ETUDE DE LA PROTEASE DE L'ADENOVIRUS DE TYPE 2
par
SIRA N'DIA YE DOUKOURE
Département de microbiologie
Mémoire présenté à la Faculté de Médecine en vue de l'obtention du grade de Maître è.s Sciences (MSc)
Acquisitions and Direction des acquisitions et Bibliographie Services Branch des services bibliographiques
395 Wellington Street Ottawa, Ontario K1AON4 395, rue Wellington Ottawa (Ontario) K1A ON4
The author has granted an
irrevocable non-exclusive licence
allowing the National Library of
Canada to reproduce, loan,
distribute or sell copies of
his/her thesis by any means and
in any form or format, making
this thesis available to interested
persans.
The author retains ownership of
the copyright in his/her thesis.
Neither the thesis nor substantial
extracts from it may be printed or
otherwise reproduced without
his/her permission.
Your file Votre référence Our file Notre reference
L'auteur a accordé une licence
irrévocable et non exclusive
permettant
à
la Bibliothèque
nationale
du
Canada
de
reproduire, prêter, distribuer ou
vendre des copies de sa thèse
de quelque manière et sous
quelque forme que ce soit pour
mettre des exemplaires de cette
thèse
à
la disposition des
personnes intéressées.
L'auteur conserve la propriété du
droit d'auteur qui protège sa
thèse. Ni la thèse ni des extraits
substantiels de celle-ci ne
doivent être imprimés ou
autrement reproduits sans son
autorisation.
ISBN 0-612-09499-5
YOU MAKE ME PROUD TO BE A MOM AND YOU SURE MAKE LIFE INTERESTING AND WONDERFUL. 1 LOVE YOU. GOD BLESS YOU.
LISTE DES FIGURES ... III LISTE DES TABLEAUX ... IV ABREVIATIONS ... V RESUME ... VII
INTRODUCTION ... 1
MATERIELS ET METHODES ... 9
A. ACTIVITE DE IA PROTEASE D'AD2 ... 9
1.1 Lysats bactériens ... 9
1.2 Protéase purifiée ... 9
2. SUBSTRAT ... 10
3. TESTS D'ACTIVITES ENZYMATIQUES ... 10
4. GELDEPOLYACRYLAMIDESDS-PAGE ... 10
5. CELLULES ET INFECTION VIRALE ... 10
6. MARQUAGE DES PROTEINES VIRAI.ES ... 11
7. TRANSFERT SUR MEMBRANE DE NITROCELLULOSR ... 11
8. IMMUNODETECTION ... 11
B. CLONAGE DE IA PROTEINE PVI. ... 11
9. PURIFICATION D'ADN VIRAL. ... 11
10.BACTERIES ET VECTEURS ... 12
11.PURIFICATION D'UN FRAGMENT DE DNA A PARTIR D'UN GEL D'AGAROSE ... 12
12. PREPARATION DES CELLULES COMPETENTES ET TRANSFORMATION DE DNA ... 12
13. SELECTION DES CLONES RECOMBINANTS PAR HYBRIDATION ... 13
LISTE DES FIGURES
Figure 1: Modèle d'une particule d'adénovirus ... .42
Figure 2: Alignement des séquences des protéases de différents sérotypes ... .43
Figures 3: Séquence en nucléotides et en acides aminés du cadran de lecture ouvert du gène pVI d'Ad2 ... 49
Figure 4: Carte génétique du vecteur d'expression pTRXFUS ... 50
Figure 5: Carte génétique du plasmide pAL-781.. ... 51
Figure 6 Carte génétique du plasmide pAL/trxA 781.. ... 52
Figure 7: Clonage de la protéine pVI. Gel SDS PAGE d'extraits bactériens ... 19
Figure 8: Clonage de la protéine pVI. Gel SDS PAGE ... 20
Figure 9: Transfert sur filtre de nitrocellulose de protéines provenant d'extraits bactériens ... 21
Figure 10. Etat de solubilité de la protéase ... 22
Figure 11: Effet du temps d'incubation sur l'activité protéolytique ... 27
Figure 12: Effet de la température sur l'activité protéolytique ... 28
Figure 13: Effet du Sodium dodécyl sulfate et de l'urée ... 29
Figure 14: Purification de l'IgG du sérum de lapin ... 30
LISTE DES TABLEAUX
Tableau 1 : Adénovirus humains ... 41
Tableau II: Classification des protéases ... .44
Tableau ID: Protéases virales ... 45
Tableau IV: Protéines adénovirales clivées par la protéase ... .46
Ad Adz Adzts1
ADN
ARN ARN-m Da DMEM DTT ABREVIATIONS Adénovirus Adénovirus de type 2Mutant ts1 de }'adénovirus de type 2 humain Acide déoxyribonucléique
Acide ribonucléique ARN messager Dalton
Milieu Eagle modifié par Dulbecco Di thiothreitol
EDTA Ethylènediamine tétraacétate
extrait brut pLPV bactéries E. coli contenant le vecteur pLPV qui exprime la protéase de type sauvage. g HzT1 lgG IYf
G
kd Leu Met MockMOI
Unitée de force centrifuge
Mutant ts1 de }'adénovirus de type 2 humain Immunoglobuline
Isopropy 1-B-thio-galactoside Kilodalton
Leucine méthionine
Extrait de cellules non-infectées
Multiplicité d'infection (multiplicity of infection)
ng nanogramme
NP-40 Nonidet P-40
PAGE-SDS Gel de polyacrylamide-sulfate dodécyl de sodium p.b. Paire de bases
PFU
pl
pLAM pLPV PMSF Pro pTP P/V pVII SDS TAETE
TP ts tsi uCi uMv/v
wtUnité de formation de plage (Plaque forming unit) Point isoélectrique
Vecteur ne comportant pas le gène de la protéase Vecteur comportant le gène de la protéase Phénylméthysulfonylfluorure
Praline
Précurseur de la protéine terminale liée à l'ADN PoidsNolume
Précurseur de la protéine virale VII Sulfate dodécyl de sodium
Tampon Tris-acétate et EDTA
Tampon Tris-HCl lOmM, EDTA 1 mM Protéine terminale (ferminal protein) Thermosensible
Mutant ts1 de !'adénovirus de type 2 humain Microcurie
Micromolaire Volume/volume Type sauvage
Le premier objectif de mon projet était de cloner le gène p VI de !'adénovirus de type 2 dans un nouveau vecteur d'expression: pTRX FUS, afin de purifier par la suite la protéine p VI et de l'utiliser comme substrat de la protéase.
Il a été impossible de détecter la protéine pVI sur gel SDS (sodium dodécyl de sulfate) PAGE, et par Western blot en utilisant l'anticorps contre !'adénovirus 2. Ceci est du au fait que la protéine p VI est dégradée au fur et à mesure qu'elle est exprimée. Des résultats similaires ont été observés par un autre laboratoire, et ce en utilisant un vecteur différent. Les virions ad2 ts1-39°C (qui contient les formes précurseures des protéines: fila, VI, VII, VIII, X, et la protéine terminale), marqués à la méthionine 35s, purifiés et fragmentés ont par la suite été utilisés comme substrat pour faire des tests d'activités enzymatiques avec la protéase d'ad2 afin de déterminer: l'effet du temps d'incubation, de la température de réaction, des dénaturants et l'Immunoglobuline G (purifié à partir du sérum de lapin contre la protéase de !'adénovirus de type 2). Nous avions trouvé que l'enzyme est très sensible au sodium dodécyl sulfate, mais par contre maintient son activité jusqu'à 45°C ou en présence de 3M d'urée. Les anticorps antiprotéases inhibent l'enzyme seulement à hautes concentrations.
La protéase virale est très basique (Pl:l0.59), et certaines évidences montrent qu'elle est associée au coeur viral. Il a déja été démontré que l'enzyme est encapsidée en même temps que le génome viral. Nous avions alors émis l'hypothèse selon laquelle l'enzyme se lie au DNA viral, et serait par la suite encapsidée sous la forme d'un complexe protéase-DNA Une série d'expérience a été effectuée pour tester cette hypothèse en tenant compte du fait que l'enzyme du mutant tsi n'est pas encapsidée. Etant donné que les enzymes wt et ts1 se sont comportées de la même façon, il n'a pas été possible de déterminer si l'enzyme wt est encapsidée sous la forme d'un complexe avec le DNA viral. Cependant les résultats qui ont été obtenus confirment l'état d'insolubilité des enzymes wt et tsi.
Les Adénovirus forment un large groupe de virus qui ont des pathologies variées chez les vertébrés. Les adénovirus causent des infections respiratoires et oculaires aiguës. Certains types spécifiques sont des agents étiologiques d'un large genre d'infection comme les cystites hémorragiques et les gastro-entérites infantiles [Ginsberg., 1988]. Des 50 sérotypes humains décrits de nos jours les sérotypes ad2 et ad5 étroitement liés entre eux, ont été intensément étudiés depuis leur découverte en 1953 [Weber., 1990] (Tableau 1 en annexe). Quelques caractéristiques des adénovirus sont d'un intérêt particulier. 1-Les adénovirus sont composés uniquement de DNA et de protéines qui se multiplient dans le noyau de la cellule.
2-Ils induisent des infections latentes dans les adénoïdes et d'autres tissus lymphoïdes chez l'homme.
3-Plusieurs adénovirus sont oncogéniques pour un certain nombre de rongeurs.
4-Ils permettent à un petit groupe de virus défectif en ADN de se reproduire Qes "virus associés aux adénovirus"). [Ginsberg., 1988]
5-La composition et l'architecture des adénovirus sont relativement bien connues, les adénovirus 2 comportent un squelette externe de forme icosahédrale constitué de 7 polypeptides majeures qui sont encodés par le virus. Les composantes sont: l'hexon II, le penton III, la fibre IV et les protéines associées à l'hexon selon des rapports stoichiométriques: IIIa, VI, VIII, IX. Ces protéines confèrent une stabilité à la capside, forment des liens avec les protéines liées à l'ADN et jouent un rôle dans l'assemblage du virion [Nermut., 1984] [Pettersson., 1984] [Philipson., 1983]. Le virion contient également des polypeptides de faibles poids moléculaires très peu caractérisés qui dérivent du clivage de précurseurs plus larges [Anderson et al., 1973] (Figurel en annexe). L'adénovirus encode une protéase responsable du clivage des précurseurs (Yeh Caï
et al.,1983]. Le génome est sous forme d'ADN double brin, linéaire d'une longueur de 36 Kpb associé à deux protéines basiques [V, VIT] et deux copies d'une protéine terminale attachées de façon covalente à chaque extrémité 5' de l'ADN. La décapsidation a lieu immédiatement après que le virion ait pénétré le cytoplasme de la cellule. Avant que la réplication virale ait lieu, la transcription précoce se fait à partir de six régions distinctes: El A, ElB, E3, E4, E2A, et EzB respectivement La transcription tardive s'effectue peu après la réplication [Ginsberg., 1988].
Les vecteurs adénoviraux sont utilisés de façon extensive pour permettre l'expression de gènes étrangers dans les cellules de mammifères [Berkner., 1992] [Gilgardi et al., 1990] pour la production des vaccins viraux, et plus récemment pour la thérapie génique [Charlton et al., 1992].
Quelques propriétés de }'adénovirus, font que ce dernier est un bon candidat pour chacune de ces applications. 1-La facilité avec laquelle le génome peut être manipulé par de simples techniques de l'ADN recombinant. 2- Les hauts niveaux de production qui peuvent être obtenus et facilement collectés et concentrés. 3- La possibilité d'obtenir de hauts niveaux d'expression d'ADN étrangers insérés. La construction d'ADN recombinant implique l'insertion d'ADN étranger habituellement en compensant des délétions d'ADN viral. Les régions précoces El A, et E3 ne sont pas essentielles à la réplication virale. En introduisant des délétions dans ces régions, on peut construire des vecteurs pouvant accueillir des inserts plus longs. [Bett et al., 1993] [Charlton et al., 1992] [Berkner., 1992] [Gilgardi et al., 1990].
Plusieurs rôles sont associés aux protéases. Il y a tout d'abord la dégradation des protéines dans le système digestif par des enzymes comme la trypsine, la chymotrypsine et la pepsine. Ces enzymes sont facilement accessibles, c'est pourquoi elles ont été parmi les premières à être intensément étudiées. En opposition à cette dégradation, la présence de protéolyse limitée dans des cellules [Holzer et Heinrich., 1980] [Bond et Butler., 1987] a
permis de mettre en évidence d'autres rôles des protéases: la participation à des fonctions de régulation comme par exemple la coagulation sanguine, l'activation du complément, la fertilisation et la production d'hormones [Kay et Dunn., 1990]. Il existe un autre rôle très important associé aux protéases: c'est le clivage du "peptide signal" des protéines importées du réticulum endoplasmique durant la traduction. Ce sont les " peptides signaux" [Damell et al., 1990]. On retrouve les protéases chez tous les organismes vivants, des bactéries jusqu'à l'homme. Ces enzymes, qui hydrolysent les liens peptidiques, ont pu être classées dans quatre grandes familles selon la composition de leurs sites actifs: les aspartate protéases, les métallo-protéases, les cystéine protéases, et les sérines protéases (Tableau II en annexe). L'acide aminé du centre réactionnel des cystéine protéases, et des sérine protéases ne diffère l'un de l'autre que par un seul atome; un atome de soufre chez la cystéine qui est remplacé par un oxygène chez la sérine. On retrouve ces deux classes de protéases dans les différents compartiments d'une cellule, et certaines d'entre elles sont excrétées, par exemple la trypsine. Les acides aminés impliqués dans le site actif de cette enzyme sont l'histidine 57, l'aspartate 102, et la sérine 195 [Sprang et al., 1987]. En ce qui concerne les cystéine protéases, la première à être caractérisée a été la papaïne, une enzyme qui a été isolée à partir d'un arbre, le Corica papaya. Le site actif de la papaïne est composé par la cystéine 25, l'histidine 159 et l'asparagine 175 [Dufour., 1988]. Les cystéine et les sérines protéases ont le même type de mécanisme d'hydrolyse à cause des acides aminés composants leur site actif, cependant la disposition de ces acides aminés n'est pas la même. La virologie moléculaire a permis de découvrir le rôle des protéases dans le processus de maturation [Krausslich et Wimmer., 1988] [Wellink et van Kammen., 1988] [Kay et Dunn., 1990]. La production de protéines biologiquement actives nécessite le clivage des polypeptides précurseurs. La réplication de plusieurs virus dépend du clivage de ces protéines précurseures. On retrouve ce genre de maturation surtout chez les virus à ARN+, c'est à dire dont le génome a la même polarité que l'ARN-m, et chez les rétrovirus. Ils produisent une polyprotéine, qui doit être clivée par une
protéase encodée par le virus, ceci libère les protéines de structure et les protéines qui jouent un rôle dans la réplication. Ces deux groupes de virus sont les mieux connus en ce qui concerne les protéases virales. On retrouve chez les virus à ARN+, des virus de plantes (comovirus, potyvirus) et des virus d'animaux (picomavirus, etc ... ). Ces virus codent généralement pour des cystéine protéases sauf chez les togavirus chez lesquels on retrouve une sérine protéase. Les rétrovirus, codent tous pour des aspartate protéases (HIV, caulimovirus, etc ... ). La protéolyse limitée nécessite que l'enzyme soit spécifique dans le choix de ses substrats. Cette spécificité est avantageuse car le virus dépend ainsi moins de l'hôte. Par contre, ceci fait que les protéases virales sont une cible de choix pour l'éventuelle synthèse d'inhibiteurs [Dreyer et al., 1989]. Le processus de maturation des virus implique le clivage des protéines précurseures par la protéolyse spécifique, il en résulte des protéines matures. C'est le cas des adénovirus, le seul virus à ADN dont la protéase a été bien caractérisée. Ce nouveau type de clivage met en évidence l'importance du rôle des protéases chez plusieurs organismes, c'est la raison pour laquelle nous voulons étudier la protéase de !'adénovirus de type 2.
La première évidence de l'existence d'une protéase encodée par le virus a été révélée avec l'étude du mutant ts1. Cette mutation a été séquencée, et localisée dans le cadran de lecture ouvert de la région distale L3. Les études sur des revértants de ts1 ont confirmé l'identité du cadran de lecture ouvert comme étant le gène de la protéase. La preuve formelle de l'identité de la protéase virale a été établie grâce à son clonage et à son expression dans E-coli. Ceci a permis par la suite la purification partielle par chromatographie d'affinité. La protéine recombinante avait la même spécificité de clivage que l'enzyme préparé des virions, et les mêmes patrons d'inhibition [Weber., 1990]. La protéase d'ad2 a également été clonée dans un autre vecteur [Anderson., 1990], ainsi que dans une cellule d'insecte qui utilise le Baculovirus [Webster et al., 1993].
Les gènes de la protéase de 12 sérotypes différents ont été séquencés, et bien que la séquence en acides aminés soit hautement conservée, des motifs typiques des protéases lui font défaut et il n'y a aucune homologie avec des protéines connues [Weber et al., 1993] (Figure2 en annexe). L'enzyme est un monomère de 24.838 Da. Il n'y a pas de modification en N terminale au niveau du poids moléculaire, comme pour plusieurs autres protéases [Anderson., 1990]. L'alignement des séquences et la mutagénèse dirigée indiquent qu'il s'agit d'une nouvelle classe de cystéine protéase (Tableau men annexe).
Il a été démontré que la protéase adénovirale est activée par un peptide possédant des liens disulfures dérivés de l'extrémité C-terminale de la protéine de structure du virus: pVI [NH2-GVQSLKRRRCF - COOH] (Tableau V en annexe). Le mécanisme d'action suggéré est un échange de liens disulfure [Webster et al., 1993]. Cependant il n'est pas encore clair comment le peptide active l'enzyme ou s1 d'autres substrats protéiques adénoviraux sont absolument nécessaires au clivage. Dans une étude indépendante, Mange! et al., 1993 ont proposé que l'ADN viral est aussi un cofacteur et qu'en absence d'ADN le clivage est réduit. Plus récemment il a été suggéré que l'ADN stabilise la protéase en une forme active dans certaines conditions d'essais [Webster et al., 1994]. La protéase adénovirale clive six protéines de la capside [PVI, PVII, PVIII, L2-79R, pTP, IIIA], et une protéine d'échafaudage [L152K] (Tableau IV en annexe). La protéase adénovirale exprimée dans des cellules d'insectes clive les protéines adénovirales, l'ovalbumine, et la protéase de baculovirus en absence du cofacteur pVIc. Donc il doit exister d'autres facteurs ou mécanismes qui jouent le rôle d'activateur [Keyvani-Amineh et al., 1995].
Des expériences de clivage in vitro ont permis d'établir que les précurseurs de !'adénovirus de type 2 sont clivés par des protéases d'autres adénovirus ce qui suggère un haut degré de conservation de la séquence du substrat et de l'enzyme [Houde et al., 1990a ]. L'alignement des séquences de protéases provenant de 12 sérotypes différents ont permis d'identifier des résidus conservés. En effet, ceci a permis d'identifier la conservation
d'un histidine: H54 et deux cystéine.s: C104 et C122 [Rancourt et al., 1994]. L'identification des résidus du site actif est cruciale non seulement pour la classification de l'enzyme mais aussi pour l'identification des inhibiteurs potentiels, ce qui peut être approprié pour la chimiothérapie des infections par adénovirus. La protéase de l'adénovirus requière des résidus cystéine pour l'activation et la catalyse, ceci a été démontré en faisant la mutagénèse de certains codons pour les résidus cystéine et sérine séparément Suite à l'expression dans E-coli des gènes de la protéase résultant de la mutagénèse, l'activité a été établie et comparée à celle de la protéase recombinante de type sauvage. Les mutants contenant des résidus sérines altérés étaient actifs. La mutation au cystéine104, a réduit l1activité à moins de 95%.
Ces résultats confirment qu'il s'agit d'une nouvelle classe de cystéine protéase et suggère que le résidu C104 est le site actif nucléophile. Le résidu C122, fait partie possiblement du mécanisme d'échange thiol disulphide [Rancourt et al., 1994].
L'activité spécifique de la protéase tel qu'établit par Weber et al., 1994 est de 12.9 nmol-1 mïn-1 nmo1-l enzyme. Le PI est de 10.59 et le pH optimum se situe entre 7 et 8 [Tihanyi et al., 1993]. La protéase adénovirale clive six protéines de la capside, et une protéine d'échafaudage. Tous les sites de clivage sont conformes au consensus proposé, soit [M 1 L] XGG-X ou [M 1 L] XGX-G. Cependant il existe des protéines de l'adénovirus qui contiennnent la séquence consensus et qui ne sont pas clivées par l'endoprotéase, peut être parce qu'elles ne sont pas accessibles à l'enzyme [Weber., 1990]. L'enzyme est aussi capable de cliver des protéines non virales [Tihanyi et al., 1993] comme l'ovalbumine et les protéines du cytosquelette, la cytokératine K18 et K7 [Chenet al., 1993]. Toutes ces protéines portent la séquence consensus de clivage.
Comme l'enzyme est inhibé par les composés suivants: N-ethylmaléimide, iodoacétate, dithiopyridine, P-chloromercuribenzoate, leupéptin et E-64 [Tihanyi et al., 1993], ceci suggère que I1enzyme est une cystéine protéase. Les inhibiteurs d'autres classes de protéases n'ont pas d'effet sur cette enzyme.
La protéase adénovirale est encapsidée durant l'assemblage du virus et l'on suppose que les protéines précurseures sont clivées pendant ou subséquemment à l'assemblage [Weber., 1990]. Récemment Rancourt et al., 1994 ont effectué des expériences qui ont permis de conclure que la maturation des jeunes virions de ts1 est arrêtée à cause de l'absence de la protéase dans ces particules. Le rôle que joue la protéase dans le processus de maturation de !'adénovirus est semblable au rôle de la protéase du virus T4. Pour T4, la protéase est activée quand elle est incorporée dans le coeur de la capside immature et elle clive subséquemment les protéines de l'enveloppe et du coeur [Showe et al., 1976]. Apparemment comme pour !'adénovirus le cas de T4 illustre deux aspects fonctionnels du clivage protéolytique durant la morphogenèse de la capside; dans un premier temps la fragmentation et l'élimination de la protéine d'échafaudage pour faire de l'espace pour le chromosome et dans un deuxième temps la déstabilisation entre les sous unités, ce qui permet le réarrangement des protéines de l'enveloppe [Steven et al., 1991] [Steven et al., 1992] un événement qui semble se tenir lors de la morphogenèse de !'adénovirus [Cotten et al., 1995] [Rancourt et al., 1995]. Ainsi la praline 137 est importante pour l'encapsidation de la protéase et non pour l'activité enzymatique [Rancourt et al., 1995].
PROJET
Le DNA de !'adénovirus code pour une protéase dont l'action est nécessaire à la maturation et au développement de virus infectieux. Des études à l'aide du mutant thermosensible adzts1 ont démontré [Anderson et al., 1973; Boudin et al., 1980; Tremblay et al., 1983; Weber et al., 1988] que la protéase clive 6 protéines précurseures: pilla, pVI, pVIl, pVIII, pX, et la pTP (protéine terminale). Le clivage de ces protéines permet la maturation du virus. Le virus d' adzts1 n'a pas d'activité protéolytique à la température non-permissive(39°C), ainsi les protéines demeurent sous la forme de précurseur et les virus produits ne sont pas infectieux. Pour cette raison, la protéase adénovirale est une cible pour le développement d'agents antiviraux spécifiques. Afin de mieux caractériser
l'enzyme, nous avons décidé d'utiliser un substrat connu de la protéase: la protéine p VI, précurseur de la protéine VI qui est reliée à l'hexon. Nous allons ainsi cloner le gène pVI de l'adénovirus de type 2 dans un vecteur: pTRX FUS. Une fois que la protéine sera exprimée et purifiée, nous allons l'utiliser pour faire des tests d'activités enzymatiques avec la protéase d'adz. Nous allons tester l'effet du temps d'incubation, de la température de réaction, des dénaturants (urée et SDS), et de l'immunoglobuline G purifiée à partir du sérum de lapin contre la protéase de l'adz. Afin de vérifier si une mauvaise localisation de la protéase d'adzts1 dans la capside par rapport à l'enzyme de type sauvage est la raison pour laquelle elle ne peut pas cliver les protéines précurseurs, nous allons commencer par vérifier l'état de solubilité de la protéase ts1, sachant que la protéase wt est insoluble, c'est à dire qu'elle précipite après une centrifugation de 35 000 rpm pendant 1 heure [Bhatti et Weber., 1979]. L'état de solubilité sera vérifié par une expérience qui consiste à incuber dans différentes conditions des noyaux de cellules infectées par ts1 ou wt durant 16 heures, et ensuite de les centrifuger pendant 5 minutes à 10 000 rpm. Les fractions du culot et du surnageant seront par la suite séparées et analysées sur gel SDS PAGE. Par la suite, nous ferons un transfert sur membrane de nitrocellulose et une immunodétection. Nous testerons l'effet de la DNAse 1, parce que l'enzyme est très basique et pourrait interagir avec le coeur viral; l'effet du cofacteur pVI-C; ainsi que l'effet combiné de la DNAse 1 et du pVI-C.
La protéine p VI n'a pas pu être exprimée, et purifiée car elle a été dégradée par des protéases inconnues de E.coli. Nous avons par la suite utilisé le virion adz ts1 fragmenté, et purifié comme substrat pour effectuer des tests d'activités enzymatiques.
MATERIEL ET METHODES
A. Activité de la protéase d'ad2
1. Sources de protéase
1.1 Lysats bactériens
La séquence codante de la protéase de !'adénovirus de type 2 a été sous clonée dans pLAM un vecteur d'expression modifié de pRIT2T. C'est un vecteur contrôle [Bourbonnière., 1990]. En ce qui concerne le vecteur pLPV qui comporte le gène de la protéase [Tihanyi et al., 1993], les plasmides recombinants .ont été transfectés et par la suite sélectionnés sur des cellules N99 CI+ qui expriment le répresseur CI lambda sauvage de façon constitutive. Les plasmides sélectionnés ont été transférés dans les cellules AR120 de le but de maximiser l'expression. AR120 est dérivé de N99 et CI+ [Mottet al., 1985]. L'induction par l'inactivation de CI+ a été faite via le mécanisme SOS lorsque les cellules atteignent une haute densité [Mottet al., 1985].
1.2 Protéase purifiée
L'enzyme a été purifiée à partir du système procaryote [pLPV/AR120], selon la méthode décrite par Karoly Tihanyi [Tihanyi., 1993]. Le vecteur pLPV comporte le gène de la protéase [Bourbonnière., 1990]. La pureté de l'enzyme a été évaluée selon la proportion de protéase détectée sur gel SDS-PAGE coloré au nitrate d'argent, [Bollag et Edelstein., 1991] par rapport aux autres protéines lorsque un échantillon concentré provenant de la dernière étape de purification a été chargé en excès sur le gel. L'apparition d'une seule bande correspondant à la protéase sur gel SDS-PAGE nous indiquait que la protéase avait une pureté de 99-100%. Il faut donc tenir compte que la technique a une limite de détection de lng. Donc les protéines dont la concentration est inférieure à log dans l'échantillon ne seront pas détectées. En générale, leur proportion par rapport à la protéase est négligeable c'est pourquoi nous n'en avons pas tenu compte. La
protéase purifiée a été utilisée dans certains essais protéolytiques à raison d'environ 600ng par réaction.
2. Substrat
Les virions d'ad2ts1 marqués à la méthionine 35s, purifiés et fragmentés, ont été utilisés comme substrat pour faire les essais de l'activité protéasique. La préparation de ces virions a été décrite précédemment [Weber., 1976]. Le marquage des protéines à l'iode125 fut réalisé selon une technique décrite par Markwell, [Markwell et Fox., 1978] avec l'utilisation de l'IODO-GEN comme agent iodant Une dialyse de 24 heures contre un tampon TE, à la température ambiante a été effectuée suite au marquage afin d'éliminer l'iode libre.
3. Tests d'activités enzymatiques
Tous les tests d'activités enzymatiques ont été effectués dans un tampon TE [Tris lOmM pH 7.0, EDTA lmM]. La solution de lyse [Boude., 1990] a été utilisée pour arrêter les réactions avant de les mettre sur gel SDS-P AGE. Les résultats ont été révélés à l'aide d'une autoradiographie des gels. L'activité enzymatique a été évaluée selon le taux de protéines p VII transformés en VIT.
4. Gel de polyacrylamide
Afin d'effectuer les gels de polyacrylamide nous avons utilisé la méthode de Maizel [Maize!., 1969] en nous servant des appareils SE 600 de Hoefer Scientific instruments. La concentration de polyacrylamide a été de 12. 5% dans tous les essais.
5. Cellules et infection virale
Des cellules de type Hep-2 [Moore et al., 1995] ont été cultivées dans des pétris de culture cellulaire [lOOmm] dans du milieu DMEM [Dulbecco et Freeman., 1959] avec
10% v/v de sérum de veau. Lorsque la couche monocellulaire atteignait 80 à 95% de confluence avec une multiplicité d'infection de 10 unités de formation de plage /cellule, nous avons procéder à l'infection. Par la suite la concentration de sérum de veau a été abaissée à 2.5% v/v. L'incubation des cellules durant l'infection a été faite à 392C.
6. Marquage des protéines virales in vivo
La méthionine - [35S] (Amersham) a été utilisée pour marquer les protéines virales
in vivo de la 22ième heure d'infection à la 24ième heure, à raison de 75uCi par millilitre [ml]. Le marquage s'est déroulé à une température de 39°C.
7. Transfert sur membrane de nitrocellulose
(Western blot)
Les protéines qui se trouvaient sur les gels de polyacrylamide SDS-PAGE ont été transférées sur membrane de nitrocellulose [Hybond-C, Amersham] par le système d'électro-transfert polyblot de la compagnie Hoefer scientific instruments [HSI]. Nous avons suivit les indications du fabricant
8. Immunodétection
L'immunodétection a été réalisée avec l'anticorps polyclonal de lapin préparé à partir de la protéase de type sauvage purifiée exprimée dans E-coli par le vecteur pLPV. Les complexes anticorps-antigènes ont été détectés avec la protéine-A iodinée par la méthode de chloramine-T. Avec cette technique on peut détecter des concentrations de protéines de 50-100 pg [Bers and Garfin., 1985].
B. Clonage de la protéine p VI
9. Purification d'ADN viral
Pour purifier l'ADN viral nous avons utilisé la méthode décrite par Houde [Houde., 1990b]. A Partir de cet ADN nous avons amplifié par PCR [Innis et al., 1990] la région
correspondant au gène qui code pour la protéine PVI, c'est à dire entre les nucléotides 35 et 800 à l'aide des amorces suivantes:
A pVI-1 :5'-CCGGATCCGGAAGACATCAACITIG-3' A pVI-2 :3'-CACTGCAGAGCTATTTAGAAGCATC-5'
Les réactions ont eu lieu dans un volume de 100 ul contenant; Tris lOmM pH 7.5, 50mM Nacl, lOmM MgCl2 et 1.5mM deoxynucléotides triphosphates [dNTP]. La dénaturation initiale du brin matrice a durée 90 secondes à 92°C. L'hybridation s'est faite à 55°C pendant une durée de 90 secondes, et la polymérisation à 72°C pendant 3 minutes. Ce cycle a été répété 30 fois.
10. Bactéries et vecteurs
Le vecteur d'expression PTRX FUS et les vecteurs parentaux proviennent de chez LaVallie et al [Biotechnology vol, 11., 1993] de même que les bactéries E.
coti
GI724.11. Purification d'un fragment d'ADN
àpartir d'un gel d'agarose
L'ADN a été digéré avec les enzymes de restriction Barn Hl et Pstl et mis sur gel d'agarose 1 %. Le morceau d'agarose contenant la bande désirée est découpé à l'aide d'un scalpel. L'ADN est extrait à l'aide du kit Sephaglas Band Prep de la compagnie "Pharmacia".
12. Préparation des cellules compétentes et transformation de DNA
Pour préparer les cellules compétentes nous avons inoculé les bactéries GI724 dans du milieu riche et nous avons incubé à 37°C pendant 24 heures. L'étalement des bactéries transformées par la méthode de chlorure de calcium [Maniatis et al., 1982] nécessite la préparation de milieu riche [lX M9 de sel, 2%
PN
de Difco Bacto-Casamino Acids, 1 %v/v Glycérol lOmM de MgCL2, O.lmM CaCL2 lOOuglml d1 ampicilline sels: Na2HP04, KH2P04, NaCL, NH4CL et l.S% de Difco Bacto - Agar. 3ul de plasmides contenant les inserts ont été mélangés à 200 ul de bactéries GI724 et le tout étalé sur pétri à 37°C pendant 24 heures.
13. Sélection des clones recombinants par hybridation
Pour faciliter la sélection des bons recombinants, la technique d'hybridation se servant de !'ADN comme sonde, a été utilisée. Suite à la transformation des bactéries Gl724, chaque colonie est ensemencée dans Sml de milieu riche. On procède à l'extraction d'ADN plasmidique par la méthode de Priefer, 1981. L'ADN est digéré par les enzymes BamHl, et Pstl, déposé sur gel agarose 1 % afin de procéder à un buvardage à la Southern [Maniatis et al., 1982]. L'ADN est par la suite dénaturé par une solution dénaturante [NaOH lON, NaCL SM] pendant 4S minutes et neutralisé avec une solution neutralisante [Tris -HCL lmM , NaCL SM pH 7.S] pendant 30 minutes. Le filtre est ensuite placé à 80°C puis pré hybridé et hybridé avec la sonde requise [PVI].
14. Préparation d'une sonde d'ADN double brin
Le système de marquage à amorces multiples a été utilisé afin de rendre !'ADN radioactif. Nous avons utilisé le système de la compagnie Amersham: 11Multiprime DNA Labelling System 11
•
C. Expression
15. Système d'expression
Le vecteur PTRX FUS (en annexe) permet la fusion transcriptionnelle des séquences codantes à l'extrémité 31 du gène trx.A de E-coli avec une séquence de DNA qui code pour 10 résidus d'acides aminés avec un site de clivage pour la protéase entérokinase. Ce
vecteur possède le promoteur pL du bactériophage Lambda, et un site de clonage multiple qui permet d'insérer entre le gène de la thioredoxin et des signaux de terminaisons de la transcription, le gène désiré, dans notre cas celui de pVI. Le plasmide pAL 781 est un vecteur parental qui comporte uniquement: le promoteur pL, un site d'attachement au ribosome et le site de clonage multiple en 3'. C'est un contrôle négatif. Le vecteur P AL/trx.A-781 exprime le gène Trx.A et il est utile pour l'expression de "Active Site Loop Insertions", ce en utilisant le site unique Rsrll dans la séquence codante de cette région. C'est un contrôle positif. Tous ces vecteurs contiennent le réplicon pUC-18 qui confère la résistance à l'ampicilline. Les bactéries GI724 contiennent une copie unique du gène répresseur du bactériophage Lambda: CI intégré dans le locus ampC. Le gène CI est sous le contrôle transcriptionnel du promoteur synthétique de Salmonella typhymurium trp situé en amont de Cl. Ainsi la transcription est sous le contrôle du niveau cytoplasmique de tryptophane.
16. Induction de l'expression
Pour induire l'expression de protéines, les bactéries GI724 contenant les plasmides recombinants ont été inoculées dans 5 ml de milieu d'induction: [1XM9 de sels, 0.2%PN Difco Bacto-Casamino Acids, 0.5% de glucose, lOmM de MgClz, 0.lmM de CaOz,. 1 OOug/ml d'ampicilline, 1 OOug/ml de tryptophane] et incubées pendant 4 heures à 30°C . Après centrifugation [3 000 rpm pendant 15 minutes] les bactéries ont été resuspendues dans Sml de tampon TE, à deux reprises. Le volume finale était de 50 ul. Afin de procéder à une purification partielle, les bactéries ont été gelées et dégelées à l'aide de la glace sèche, puis soniquées à 40 cycles [ 4xl 0 secondes]. Après centrifugation [3000 rpm pendant 20 minutes]. Le surnageant a été recueilli et analysé sur des gels SDS polyacrylamides.
17. Etat de solubilité de la protéase
Les extraits des noyaux qui ont été obtenus par la méthode de Houde, [Houde., 1990] ont été incubés en présence de DNase, de pVI-C, de DNase et de pVI-C durant 16 heures à 40°C. Ils ont été ensuite centrifugés à 10 000 rpm durant 5 minutes. Les surnageants ont été séparés des culots. Ces derniers ont été resuspendus dans des volumes de tampon [Tris lOmM pH7.0, lmM EDTA] égal au volume du surnageant [50 ul].
Les échantillons des culots et des surnageants ont ensuite été mis sur gel de polyacrylamide SDS PAGE (12%) et transférés sur une membrane de nitrocellulose. La présence de protéase dans ces échantillons a été révélée par une immunodétection.
18. Purification de l 'IgG contre Adénovirus de type 2
L'anticorps polyclonal de lapin a été obtenu par immunisation avec la protéase de type sauvage exprimée dans E. coli (pLPV), et purifiée. La fraction IgG a été purifiée sur colonne de DEAE Affi-Gel blue( kit provenant de la compagnie Bio-Rad).
Les résultats ont été divisés en trois sections. La première regroupe des dollllées concernant le clonage de la protéine p VI. Dans la seconde partie, des essais ont été réalisés afin d'étudier l'état de solubilité de la protéase de !'adénovirus de type 2. Dans la troisième partie, il s'agit de tests d'activités enzymatiques avec la protéase adénovirale.
A. Clonage de la protéine p
VI.
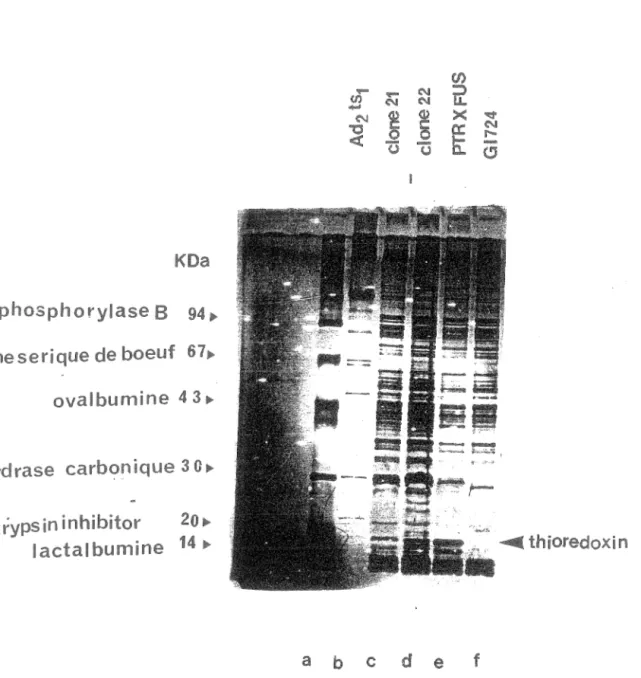
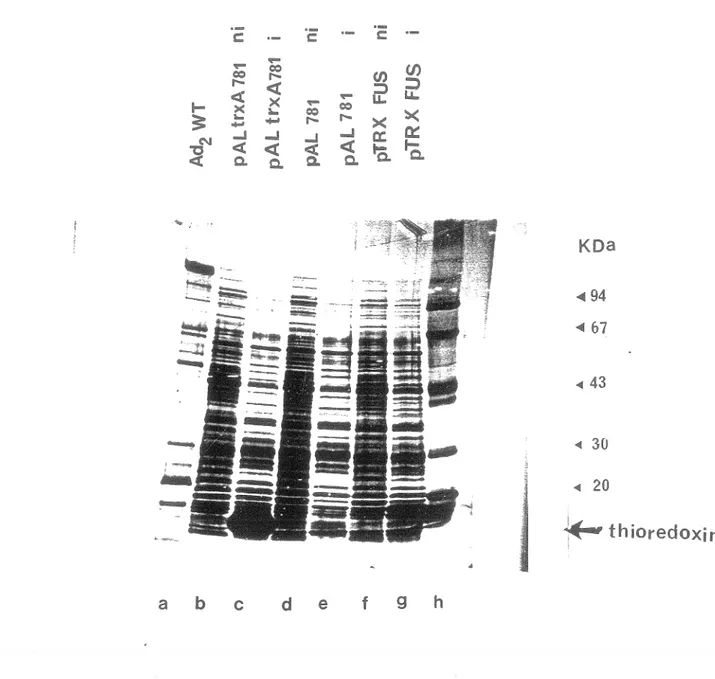
La protéase adénovirale est nécessaire à la maturation et au développement de virus infectieux. Chez le virus ad2 ts1, les protéines illa, VI, VII, VIII, X et la protéine terminale s'accumulent sous forme de précurseur et les virus produits ne sont pas infectieux. [Anderson et al., 1973; Boudin et al., 1980; Tremblay et al., 1983; Weber et al.,1988; Hallllan et al.,1983; Mirza et Weber., 1980]. Ainsi, le mutant tsl a été important dans l'étude de la protéase, et indirectement dans le but ultime de synthétiser des agents antiviraux spécifiques. Afin de mieux caractériser cette protéase, nous avions décidé de cloner la protéine p VI qui est un substrat COilllU de la protéase. La protéase d'ad2 clive la protéine pVI aux extrémités N-terminale et C-terminale tel que démontré sur la figure 3 (en allllexe, les sites de clivage sont indiqués par des flèches verticales). Le gène pVI a été inséré dans le vecteur pTRX FUS, puis la transformation a été faite dans les bactéries GI724 (section 12 M. et Méthodes). Le vecteur pTRX FUS est représenté sur la figure 4 (en annexe), les figures 5 et 6 (en annexe) représentent les vecteurs parentaux du vecteur pTRX FUS. L'induction s'est déroulée pendant 4 heures à 30°C suite à l'ajout de tryptophane (section 16). Les extraits bactériens ont été mis sur gel SDS PAGE. Sur la figure 7 on peut voir les pistes suivantes; piste c: clone 21, piste d: clone 22, piste e: vecteur pTRX FUS (contrôle), piste f: bactéries GI724 (contrôle). Le poids moléculaire de la protéine p VI est d'environ 27 KDa, le poids moléculaire de la thioredoxin est de llKDa. Sur la figure 7, on ne voit pas une protéine majeure qui correspondrait au poids moléculaire de la protéine recombinante soit 40 KDa. Les clones 21 et 22 ont été identifiés
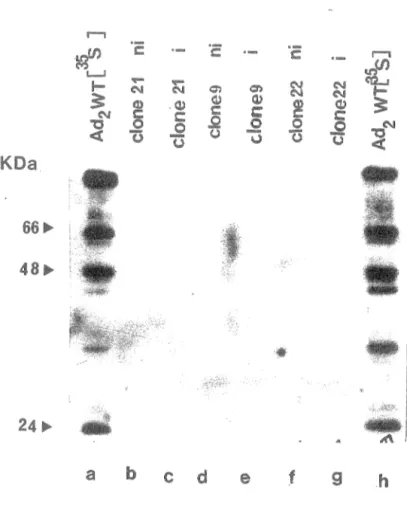
comme positifs après buvardage à la Southern en utilisant la sonde p VI radioactive. Sur cette figure, se sont les fractions du surnageant qui ont été mises sur gel, et sur les pistes c et d on voit les bandes correspondant à la thioredoxin. Ceci suggère que la protéine p VI a été dégradée. Les fractions du culots ont également été mises sur gel PAGE SDS afin de vérifier si la protéine recombinante n'aurait pas précipitée, ce qui n'était pas le cas (résultat non présenté). Nous avons également contrôlé l'état des vecteurs. Sur la figure 8, se sont les extraits bactériens non induits et induits au tryptophane pendant 4 heures à 30°C qui ont été mis sur gel SDS PAGE. Sur la piste c on peut voir la bande correspondant à la thioredoxin (pAL trxA 781 induit), ainsi que sur la piste g (pTRXFUS induit). Ceci prouve qu'il n'y avait aucun problème à ce niveau. Nous avions aussi émis l'hypothèse selon laquelle la protéine pourrait être synthétisée mais pas en quantité suffisante pour être détectée. Les protéines provenant de bactéries non induites et induites au tryptophane pendant 4 heures (section 16), ont été transférées sur filtre de nitrocellulose (section 8), et mises en présence d'un sérum contenant des anticorps dirigés contre la protéine p VI . Sur la figure 9 on voit qu'il n'y a aucune bande révélée sur les pistes b, c, d, e, f, et g. Matthews et al., 1994 sont parvenu a cloner la protéine pVI dans un vecteur d'expression pRSETA [Invitrogen], en utilisant la souche E. coli BL21 (DE3) [Novagen]. Dans une communication personnelle, ils nous ont fait savoir que la protéine p VI est très toxique pour E. coli et qu'elle est rapidement dégradée par des protéases non identifiées de la bactérie. Etant donné que le gène p VI est bel et bien présent dans notre vecteur, et que la partie thioredoxin de la protéine de fusion a été détectée dans notre système, la dégradation de p VI reste la seule explication pour l'absence de p VI détectable. Nous avons donc abandonné l'idée d'utiliser pVI comme substrat. Toutes les expériences subséquentes ont été faites avec le substrat qui a déjà fait ses preuves, soit la protéine virale p VII provenant du mutant tsl.
B. Localisation et état de solubilité de la protéase
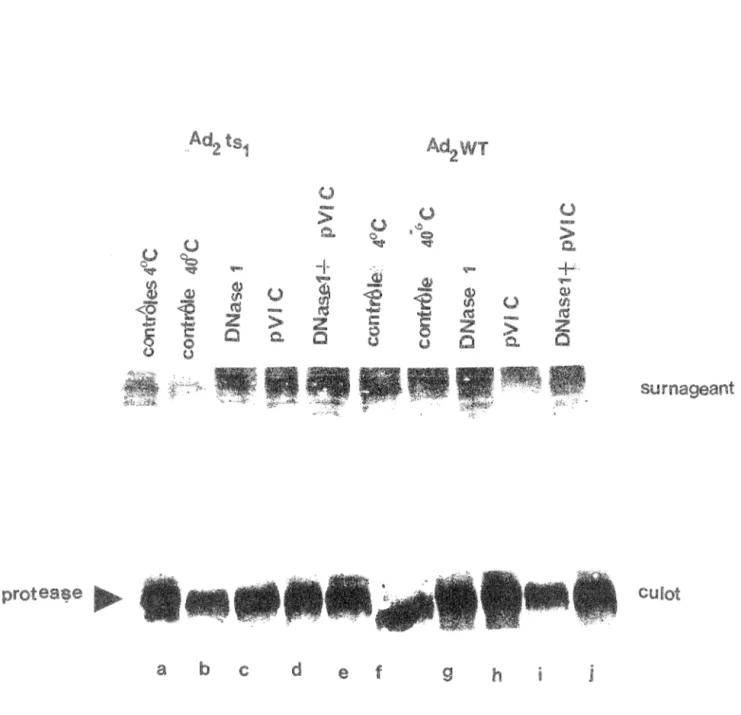
La déficience entraînée par la mutation pour le virion ts1 n'est pas au niveau de l'interaction entre le cofacteur pVI-C et l'enzyme (Labrecque., 1993). Nous avions posé comme hypothèse que la mutation pourrait avoir causé une mauvaise localisation de l'enzyme d'où son incapacité à rencontrer le cofacteur pVI-C nécessaire à son activité dans les noyaux. Dans cet optique, nous avons commencé par vérifier l'état de solubilité de la protéase ts1. Sachant que la protéase wt est insoluble puisqu'elle ne reste pas en solution après une centrifugation de 35 000 rpm pendant 1 heure [Bhatti et Weber, 1979]. L'état de la solubilité a été vérifié par une expérience qui consiste à incuber dans différentes conditions des noyaux de cellules infectées par les virions ts1 ou wt, durant environ 16 heures et ensuite les centrifuger à 10 OOOrpm pendant 5 minutes. Les culots et les surnageants ont été séparés et mis sur gel PAGE SDS. Un transfert sur membrane de nitrocellulose et une immunodétection ont suivit (section 17). Nous avons testé l'effet de la DNAsel (pistes cet h) parce que l'enzyme est très basique et est réputé d'interagir avec le DNA ou complexe DNA-protéine (core), l'effet du peptide pVIC (pistes d et i), ainsi que l'effet combiné de la DNAsel et du cofacteur pVIC (pistes e et j). Sur la figure 10 on voit l'apparition de la bande correspondant à la protéase, cependant il n'y a aucune différence détectable concernant la localisation de la protéase. Labrecque., en 1993 avait effectué les mêmes expériences, et selon ses résultats lorsque l'on ajoute la DNasel et le pVI-C aux noyaux des cellules infectées par adz wt ou adz ts1 on observait une solubilisation. Ces expériences ont été répétées afin de nous assurer que les résultats qu'elle avait obtenus étaient reproductibles, ce qui n'est pas le cas. Notre résultat suggère que, d'une part la protéase de ts1 possède la même propriété d'insolubilité que celle du type sauvage, et d'autre part que la liaison avec le complexe insoluble est plus forte qu'avec le complexe DNAviral-protéine (core).
KDa phosphorylase B 9411> :mimineserique de boeuf ovalbumine 4 31?> in inhibitor !actai mine
-"'"'
N ~ © X "'1' "O i;: c: iN <t 0 05:
~~ u u (!j a b c d e fFigure 7: Clonage protéine Les extraits bactériens induits pendant 4 à 30°C ont été analysés sur gel SDS PAGE, et comparés" Piste c: clone 21, piste d: clone22, piste e: vecteur pTRX FUS (contrôle), piste f: bactéries GI724 (contrôle )o Les protéines du mutant ts1 ont été utilisées comme marqueur de moléculaire ainsi
c: c
--
cc cc ... ... <( <( ><-....
>< :a.. cc3=
-
...
-
... ...1 ...1 ...1 N~
<( a. <(o. a.
<( pVll .,_ ab
c d e·-
c: Cl)"'
::::>~
-
u. cc ... :>(><
et: ...1a:
<("ô.
'ë.
o.f
g h KDa ... 94 ... 67 .... 43 .... 30 .... 20~
th ioredoxi nFigure 8: Clonage de la protéine pVI. Les extraits bactériens non induits (ni) ou induits (i) au tryptophane pendant 4 heures à 30°C ont été analysés sur gel SDS PAGE, et comparés. Les protéines du virus ad2 wt ont été utilisées comme marqueur de poids moléculaire (a) et des marqueurs de poids moléculaires (h). Piste b: pALtrxA 781 ni, piste c: pALtrxA 781 i, piste d: pAL 781 ni, piste e: pAL 781 i, piste f: pTRX FUS ni, piste g: pTRX FUS i.
r-i u:{.l'J ~ ·2 c: r-ï ~·t;i li:"}') ~ ~
....
~ N ~ N lNJ :::Il 11';}") N!-3:
«iJIw
(!.l t'll.! ~w
~ N c c: !:: c; c: "O 0 0J2
0 J2 N <le u ü 0u
t,j 0 u "'C ~ KDa ac
d fg
hFigure 9: Transfert sur filtre de nitrocellulose de protéines provenant de bactéries non induites (ni) et au tryptophane et mises en présence d'un contenant des anticorps dirigés contre la protéine p v'"t Les protéines ont été révélées par autoradiographie suite à formation complexe entre les anticorps et fa protéiI1e A [1251 ]. Piste clone 21(ni), piste c: 21 piste d: 9 (ni), e: clone 9 (i), piste f: 22 (ni), piste g: done Les protéines ont utilisées comme marqueur de
,jl)d t
c.
'2 Si . .. 1 Ad2VVîu
~ 1t_lu
>
u
<D ~ ~ 0 'Q>
~ ~o.
()· ~-
-j~ ijj-
+
1111w
~ JJ2 """" ©w
~1?
w
@(J'I tri
<E
<0
<E
~ ~ -!""' lj'J Illt:
flS l\S....
2>
!::..
,.,,..-
z
2>
z
5
b Clc.
a
!;) 0c
c
0 u ua.
0 u surnageant culota
be
f g hFigure 10: Etat de solubilité de protéase. Les extraits nucléaires des cellules infectées par ad2 et ad2 wt ont incubés à 4 °C (pistes a et f) et à 40°C (pistes b à e et g à j), puis soumis aux traitements suivants: originaux (pistes b et g), en présence de DNase I (pistes cet h), de pVI-C de et de pVI-C (pistes e et échantillons ont été par la suite centrifugés rpm, 5min ). Les culots et surnageants ont été mis sur SDS PAGE L1autoradiognmme produit par fimmunodétection est présenté sur cette
C. Tests d'activités enzymatiques
Puisque nous n'étions pas parvenu à exprimer et purifier la protéine p VI, nous avons décidé d'utiliser comme substrat les virions ad2 ts1 marqués à la méthionine 35s purifiés et fragmentés. Chez le virus ad2 ts1, les protéines illa, VI, VII, VllI, X et la protéine terminale s'accumulent sous leur forme précurseure et les virus produits ne sont pas infectieux. [Anderson et al., 1973; Boudin et al., 1980; Tremblay et al., 1983; Weber et al.,1988; Hannan et al.,1983; Mirza et Weber., 1980]. L'ad2 contient 1070 copies de la protéine VII [Anderson et al., 1973]. Nous avons évalué l'activité protéolytique selon le taux de protéines p VII transformé en VII. Toujours dans le but de mieux caractériser la protéase de l'Adénovirus de type 2, nous nous sommes proposé d'effectuer des tests d'activités enzymatiques: pour étudier l'effet du temps d'incubation, de la température de réaction, des dénaturants, et des anticorps anti-protéase.
1- Effet du temps d'incubation
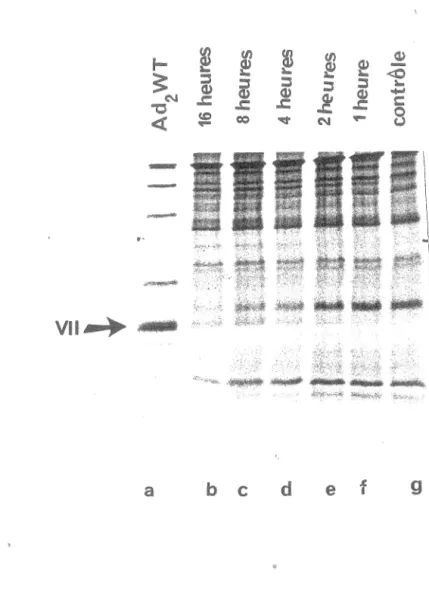
Les virions d'ad2 ts1 marqués à la méthionine 35s purifiés et fragmentés (section 2) ont été incubés à 37°C avec l'extrait brut bactérien pLPV (section 1.1) pour des temps déterminés (piste f: 1 heure, piste e: 2 heures, piste d: 4 heures, piste c: 8 heures, piste b: 16 heures). La piste g représente le contrôle sans protéase. Sur la figure 11 on peut déterminer l'efficacité de clivage selon le taux de conversion de la protéine p VII en VII. Le pourcentage de clivage le plus élevé est obtenu après 16 heures d'incubation dans des conditions standards (piste b 80%, pourcentage évalué par densitométrie). Les expériences indiquent que l'enzyme perd son activité après 16 heures d'incubation parce que: 1- Il n'y a plus de clivage. 2- Il reste encore 20% de substrat. 3- En ajoutant l'enzyme à nouveau à 16 heures le substrat résiduel est clivé. Donc nous avons adopté une incubation de 16 heures pour une réaction standard.
2- Effet de la température de réaction
Les virions d'ad2 ts1 marqués à la méthionine 35s purifiés et fragmentés (section 2) ont été incubés avec l'extrait brut pLPV (section 1.1) à différentes températures (piste b: 4°C, piste c: 25°C, piste d: 33°C, piste e: 37°C, piste f: 39°C, piste g: 45°C). En comparant le taux de clivage de la protéine pVII sur la figure 12 on peut voir que le clivage est très effectif à 37°C (95% ), donc après 16 heures d'incubation à 37°C il ne reste plus que 5% de substrat (piste e). Cependant après 16 heures d'incubation à 45°C il y a encore 80% de clivage (piste g).
3- Effet des dénaturants
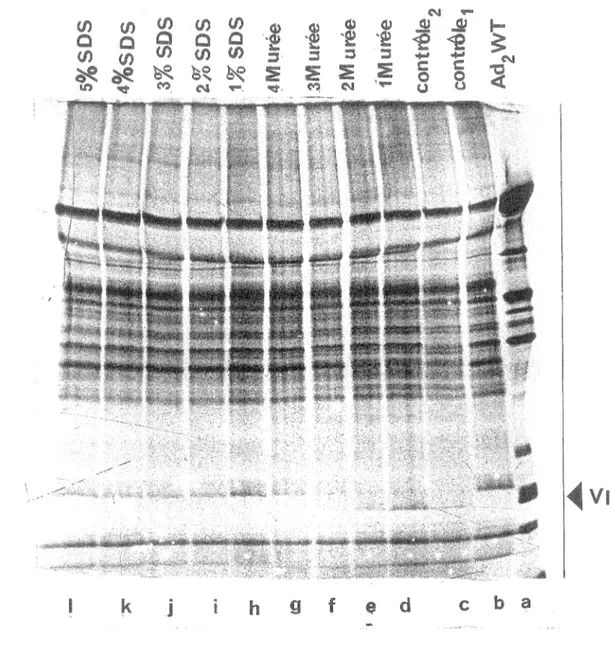
Lors des différentes étapes de la purification de la protéase d'ad2 de l'extrait brut pLPV, divers produits chimiques sont utilisés dont l'urée. Nous avions alors observé que l'activité de l'enzyme diminuait considérablement avec le temps après purification, dans certains cas l'enzyme était inactive. Cette perte d'activité était peut être causée par l'effet de ces produits. Pour vérifier si l'urée résiduel pouvait inhiber l'enzyme, nous avons mesuré l'activité en présence de différentes concentrations d'urée. Les virions d'ad2 ts1 marqués à la méthionine 35s, purifiés et fragmentés (section 2), ont été incubés avec la protéase purifiée de l'extrait brut bactérien pLPV (section 1.2) et mis en présence de différentes concentrations d'urée et de sodium dodécyl sulfate (lM urée: piste d, 2M urée: piste e, 3M urée: piste f, 4M urée: piste g; et 1 % SDS: piste h, 2% SDS: piste i, 3% SDS: piste j, 4% SDS: piste k, 5% SDS: piste 1). La piste b représente un contrôle sans enzyme, la piste c représente un contrôle avec substrat et enzyme. La température d'incubation a été maintenue à 37°C. Sur la figure 13, on voit qu'en présence d'urée le taux de clivage de la protéine pVII augmente jusqu'à lM d'urée à des concentrations d'urée plus élevé le taux de clivage diminue. Cette expérience ne permet pas de distinguer entre un effet stimulateur sur l'enzyme, et un effet positif sur la disponibilité ou la conformation du substrat pVTI.
D'autres expériences effectuées dans le laboratoire (voir discussion) nous indiquent que l'effet stimulateur de l'urée se manifeste via le substrat, et non via l'enzyme. Certaines protéases telle que V8, sont peu affectées par des dénaturants ioniques comme le SDS. Cette propriété de résistance à la dénaturation nous indique des choses intéressantes sur la structure de l'enzyme, puisque le SDS brise les liens non-covalents. De plus, elle permet de faire des expériences importantes tel que le clivage avec VS, pour étudier l'identité des protéines. Nos résultats indiquent clairement que la protéase d'adénovirus est très sensible au SDS (Figure 13 pistes h et l). Des expériences précédentes, ont déjà démontrées que p VTI dénaturé par le SDS est bien clivé par l'enzyme après avoir enlevé ce dénaturant. Donc l'effet de l'inhibition observé ici est exercé sur l'enzyme, et non pas sur le substrat.
4.1-Puriflcation de l'immunoglobuline G à partir du sérum de lapin
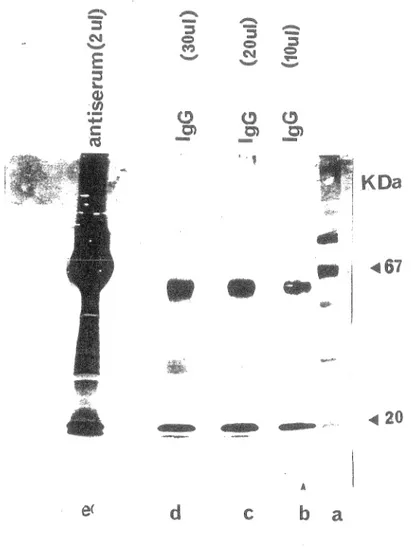
Lorsque !'adénovirus infecte un organisme, celui ci produit des anticorps. L' immunoglobuline G est prédominante dans le sang, la lymphe, le liquide cérébro-spinal et le liquide péritonéal. L'on peut alors se poser la question à savoir si l'IgG affecte l'activité de la protéase. Ainsi, nous avons purifié les IgG de l'antisérum contre la protéase d'adz par chromatographie d'affinité (section 18), afin de tester par la suite l'effet des IgG sur l'activité de la protéase. Sur la figure 14 on voit que sur les pistes b, c, et d apparaissent deux bandes. La première a un poids moléculaire de 50 000 Da, la deuxième 20 000 Da. Il s'agit respectivement des chaînes lourdes et légères de l'immunoglobuline. La concentration a été évaluée à 225ug/ml (par la méthode de Bradford), et un volume total de lml a été recueilli.
Vil (fJ
m
iv.ri i:!I 1- w ~IDf
f!:' @,1 ;\,. """ <Os::
:Jw
:;s ::î :J ::i l..m © {l,j © ~ ~'11 ~.c
ml:: 41 J:: t:: "C . .k 0 ~-
·t!!) œi ~· IJ:'cl-
t:J ~ "'1c'll'·r_:,,~•1illli> a bc
d e fg
Figure 11: Effet du temps d'incubation sur l'activité protéolytique. Les virions d1ad2 ts1 marqués à
la méthionine 35s purifiés et fragrnentés ont été incubés à 37°C avec l'extrait brut bactérien pLPV pour des temps détenninés (piste f: 1 heure, piste e: 2 heures, piste 4 heures, piste c: 8 heures, piste b: 16 heures). piste g représente contrôle sans Dn)re:asie. Les protéines wt ont été utfüsées comme marqueur de poids moléculaire.
Figure
a b
d
f
gh
Effet de la température sur factivité protéolytique. Les virions d'ad2 ts1 marqués à la méthionine 35s purifiés et fragmentés ont été incubés pendant 16 heures avec l'extrait brut pLPV à différentes températures. Piste 4 °C, piste c: piste 33"C, piste e: 37°C, f: piste 45°C. Les protéines d'adz wt ont utilisées comme marqueur de poids moléculaire.
1!!
12
(/') U".11 U) ~ ';;:î
-
=' """"'..
-
N~~ ~
:E:
,_
~c:
y 0 c: 0 u~
k
h g f
e
d
c
ba
Figure 13: Effet du Sodium dodécyl sulfate et de furée sur l'activité protéolytique. Les virions d'ad2 ts1 marqués à la méthionine 35s, purifiés et fragmentés, ont été incubés avec la protéase purifiée de l'extrait brut bactérien pLPV pendant 16 heures et mis en présence de différentes concentrations d'urée et de sodium dodécyl sulfate . lM urée (piste 2!v! urée (piste e ), urée (piste 41vI urée (piste et SDS (piste h), 2% SDS (piste 3% SDS (piste 4% SDS (piste k), SDS piste b représente u.n contrôle sans enzyme, piste c représente un contrôle avec et elJlZ)'me" Température d'incubation:
-"'
-"5 :li -::;: "= ~ N e 0 :::.1·-
1~ ,_, l:":l1 N QI 'Ili""' ri::·-
-::J11-œ
,!!l If.!) C) (!)""""
.rd s::: ~ C).si
' ~KDa
e<
d
c
b
a
Figure 14: Purification des IgG du sérum de lapin. Les IgG du sérum de lapin ont été purifiées sur une DEAE A..ffi-Gel blue (Kit la compagnie Bio RAD) et mis sur gel PAGE-SDS. Piste e: 2ul d1antisérum, piste b: 10 ul de l'échantillon purifié,, c: ul
l1échantillon purifié, piste ul de l'échantillon purifié. Des marqueurs de moléculaires ont été utfüsés (piste
4.2- Effet de l'lgG purifié sur l'activité de la protéase
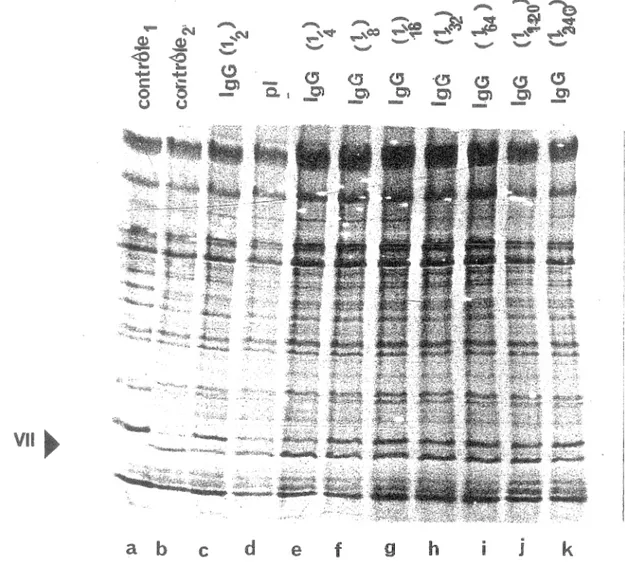
Sur la figure 15, il s'agit des virions d'ad2 ts1 marqués à la méthionine 35s, purifiés et fragmentés (section 2) qui ont été incubés à 37°C pendant 16 heures avec la protéase purifiée (section 1.2) et mis en présence des IgG qui ont été purifiées (section 18). La protéase a été incubée au préalable avec l'IgG pendant 30 minutes à 4°C. Contrôle sans protéase: piste a, contrôle substrat et protéase: piste b. Sur les pistes e, f, i, j, k, l'IgG a été dilué au préalable (piste e: dilution 1 \4, piste f: dilution 1 \8, piste g: dilution 1 \16, piste h: dilution 1\32, piste i: dilution 1\64, piste j: dilution 1\120, piste k: dilution 1\240). Sur la piste d le sérum préimmunitaire a été ajouté. On peut voir qu'il y a une perte d'activité de 80% en présence d'IgG dilué 1\2 (piste c). Cependant lorsque l'on dilue l'IgG à 1\4 il n'y a plus d'effet car le taux de protéines pVII (non clivé) est le même que le taux de protéines VII (pistes e à k ). Le sérum préimmunitaire n'avait aucun effet sur l'efficacité de clivage (piste d). L'inhibition marginale (titre 1 \2) de l'activité protéasique par les anticorps anti-protéase a été confirmée par un test in vitro tout à fait différent de celui utilisé ici, par W
""""'
a
"""""' Q-
-
"');!
~ f;f_J 'w;;;jf- -
-"'i.!
~-
i:\i_,oey-
'J,p.'l,,~ ~_..,r
~~ il'l.l Gll-·
~"""'-
~-
- -
-<O :!.,, ·~ ~ C)-
!'.:*"'
i:::a
~!$ (!j ·(31 C:J (:J ~ 0 0 n 1 ~,_pi
C'l tJ! t'Jj .,gi u u ~a
b
d
e
h
Jk
figure , Effet de llgG sur foctivité protéolytique. Les virions d1ad2 ts1, marqués à la méthionine
et fragmentés ont été incubés à pendant 16 heures avec la protéase purifiée et mis en présence l'IgG purifié protéase a été incubée au préalable avec pendant 30 à 4 °C). Contrôle sans protéase: contrôle substrat et protéase: piste b. Pistes c: 1 \2, piste e: dilution 1
dilutionl\16, h: piste ï: dilution 1\64,
A. CLONAGE DE LA PROTEINE PVI
Il est important de bien connaître les propriétés de la protéase adénovirale pour pouvoir synthétiser des agents antiviraux spécifiques. Afin de mieux caractériser la protéase adénovirale nous avions décidé d'utiliser un substrat simple et connu de la protéase d'adz: la protéine pVI (protéine précurseure de la protéine VI). La protéine VI en tant que dimer de trimères est associée aux hexons péripentonaux à l'intérieur de la capside. Le clivage de la protéine p VI en iVI (forme intermédiaire de VI) puis VI, augmente progressivement l'affinité d'attachement à l'hexon [Matthews et al., 1994]. La protéine pVI est clivée aux extrémités C-terminale et N-terminale par la protéase. Labrecque en 1993 a effectué des expériences avec la protéine ovalbumine, qui contient un site de clivage spécifique pour la protéase. Ce substrat est simple et peu coûteux cependant la protéine n'est pas spécifique au virus et son clivage a été peu efficace, c'est pourquoi nous avions décidé de cloner le gène pVI.
Les résultats du transfert sur filtre de nitrocellulose effectué avec l'anticorps polyclonal de lapin démontre l'absence de la protéine de fusion p VI (Figure 9), même si par buvardage à la Southern nous avions pu démontrer la présence de l'insert (résultat non présenté). Ceci peut s'expliquer par le fait que la protéine p VI pourrait être synthétisée mais par la suite dégradée par une ou plusieurs protéases bactériennes. En effet Matthews et al., 1994 ont eu le même problème avec l'expression de p VI. Ils nous ont donné des renseignements importants concernant le clonage de la protéine p VI dans une communication personnnelle: la protéine pVI est très toxique pour E.coli, ce qui explique pourquoi il est très difficile de cloner le gène p VI. Afin de contourner ce problème de toxicité, ils ont utilisé le vecteur d'expression pRSETA (Invitrogen) et la souche E.coli BL21(DE3) pLysE (Novagen). La protéine p VI dans ce cas, est fusionnée à un ensemble de 42 acides aminés qui inclue les six résidus histidine qui ont une forte affinité pour des billes de résine
chargées de Nickel. Ceci a permis la purification de la protéine à partir de E.coli. La transcription s'effectue à partir d'un promoteur T7, ce qui explique l'utilité de la souche BL21(DE3). Cette souche code pour le gène polymérase T7 sous le contrôle d'un promoteur lac. La souche BL21(DE3) pLysE produit la protéine lysozyme à de très hauts niveaux à partir d'un plasmide (pLysE) qui contient également un marqueur de résistance au chloramphénicol. Le lysozyme s'attache au polymérase T7 produit à un faible niveau par BL21(DE3), et l'inactive. Cette production de la polymérase T7 avant l'induction à l'IPTG est due à la "faiblesse" du promoteur lac. Ceci a permis de réprimer le promoteur T7. Après l'induction par l'IPTG, le taux de la polymérase T7 augmente rapidement et dépasse le taux de production du lysozyme T7, permettant l'expression de la protéine p VI. Les bactéries doivent être recueillies 30 à 60 minutes après induction, parce que p VI est très toxique pour E.coli. Après centrifugation, les bactéries doivent être lysées par choc thermique à l'aide de la glace sèche et de l'éthanol. La purification doit être faite en présence de 6M de guanidine hydrochloride, afin d'empêcher la dégradation de la protéine par des protéases de E.coli. De plus, à cause d'un problème de la solubilité il est difficile d'obtenir une concentration de la protéine supérieure àlO ug/ml.
B. ETAT DE SOLUBILITE DE LA PROTEASE (ETUDE DU PHENOTYPE tsi) La déficience de la protéase tsi, résulte de la substitution d'une Praline en Leucine de l'acide aminé 137 à l'intérieur du cadre de lecture de l'unité de transcription tardive L3 qui code pour une protéine de 23 K.Da: la -protéase adénovirale. Afin de mieux comprendre le rôle de la protéase wt dans l'infectivité du virus, il est nécessaire d'étudier le phénotype ts1. Labrecque en 1993, a effectué des expériences in vitro qui ont permis de récupérer l'activité de la protéase ts1 à l'aide du cofacteur pVI-C. Ceci démontre que la mutation à l'intérieur de l'enzyme ne l'empêche pas d'interagir avec son cofacteur. Nous savons que la protéase wt est insoluble dans les noyaux des cellules infectées, c'est pourquoi nous avons vérifié si c'était le cas de la protéase ts1. A 10 000 rpm, durant 5
minutes, les protéases de ts1, et wt sont demeurées dans les culots c'est à dire sous forme insoluble et ce, malgré l'ajout du cofacteur pVI-C (Figure 10 pistes d et i) ou, de DNase I (Figure 10 pistes c et h) dans des réactions séparées. Ces résultats ne nous permettent donc pas d'établir une différence entre les protéases wt et ts1. Rancourt et al., 1995 ont démontré par des expériences que l'enzyme recombinante ts1 n'est pas thermosensible de façon significative in vitro: cependant l'enzyme purifié, tout comme l'enzyme in vivo,
nécessite le peptide pVI-C. Donc par des expériences in vivo et in vitro ils ont démontrés que la déficience de la protéase ts1 est causée par un empêchement de l'activation de l'enzyme et de l'encapsidation à la température non permissive. La protéase thermosensible serait synthétisée et transportée au noyau afin d'être associée avec des particules incomplètes dans une manière semblable à la protéase wt. Il semblerait que la maturation des particules adénovirales se déroule dans des particules incomplètes et dans les jeunes virions qui contiennent un génome complet mais des protéines immatures, la maturation se poursuit et il en résulte des virions infectieux suite à de nombreux clivages par la protéase virale. La maturation des jeunes virions ts1 est arrêtée à cause de l'absence de protéase dans ces particules [Rancourt et al., 1995].
C. TESTS D'ACTIVITES ENZYMATIQUES
Afin de caractériser la protéase adénovirale des tests d'activités enzymatiques ont été effectués. Nous avons testé l'activité de la protéase adénovirale selon le temps d'incubation et l'activité optimale est obtenue après 16 heures d'incubation (Figure 11). Ce résultat ne remet pas en question les tests d'activité effectués dans le laboratoire puisque jusqu'ici les essais avaient une durée de 16 heures à 37°C. Selon le test effectué à 37°C le pourcentage de clivage est de 95% (Figure 12). Les stratégies de purification de la protéase adénovirale implique la solubilisation de la protéine avec de l'urée ce qui permet son repliement [Tihanyi et al., 1993]. D'après les résultats de la figure 13, les concentrations d'urée de 3M - 4M inhibent l'activité enzymatique. Cependant les faibles
concentrations d'urée, jusqu'à lM semblent augmenter le clivage. Cette expérience ne permet pas de distinguer entre un effet stimulateur sur l'enzyme, et un effet positif sur la solubilisation du substrat pVII pour le rendre plus accessible à l'enzyme. D'autres expériences qui ont été faites au préalable, nous laissent croire que ce serait plutôt le deuxième cas. En présence d'une aussi faible quantité de SDS que 1 %, on n'observe pas de clivage. Ceci s'explique bien car le sodium de dodécyl sulfate est un détergent ionique et donc dénature la structure de l'enzyme de façon irréversible l'enzyme. Les résultats de la figure 15 nous permettent de conclure que l'immunoglobuline G purifié de l'antisérum de lapin contre la protéase de l'adénovirus de type 2 n'inhibe pas fortement l'enzyme. L'on peut alors émettre l' hypothèse suivante: I1IgG se fixe à la protéase mais pas au niveau du site actif. Ainsi en présence d'IgG il y aurait un encombrement stérique et la protéase ne serait plus en mesure d'accéder au substrat. Dans notre laboratoire, des expériences de caractérisations fonctionnelles ont été effectuées en utilisant des substrats fluorogéniques qui comportent les séquences consensus de clivage de la protéase. Il y a plusieurs avantages attribués à cette technique: la précision, la sensibilité et la rapidité comparé à l'utilisation du substrat radioactif pour lequel les résultats sont obtenus 24 heures après. En utilisant les substrats fluorogéniques, par spectrophotométrie on détecte l'intensité de fluorescence, ainsi il a été possible de conclure qu'il y a une inhibition de l'activité en présence d'urée [Diouri et al., 1995 en préparation].
En conclusion, l'utilisation du virion adz ts1 purifié et fragmenté, comme substrat nous a permis de savoir que l'enzyme est très sensible au SDS mais par contre maintient son activité jusqu'à 45°C ou en présence de 3M d'urée. Les anticorps antiprotéases inhibent l'enzyme seulement à hautes concentrations. L'étude du phénotype ts1 nous a permis de savoir que les enzymes wt et ts1 sont insolubles. Cependant, il faudra tenté d'élaborer d'autres expériences pour identifier le mécanisme impliqué dans le défaut d'encapsidation de la protéase du virus ts1.