en quatre grades cliniques selon leur apparence histologique basée sur les 4 points suivants : la densité cellulaire, les mitoses, la prolifération microvasculaire et la nécrose (pour revues, Schwartzbaum et al., 2006 ; Buckner et al., 2007) (Tableau I).
Il existe une autre classification fréquemment utilisée en France, la classification de l’Hôpital Sainte Anne. Cette classification repose sur l’examen anatomo-pathologique et sur l’analyse des images obtenues par résonnance magnétique. Elle permet de distinguer trois catégories : les oligodendrogliomes et les oligoastrocytomes de grade A (caractérisés par l’absence d’hyperplasies endothéliales et de prise de contraste), les oligodendrogliomes et les oligoastrocytomes de grade B (caractérisés par une hyperplasie et/ou prise de contraste) et les glioblastomes (Daumas-Duport et al., 1997a, b).
Le glioblastome multiforme (GBM), la forme la plus agressive des gliomes, est un astrocytome classé en grade IV selon l’OMS puisqu’il présente les quatre caractéristiques histologiques citées précédemment. Il représente environ 50% de toutes les tumeurs de type gliales et est associé à un mauvais pronostic. La médiane de survie est d’environ un an en raison de la résistance du GBM aux interventions thérapeutiques, due au caractère complexe de la tumeur elle-même (pour revue, Holland, 2000).
~ 6 ~
B. Incidence et facteurs de risque
1. Incidence
Le GBM reste une maladie rare puisque son incidence est d’environ 3 pour 100 000 par an. Bien que cette tumeur puisse se développer à tout âge, elle est souvent diagnostiquée chez des patients âgés de plus de 50 ans (pour revues, Oghaki et Kleihues, 2005 ; Krex et al., 2007). Les femmes ont un plus faible risque de développer un GBM comparé aux hommes (l’incidence chez les hommes est 1,5 à 2 fois plus élevée que chez les femmes). Cette différence semble être due aux hormones sexuelles puisque chez la femme, le risque augmente à partir de la ménopause, suggérant un rôle protecteur des oestrogènes (McKinley et al., 2000). Une étude effectuée sur des rats a démontré ce rôle protecteur des oestrogènes. En effet, des femelles ovariectomisées et des rats (mâles et femelles) traités avec de la progestérone vivent moins longtemps que des rats traités avec des oestrogènes et des femelles traitées avec un placebo, après implantation des cellules U87 de GBM humain (Plunkett et al., 1999).
2. Facteurs de risque
Le GBM apparaît de façon sporadique et pendant longtemps aucune prédisposition génétique n’a pu être établie. De nombreuses études ont alors évalué l’impact de nombreux facteurs exogènes tels que le tabac, l’alimentation, les rayonnements ionisants, les téléphones cellulaires, les champs électromagnétiques, le statut socio-économique, mais également les facteurs d’origine médicale tels que l’allergie, le statut immunologique et les infections virales. Cependant, aucun lien spécifique entre ces facteurs et l’apparition du GBM n’a pu être établi, exceptée l’association avec l’exposition aux rayonnements ionisants (pour revue, Wrensch et al., 2002).
a. Les rayonnements ionisants
Les radiations thérapeutiques constituent le principal facteur environnemental associé à un fort facteur de risque de développer une tumeur du cerveau. La première évidence de cette association provient d’une étude réalisée sur le suivi d’enfants israéliens ayant reçu des radiations thérapeutiques pour soigner le tinea capitis (la teigne, une maladie de la peau causée par une infection fongique qui touche le cuir chevelu, les cils et les sourcils. En Israël,
1ère Partie – Etude bibliographique
Chapitre 1: Le glioblastome multiforme
~ 7 ~
cette maladie était traitée par irradiation de la fin des années 40 jusqu’aux années 60). Par la suite, des études ont montré que des enfants ayant reçu des irradiations pour soigner la leucémie lymphoblastique aigüe (LLA) développaient des tumeurs 7 à 9 ans après irradiation (pour revues, Oghaki et Kleihues, 2005 ; Bondy et al., 2008).
Une étude réalisée sur les survivants de la bombe atomique d’Hiroshima a montré une haute incidence de méningiomes. Certaines études n’ont montré aucune augmentation du risque de développer des tumeurs du cerveau après une exposition in utero tandis que d’autres études ont montré une augmentation de 20 à 60% du facteur de risque chez l’enfant à la suite d’une exposition prénatale à ces radiations (pour revues, Wrensch et al., 2002 ; Oghaki et Kleihues, 2005 ; Bondy et al., 2008).
La possible implication des techniques de diagnostic, telles que les rayons-X utilisés par les dentistes, dans la formation des gliomes n’est pas clairement démontrée (Ryan et al., 1992).
Finalement, l’association entre la formation des gliomes et l’exposition aux radiations ionisantes ne semble être établie que pour quelques cas. C’est pourquoi, un autre facteur de risque, la prédisposition génétique, a été étudié.
b. La prédisposition génétique
Il semblerait que chez certains individus l’augmentation du facteur de risque de développer un gliome soit une conséquence de l’héritage de multiple variants génétiques. Une étude comparative de génomes entre des patients atteints de gliomes et des sujets sains a été réalisée. Cette analyse menée aux Etats-Unis d’Amérique et au Royaume Uni, a permis à Shete et ses collaborateurs d’identifier 5 loci associés à une prédisposition au gliome : 5p15.33 (TERT), 8q24.21 (CCDC26), 9p21.3 (CDKN2A-CDKN2B), 20q13.33 (RTEL1) et 11q23.3 (PHLDB1). TERT est la transcriptase inverse de la télomérase, essentielle pour le maintien des télomères et l’immortalisation des cellules. CCDC26 code pour un modulateur de l’acide rétinoïque. CDKN2A code pour la protéine p16(INK4A), un régulateur négatif des kinases dépendantes des cyclines (CDK), et pour p14(ARF1), un activateur de p53. RTEL1 maintient la stabilité génomique en supprimant directement la recombinaison homologue et PHLDB1 n’a pour le moment aucun rôle défini (Shete et al., 2009). Une autre équipe a également confirmé l’association de variants génétiques localisés au niveau de 2 loci,
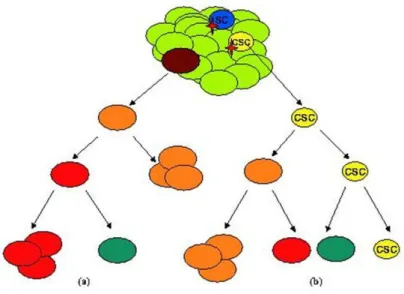
autre hypothèse du développement tumoral serait l’évolution clonale de cellules souches cancéreuses obtenues par la transformation de cellules souches normales résultant de l’accumulation de modifications génétiques (mutations au niveau d’oncogènes ou de gènes suppresseurs de tumeur) et épigénétiques (méthylation anormale, modification des histones).Vert clair : cellules niches, bleu : cellules souches, jaune : cellules souches cancéreuses (CSC), marron, orange, rouge et vert foncé : cellules accumulant des altérations génétiques. D’après Gil et al., 2008.
Figure 2 : Le GBM humain exprime in vitro des marqueurs d’astrocytes. (a) Deux neurosphères dérivées
d’un GBM humain indiquent la diversité des types cellulaires générés à l’intérieur et à l’extérieur des sphères (la tubuline β-III est montrée en vert et la GFAP en bleu). (b) Une unique cellule tubuline β-III positive à partir d’une de ces sphères montre une morphologie qui évoque le comportement migratoire comme observé dans les cellules invasives. Ces neurosphères dérivées de GBM expriment également la tenascine C, en plus d’autres marqueurs associés aux cellules souches astrocytaires normales. Les barres d’échelle représentent (a)=100µm et (b)=10µm. Modifiée d’après Silver et Steindler, 2009.
1ère Partie – Etude bibliographique
Chapitre 1: Le glioblastome multiforme
~ 8 ~
CDKN2B et RTEL1, et le risque de développer un gliome de haut grade (Wrensch et al, 2009).
C. Origine
Les cellules à l’origine du GBM ont été un sujet de discorde pendant plusieurs décennies. L’hétérogénéité génétique mais aussi la présence de plusieurs populations de cellules au sein du GBM (d’où le terme « multiforme ») rendent le débat sur son origine encore ouvert (Shapiro JR et Shapiro WR, 1985). Ainsi deux hypothèses ont été proposées : la dédifférenciation des astrocytes et la présence de cellules souches cancéreuses (Figure 1).
La première repose sur de nombreuses observations indiquant que la tumorigénèse est un processus multi-étapes accompagnées d’altérations génétiques qui conduisent à la transformation progressive de cellules normales en cellules hautement malignes. Six altérations sont essentielles pour la progression tumorale : (1) l’indépendance vis-à-vis des facteurs de croissance, (2) l’insensibilité aux signaux inhibant la prolifération cellulaire, (3) l’affranchissement de l’apoptose, (4) un potentiel réplicatif illimité, (5) la capacité de stimuler l’angiogénèse et (6) la capacité d’envahir d’autres tissus et de métastaser (pour revue, Hanahan et Weinberg, 2000).
La deuxième hypothèse a été décrite pour la première fois par Rudolf Virchow au 19ème siècle. En se basant sur des similarités histologiques entre le développement des cellules embryonnaires et certaines cellules cancéreuses (c’est-à-dire leur capacité de proliférer et de se différencier), Rudolf Virchow propose que le cancer résulte de l’activation de cellules « dormantes » présentes dans les tissus. Par la suite, les progrès techniques en biologie moléculaire ont permis l’identification de cellules souches dites cancéreuses dans plusieurs types de tumeurs (pour revues, Gil et al., 2008 ; Silver et Steindler, 2009).
1. Théorie de la dédifférenciation des astrocytes
Les quelques similarités morphologiques, les marqueurs moléculaires communs (figure 2), et le fait que les astrocytes sont des cellules capables de proliférer dans le cerveau mature, font que ces cellules ont longtemps été considérées comme étant à l’origine du GBM (pour revue, Quigley et al., 2007). Pour que les astrocytes matures puissent être des progéniteurs, ces cellules doivent se dédifférencier afin de produire des cellules malignes,
~ 9 ~
multipotentes qui sont retrouvées dans les gliomes humains. Il a été démontré in vitro et in vivo que l’activation dans des astrocytes corticaux d’oncogènes et la perte de suppresseurs de tumeurs produisent des tumeurs dans des modèles animaux, avec une histologie semblable au GBM (pour revue, Stiles et Rowitch, 2008).
Ainsi, la perte d’INK4A/Arf associée à une activation de KRas et d’Akt dans des astrocytes matures entraîne la formation de tumeurs ayant une morphologie proche du GBM. Dans ce modèle, il apparaitrait que la perte d’INK4A/Arf induit une dédifférenciation des astrocytes, les rendant alors sensibles à la transformation par un stimulus oncogénique tel que KRas activé (Urhbom et al., 2002).
De plus, Bachoo et ses coll. ont pu mettre en évidence la formation de tumeurs à partir d’astrocytes. En se basant sur les mécanismes moléculaires impliqués dans le GBM, ces investigateurs ont montré que les trois facteurs principaux, les suppresseurs de tumeur p16INK4A et p19ARF et le récepteur de l’epidermal growth factor (EGFR), participent à la génèse des gliomes. Les astrocytes obtenus à partir de souris knockout (KO) pour p16INK4A et p19ARF (Ink4a/Arf-/-), et surexprimant le récepteur de l’EGF, subissent une dédifférenciation vers un phénotype de type cellule progénitrice multipotente. L’implantation orthopique de ces astrocytes Ink4a/Arf-/- et surexprimant l’EGFR, induit la formation de gliomes de haut grade (Bachoo et al., 2002).
Cependant, la découverte des cellules souches au niveau du cerveau adulte a entrainé de nouvelles recherches sur l’origine des GBM qui ont confirmé la présence de cellules évoquant les progéniteurs neuronaux pluripotents dans ces tumeurs.
2. Théorie des cellules souches
La première évidence de l’implication de cellules souches tumorales dans l’initiation du cancer a été démontrée dans la leucémie myéloïde chronique (LMC) (Fialkow et al., 1967). En 1997, Bonnet et Dick isolent chez des patients atteints de leucémie myéloïde aigüe (LMA) une sous population de cellules immatures caractérisées par la présence d’un marqueur de surface spécifique CD34 (CD34+) et l’absence du marqueur CD38 (CD38-). Après transplantation de ces cellules CD34+/CD38- chez des souris NOD/SCID, des tumeurs se développent. Au contraire, les cellules CD34+/CD38+, phénotype typique des cellules leucémiques, sont incapables d’initier le développement de tumeurs chez les souris
Figure 3 : Anatomie et fonctionnement de la zone sous ventriculaire et de la zone sous granulaire chez les rongeurs et l’homme. (a) Coupe sagittale à travers le ventricule latéral qui montre l’aire de neurogénèse chez
l’adulte, la zone sous ventriculaire (SVZ). Cette région délimite les ventricules latéraux et est composée de trois principaux types de cellules. Les cellules souches astrocytaires (B) sont capables de s’autorenouveler et d’engendrer des progéniteurs neuronaux à division rapide, les cellules de type C, qui à leur tour produisent des neuroblastes, les cellules de type A. Les cellules de type A migrent vers le bulbe olfactif où elles s’intègrent comme nouveaux inter neurones. (b) Une autre aire neurogénique chez l’adulte est trouvée dans la zone sous granulaire (SGZ), localisée dans le gyrus denté de l’hyppocampe. Une hiérarchie cellulaire, quelque peu similaire à celle de la SVZ, est aussi observée sans la SGZ dans laquelle les vrais cellules souches sont les cellules souches astrocytaires, qui produisent les précurseurs intermédiaires qui éventuellement donnent lieu aux neurones granulaires. Ces neurones s’intègrent dans la couche granulaire. Modifiée d’après Vescovi et al., 2006.
Flux de migration rostrale
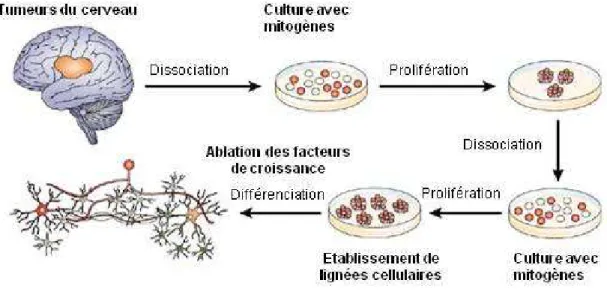
Figure 4 : Isolement et perpétuation de cellules souches de tumeurs du cerveau en culture. La technique de
formation de neurosphères est un système de culture en milieu sans sérum qui permet l’isolement et la propagation de cellules souches dérivées du système nerveux central (SNC). Les précurseurs adultes sont dissociés et placés dans un milieu de culture qui contient les facteurs mitogéniques des cellules souches ; l’epidermal growth factor et/ou le fibroblast growth factor 2. A cause du manque de sérum et de la faible densité, de nombreuses cellules meurent, excepté celles qui se divisent en réponse aux facteurs mitogéniques. Ces cellules prolifèrent pour former des clusters flottants appelés neurosphères. Ces neurosphères peuvent être dissociées en une suspension de cellules et être replacées dans du milieu frais pour former des neurosphères secondaires. Ce processus peut être répété. Après avoir ôté les facteurs mitogéniques, les cellules issues des cellules en prolifération peuvent se différencier en neurones, astrocytes et oligodendrocytes, les trois principaux types cellulaires qui sont trouvés dans le SNC adulte des mammifères.D’après Vescovi et al., 2006.
1ère Partie – Etude bibliographique
Chapitre 1: Le glioblastome multiforme
~ 10 ~
NOD/SCID. La découverte de cette sous population de cellules CD34+/CD38- est la première preuve de l’existence de cellules souches cancéreuses dans la leucémie et est le début de la recherche de la présence de ce type de cellules dans les tumeurs solides (Bonnet et Dick, 1997).
Ce concept a été étendu au GBM et aux autres tumeurs du cerveau, suite à la découverte dans les années 90 de la persistance de la neurogénèse et du développement de cellules matures dans le cerveau adulte dans des zones localisées : le gyrus denté au niveau de l’hippocampe et la zone sous ventriculaire (SVZ) au niveau du ventricule latéral, balayant ainsi l’idée que le cerveau a une structure constante après la naissance et qu’aucun nouveau neurone ne peut être généré à l’âge adulte (figure 3 ; pour revue, Ma et al., 2009). Au niveau de la SVZ, ces cellules souches neurales sont organisées selon une hiérarchie. Les cellules quiescentes de type B qui expriment un marqueur astrocytaire, la glial fibrillary acidic protein (GFAP), et qui présentent des traits morphologiques des astrocytes, sont capables de répondre aux facteurs de croissance tels que l’EGF et le plateled derived growth factor (PDGF). Les cellules de type B traitées avec des facteurs mitogéniques donnent lieu aux cellules à division rapide de type C, qui dans la plupart des cas forment les neuroblastes (cellules de type A) qui migrent vers le bulbe olfactif en suivant le flux de migration rostrale. Cependant, les cellules de type C peuvent aussi donner lieu aux oligodendrocytes durant le développement normal du cerveau (figure 3, pour revue Alvarez-Buylla et al., 2001). Ces cellules souches neurales définies comme ayant des capacités d’auto-renouvellement et étant capables de générer les multiples cellules matures neurales incluant les neurones, les astrocytes et les oligodendrocytes, ont été mises en évidence par leur capacité à former des neurosphères in vitro (figure 4).
En utilisant cette stratégie, de nombreuses équipes ont ainsi pu isoler des cellules souches cancéreuses à partir de GBM (Ignatova et al., 2002 ; Yuan et al., 2004 ; Galli et al., 2004 ; Günther et al., 2008). Après transplantation dans des souris SCID, ces cellules souches cancéreuses sont capables de former des tumeurs ayant les mêmes traits histologiques que le GBM humain, à savoir : (1) la présence de plusieurs aires de nécrose entourées par des structures typiques de pseudo-palissade, (2) une prolifération vasculaire élevée, (3) une immunoréactivité à la GFAP et (4) des mitoses multiples. Ces tumeurs présentent également des capacités de migration et d’invasion typiques du GBM humain. De plus, des cellules souches cancéreuses peuvent être à nouveau ré-isolées à partir de ces tumeurs et peuvent
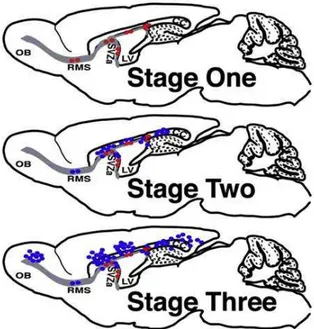
Figure 5 : Cellules souches p53∆5-6 de la SVZ pour et les précurseurs des gliomes. Localisation anatomique
des précurseurs p53∆5-6 durant les étapes précoces du développement des gliomes. Les cellules souches de type B
p53∆5-6 (les points rouges), localisées au niveau de la zone sous ventriculaire (SVZ) accumulent des altérations
génétiques et donnent lieu aux souches de type C p53∆5-6 (les points bleus). Ces cellules souches de type C
peuvent migrer vers le corps calleux (CC) ou vers le bulbe olfactif (OB) en empruntant la voie de migration rostrale (RMS) pour former des tumeurs de type glioblastome. LV : ventricule latéral. Modifiée d’après Wang et
al., 2009.
Figure 6 : Zones nécrotique et pseudo-palissadique du GBM. (a) Les pseudo-palissades sont caractérisées par
une accumulation de cellules tumorales entourant la zone nécrotique centrale. (b) L’hyperplasie microvasculaire (flèche) est une forme d’angiogénèse induite par les cellules pseudo-palissadiques hypoxiques et est usuellement présente dans les régions adjacentes à la nécrose. D’après, Brat et Van Meir, 2004.
1ère Partie – Etude bibliographique
Chapitre 1: Le glioblastome multiforme
~ 11 ~
après transplantation, donner lieu à de nouvelles tumeurs, ce qui démontre l’auto-renouvellement in vivo (Galli et al., 2004).
Ces cellules souches cancéreuses proviendraient de la transformation des cellules progénitrices de type B se trouvant dans les aires du cerveau adulte où persiste la neurogénèse, la SVZ. Dans de nombreux modèles animaux, la SVZ est le site où se développent initialement les gliomes (pour revues, Sanai et al., 2005 ; Stiles et Rowitch, 2008). Par utilisation du sytème Cre/lox, il est possible de produire des modifications génétiques dans des cellules cibles spécifiques. Cette stratégie permet de générer des souris transgéniques qui expriment l’enzyme Cre (une recombinase qui agit lorsqu’elle reconnait dans un segment d’ADN une séquence de 34 paires de bases appelée loxP) sous le contrôle d’un promoteur spécifique de la cellule cible. L’utilisation de ce système à permis de générer un modèle de souris hGFAP-Cre, p53flox/flox, où Cre est sous le contrôle du promoteur de la GFAP humaine et où la protéine p53 est délétée dans les exons 5 et 6. Ces souris hGFAP-cre, p53flox/flox ont permis de démontrer que (1) les cellules souches de type B sont les cellules les plus susceptibles d’accumuler des altérations génétiques, (2) les cellules progénitrices de type C Olig2+ sont les cellules impliquées dans les étapes précoces du développement des tumeurs gliales, (3) la majorité des cellules progénitrices de type C Olig2+ migre vers le corps calleux et une minorité migre en tant que neuroblastes, ou cellules de type A, vers le bulbe olfactif grâce au flux de migration rostrale, et (4) les cellules progénitrices de type C peuvent accumuler d’autres altérations pour former des gliomes de haut grade avec un haut degré d’hétérogénéité (figure 5 ; Wang et al., 2009).
Ainsi, bien que l’initiation du développement tumoral ait lieu au niveau de la SVZ, la migration des progéniteurs de type C anormaux pourrait expliquer la localisation du GBM au niveau du parenchyme au moment du diagnostic (pour revues, Sanai et al., 2005 ; Vescovi et al., 2006 ; Stiles et Rowitch, 2008).
D. Anatomie du GBM
Le GBM est une tumeur très hétérogène comprenant une zone nécrotique au niveau du cœur de la tumeur, puis une zone pseudo-palissadique, une zone tumorale et une zone macroscopiquement normale (figure 6). Cette structure tumorale est due à la croissance rapide du GBM. Les cellules pseudo-palissadiques, qui entourent la nécrose, sont en hypoxie et
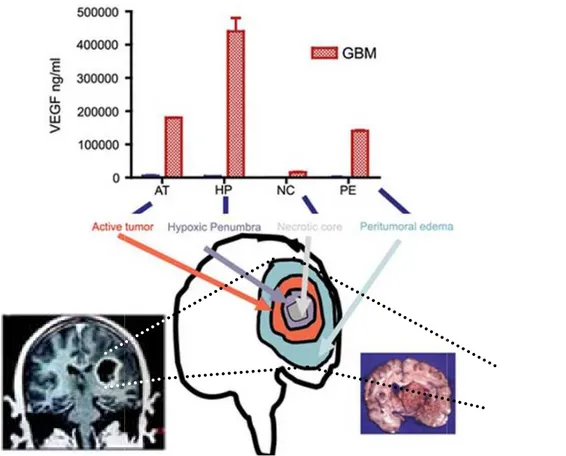
la localisation stéréotaxique intraopérat
dans ces différentes zones. Les zones sont la zone tumorale active (AT) nécrotique (NC) et l’œdème péritumoral (P)
VEGF. Modifiée d’après Jensen, 2009.
Figure 8 : Mécanismes potentiels de la formation de la
induite par l’hypoxie. (1) Une possibilité est que les cellules tumorales se tr
vaisseau sanguin deviennent hypoxiques (cellules bleues) et migrent vers les vaisseaux périphériques, laissant une zone centrale nécrotique (zone en bleu clair)
d’un vaisseau sanguin peut mener à la formation d’une zone hypoxique, suivi de la migration des cellules vers des vaisseaux viables. D’après, Brat et Van Meir, 200
opératoire basée sur l’image préopératoire afin de mesurer la quantité de VEGF Les zones sont la zone tumorale active (AT), la zone hypoxique (HP), la zone péritumoral (P). La zone hypoxique est la zone qui sécrète de très grande quantité de d’après Jensen, 2009.
: Mécanismes potentiels de la formation de la zone pseudo-palissadique lors de
Une possibilité est que les cellules tumorales se trouvant à une grande distance vaisseau sanguin deviennent hypoxiques (cellules bleues) et migrent vers les vaisseaux périphériques, laissant
(zone en bleu clair). (2) Alternativement, l’occlusion vasculaire ou le collapsus d’un vaisseau sanguin peut mener à la formation d’une zone hypoxique, suivi de la migration des cellules vers des vaisseaux viables. D’après, Brat et Van Meir, 2004.
afin de mesurer la quantité de VEGF , la zone hypoxique (HP), la zone de très grande quantité de
palissadique lors de la migration
ouvant à une grande distance d’un vaisseau sanguin deviennent hypoxiques (cellules bleues) et migrent vers les vaisseaux périphériques, laissant lusion vasculaire ou le collapsus d’un vaisseau sanguin peut mener à la formation d’une zone hypoxique, suivi de la migration des cellules vers
1ère Partie – Etude bibliographique
Chapitre 1: Le glioblastome multiforme
~ 12 ~
expriment une forte quantité de facteurs angiogéniques tels que le vascular endothelial growth factor (VEGF). Le VEGF a été trouvé à des concentrations 200 à 300 fois plus élevées dans le liquide kystique du GBM que dans le sérum (figure 7). Le VEGF se fixe sur ses récepteurs de haute affinité, le VEGFR-1 et le VEGFR-2, qui sont surexprimés dans les cellules endothéliales des gliomes de haut grade, ce qui conduit à l’angiogénèse dans les régions hypoxiques adjacentes aux régions pseudo-palissadiques. L’angiogénèse dans les GBM est caractérisée par de nombreuses cellules endothéliales se divisant rapidement (pour revue, Brat et Van Meir, 2004) et ayant une morphologie différente des cellules endothéliales normales. Ces cellules endothéliales ont une apparence plate, avec un large noyau, un cytoplasme abondant et de multiples nucléoles. Ceci entraîne la formation de vaisseaux sanguins désorganisés, tortueux, dilatés, perméables et hémorragiques (pour revue, Charalambous et al., 2006).
La zone pseudo-palissadique présente des cellules qui prolifèrent peu et possède un niveau d’apoptose élevé, comparé à la zone tumorale adjacente. Cependant, les cellules pseudo-palissadiques sont les cellules qui migrent le plus. Deux mécanismes peuvent expliquer la migration induite par l’hypoxie, menant ainsi à la formation de la zone pseudo- palissadique entourant la zone nécrotique. Une possibilité serait que les cellules se trouvant à une grande distance d’un vaisseau sanguin, deviennent hypoxiques à un stade critique de la croissance tumorale. Ces cellules hypoxiques migreraient vers les vaisseaux les plus proches, laissant ainsi une zone centrale composée de cellules qui ne migrent pas et qui peuvent éventuellement devenir nécrotiques. Une deuxième possibilité serait que des vaisseaux sanguins se trouvant dans la zone de la tumeur subiraient un collapsus, menant ainsi à une zone hypoxique perivasculaire et à la migration des cellules loin de la zone d’occlusion (figure 8 ; pour revue, Brat et Van Meir, 2004). Ainsi, la formation de la zone pseudo palissadique peut expliquer la nécrose, la forme de mort cellulaire la plus rencontrée dans le GBM.
E. Diagnostic
Les symptômes du GBM, comme pour d’autres tumeurs du cerveau, incluent les maux de tête, des vertiges, des nausées, des déficits neurologiques ou des troubles cognitifs (pour revue, Grahovac, 2009). Cependant ces symptômes ne sont pas spécifiques et peuvent avoir
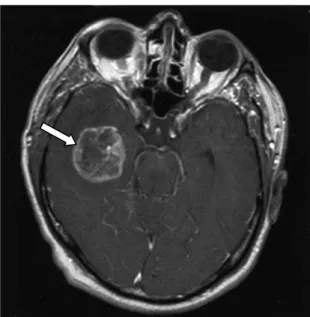
Figure 9 : Image de résonance magnétique (IRM) après injection de gadolinium, un agent contrastant, montrant l’aspect typique d’un glioblastome multiforme (indiqué par les flèches blanches). D’après, Buckner
1ère Partie – Etude bibliographique
Chapitre 1: Le glioblastome multiforme
~ 13 ~
lieu dans d’autres maladies. C’est pourquoi le recours à l’imagerie par résonance magnétiques (IRM) permet un meilleur diagnostic. Les vaisseaux sanguins nouvellement formés dans le GBM étant désorganisés, permettent une nette visualisation de l’agent contrastant, le gadolinium injecté par voie intraveineuse. Ainsi, la présence d’une masse tumorale est facilement décelée par IRM, avec un signal de faible intensité en son centre et un signal de forte intensité correspondant à un œdème en périphérie de la tumeur (figure 9 ; pour revues, Buckner et al., 2007 ; Kumar et al., 2008). Si une masse tumorale est révélée par IRM, un examen anatomopathologique est également réalisé sous forme d’une biopsie et /ou d’une exérèse chirurgicale. Cette approche histologique permet de déterminer le grade tumoral et le niveau de différenciation en se basant sur les critères de l’OMS. Cependant, cette approche reste subjective avec des variabilités inter-observateur dues à l’hétérogénéité et à la croissance diffuse des gliomes en général (pour revue, Kumar et al., 2008).
2. Les données biologiques
Pour identifier les altérations génétiques dans le GBM, 20 661 gènes codant pour des protéines ont été séquencés dans une étude récente pour déterminer la présence d’amplifications et de délétions, et afin d’en définir le profil d’expression (Parsons et al., 2008). Ceci a permis de confirmer l’implication de l’amplification de l’EGFR, les altérations de p16INK4A, de p53, de la « phosphatase and tensin homologue deleted on chromosome 10 » (PTEN) et la perte d’hétérozygotie (LOH) du chromosome 10q dans le GBM.
A. Amplification du récepteur de l’EGF
1. Généralités
L’EGFR, aussi connu sous le nom d’ErbB1 ou Her1, a été le premier membre de la famille des récepteurs humains de l’EGF à avoir été découvert. Il fait partie de la superfamille des récepteurs tyrosine kinases (RTK) puisqu’il contient dans son domaine cytoplasmique plusieurs résidus tyrosine qui sont phosphorylés par suite de la liaison du ligand, permettant l’activation du récepteur. Cela entraîne l’activation des substrats en aval et la transcription de
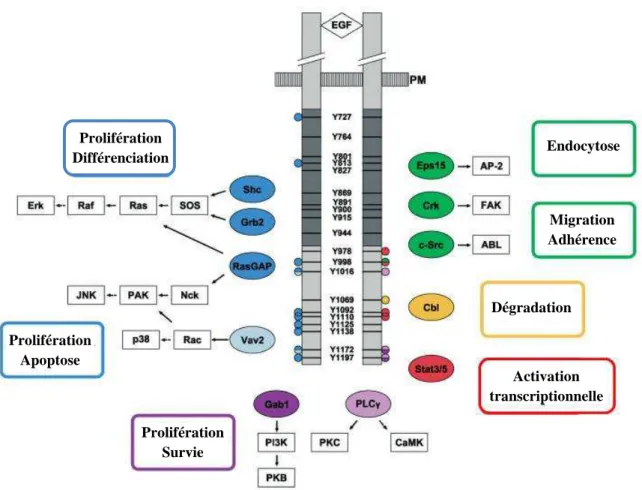
Figure 10 : Principales voies de signalisation de l’EGFR. Les sites de phosphorylation au niveau des résidus
tyrosine sur l’homodimère EGFR activé par l’EGF sont indiqués par des traits noirs. Les sites de liaison connus sont indiqués par des cercles colorés, les couleurs correspondantes indiquent les interactions directes des partenaires avec les fonctions biologiques associées. Le domaine kinase est coloré en gris foncé. PM : membrane plasmique. Les protéines clés permettant d’activer la cascade de signalisation des mitogen activated protein kinases (MAPKs) sont le growth factor receptor bound protein 2 (Grb2) et Src homology 2 domain containing transforming protein (Shc). D’autres MAPKs activées sont c-Jun terminal kinase (JNK) et p38. La protéine kinase C (PKC) peut être activée par la protéine phospholipase Cγ (PLCγ), et la protéine kinase B (PKB) par la phosphatidylinositol-3-kinase (PI3K). Les protéines STATs (signal transducer and activator of transcription) sont activées par interaction directe avec l’EGFR. Les kinases associées à la régulation de la motilité et à l’adhérence telles que la focal adhesion kinase (FAK) ou Abelson proto-oncogene homolog (Abl) sont activées par interaction avec des partenaires comme v-src v-jun sarcoma virus 17 oncogene homolog (c-Src) ou v-crk sarcoma virus CT10 oncogene homolog (Crk), respectivement. D’après Morandell et al., 2008.
Prolifération Différenciation Prolifération Survie Prolifération Apoptose Activation transcriptionnelle Dégradation Migration Adhérence Endocytose
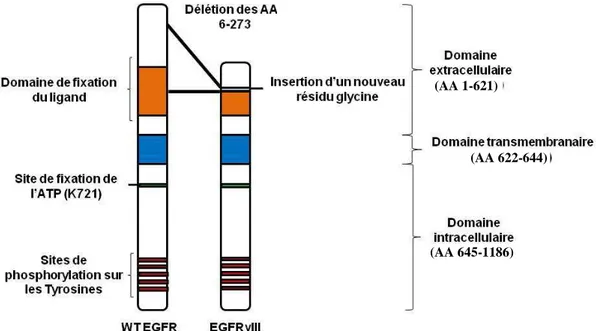
Figure 11 : Représentation schématique du récepteur de l’epidermal growth factor (EGFR)vIII. Le variant
EGFRvIII est caractérisé par une délétion des exons 2-7 du gène du type sauvage (WT) de l’EGFR. Ceci entraine l’absence des acides aminés (AA) 6 à 273 du domaine extracellulaire de la protéine sauvage, permettant la formation d’un variant du récepteur constitutivement actif qui ne peut pas fixer le ligand. L’EGFRvIII contient aussi un nouveau résidu glycine inséré dans la zone de jonction. D’après Gan et al., 2009.
(AA 1-621)
(AA 622-644)
~ 14 ~
gènes contrôlant de nombreuses fonctions cellulaires telles que la division cellulaire, la survie et la mort cellulaire, la migration et l’invasion (figure 10 ; pour revue, Morandell et al., 2008). L’amplification de l’EGFR a lieu dans environ 40% des GBM et cette amplification est souvent associée à des altérations structurales. Sept variants majeurs mutés de l’EGFR ont été identifiés, mais le plus commun dans le GBM est le variant III (EGFRvIII), aussi appelé de2-7EGFR ou deltaEGFR. Ce variant est présent dans 20 à 50% des GBM présentant une amplification EGFR (pour revue, Oghaki, 2005). La délétion des exons 2 à 7 entraîne la biosynthèse d’une protéine avec une nouvelle conformation du domaine extracellulaire, laissant les domaines transmembranaire et intracellulaire intacts (figure 11). Ce variant, bien qu’incapable de fixer ses ligands, l’EGF et le transforming growth factor α (TGFα), est constitutivement actif, phosphorylant ainsi les protéines effectrices même en absence de liaison du ligand (pour revues, Nicholas et al., 2006 ; Gan et al., 2009).
2. Implication de l’EGFR dans le GBM
La présence d’EGFRvIII dans le GBM favorise la tumorigénicité au travers de multiples mécanismes. La surexpression d’EGFRvIII dans une lignée cellulaire de GBM humain, les cellules U87, entraîne une croissance tumorale in vivo et in vitro plus importante comparée à la lignée parentale (U87-mock) ou la lignée parentale surexprimant l’EGFR non muté (U87.wt EGFR). Cette croissance tumorale semble être le résultat d’une augmentation de la prolifération cellulaire et d’une réduction de l’apoptose (Nagane et al., 1996). La principale voie de signalisation activée par l’EGFR étant la voie de la phosphatidylinositol-3-kinase (PI3K), il a été montré que l’activité de la PI3K est augmentée dans les cellules U87 surexprimant EGFRvIII (U87.∆EGFR). Cette augmentation de l’activité de la PI3K entraîne la phosphorylation d’Akt et une réduction du niveau d’expression de p27KIP1, une protéine régulatrice du cycle cellulaire qui inhibe la transition G1/S. La diminution de l’expression de p27KIP1 dans les cellules U87.∆EGFR permet une activité plus élevée du complexe CDK2/cycline A que dans les cellules U87.mock, menant ainsi à l’hyperphosphorylation de la protéine RB et à la progression du cycle cellulaire (Narita et al., 2002). Dans d’autres cellules de GBM, les cellules U373, la présence d’EGFRvIII favoriserait la croissance tumorale in vitro et in vivo, surtout dans des conditions hypoxiques. Dans ces conditions où le manque de nutriments et où le stress cellulaire sont des phénomènes majeurs, la présence d’EGFRvIII
Figure 12 : Organisation du locus INK4A/ARF. Les régions codantes de p16INK4a sont en noir, et celles de p14ARF sont en gris. Les premiers exons alternatifs sont transcrits à partir de promoteurs différents (flèches).
~ 15 ~
constituerait un avantage pour la croissance ou favoriserait la survie des cellules tumorales (Weppler et al., 2007).
La présence d’EGFRvIII confère également une radiorésistance aux cellules de GBM. Les cellules U87.∆EGFR montre une hyperactivation de la sous unité catalytique de la protéine kinase dépendante de l’ADN (DNA-PKc), une protéine impliquée dans la réparation des cassures doubles brins induites par les radiations ionisantes. Après une exposition aux radiations ionisantes, cette hyperactivation de la DNA-PKc permet une réparation des cassures doubles brins avec une cinétique plus rapide dans les cellules U87.∆EGFR que dans les cellules parentales U87.mock, conférant ainsi aux cellules U87.∆EGFR une radiorésistance. Cette hyperactivation de la DNA-PKc semble être le résultat de l’activation de la voie PI3K/Akt. En effet, l’inhibition de PI3K par le LY294002 empêche la réparation rapide des cassures doubles brins conférée par la surexpression d’EGFRvIII et l’hyperactivation d’Akt mime les effets de la surexpression d’EGFRvIII sur la réparation des cassures doubles brins dans les astrocytes. Ces mécanismes de résistance aux radiations ionisantes restent performants in vivo dans un modèle de gliome orthotopique chez la souris (Mukherjee et al., 2009).
B. Les altérations de INK4A/ARF
1. Généralités
Le locus INK4A/ARF, aussi connu sous le nom de CDKN2A, code pour deux protéines distinctes obtenues par l’utilisation alternative de deux promoteurs distincts. La protéine p16INK4A (p16), une protéine inhibitrice des CDK, est générée par un ARN messager (ARNm) comprenant l’exon 1α et les exons 2 et 3, tandis que la protéine p14/p19ARF (ARF ; p14ARF chez l’homme et p19ARF chez la souris), qui est impliquée dans la régulation de la fonction de p53, correspond à l’exon 1β et aux exons communs 2 et 3 (figure 12). Ces deux protéines produites à partir du locus INK4A/ARF, ayant des séquences en acides aminés différentes, sont toutes les deux capables de contrôler le cycle cellulaire en régulant deux composants suppresseurs de tumeur : pRB et p53 (pour revue, Ivanchuk et al., 2001).
p16 se fixe sur les CDK4/6 pour inhiber leur activité kinase et ainsi arrêter la progression du cycle cellulaire en G1. Le rapide recyclage de la cycline D permet à p16
Figure 13 : Voies de signalisation p53 et pRB. P16INK4A est impliquée dans la voie pRB où elle se lie à CDK4
pour inhiber la phosphorylation de pRB et ainsi inactiver E2F1ce qui bloque l’entrée en phase S. P14ARF (p19ARF
est l’homologue chez la souris) forme un complexe avec MDM2 pour stabiliser p53 en empêchant sa
dégradation et favoriser l’apoptose via Bax ou l’arrêt du cycle cellulaire via p21. p14/p19ARF et p53 sont des
~ 16 ~
d’entrer en compétition avec la cycline D pour s’associer aux CDK4/6, entrainant la formation d’un complexe CDK4/6 inactif contenant p16 au lieu d’un complexe CDK4/6 actif avec la cycline D. L’inhibition de l’activité kinase de CDK4/6 empêche la phosphorylation de pRB menant à la restriction de la transactivation par E2F de l’expression de gènes importants pour l’entrée en phase S (figure 13 ; pour revue, Ohtani et al., 2004).
ARF est capable d’induire l’arrêt du cycle cellulaire au niveau de la transition G2/M aussi bien de la transition G1/S. Cet arrêt du cycle cellulaire par ARF est dépendant de p53. ARF interagit avec MDM2, une ubiquitine ligase responsable de l’ubiquitinylation et de la dégradation de p53. Cette interaction permet le passage de MDM2 du noyau vers le nucléole. La séquestration et l’inhibition de MDM2 permettent une stabilisation de l’expression de p53, qui peut agir comme facteur de transcription, induisant des gènes cibles tels que p21 et l’arrêt du cycle cellulaire (figure 13 ; pour revue, Gallagher et al, 2006).
2. Délétion du locus INK4A/ARF dans le GBM
La délétion du locus INK4A/ARF est rencontrée dans environ 60% des GBM et constitue une des plus fréquentes mutations dans ces tumeurs. Les premières études réalisées concernent principalement p16. L’analyse de l’expression de p16 dans des gliomes de différents grades a suggéré que la perte de p16 a lieu lors de la progression tumorale au niveau de la transition du grade II au grade III (Nishikawa et al., 1995). La perte de p16 confère un avantage pour la croissance tumorale puisque l’introduction de l’ADNc de p16 dans des lignées de GBM ne produisant pas de protéine p16 endogène, inhibe la croissance cellulaire (Arap et al., 1995). Cette inhibition de la croissance cellulaire passe par une inhibition de la prolifération consécutive à l’arrêt du cycle cellulaire en phase G1, et non à une augmentation de l’apoptose (Uhrbom et al., 1997). L’effet suppresseur de tumeur de p16, à la suite de sa réexpression, passerait également par sa capacité à inhiber l’invasion de cellules de GBM en réduisant l’activité protéolytique de la matrix metalloproteinase-2 (MMP2) (Chintala et al., 1997).
Des travaux ont démontré que la perte d’Ink4a/Arf est nécessaire pour induire des lésions de type GBM à partir de progéniteurs neuronaux ou d’astrocytes en coopération avec l’activation d’EGFR, ou avec l’activation de Ras (Uhrbom et al., 2002) Cependant, malgré le rôle suppresseur de p16 et ARF, ces deux protéines auraient des fonctions bien distinctes
1ère Partie – Etude bibliographique
Chapitre 1: Le glioblastome multiforme
~ 17 ~
selon le type cellulaire utilisé comme cellule d’origine des GBM (les astrocytes ou les progéniteurs neuronaux). La perte d’ARF rend les astrocytes plus sensibles à la transformation tumorale induite par l’activation de Ras tandis que la perte de p16 permet la formation de lésions typique de GBM uniquement à partir des progéniteurs neuronaux surexprimant Ras (Uhrbom et al., 2005).
La délétion du locus INK4A/ARF est souvent associée à l’amplification d’EGFR dans le GBM. La perte du locus INK4A/ARF favorise l’activation soutenue des voies de signalisation en aval d’EGFR (Lachat et al., 2004), telles que la voie PI3K/AKT/mTOR ou la voie ras/raf/MEK/ERK, permettant ainsi la formation de GBM de novo (Zhu et al., 2009).
C. Les altérations de p53
Bien que p14ARF soit intimement liée aux fonctions de p53, aucune corrélation n’a été observée entre les délétions du locus INK4A/ARF et les mutations de p53 dans le GBM. Au cours de la gliomagénèse, un stress oncogénique pourrait créer une pression de sélection pour muter soit p14ARF soit p53 (Fulci et al., 2000). Au niveau du GBM, les mutations de p53 sont principalement des mutations hétérozygotes. Les cellules de GBM auraient la capacité de développer un (des) mécanisme(s) qui permet(tent) de favoriser l’expression de p53 mutée non fonctionnelle soit (1) par l’extinction de la transcription de p53 sauvage, soit (2) par la dégradation de l’ARNm de p53 sauvage, ou soit (3) par la surexpression sélective de l’ARNm de p53 mutée. Un des mécanismes impliqués dans l’extinction de l’ARNm de p53 sauvage est la méthylation du promoteur p53 (Szybka et al., 2008). Néanmoins, l’extinction de l’activité de la protéine p53 sauvage serait également due à un mécanisme autre que les changements épigénétiques au niveau du promoteur p53. En effet, la protéine p53 peut être régulée par le receptor interacting protein 1 (RIP1). RIP1 est une protéine intervenant dans la voie de signalisation du facteur nucléaire κB (NF-κB). Elle est absente dans les gliomes de bas grades (les grades II et III) et voit son expression augmentée dans environ 30% des GBM. La présence de RIP1 dans les cellules de GBM entraîne l’activation de NF-κB, menant à la surexpression de mdm2 et à l’inhibition de p53 (Park et al., 2009).
Bien que la perte de p53 dans des souris KO p53-/- n’induise pas spontanément la formation de tumeurs de type gliales, elle induit une hyperplasie cellulaire au niveau de la SVZ adulte, caractérisée par des groupes de cellules souches GFAP+, de glie mature et de
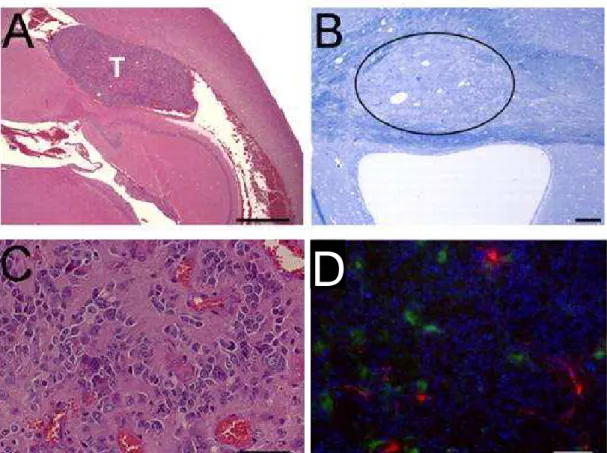
Figure 14 : Les tumeurs de type glioblastome sont détectées dans les zones périventriculaires chez des souris adultes p53-/- qui ont été exposées au mutagène ENU. A, Micrographie d’une section sagittale marquée
avec l’hématoxyline/éosine montrant une grosse masse tumorale (T) dans la zone périventriculaire. Barre d’échelle, 500µ m. B, Section marquée avec du bleu toluidiné montrant une masse tumorale envahissant le corps calleux et déplaçant les axones myélinés (cercle). Barre d’échelle. 75µ m. C, Fort grossissement de la tumeur, montrant des cellules anaplasiques, une vaste microvascularisation et des hémorragies typiques des glioblastomes. Barre d’échelle, 50µm. D, Analyse immunohistochimique de la masse tumorale montrant la présence de cellules nestine+ (vert) et GFAP+ (rouge). Le DAPI (bleu) est utilisé comme co-marqueur nucléaire. Barre d’échelle, 200µm. D’après Gil-Perotin et al., 2006.
1ère Partie – Etude bibliographique
Chapitre 1: Le glioblastome multiforme
~ 18 ~
neuroblastes. Cette perte de p53 confère une augmentation de la prolifération des cellules progénitrices de type B, C et A et favorise la progression des cellules de type C vers un phénotype différencié. A la suite d’une exposition prénatale à l’éthylnitrosourée (ENU, un agent alkylant endommageant l’ADN), les souris p53-/- développent des tumeurs de type GBM au niveau de la SVZ adulte (figure 14). Ceci est une conséquence de la stimulation de l’auto-renouvellement et de la division cellulaire plus rapide des cellules progénitrices, associée à une expansion et une différenciation détériorée des progéniteurs multipotents. Ces effets sont similaires à ceux observés in vitro dans les cellules p53-/- surexprimant Ras sous une forme constitutivement active (Gil-Perotin et al., 2006). En association avec la perte de PTEN, la perte de p53 dans les cellules souches progénitrices entraîne une augmentation de l’expression de MYC, un facteur transcriptionel connu pour son rôle dans la progression du cycle cellulaire et l’apoptose mais aussi dans l’auto-renouvellement des cellules souches et de la différenciation au cours du développement et des processus oncogéniques. Cette augmentation de l’expression de MYC empêche la différenciation des cellules souches et favorise la prolifération et la capacité d’auto-renouvellement de ces cellules, entraînant la formation de gliomes de haut grade présentant des similitudes clinique, pathologique et moléculaire avec le GBM primaire chez l’homme (Zheng et al., 2008).
D. Les altérations de PTEN
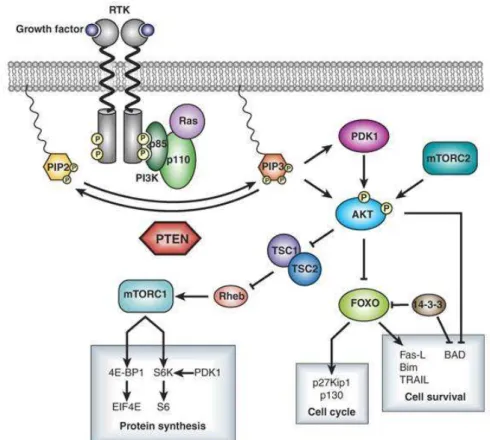
PTEN, aussi connu sous le nom de « mutated in multiple advanced cancers » (MMAC) ou « transforming growth factor β-regulated and epithelial cell enriched phosphatase 1 (TEP1) », déphosphoryle in vitro ses substrats au niveau des résidus sérine, thréonine et tyrosine. Elle possède également une activité phosphatase pour le phosphatidylinositol-3,4,5-triphosphate (PIP3), libérant le phosphate se trouvant en position 3 du cycle inositol, pour générer le phosphatidylinositol-4,5-bisphosphate (PIP2), bloquant ainsi directement la voie de signalisation de la PI3K (figure 15 ; pour revue, Endersby et Baker, 2008).
Les mutations de PTEN ont lieu dans 25% des GBM. Les principales mutations rencontrées dans le GBM sont les mutations non-sens et faux-sens, des délétions et des insertions de codons stop menant à la traduction d’une protéine tronquée. La plupart de ces mutations sont localisées dans l’ensemble des exons, tandis que les mutations faux-sens
phosphatase de PTEN réverse cet évènement et contrecarre directement les signaux de la PI3K. Les sérine/thréonine kinases AKT et PDK1 sont recrutées à la membrane par fixation de leur domaine PH au PIP3. Suite au recrutement à la membrane, AKT est activée par phosphorylation par PDK1 et mTORC2. AKT phosphoryle de nombreuses cibles pour transmettre des signaux de croissance, de prolifération et de survie. D’après Endersby et Baker, 2008.
Figure 16 : Représentation graphique des mutations de PTEN rencontrées dans le GBM (en bleu) dont certaines sont également détectées dans des harmatomes, des malformations tissulaires d’aspect tumoral (en violet). PTEN contient 9 exons (gris) et code une protéine de 403 acides aminés. Les domaines de PTEN
incluent une région de fixation au PIP2 (PBR, rouge), un domaine phosphatase (vert), un domaine C2 (jaune), avec une queue C-terminale contenant deux domaines PEST pour la dégradation (orange) et une séquence consensus permettant l’interaction avec les motifs PDZ de nombreuses protéines (bleu). Les substitutions d’acide aminé (mutations faux sens) sont représentées par un cercle, les mutations non sens par des étoiles et les mutations impliquant des insertions ou des délétions par des rectangles. Modifiée d’après Endersby et Baker, 2008.
1ère Partie – Etude bibliographique
Chapitre 1: Le glioblastome multiforme
~ 19 ~
menant au changement d’un acide aminé sont principalement rencontrées au niveau des exons 1 à 6, correspondant au domaine phosphatase (figure 16 ; pour revues, Knobbe et al., 2002 ; Oghaki, 2005). Bien que la méthylation du promoteur de PTEN ait été décrite dans quelques tumeurs et lignées de GBM, le lien avec la régulation de l’expression de PTEN reste encore à démontrer (Baeza et al., 2003 ; Wiencke et al., 2007).
Bien qu’étant associée à de nombreuses fonctions cellulaires telles que la prolifération, la migration, l’invasion et l’angiogénèse, la fonction la plus décrite de PTEN dans le GBM est son implication dans la régulation de la migration cellulaire et l’invasion. La réexpression de PTEN dans les cellules U87 de GBM entraîne une inhibition de la migration, de l’invasion et de la prolifération cellulaire par déphosphorylation directe de la focal adhesion kinase (FAK), chacun de ces effets pouvant-être antagonisé par la surexpression de FAK. La déphosphorylation de FAK est suivie de la déphosphorylation de sa protéine effectrice p130 Crk-associated substrate (p130Cas). Contrairement à FAK, la surexpression de p130Cas restaure sa propre phosphorylation mais pas celle de FAK, ce qui permet d’antagoniser les effets de PTEN sur la migration et l’invasion mais pas sur la prolifération cellulaire. Ceci suggère que PTEN peut réguler la progression tumorale au travers de différents effets qui sont médiés par des voies de signalisation distinctes qui divergent au niveau de FAK (Tamura et al., 1999). La surexpression de PTEN est également associée à une diminution de l’activité GTPasique des protéines Cdc42 et Rac, des petites protéines de fixation au GTP de la famille Rho impliquées dans le remodelage du cytosquelette d’actine, et à une diminution de l’activité enzymatique des métalloprotéinases MMP-2 et MMP-9. Ainsi, en empêchant la protéolyse de la matrice extracellulaire par les MMP et en diminuant la capacité à migrer des cellules de GBM, PTEN inhibe l’invasion cellulaire (Furukawa et al., 2006). Afin d’être plus proche des conditions in vivo, la migration des cellules après réexpression de PTEN a été étudiée sur des composants de la matrice extracellulaire tels que la vitronectine, qui se lie à l’intégrine αvβ3. Sur vitronectine, l’activité phosphatase de PTEN inhibe indirectement l’activité de Rac1 par inhibition de l’activité de FYN, une protéine kinase de la famille SRC (SFK). Bien que la réexpression de PTEN entraîne une inhibition de l’activité d’Akt, l’inhibition de la migration cellulaire sur vitronectine ne semble pas impliquer cette voie, puisque la surexpression d’Akt n’a pas d’effet sur la migration cellulaire dans ces conditions. Ceci suggère que la migration des cellules de GBM n’est pas contrôlée par l’activité lipide phosphatase mais par l’activité protéine phosphatase de PTEN (Dey et al., 2008).
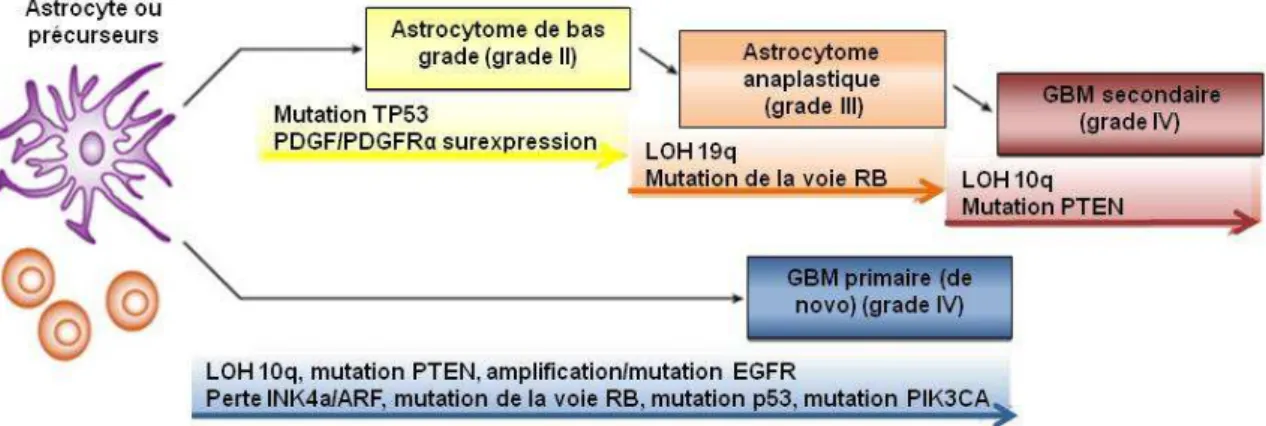
Figure 17 : Altérations génétiques impliquées dans le développement du GBM. Les analyses d’échantillons
de tumeur primaire ont montré des altérations fréquentes de voies de signalisation permettant l’initiation et la progression du GBM. La perte de régulation par p53 (par les mutations, amplification de MDM2 ou délétion de ARF) et la perturbation de la voie RB (telle que la délétion de CDKN2A (INK4a) ou la perte de RB1) sont fréquemment observées. L’activation de voies de signalisation par des facteurs de croissance est également commune. Les voies principalement activées sont la voie du platelet-derived growth factor (PDGF) dans le GBM secondaire et la voie d’EGF dans le GBM primaire. Dans le GBM secondaire, il est clair que la mutation de p53 est impliquée dans l’initiation tumorale, tandis que la mutation PTEN a lieu à une étape plus tardive et semble être impliquée dans l’acquisition du phénotype aggressif et invasif. A noter que la LOH 10q a lieu fréquemment dans le GBM primaire et secondaire (70 et 65% respectivement). Les mutations permettant l’activation de PIK3CA, qui code pour p110α la sous unité catalytique de PI3K, sont aussi observées dans le GBM primaire. D’après Endersby et Baker, 2008.