Targeting steroidogenic enzymes and cyclin-dependent
kinases in breast cancer cells: novel attempt to control
cell proliferation and cell cycle
Thèse
Xiaoye Sang
Doctorat en médecine moléculaire
Philosophiæ doctor (Ph. D.)
Targeting steroidogenic enzymes and
cyclin-dependent kinases in breast cancer cells:
novel attempt to control cell proliferation and
cell cycle
Thèse
Xiaoye Sang
Sous la direction de:
Résumé
Les enzymes stéroïdogènes telles que les 17β-hydroxystéroïdes déshydrogénases réductrices (17β-HSD) et la 11β-hydroxystéroïde déshydrogénase 2 (11β-HSD2) jouent un rôle important dans la progression du cancer du sein à récepteurs d'oestrogènes positifs (ER +) en régulant les stéroïdes sexuels et les glucocorticoïdes. Par exemple, la stéroïde sulfatase (STS) est impliquée dans la production de E2. Le STX64, un inhibiteur de STS, a provoqué une réduction de tumeur dans un essai animal. Malgré le succès de l'essai clinique de phase І, les résultats de l'essai de phase II n'étaient pas significatifs. La plupart des études dans le domaine se sont concentres sur la fonction et la régulation individuelle des enzymes stéroïdogènes. Cependant, la régulation mutuelle de ces enzymes et la modulation induite sur les récepteurs des œstrogènes et des androgènes pour la promotion du cancer du sein ne sont pas encore claires.
Dans cette étude, les cellules MCF-7 et T47D ont été utilisées pour déterminer le mécanisme d'inhibition de STS et les régulations mutuelles d'importantes enzymes stéroïdogènes. Nous avons démontré que l'inhibition du STS diminuait les niveaux d'estradiol et de DHT, entraînant une limitation de l'effet cellulaire. Ceci est différent de l'inhibition de la 17β-HSD7, qui diminue l'estradiol tout en augmentant la concentration de DHT, démontrant ainsi le double contrôle du cancer du sein. La 17β-HSD7, la STS et la 11β-HSD sont étroitement corrélées. L'inhibition de l'un d'entre eux peut réduire l'expression de l’autre, amplifiant ainsi le rôle de l'inhibition. De plus, l'inhibition de 17β-HSD7 augmente l'expression des gènes récepteurs d’androgènes, qui sont considérés comme un meilleur marqueur dans le pronostic du cancer du sein ER +, et qui maintiennent le niveau du gène des récepteur
ostrogénique α.
D'autre part, le cancer du sein triple négatif (TNBC) est un sous-type très agressif de tumeur qui pose un défi clinique à la résistance potentielle à la chimiothérapie. Les kinases dépendantes des cyclines (CDK) sont des cibles thérapeutiques
impliquées dans des événements de carcinogenèse en raison de leur implication dans les phases du cycle cellulaire. Le facteur 1 d'interaction CR6 (CRIF1) a été signalé agissant comme un régulateur négatif du cycle cellulaire par interaction avec CDK2. Deux inhibiteurs sélectifs de l'interface CRIF1-CDK2 ont été utilisés pour étudier les effets antiprolifératifs sur les lignées de cellules TNBC de MDA-MB-231 et MDA-MB-468. Il est révélé que le traitement des articulations avec des inhibiteurs sélectifs de l’interface CRIF1-CDK2 permet de réduire considérablement la résistance aux effets anti-proliférants induits par Paclitaxel (Taxol). Il a été indiqué que le traitement d'association d'inhibiteurs de l'interface CRIF1-CDK2 4 μM et de Taxol pourrait encore diminuer la prolifération jusqu’à 44%, ce qui est nettement plus significatif que la réduction de prolifération induite par Taxol (réduction de 18,8%)
dans la lignée cellulaire TNBC MDA-MB-468.
Notre découverte fournit une base pour améliorer d’avantage le traitement endocrinien du cancer du sein ER +. Par exemple, le ciblage combiné du 17β-HSD7 et du STS peut être un nouveau traitement prometteur pour sélectionner les enzymes stéroïdiennes cibles.. De plus, l'inhibition combinée par les inhibiteurs de l'interface CRIF1-CDK2 pourrait réduire considérablement la résistance à la chimiothérapie, laissant ainsi un espoir de vaincre la résistance à la chimiothérapie par TNBC.
Abstract
Steroidogenic enzymes such as reductive 17β-hydroxysteroid dehydrogenases (17β-HSDs) and 11β-hydroxysteroid dehydrogenase 2 (11β-HSD2) play an important role for the progression of estrogen receptor positive (ER+) breast cancer by regulating sex-steroids and glucocorticoids. For example, Steroid sulfatase (STS) is involved in the E2 production. STX64, an STS inhibitor induced tumor reduction in an animal assay. Despite success in phase І clinical trial, the results of phase II trial were not significant. Most studies in the domain focused on the function and individual regulation of steroidogenic enzymes. However, mutual regulation of these enzymes and the induced modulation on the estrogen and androgen receptors for breast cancer promotion have not yet been clear.
In this study, MCF-7 and T47D cells were used to determine the mechanism of STS inhibition and the mutual regulations of important steroidogenic enzymes. We demonstrated that inhibition of STS decreases both estradiol and DHT level, leading to a limitation in the cell effect. This is different from 17β-HSD7 inhibition, which decreases estradiol while increaseing DHT concentration, demonstrating a dual control of breast cancer. 17β-HSD7, STS and 11β-HSD are mutually closely correlated. Inhibition of one of them can reduce the expression of another, thereby amplifying the role of the inhibition. Furthermore, inhibition of 17β-HSD7 increases the expression of androgen receptor gene which is considered as a marker for better prognosis in ER+ breast cancer, while maintaining estrogen receptor α gene level.
On the other hand, triple negative breast cancer (TNBC) is a highly aggressive subtype of breast cancer that imposes a clinical challenge to the potential resistance to chemotherapy. Cyclin-dependent kinases (CDKs) are therapeutic targets implicated in carcinogenesis events due to their involvement in the cell cycle phase. CR6-interacting factor 1 (CRIF1) was reported to act as a cell cycle negative regulator through interaction with CDK2. Two selective CRIF1-CDK2 interface inhibitors were used to study the anti-proliferative effects on the TNBC cell lines of MDA-MB-231 and MDA-MB-468. It is revealed that the resistance of anti-proliferative
effects induced by Paclitaxel (Taxol) can be significantly reduced by the joint treatment with selective CRIF1-CDK2 interface inhibitors. It was indicated that a
combined treatment of 4 μM CRIF1-CDK2 interface inhibitors and Taxol could further decrease proliferation to approximately 44%, which is markedly more significant than the proliferation reduction induced by Taxol (18.8% reduction) in TNBC cell line MDA-MB-468.
Overall, our findings provide a base for further improving the endocrine therapy of ER+ breast cancer, e.g., for selecting the target steroid enzymes, and the combined targeting of 17β-HSD7 and STS can be a promising new treatment. In addition, combined inhibition by CRIF1-CDK2 interface inhibitors can significantly reduce the resistance of chemotherapy, thus holding a promise for overcoming the resistance in response to chemotherapy in TNBC.
Table des matières
Résumé ... ii
Abstract ... iv
Table des matières ... vi
Liste des figures ... viii
Liste des figures, tableaux, illustrations ... ix
Liste des abréviations, sigles, acronymes ... x
Remerciements ... xiii
Avant-propos ... xv
Introduction... 1
Chapitre 1 Steroid sulfatase inhibition success and limitation in breast cancer clinical assays: an underlying mechanism ... 31
1.1 Résumé ... 31
1.2 Abstract ... 32
1.3 Introduction ... 33
1.4 Materials and Methods ... 35
1.5 Results ... 39
1.6 Discussion ... 46
1.7 Conclusion ... 49
1.8 References ... 49
Chapitre 2 Mutual regulations and breast cancer cell control by steroidogenic enzymes: dual sex-hormone receptor modulation upon 17β-HSD7 inhibition ... 70
2.1 Résumé ... 70
2.2 Abstract ... 72
2.3 Introduction ... 73
2.4 Materials and Methods ... 75
2.5 Results ... 78
2.6 Discussion ... 84
2.7 Conclusion ... 88
Chapitre 3 Targeting the lethal triple negative breast cancer: CRIF1-CDK2
interface inhibitors enhance Taxol inhibition of cancer ... 111
3.1 Résumé ... 111
3.2 Abstract ... 113
3.3 Mini communication body ... 114
3.4 Supporting information ... 119
3.5 References ... 123
Conclusion... 137
Bibliographie ... 142
Annexe A Highlights and graphic abstracts of published papers ... 157
4.1 Steroid sulfatase inhibition success and limitation in breast cancer clinical assays: an underlying mechanism ... 157
4.2 Mutual regulations and breast cancer cell control by steroidogenic enzymes: dual sex-hormone receptor modulation upon 17β-HSD7 inhibition ... 158
Liste des figures
Figure 1. Normal breast tissue ……….……2 Figure 2. Ten leading cancer types for the estimated new cancer cases and deaths
by sex………...……...…3
Figure 3. Incidental and mortality of breast cancer in Canada based on Canadian
cancer statistics ………...…5
Figure 4. Patient outcome based on breast tumor intrinsic subtypes …………...7 Figure 5. Dehydroepiandrosterone (DHEA) as being the unique source of sex
steroids in women after menopause……… ..………...9
Figure 6. Human steroidogenic and steroid-inactivating enzymes in peripheral
intracrine tissues………....…10
Figure 7. Illustration of dual function of Estradiol as growth factor and procarcinogen
in initiation and progression of breast cancer………...…11
Figure 8. Different mechanisms of estrogen dependence for hormone related breast
cancer in premenopausal and in postmenopausal women...………...…13
Figure 9. Schematic illusion of steroidogenesis (estrogen/androgen) in intracrine
tissues in postmenopausal women ……….…14
Figure 10.The role of STS in ER+ breast cancer……….………17 Figure 11. 11β-hydroxysteroid dehydrogenase (11β-HSD) .……….…21 Figure 12. Model of the human mammary epithelial hierarchy linked to cancer
subtypes ……….…23
Figure 13.Recurrence and survival of TNBC…………...….………24
Liste des figures, tableaux, illustrations
Table 1. Estimated New Female Breast Cancer Cases and Deaths by Age, US,
Liste des abréviations, sigles, acronymes
3α-diol 3α-androstanediol
3β-diol 5α-androstane-3β, 17β-diol
3β-HSD 3β-hydroxysteroid dehydrogenase
4-dione 4-androstene-3,17-dione
5-diol 4-androstene-3β,17β-diol
11β-HSD2 11β-hydroxysteroid dehydrogenase 17β-HSDs 17β-hydroxysteroid dehydrogenases
ABC ATP-binding cassette
Adione 5α-androstane-3,17-dione
ADT espiandrosterone
ALDH1 aldehyde dehydrogenase 1
ARE androgen response element
ACTH adrenocorticotropic hormone
AI aromatase inhibitor
AR Androgen Receptor
BC breast cancer
BRCA1 breast cancer type 1 susceptibility protein
CDKs Cyclin-dependent kinases
CRH corticotropin releasing hormone
CRIF1 CR6-interacting factor 1
DBD DNA binding domain
DHEA dehydroepiandrosterone
DHEA-S dehydroepiandrosterone sulfatase
DHT Dihydrotestosterone
E1 estrone
E1-S estrone-sulfate
E2 estradiol
E2-S estradiol sulfate
EMT epithelial to mesenchymal transition
ER estrogen receptor
ER+ estrogen receptor positive
ERE estrogen-responsive element
ERG estrogen-responsive gene
ESR1 estrogen receptor α gene
F0922-0913 CRIF1-CDK2 interface inhibitor F1142-3225 CRIF1-CDK2 interface inhibitor
FBS fetal bovine serum
GnRH gonadotropin releasing hormone
GSH glutathione
GST Glutathione-S-transferase
HER2 human epidermal growth factor receptor 2
IL-6 interleukin-6
IGF insulin growth factor
INH1 17β-HSD type 1 inhibitor
INH80 17β-HSD type 7 inhibitor
LBD ligand binding domain
LH luteinizing hormone
NF1 nuclear factor
NF-κB kappa-light-chain-enhancer of activated B cells
nM nanomolar
NTD N terminus domain
OS osteosarcoma
PBLs peripheral blood lymphocytes
PI propidium iodide
PR progesterone receptor
QTR-PCR quantitative Real-time PCR
SERMs selective estrogen receptor modulators
STS steroid sulfatase
TNBC triple negative breast cancer
TNF-α tumor necrosis factor-α
Remerciements
I would like to express my great thanks and sincere gratitude to my director of research, professor Sheng-Xiang Lin, for his meticulous guidance and helpful support to my research and my life during my PhD studies in Laval University. Thanks to his enlightening discussions and timely supports on my project, I can overcome all the difficulty and publish two first-authored papers and prepare a third before presenting this thesis.
I also greatly appreciate the inspiration that Professor Sheng-Xiang Lin offered me during the studies, this inspiration provided me with more original thinking and creative ideas. I also admire his vision and experience in science, which led me to have more knowledge of breast cancer research development. I will certainly benefit from that in my future career. I also sincerely appreciate the following persons and organisations for supporting my PhD studies.
I would like to thank all the members in Dr.Lin‘s lab. I express my great thanks to Dre. Ming Zhou for helping me start the cell culture studies when I began the PhD program; I thank to Dre. Hui Han and Dr. Tang Li, who showed me the important skills in cell studies and being involved in my papers. I also appreciate the inspiring discussion in cell cycle studies with Ruixuan Wang and Preyesh Stephen. I am grateful for the discussion of calculation methods with Wenfa Zhang, for the help of ordering materials by Jean-François Thériault.
I would like to thank Dr. Donald Poirier for providing important inhibitors used for my experiment and the helpful suggestions for the writing of my paper.
I would like to thank the professional groups who work at the platforms at the Centre Hospital University (CHU) de Quebec Research Center (CHUL) and Laval
University. Thanks to Mr. Martin Thibault and Mr. Ronald Maheux for the professional advice on the image analysis. Thank to Dr. Alexandre Brunet for the advice of analyzing data of cell cycle and cell apoptosis. I really appreciate all the advice and help that they have provided me, helping me to analyze my data more accurately and quickly.
Finally, I am very grateful to my family, my parents and friends, who gave me strong mental support and great understanding during my studies. Thanks to my boyfriend, Dominic Faucher who provides me a lot of love and help in learning French. Because of their support, my studies are completed with more pleasure.
Avant-propos
I fulfilled my doctoral study in the Centre de Recheche en Endocrinologie et Népherologie, CHU de Quebec Research Center Affiliated to Laval University. My studies involved several aspects of molecular and cellular biology using a variety of assessments mainly at the cell level. In particular, I studied the cell mechanism of steroid sulfatase and mutual regulations of steroidogenic enzymes in hormone-dependent breast cancer and how to reduce the chemotherapy resistance in triple negative breast cancer by regulating Cyclin-dependent kinase 2 (CDK2) and cell cycle with interface inhibitors of and CDK2 and CR6-interacting factor 1 (CRIF1).
The thesis is written in English, except for the summary as well as the abstract of each article, which are in French. Two articles have been published in Journal of Steroid Biochemistry and Molecular Biology. The last paper is in preparation for submission and publication.
In the introduction, several important steroidogenic enzymes in hormone-dependent breast cancer were reviewed, including steroid sulfatase, aromatase, 17β-HSD1, 17β-HSD7 and 11β-HSD2. Moreover, because triple negative breast cancer lacks efficient therapy, the CDKs and CRIF1 were also discussed as they could be the potential targets in decreasing the chemotherapy resistance in triple negative breast cancer. The hypothesis and objectives were described of the end of this chapter.
Chapter Ⅰ: I am the first author of this article entitled “Steroid sulfatase inhibition success and limitation in breast cancer clinical assays: An underlying mechanism” In this article, I demonstrated the cell mechanism of steroid sulfatase, addressing a possible explanation for the limited results of the sulfatase inhibitor (STX64) in the phase II clinical study.
List of co-authors: Xiaoye Sang, Hui Han, Donald Poirier, Sheng-Xiang Lin
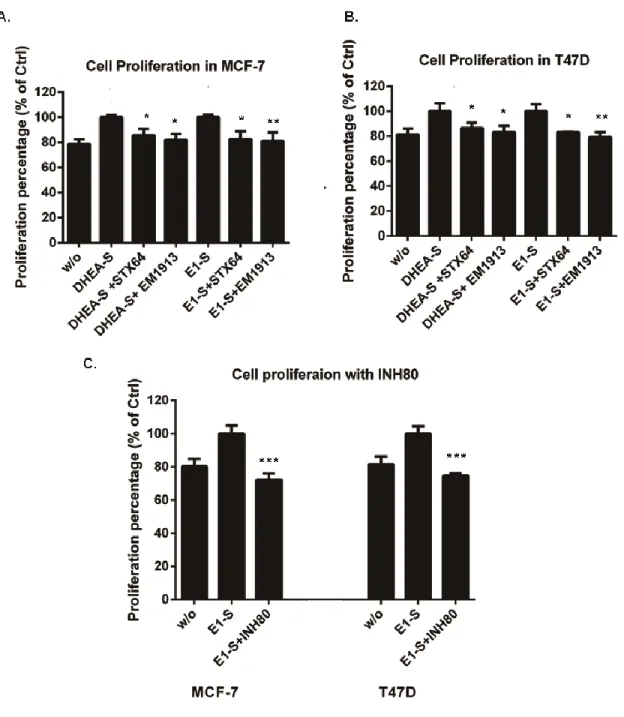
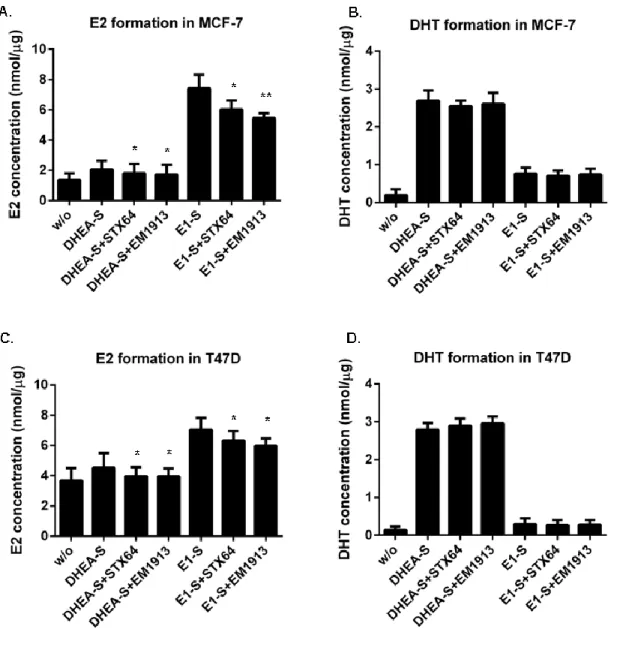
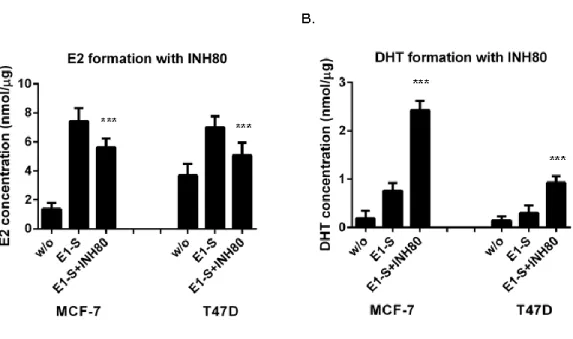
Xiaoye Sang performed most of the research works and wrote this paper; Hui Han carried out the experiments include: EM1913 and STX64 suppressed cell proliferation providing DHEA-S; and EM1913 and STX64 decreased E2 and DHT biosynthesis in MCF-7 and T47D; Donald Poirier gave some important advices and supplied enzyme inhibitors;Sheng-Xiang Lin supervised the research and finalized the manuscript.
Published in Journal of Steroid Biochemistry and Molecular Biology, Vol. 183, 80-93 (2018). doi: 10.1016/j.jsbmb. 2018.05.009. Recieved 14 November 2017; Accepted 23 May 2018; Avaliable online 24 May 2018.
Chapter Ⅱ: I am the first co-first author of this article entitled “Mutual regulations and breast cancer cell control by steroidogenic enzymes: dual sex-hormone receptor modulation upon 17β-HSD7 inhibition”. In this article, we demonstrated that important steroidogenic enzymes transcriptionally regulated by Estrogen Receptor α, can be mutually closely correlated.
List of co-authors: Xiaoye Sang1, Hui Han1, Tang Li, and Sheng-Xiang Lin.
Xiaoye Sang completed most of the experimental work and wrote the paper. Hui Han conducted the experiments for 17β-HSD7 mRNA expression and the regulation for E2 and DHT concetration. Tang Li searched for the information of cistrome database. Sheng-Xiang Lin supervised the whole work and finalized the manuscript.
Published in Journal of Steroid Biochemistry and Molecular Biology, Vol. 193, in press (2019). Doi: 10.1016/j.jsbmb.2019.105411. Recieved 5 March 2019; Accepted 13 June 2019; Avaliable online 15 June 2019.
Chapter Ⅲ: I am the first author of the communication paper entitled “Targeting the lethal triple negative breast cancer: CRIF1-CDK2 interface inhibitors enhance Taxol inhibition of cancer cells”. In this paper, two selective CRIF1-CDK2 interface inhibitors were used to study the anti-proliferative effects on the triple negative breast cancer (TNBC) cell lines, they further significantly decreased cell proliferation induced by Taxol in TNBC cells, improving the sensitivity to Taxol in TNBC cell lines. This finding suggests new targets to reduce the resistance of chemotherapy in TNBC. This paper is prepared for submission.
List of co-authors: Xiaoye Sang, Sheng-Xiang Lin.
Xiaoye Sang conducted all the experiments and wrote the manuscript. Sheng-Xiang Lin supervised the research and finalized the manuscript.
In the conclusion, I made the comparison of effects of sulfatase and 17β-HSDs inhibitions, demonstrating that 17β-HSD7 can be an important target for the treatment of hormone dependent breast cancer because the inhibition of 17β-HSD7 can increase androgen receptor expression. In the TNBC, the use of CRIF1-CDK2 interface inhibitors can increase the cell sensitivity to Taxol.
The references of introduction and conclusion are listed after the conclusion. References of each publication are listed after the main text body of each article.
Introduction
1 Breast cancer
1.1 General Concept
Breast cancer starts when cells in the breast begin to grow out of control. These cells usually form a tumor that can be seen on an x-ray or felt as a lump. The tumor is malignant (cancer) if the cells invade surrounding tissues or spread (metastasize) to distant areas of the body.
Breast cancer can be initialed from different parts of the breast. Most breast cancers start in the ducts that carry milk to the nipple (ductal cancers). And some begin in the glands which produce breast milk (lobular cancers). Other types of breast cancer can also be seen but they are less common. Most breast tumors are carcinomas, which arise from breast epithelial elements. Breast cancers can be divided into two major types, in situ carcinomas and invasive (or infiltrating) carcinomas (American cancer society, https://www.cancer.org/cancer/breast-cancer/about/what-is-breast-cancer.html).
The former type may arise in either ductal or lobular epithelium, without invasion of the underlying basement membrane that would constitute extension beyond epithelial boundaries. However, when there is extension of the ductal or lobular malignancy beyond the basement membrane that constitutes the epithelial border, then the malignancy is considered invasive (or infiltrating) ductal or lobular carcinoma. The potential for metastases and ultimately death occurs in invasive disease [1].
Figure 1: Normal breast tissue (American Cancer Society, https://www.cancer.org/cancer/breast-cancer/about/what-is-breast-cancer.html)
1.2 Epidemiology of Breast cancer
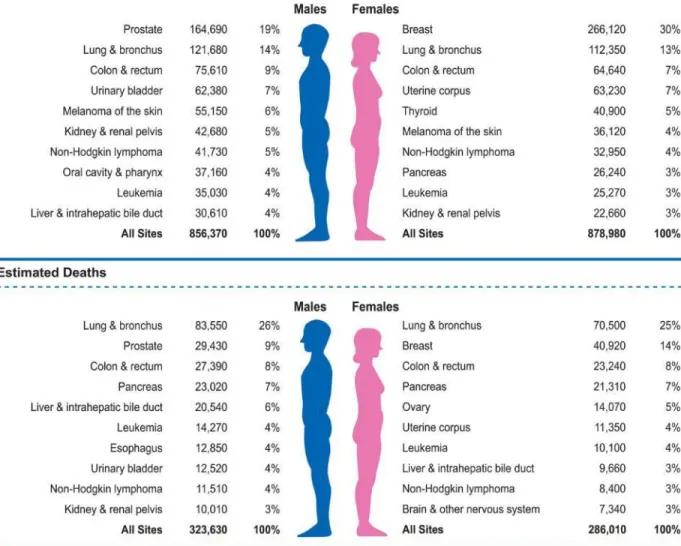
Breast cancer is the most common cancer in women, comprising about one third of all malignancies in females. It is the second cause of cancer mortality, second after lung cancer, and it is also the leading cause of death for American women between the ages of 40 and 55 (Figure 2) [2,3].
Figure 2: Ten leading cancer types for the estimated new cancer cases and deaths by sex, United States, 2018 (Siegel et al, 2018)
The breast cancer incidence rate in Canadian women increased through the early 1990s but decreased in the early 2000s. Breast cancer mortality rate in women have reduced in every age group since the mid-1980s, which reflects the improvement of diagnosis and treatments for this type of cancer [4].
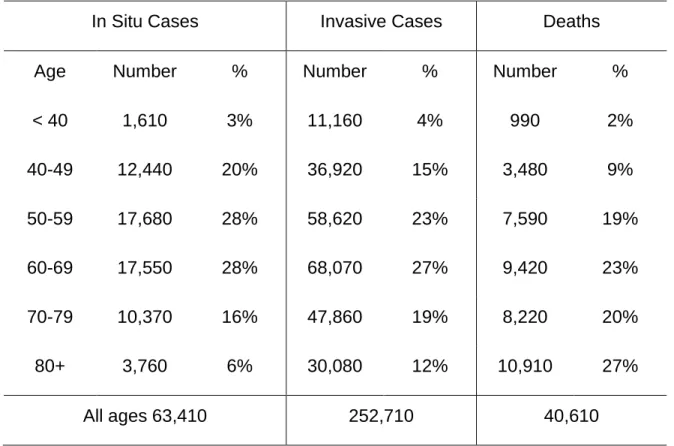
According to statistics from the American Cancer Society, based on the estimation in 2017, about 252,710 women in Untied States were expected to develop breast
cancer during their lifetime. Among them, approximately 40,610 women would die from it (Table 1) [5].
Table 1. Estimated New Female Breast Cancer Cases and Deaths by Age, US, 2017
In Situ Cases Invasive Cases Deaths
Age Number % Number % Number %
< 40 1,610 3% 11,160 4% 990 2% 40-49 12,440 20% 36,920 15% 3,480 9% 50-59 17,680 28% 58,620 23% 7,590 19% 60-69 17,550 28% 68,070 27% 9,420 23% 70-79 10,370 16% 47,860 19% 8,220 20% 80+ 3,760 6% 30,080 12% 10,910 27% All ages 63,410 252,710 40,610
Estimates are rounded to the nearest 10. Percentage may not sum to 100 due to rounding (American Cancer Society, Inc, Surveillance Research, 2017)
Similarly, the Canadian Cancer Society also reported that 26,300 women were diagnosed with breast cancer, which accounts for 25% of all new cancer cases for females in 2017. Moreover, 5,000 women died from breast cancer, which represents 13% of all female cancer mortality in 2017 [4]. Female breast cancer is the most common form of cancer for women from 55-64 years old. The median age at diagnosis is 61 [6].
Figure 3 Incidental and mortality of breast cancer in Canada based on Canadian cancer statistics (http://www.cancer.ca/en/cancer-information/cancer-type/breast/statistics/?region=bc)
1.3 The subtypes of Breast Cancer
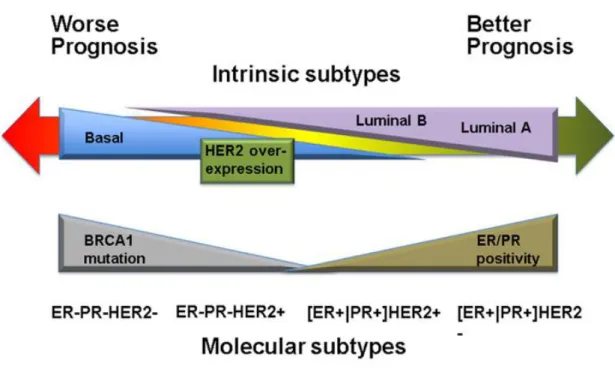
Based on the genes that a cancer expresses, there are several main intrinsic or molecular subtypes of breast cancer (Figure 4).
1.3.1 Luminal tumors
The luminal-like tumors express hormone receptors, with expression profiles reminiscent of the luminal epithelial component of the breast [7]. Two subtypes exist within luminal-like tumors, which are luminal A and luminal B. Luminal A represents tumors that are positive for the estrogen receptor (ER) and progesterone receptor (PR) but negative for human epidermal growth factor receptor 2 (HER2). Luminal B represents the tumors with ER or PR positivity and HER2 positivity [8].
Luminal tumors are the most common subtypes among breast cancer, with luminal A being the majority. In the Carolina Breast Cancer Study, luminal breast tumors account for 64.3% of all breast cancer patients, where luminal A cancers represent 54.3% [9]. In general, the luminal subtypes carry a good prognosis, especially for luminal A tumors, which could be adequately treated with endocrine therapy [10]. While luminal B tumors, which are more proliferative may benefit more from the combined therapeutic strategy of chemotherapy and hormonal treatment.
1.3.2 HER2 over-expression tumors
The intrinsic HER2 over-expression tumors are hormone-receptor negative and HER2 positive [9]. This kind of breast cancer tends to proliferate faster than luminal cancers and can have a worse prognosis [10–12]. However, they could be successfully treated with targeted therapies aiming at the HER2 protein [13,14].
The basal subtype is composed of ER negative, PR negative and HER2 negative tumors. This type of cancer is more common in women with BRCA1 gene mutations and this type of cancer is more frequent among younger and African-American women [15]. Basal tumors account for 60% to 90% triple negative cases [16]. They are of particular interest because they follow aggressive clinical course and currently lack any form of standard target systemic therapy.
Similarly, triple negative breast cancer is also defined by the lack of ER, PR and HER2 expression. And it is also an aggressive subtype because of a shortage of therapeutic targets. However, the basal subtype is defined by a distinct gene expression signature characterized by strong expression of basal makers such as cytokeratin 5,6 and 17 and also encompasses a various tumors [17]. Not all basal tumors are triple-negative, and conversely, not all triple negative breast cancer consist of the basal-like subtype [18].
2 Estrogen-dependent breast cancer
2.1 General Concept
Estrogen-dependent breast cancer is characterized by the expression of ERs, so it is also called ER positive (ER+) breast cancer. The initiation and progression of the carcinoma are directly associated with estrogen, especially estradiol (E2). Estrogen- dependent breast cancer refers to the Luminal subtype by molecular subtype classification. Most of breast cancers, approximately 60% in premenopausal women and 75% in postmenopausal women, are estrogen-dependent breast cancers [19,20].
2.2 The development of intracrinology and the source of sex steroids in women after menopause
A particularly remarkable and highly sophisticated achievement of evolution is intracrinology, a mechanism allowing specific and peripheral production of sex steroids for a strictly local action, without significant release of active hormone into the circulation. This mechanism was named introcrinology in 1988 [21] according to observations obtained in the early 1980s in men castrated for prostate cancer [22] and it showed that a significant proportion of the androgens present in the human prostate is produced from dehydroepiandrosterone (DHEA) itself. In fact, after menopause, DHEA has been the unique source of sex steroids in women [23] (Figure 5).
Approximately 80% of circulating DHEA originates from the adrenals while the other 20% is produced from the ovary [24]. Accordingly, after menopause, all estrogens and all androgens are synthesized from local DHEA in peripheral target tissues (Figure 6). The amount of sex steroids produced depends on the level of steroidogenic enzymes specifically expressed in each cell type of each peripheral tissue [23]. And evolution has continuously provided the peripheral tissues with the enzymes locally producing and inactivating sex steroids.
Figure 5: dehydroepiandrosterone (DHEA) as the unique source of sex steroids in women after menopause. GnRH, gonadotropin releasing hormone; LH, luteinizing hormone; CRH, corticotropin releasing hormone; ACTH, adrenocorticotropic hormone (Labrie et al, 2013)
In the presence of these enzymes expressed in each cell type of human peripheral tissue, coupled with the availability of the precursor DHEA, all the elements are in place for delivery of the required amounts of sex steroids to the cells in each tissue. This compensates and minimize the negative impact of reduced estradiol secretion by the ovary at menopause [23]. Major and essential progress in this area has been made by elucidation of the structure of practically all the tissue-specific genes that encode the human steroidogenic enzymes responsible for the transformation of DHEA into estrogens and androgens (Figure 6) [25,26].
Figure 6: Human steroidogenic and steroid-inactivating enzymes in peripheral intracrine tissues. 4-dione, androstenedione; A-4-dione, 5α-androstane-3,17-dione; ADT,epiandrosterone; E1, estrone; E1-S, estrone sulfate; E2, 17β-estradiol; E2-E1-S, estradiol sulfate; 5-diol, androst-5-ene-3α, 17β-diol-FA, 5-diol fatty acid; 5-diol-s, androst-5-ene-3alpha,17beta-diol sulphate; HSD, hydroxysteroid dehydrogenase; testo, testosterone; RoDH-1, Ro dehydrogenase 1; ER, estrogen receptor; AR, androgen receptor; ugt2b28, uridine glucuronosy transferase 2B28; Sult2B1, sulfotransferase 2B1; UGT1A1, uridine glucuronosyl transferase 1A1 (Labrie et al, 2012).
2.3 E2 and its role in ER+ breast cancer
Epidemiological evidence indicates that prolonged exposure of the mammary glands to high levels of E2 is a high-risk factor of ER+ breast cancer. In this process, E2 plays a dual role in breast carcinogenesis, as a growth factor as well as a pre-carcinogen (Figure 7) [27,28]. In addition, E2 stimulates prolactin secretion and production of local growth factors, implicated in cancer promotion [29–32].
Figure 7: Illustration of the dual function of Estradiol as a growth factor and procarcinogen involved in the initiation and progression of breast cancer (Yang et al, 2007).
2.4 Estrogen Receptors (ERs) and their roles in ER+ breast cancer
ER belongs to a superfamily of nuclear receptors, including those of other steroid hormones, thyroid hormones, vitamin D and retinoic acid [33]. Classically, two molecular forms of ER have been described: ERα and ERβ. The expression of these two subtypes of ERs is highly related to specific tissues. ERβ is more expressed in prostate, bone, ovaries (granulosa cells), lungs, and in various parts of the central and peripheral nervous system. While ERα is predominantly expressed in the pituitary gland, ovaries (thecal and interstitial cells), uterus, liver, kidneys and the mammary glands [34–36]. Both ERα and ERβ play vital roles in the malignant
progression of breast cancer by comprehensively regulating down-stream estrogen response genes.
The ERα comprises a N-terminal AF1 domain, a DNA-binding domain, and a C-terminal ligand-binding region that contains an AF2 domain [37]. Upon the binding of estrogen to ERα, the ligand-activated ERα translocates to the nucleus, binds to the responsive elements in the target gene promoter, and stimulates gene transcription [38,39]. ERα is also the major ER subtype in the mammary epithelium and plays a critical role in mammary gland biology as well as in breast cancer progression [40,41].
ERβ, similar to ERα, also functions as a transcription factor that mediates various physiological responses to estrogen signaling [42]. However, the physiological consequences of ERβ-mediated transcriptional regulation are distinct from those of ERα. A number of studies suggest that an increase in ERβ has anti-proliferative functions [43–45]. Reduced expression of ERβ was reported in invasive breast cancer [46], and ERβ expression is associated with less invasive and proliferating tumors [47].
2.5 E2 formation in ER+ breast cancer
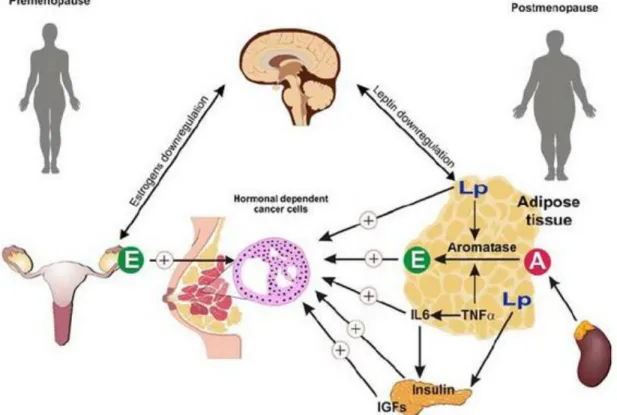
In premenopausal women, E2 is mainly synthesized by the gonads such as the ovary and is released in circulation. After the menopausal, the levels of circulating hormones are very low [48], and the E2 is predominantly produced from adrenal precursors such as DHEA, 4-androstene-3,17-dione (4-dione) and estrone-sulfate (E1-S) by several steroidogenic enzymes [49].
Figure 8: Various mechanisms of estrogen dependence for hormone-related breast cancer in premenopausal and in postmenopausal women (Abbreviations: IGF, insulin growth factor; IL-6, interleukin-6; TNF-α: tumor necrosis factor-α, E: Estradiol, A: Androgen) (Maccio et al, 2010)
In postmenopausal women, two pathways are important for local E2 production in target tissues. The “sulfatase pathway”, in which biological inactive steroid sulfates are the source for E2 (E1-S or DHEAS to E1 or DHEA), and the “aromatase pathway”, which transfers androgens into estrogens [50]. For both pathways, 17β-Hydroxysteroid dehydrogenases (17β-HSDs) (type1 and type7) were integrated and convert estrone (E1) into potent E2 [51]. With the up-regulation of some steroidogenic enzymes, an estrogen-rich microenvironment was generated to sustain the tumor growth and proliferation [52,53]. Therefore, steroid sulfatase (STS), aromatase and reductive 17β-HSD1 and 7 responsible for the last step of E2
biosynthesis are primary targets for the blockade of E2 production. Here, the following figure (Figure 9) concentrates on sex hormone metabolism.
Figure 9: Schematic illusion of steroidogenesis (estrogen/androgen) in intracrine tissues in postmenopausal women (Aka JA, et al. 2009)
3 Steroidogenic enzymes in E2 formation and DHT degradation in ER+ breast cancer
3.1 Aromatase
The aromatase enzyme is a member of the cytochrome P450 and is the product of the CYP19A1 gene [54]. It functions to catalyze the rate-limiting and final step of estrogen biosynthesis: the aromatization of androgens to estrogens [55]. For example, the aromatase enzyme catalyzes C19 steroid into estrogens, usually it catalyzes 4-dione into E1 and testosterone (Testo) into E2 [56]. Most aromatase expression is found in adipose fibroblasts rather compared to in epithelial cells [57].
Breast cancer tissues have been shown to express aromatase and produce higher levels of estrogens than normal breast cells. This is one of the main reasons that
aromatase has induced a high level of interest for the treatment of ER+ breast cancer [58]. In addition, aromatase is reported to be mainly expressed in fibroblasts but it has also some low-expression in malignant epithelial cells [59,60].
Currently, several aromatase inhibitors (AIs) have been used in clinical treatments which have been approved by the US Food Drug Administration (FDA), including Anastrozole, Letrozole and Exemestane. These inhibitors are approved for postmenopausal women with ER+ breast cancer in both the adjuvant and metastatic setting. All these AIs appear to have comparable efficacy and there is evidence that Letrozole is more potent in reducing plasma estrogen levels [61].
Although AIs provide an effective treatment, their benefit is limited by the occurrence of resistance which can occur in a significant number of patients in the adjuvant setting and is inevitable in metastatic breast cancer [55]. Therefore, further studies are needed to identify strategies that prolong or avert AI resistance.
3.2 Steroid sulfatase (STS)
STS is mainly expressed in normal and malignant breast tissues [49]. It is a member of the family of arylsulfatases in the sulfatase superfamily, whose
membres catalyze the hydrolysis of sulfate ester bonds in various endogenous and exogenous substrates [62]. STS has a central role in the formation of active sex steroid hormones, as it is the key enzyme catalyzing E1-S into E1 as well as dehydroepiandrosterone sulfatase (DHEA-S) into DHEA [49,63] (Figure 10).
E1-S can have an important contribution to the supply of E1 to target tissues. In fact, after the synthesis of E1 from 4-dione by aromatase, a number of sulfotransferases will easily add sulfate to generate E1-S [64]. The concentration of E1-S is 10-20 times higher than that of unconjugated estrogens and E1. And the half-life of E1-S in plasma (10-20 hours) is significantly longer than that for E1 and E2 (20-30mins).
Therefore, E1-S acts as a reservoir for the formation of active estrogens via the STS action [65]. Then the E1, the product of this catalysis is reduced to E2 by 17β-HSD1 and 17β-HSD7.
DHEA-S, largely secreted by the adrenal cortex [66], is converted to DHEA by STS activity, which can be further reduced to 4-dione by 3β-HSD1 in peripheral tissues (Figure 6). And 4-dione can bind to ER to stimulate the growth of ER+ BC cells [67,68]. 4-dione is considered the second most active natural estrogen, with significantly less affinity to ERα than E2, although it can be potent in postmenopausal women because of its high concentration in plasma and tissues [69,70]. Furthermore, it is suggested that STS mRNA expression has been detected in breast tumors showed a much higher level than that of non-malignant breast tissues [71].
It is also suggested that STS activity is synergistically increased by cytokines IL-6 and TNFα in BC cells, which also facilitate STS as a key regulator of in situ estrogen formation [72]. Therefore, STS is proposed to play an important role in regulating hormones and was considered as having therapeutic potential for the treatment of hormone-dependent breast cancer.
The development of STS inhibitors began in the 1990s [73–77], and was further promoted during the decade of 2000 [78–81]. One STS inhibitor STX64 (also known as 667 Coumate) was shown to be a potent STS inhibitor in Phase Ⅰ trail [82]. However, this success was not repeated in clinical phase Ⅱ [83,84]. As a main pathway for E2 production, it is important to better clarify the STS mechanism and identify the possible reason for the insignificant results of STX64 in clinical phase Ⅱ.
Figure 10: The role of STS in ER+ breast cancer (MJ Reed, 2005)
3.3 17β-HSD1
In breast cancer cells, 17β-HSD1 is a principle enzyme converting E1 into the most potent estrogen E2. E1 has a low affinity for the estrogen receptor, however, when it is converted into E2, the affinity for the estrogen receptor is high [85]. 17β-HSD1 also catalyzes DHEA into 4-androstene-3β,17β-diol (5-diol), which is a weaker estrogen than E2 and is more important for postmenopausal women [86].
It has been suggested that high 17β-HSD1 mRNA expression can be observed in breast carcinoma specimens from patients and high expression of the enzyme correlates with a weak prognosis for breast cancer [87–89]. Inhibition of 17β-HSD1 activity thus constitutes a significant approach to decrease E2 level with the aim of preventing breast tumors growth [90–93]. Despite the high expression of 17β-HSD1, this enzyme cannot be regulated by E2 [94].
3.4 17β-HSD7
It has been shown that 17β-HSD7 is expressed in the ovary, placenta, mammary gland, and also in the brain and liver [95,96]. It is a 32-kDa microsomal protein involved in estradiol synthesis [97]. 17β-HSD7 was first identified as a prolactin receptor-associated protein in the rat [97]. Detection of a high expression in corpus luteum of pregnant mice suggested a role in E2 production [98].
Our lab performed profound functional study of 17β-HSD7 in estrogen-dependent breast cancer with an emphasis on demonstrating its role in sex hormone biosynthesis and breast cancer stimulation [99,100]. 17β-HSD7 not only catalyzes E1 into E2, but it can also inactivate dihydrotestosterone (DHT) to the weak estrogen 3β-diol, thereby making it a dual intracrine regulator for the most potent estrogen and androgen [95].
17β-HSD7 is an enzyme characterized as being feed-forward regulated by E2 [101]. Such strong positive regulation mechanism increases local E2 production, leading to the growth of estrogen-dependent breast cancers [102]. The overexpression of 17β-HSD7 in tumor tissues can be as high as 3.5 fold compared to normal tissues, which also contributes to intratumoral estrogen deposition and is correlated with an increasing ratio of E2 to E1 in breast carcinoma compared with normal tissue [103].
Therefore, the high expression and regulation pattern of 17β-HSD7 suggests that it is an important regulator of intraturomal estrogen deposition. The original inhibitors used in this article towards 17β-HSD7 were developed by Dr Donald Poirier [104].
4 Androgen and androgen receptor in ER+ breast cancer
Androgens are synthesized from adrenal precursors such as DHEA or 4-androstenedione (4-dione). Moreover, 4-dione and Testo can also be direct precursors of estrogens. It is suggested that the increasing circulating level of androgens including DHEA, 4-dione and Testo imposes a higher risk of breast cancer because these androgens can be directly or indirectly converted into estrogens in breast cancer [19]. The most potent androgen DHT displays 5-10 times higher affinity towards the androgen receptor (AR) compared to Testo [70].
In breast cancer, AR is more widely detected than ER or PR. AR has also emerged as a useful marker for further refinement of breast cancer subtype classification [105,106]. Among the subtypes, 88% of ER+, 59% of HER2+, and 32% of triple-negative breast cancers were positive for AR expression [107].
In addition, a systematic meta-analysis indicated that co-expression of AR and ER in breast carcinoma is associated with a better prognosis and outcome [108]. The AR gene is located on the X chromosome at q11 and contains eight exons encoding for a DNA binding domain (DBD), a N terminus domain (NTD), a hinge region and a ligand-binding domain (LBD) [109]. The DBD contains two zinc finger motifs that recognize consensus androgen response elements (AREs) and anchoring of the AR to recognized sequences [110].
In contrast to E2, which predominantly contributes to breast cancer development through ER, DHT exerts anti-proliferative effects via the AR by activating p21waf1/cip1
and inhibiting Cyclin D1 in vitro [111,112]. Furthermore, the metabolites of DHT such as 5α-androstane-3β,17β-diol (3β-diol) have weak estrogenic potency towards MCF-7 cells through binding to ER[MCF-70], and this is thought to be a possible mechanism of aromatase inhibitor resistance [113,114]. Therefore, reducing E2 and restoring DHT has been suggested to be beneficial to ER+ breast cancer treatment.
5 The role of 11β-HSD2 in ER+ breast cancer
Glucocorticoids are an important component of treatment regimens in hematological malignancies because of their pro-apoptotic properties. They are also used as co-treatment in chemotherapy co-treatments of several cancers including breast cancer [115]. In breast cancer, glucocorticoids can play a very complex role [115]. Studies have shown that glucocorticoids modulate aromatase activity and can affect the estrogen synthesis rate in breast carcinoma cells [116].
11β-Hydroxysteroid dehydrogenase enzymes (11β-HSD) regulate the ratio of active endogenous glucocorticoids to their inactive keto-metabolites, thus controlling the access of glucocorticoids to their receptors [117].
HSDs involve two main isoforms which are HSD1 and HSD2. 11β-HSD1 is expressed in a wide array of tissues, with the highest levels in the liver, from which it was purified originally [118]. It mainly acts as an oxidoreductase, increasing the concentration of active glucocorticoids [117].
11β-HSD2 is expressed in mineralocorticoid target cells. It catalyzes exclusively the oxidation of 11β-hydroxyglococorticoids, using NAD+ as a cofactor [118] (Figure 11). In addition, inhibition of 11β-HSD2 activity in breast cancer cells in vitro has been shown to enhance the antiproliferative effect of glucocorticoids [119]. Therefore, it is important to understand the regulation of 11β-HSD2 in ER+ breast cancer.
Figure 11: 11β-hydroxysteroid dehydrogenase (11β-HSD) (Seckl et al., 2004)
6 Endocrine therapy for ER+ breast cancer
For more than half a century, human endeavours against breast cancer that had significant success were those involving the development of selective estrogen receptor modulators (SERMs) and aromatase inhibitors [120]. However, side effects and resistance limit their effectiveness [121].
The improvements in adjuvant therapy and screening have led to a reduction in breast cancer mortality in the past few decades. Mortality from breast cancer had remained nearly the same from 1975 to 1990, but then decreased by 40% from 1990 to 2006 [122].
More recently, the dual control of estrogens and androgens have demonstrated the potential for further improvements of breast cancer therapy [100,122].
7 Epidemiology and treatment of triple negative breast cancer
Triple negative breast cancer (TNBC) is characterized by tumors that do not express ER, PR and HER-2 genes, representing an important clinical challenge because such cancer does not respond to endocrine therapy or other available agents. Moreover, these tumors are associated with a poor prognosis.
TNBC can be further classified in several subgroups. The first subtype is basal-like, where tumors have some characteristics of breast myoepithelial cells. These tumors have a high proliferation potential and they are associated with very poor prognosis [123]. Another subgroup is claudin-low, which presents the epithelial to mesenchymal transition (EMT), as well as stem cell-like and tumor-initiating cell features [124]. This subtype is also associated with a poor prognosis.
7.2 The cell origin differentiation of ER positive cells and TNBC cells
Breast cancer is considered to be a heterogeneous disease and the presence of distinct molecular entities, known as ‘intrinsic’ subtypes such as ER positive breast cancer or TNBC, suggest the existence of multiple ‘cells of origin’. It has been proposed that luminal tumors arise from luminal progenitor cells, while basal tumors originated from the basal or stem cell compartment [10,125,126].
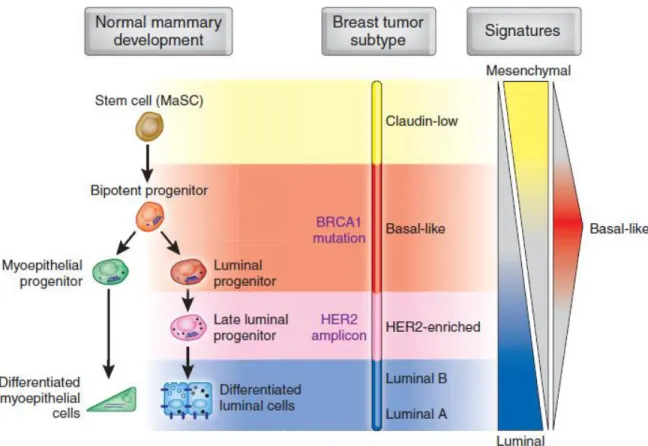
However, comparisons of the transcriptional signatures with gene expression profiles of cell subsets isolated from mammary tissue indicate that different molecular subtypes of breast cancer may arise from cells at various stages of differentiation along the mammary epithelial hierarchy (Figure 12) [127].
In TNBC, claudin-low tumors resemble those of mammary stem cells, while basal tumors display gene expression profiles similar to bipotent or early luminal progenitors [128–130]. In addition, luminal tumors are closest to the differentiated luminal cell compartment [127]. This scheme is complicated by the possibility that tumor cells could be transferred from their original configuration, losing specific
markers of the cell of origin and taking on others [131]. A prime example of this is EMT, in which transformed epithelial cells switch to a mesenchymal phenotype with an expression profile resembling that of stem cells [132].
Figure 12: Model of the human mammary epithelial hierarchy linked with cancer subtypes (Prat A et al, 2009)
7.3 The incidence rate and current treatment of TNBC
Higher rates of TNBC have been detected in younger women, and this can be associated with a greater probability of BRCA1 expression [133]. The history of TNBC has been redefined over the last few years. Luminal A metastatic hormone receptor positive breast cancer typically causes late bone metastases, whereas TNBC is more likely to cause early visceral metastases. Basically, in the early-stage setting, TNBC is associated with earlier versus later events and a shorter period from the time of recurrence until death (Figure 13) [133].
Figure 13: Recurrence and survival of TNBC (Kim K et al, 2009)
Current standard treatment for TNBC include many chemotherapy agents such as anthracyclines, taxanes, ixabepilone, and platimum agents as well as selected biological agents [134]. Paclitaxel (Taxol) is a microtubule-stabilizing drug that is approved by the Food and Drug Administration for the treatment of ovarian, breast, and lung cancer. It has been recognized to induce mitotic arrest, resulting in cell death in a subset of arrested population [135]. However, recent evidence demonstrates that over 50% of TNBC patients become resistant to chemotherapy typically after 6-10 months of treatment [136].
7.4 The chemoresistance in triple negative breast cancer
It is well established that TNBC is an aggressive subtype of breast cancer despite having a good initial response to chemotherapy. Although TNBCs are generally very sensitive to chemotherapy initially, early complete response (CR) does not correlate with overall survival [137]. In fact, the risk of relapse for TNBC patients in the first 3-5 years is significantly higher than for women suffering from hormone positive breast cancer [133,138]. Clinically, this makes it particularly challenging to find the optimal survival for these women.
Chemoresistance can be attributable to the fact that although cytotoxic chemotherapy aims to kill cancer cells through apoptosis, tumour cells have the ability to maintain viability following chemotherapeutic exposure by undergoing alternative cellular fates such as cellular senescence, therapeutic induced senescence (TIS) and autophagy [137].
Standard chemotherapy remains the backbone of systemic TNBC treatment even amidst advances in cancer treatment, because neither hormonal therapy or anti-HER2 agents are effective against TNBC because of the absence of a target [139]. At present, the cure for cancer continues to escape oncologists due in large part to chemoresistance, which accounts for about 90% of drug failures in metastatic cancers [140].
Tomassi et al, 2007 and Paradioso et al., 2005 have found that in advanced breast cancer, overexpression of βⅢ correlated with the resistance and progression of disease in patients given first-line Taxol (paclitaxel) chemotherapy [141,142].
Other mechanisms of chemoresistance in TNBC include ATP-binding cassette (ABC) transporters, mutations in DNA repair enzymes such as topoisomerase, alterations in genes involved in apoptosis, aldehyde dehydrogenase 1 (ALDH1) and glutathione (GSH)/ Glutathione-S-transferase (GST) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathways (Figure 14) [137].
Figure 14: Mechanisms of Chemoresistance in TNBC. (A) ABC transporters efflux chemotherapeutics out of cancer cells; (B) Overexpression of β-tubulin III subunit induces paclitaxel resistance; (C) Mutations in DNA repair enzymes and enzymes altering drug sensitivity; (D) Alterations in genes involved in apoptosis prevent chemotherapy-induced apoptosis; (E) ALDH1 and glutathione (GSH)/Glutathione-S-transferase (GST) mediate chemotherapeutic inactivation/detoxification; (F) Role of NF-κB signaling pathway in chemoresistance (O’Reilly et al, 2015).
8 Role of Cyclin-dependent kinases (CDKs) and CR6-interacting factor 1 (CRIF1) in cell cycle arresting (cytostatic effect)
Cyclin-dependent kinases (CDKs) are serine/threonine kinases and their activity depends on a regulatory subunit: a cyclin, that provides domains essential for enzymatic activity [143]. CDKs play important roles in the control of cell division and modulate transcription in response to several extra- and intracellular cues [136].
In breast cancer, CDK4 and CDK6 interact with cyclin D, leading to the promotion of cell cycle progression from the G0 to G1 phase [144]. In recent years, new therapeutic options selectively targeting CDK 4 and 6 have shown promising clinical activity in breast tumors. For example, since 2015, three CDK4/6 inhibitors that are palbocicib, ribocicib and abemacicib have been approved for the treatment of hormone-receptor positive and HER2 negative metastatic breast cancer. These three drugs are used respectively, in combination with endocrine therapy, significantly prolonged progression-free survival [145]. However, these inhibitors do not bring significant results for TNBC treatment [146,147].
In addition, CDK2 plays an important role in regulating multiple events of cell cycle including centrosome duplication, DNA synthesis, G1-S transition and the modulation of G2 progression [148–151]. Accumulating evidence has indicated that CDK2 is functionally associated with hyperproliferation in multiple cancer cells and could be regarded as a potentially therapeutic target for cancer treatment [148]. One study showed that inhibition of CDK2 kinase activity reduced tumor proliferation by selectively targets the CD44+/CD24-/Low stem-like cells in TNBC when combined with
chemotherapy [152].
CR6-interacting factor 1 (CRIF1) is a newly discovered cell cycle negative regulatory factor. A previous study found that CRIF1 may play a regulatory role in microenvironment-induced leukemia cell cycle arrest through interacting with CDK2 as an CDK2 inhibitor [153]. And the CRIF1-CDK2 interface inhibitor could selectively promote G2/M arrest and apoptosis related to CDK2 over-activation in
osteosarcoma(OS) cell lines, which improves the cell sensitivity to radiotherapy [153].
On the other hand, there is accumulating evidence suggesting that key negative regulators of CDKs may represent the therapy targets in TNBC, such as WEE1 targeting CDK4/6 [154]. However, many CDK inhibitors have shown disappointing results in clinical trials because of their low specificity and selectivity to target specific CDKs enzymes [155]. Therefore, in this study, we used the CRIF1-CDK2 interface inhibitor to target CRIF1, indirectly regulating CDK2 and cell cycle checkpoints in TNBC.
9 Breast cancer cell lines
In vitro studies confirmed that MCF-7 and T47D cell lines are receptors positive, including the androgen, estrogen, and progesterone receptors [156].
The MCF-7 cell line was derived from a 69-year-old Caucasian women with breast malignant tumor [157]. This cell line was established in 1975 as a human BC cell line expressing estrogen, androgen, progesterone and glucocorticoid receptors [158]. This cell line is an excellent and basic model for studying the mechanisms of steroids in BC cells, especially for estrogens.
The T47D cell line is originated from a 54-year-old female patient with an infiltrating ductal breast carcinoma [159]. Both MCF-7 and T47D cells are ER+ cell lines and which possess similar characteristics [160,161]. However, 17β-HSD1 expression is much higher in T47D than that in MCF-7 (697 thousand copies of mRNA/μg vs. 87 thousand copies of mRNA/μg) [160].
On the other hand, cell lines MDA-MB-231 and MDA-MB-468 have been identified as typical TNBC cell lines [162]. MDA-MB-231 was established in the 1970s. It was derived from a 51-year-old patient with a mastectomy for an inner quadrant tumor [163,164]. MDA-MB-468 was isolated from a pleural effusion in a black female patient aged 51 in 1977 [164]. Both of these cell lines are basal cell lines. The difference is that MDA-MB-231 was basal B TNBC cell line and MDA-MB-468 was basal A TNBC cell line[165]. Both of them are widely used in the studies of TNBC.
MCF10A is a spontaneously immortalized cell line derived from benign breast tissue of women who suffered from fibrocystic disease[165]. It has been demonstrated to be a human mammary epithelial cell line which is widely used as an in vitro model for the study of normal breast cell function and transformation[166].
10 Working hypothesis and research objectives
10.1 Hypothesis
10.1.1 The negative result of STX64 in phase Ⅱ clinical trials is related with the mechanism of STS inhibition. Single blockade of the STS pathway decreases both E2 and DHT concentrations, inducing a modest inhibition of ER + BC cell proliferation. Combined inhibition of STS with other 17β-HSDs such as 17β-HSD7 would be a more beneficial strategy through the dual regulation of estrogen and androgen.
10.1.2 17β-HSD7, 11β-HSD2 and STS are mutually positively correlated through E2, inhibition of one of them could reduce the expression of another.
10.1.3 A CRIF1-CDK2 interface inhibitor can indirectly regulate CDK2 through targeting CRIF1, which results in the cell cycle arrests and improves cell sensitivity to chemotherapy in TNBC.
10.2 Objectives
Objective 1: Using the two ER+ breast cancer cell lines MCF-7 and T47D, to clarify observations from several studies of steroid metabolism, addressing the modest results of the sulfatase inhibitor (STX64) in the phase II clinical study. We also aim to provide a potential combined inhibition of STS with other 17β-HSDs, which would be a more beneficial strategy through the dual regulation of estrogen and androgen synthesis.
Objective 2: To characterize the possible mutual regulations between steroid enzymes in ER+ breast cancer, such as 17β-HSD7, STS and 11β-HSD2. Considering that these enzyme genes contain estrogen responsive elements on their promoters, they can be regulated by E2 as well as steroidogenic enzymes involved in E2 production. Two typical ER+ cell lines MCF-7 and T47D are treated by inhibitors of 17β-HSD7, STS, aromatase and 17β-HSD1 respectively, then mRNA levels of 17β-HSD7, STS, 11β-HSD2, estrogen receptors α (ERα) and androgen receptor (AR) are determined by Q-PCR.
Objective 3: As chemotherapy is the standard treatment for TNBC and the inhibitors targeting CDK4/6 lack selectivity to target specific CDK enzyme subtypes, our study indirectly targets CDK2 by making use of its cell cycle regulator CRIF1, through which we expect to overcome chemotherapy resistant TNBC. Two selective CRIF1-CDK2 interface inhibitors are used to study the anti-proliferative effects on the typical TNBC cell lines of MDA-MB-231 and MDA-MB-468. The cell cycle, cell proliferation and cell apoptosis are analyzed to determine whether CRIF1 can be potential targets for reducing chemotherapy resistance in TNBC
Chapitre 1 Steroid sulfatase inhibition success
and limitation in breast cancer clinical assays: an
underlying mechanism
1.1 Résumé
La stéroïde sulfatase est détectable dans la plupart des cancers du sein hormono-dépendants. Le STX64, un inhibiteur de STS, a provoqué une réduction tumorale dans les tests sur des animaux. Malgré le succès de l'essai clinique de phase І, les résultats de l'essai de phase II n'étaient pas significatifs. Les cellules épithéliales du cancer du sein (MCF-7 et T47D) ont été traitées avec deux inhibiteurs de STS (STX64 et EM1913). La prolifération cellulaire, le cycle cellulaire et les concentrations d'estradiol et de 5α-dihydrotestostérone ont été mesurées pour déterminer le mécanisme endocrinologique de l'inhibition de la sulfatase. Des comparaisons ont été faites avec les inhibitions des 17β-hydroxystéroïdes déshydrogénases (17β-HSD) réductrices. Des études de prolifération ont montré que la synthèse de l'ADN dans les cellules a eu une réduction d’environ 20%, accompagnée d'une augmentation allant jusqu'à 6,5% dans les cellules en phase G0 / G1 et de la réduction de l'expression de la cycline D1. Les concentrations d’estradiol et de 5α-dihydrotestostérone ont été réduites de 26% et 3% respectivement. Cependant, la supplémentation en 5α-dihydrotestostérone a entraîné une augmentation significative (jusqu’au 35,6%) de l'effet anti-prolifératif de l'inhibition de la sulfatase. Cette étude a clarifié le contrôle des hormones sexuelles par la sulfatase en cancers du sein, suggérant que les différents rôles de l'œstradiol et de la 5α-dihydrotestostérone peuvent entraîner une réduction de l'effet de l'inhibition de la sulfatase par CYP19A1, rapport à celle de la 17β-HSD7. Ceci suggère que le traitement combiné d'inhibiteurs de sulfatase avec des inhibiteurs des 17β-HSDs réductrices tels que l'inhibiteur de la type7 pourrait être prometteur pour le cancer du sein hormono-dépendant.
1.2 Abstract
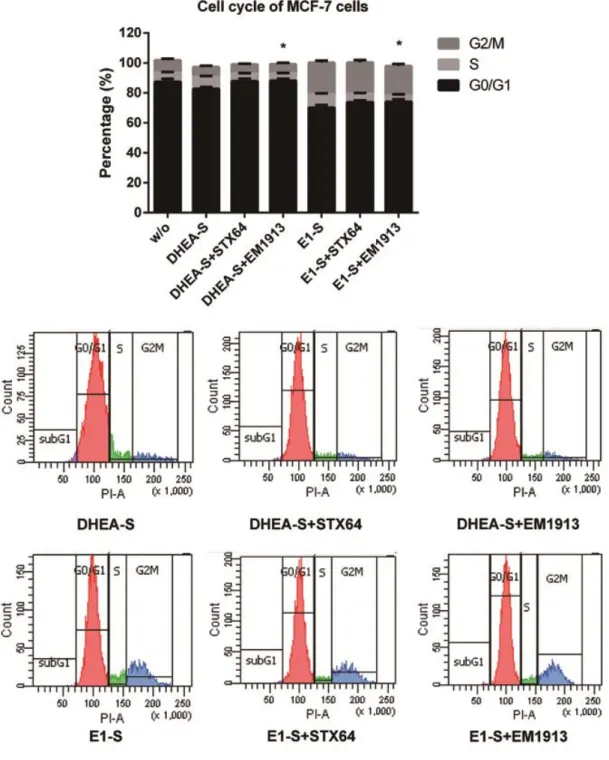
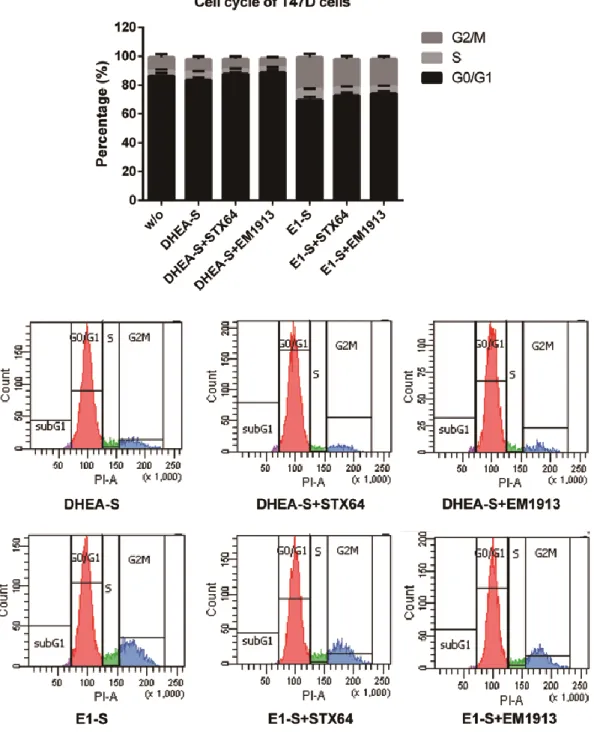
Steroid sulfatase is detectable in most hormone-dependent breast cancers. STX64, an STS inhibitor, induced tumor reduction in animal assay. Despite success in phase І clinical trial, the results of phase II trial were not that significant. Breast Cancer epithelial cells (MCF-7 and T47D) were treated with two STS inhibitors (STX64 and EM1913). Cell proliferation, cell cycle, and the concentrations of estradiol and 5α-dihydrotestosterone were measured to determine the endocrinological mechanism of sulfatase inhibition. Comparisons were made with inhibitions of reductive 17β-hydroxysteroid dehydrogenases (17β-HSDs). Proliferation studies showed that DNA synthesis in cancer cells was modestly decreased (approximately 20%), accompanied by an up to 6.5% in cells in the G0/G1 phase and cyclin D1 expression reduction. The concentrations of estradiol and 5α-dihydrotestosterone were decreased by 26% and 3% respectively. However, supplementation of 5α-dihydrotestosterone produced a significant increase (approximately 35.6%) in the anti-proliferative effect of sulfatase inhibition. This study has clarified sex-hormone control by sulfatase in BC, suggesting that the different roles of estradiol and 5α-dihydrotestosterone can lead to a reduction in the effect of sulfatase inhibition when compared with 17β-HSD7 inhibition. This suggests that combined treatment of sulfatase inhibitors with 17β-HSD inhibitors such as the type7 inhibitor could hold promise for hormone-dependent breast cancer.
1.3 Introduction
Breast cancer (BC) is the most frequently occurring cancer in women, accounting for approximately 10% of cancer incidence worldwide. Furthermore, it is diagnosed in one million women each year [1,2].
A large proportion of BCs are initially estrogen-dependent [3], including 60% of BC cases in premenopausal women and 75% of cases in postmenopausal women [4]. Estrogens are synthesized from precursor steroids by steroidogenic enzymes in peripheral tissues in an intracrine manner in postmenopausal women [5,6] . Estradiol (E2), the most potent estrogen, is suspected to play an important role in carcinogenesis by stimulating cell proliferation through binding to estrogen receptors (ER) and activating down-stream transcriptions [7].
Following menopause, E2 is predominantly produced from adrenal precursors such as dehydroepiandrosterone (DHEA), 4-androstene-3,17-dione (4-dione) and estrone-sulfate (E1-S) by several steroidogenic enzymes [8]. Two pathways are important for local E2 production in target tissues: the “sulfatase pathway,” in which biologically inactive steroid sulfates are the source for E2 (E1-S to E1 and DHEA-S to DHEA), and the “aromatase pathway”, which transforms androgens into estrogens [9]. 17β-HSDs are integral to both pathways and convert estrone (E1) into potent E2 [10–12]. A consequence of the up-regulation of steroidogenic enzymes is the generation of an estrogen-rich microenvironment that sustains tumor growth and proliferation [13,14]. Therefore, steroid sulfatase (STS), aromatase and reductive 17β-HSDs involved in the last step of E2 biosynthesis are primary targets for the blockade of E2 production [15].
Androgens are synthesized in breast cancer from adrenal precursor such as DHEA or 4-dione, which are the metabolites in the sulfatase pathway. In contrast with E2, which predominantly contributes to the breast carcinoma growth through binding to
the ER, the non-aromatic androgen dihydrotestosterone (DHT), displayed 5~10 times higher affinity towards androgen receptor (AR) than Testosterone [16] and exerted anti-proliferative effects via activating P21waf1/cip1 or inhibiting Cyclin D1 in
vitro [17,18]. Furthermore, about 80% of ER positive breast cancers were found to co-express AR [19]. A systematic meta-analysis demonstrated that co-expression of AR in female breast tumour is associated with a better prognosis and outcome [20].
According to extensive studies, STS activity can be detected in most breast tumors [21]. This enzyme belongs to the family of arylsulfatases of the sulfatase superfamily, which catalyzes hydrolysis of the sulfate bond present in various endogenous and exogenous substrates [22]. Steroid sulfatase performs a central role in the formation of active sex steroid hormones, as it is the key enzyme hydrolyzing steroid sulfates, including E1-S into E1 and DHEA-S into DHEA [23,24] . Therefore, STS was proposed to play an important role in regulating hormones and was considered a potential target for the treatment of hormone-dependent BC.
The development of STS inhibitors began in the nineties [25–31] and accelerated during the decade following 2000 [32–35]. Most compounds were in preclinical testing but one inhibitor was scheduled for clinical phase І trial. STX64 (also known as Irosustat, Table 1) belongs to the first-generation of STS inhibitors and was shown to be a potent STS inhibitor in placenta (in vitro). It also blocked the ability of E1-S to stimulate the growth of carcinogen-induced mammary tumors in ovariectomized mice [36]. The results from phase І trial have shown it is possible to inhibit STS activity by over 90% in peripheral blood lymphocytes (PBLs), which have a high level of STS activity [37]. However, only modest effects of sulfatase inhibition were obtained in the phase II clinical study [38–41].
This study helps to clarify observations from several investigations of steroid metabolism in ER+ BC cell lines and addresses a possible explanation for the modest results of the sulfatase inhibitor (Irosustat) in the phase II clinical study.
When sulfatase is inhibited, both E2 and DHT may be decreased in BC cells as DHEA-S is the common intracrine source for estrogens and androgens in post-menopausal women. Thus, it is essential to verify the cellular effects of the STS inhibitor on sex-hormone levels and cell cycle progression. Furthermore, while the critical role of DHT inactivation by 17β-HSD7 has been demonstrated by our laboratory in both in vitro and in vivo studies [42], it is fundamentally important to understand the impact of the cellular DHT concentration with regard to the cell cycle and cell proliferation [17,18]. Such comparative contributions will be analyzed in light of the in vitro study with the respective enzyme inhibitors and may help us to design the best strategy for therapeutic targeting of estrogen-dependent BC.
1.4 Materials and Methods
1.4.1 Inhibitors and chemicalsTwo STS inhibitors were used in this study: the non-steroidal STS inhibitor STX64 or Irosustat (IC50: 8 nM) [43] was purchased from Sigma-Aldrich Canada (Oakville,
ON, Canada). The 17β-HSD7 inhibitor INH80 (IC50: 200nM) [44,45] and steroidal
inhibitor EM1913 (IC50: 0.024 nM) without estrogenicity were synthesized in the
Laboratory of Medicinal Chemistry (CHU de Québec-CHUL) as reported [46]. Stock solutions were prepared by dissolving the compounds in ethanol. Dilution to the desired concentrations was performed using culture medium (Table 1.1). The culture medium for MCF-7 was Dulbecco’s Modified Eagle’s Medium-Low glucose which was purchased from Sigma-Aldrich Canada (Oakville, ON, Canada). The medium for T47D was Gibco RPMI Medium 1640 (1×), purchased from Thermo Fisher scientific Canada.
1.4.2 Cell Culture
The ER+ cell lines MCF-7 and T47D, both of which express STS, were obtained from the American Type Culture Collection, and propagated according to a previously described protocol [47]. Both culture media were phenol red-free. Fetal
![[PDF] Cours Pascal les procédures et les fonctions pdf | Formation informatique](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)