© Hugo Boutin, 2019
Effet de l'acide de Lewis sur l'activation C-H par des
paires de Lewis frustrées
Mémoire
Hugo Boutin
Maîtrise en chimie - avec mémoire
Maître ès sciences (M. Sc.)
ii
Effet de l’acide de Lewis sur l’activation C-H par
des paires de Lewis frustrées
Mémoire
Hugo Boutin
Sous la direction de :
iii
Résumé
Le domaine de recherche principal du groupe Fontaine porte sur le développement de catalyseurs sans métaux de type paire de Lewis frustrée (FLP). La formation de dimères entraine une perte de réactivité en raison du coût énergétique du bris du dimère. L’ajout de groupements alkyles à l’acide de Lewis de FLPs du type 1-BH2 -2-NR2-C6H4 permet d’augmenter l’encombrement stérique de la molécule ambiphile, et ainsi déstabiliser la formation de dimères. Ces molécules ambiphiles (1-B(R)H-2-NR2-C6H4) sont cependant instables et nécessitent d’être stabilisées par l’ajout d’une base de Lewis. La réactivité de ces adduits a pu être évalué pour la borylation catalytique du N-méthylpyrrole. La forme dimérique des molécules ambiphiles semble prévenir leur dégradation.
La sensibilité des aminoboranes aux réactifs protiques est connue. L’étude de la réactivité entre les thiols et l’aminoborane [1-BH2-2-NMe2-C6H4]2 a mené à la découverte d’un catalyseur efficace pour la borylation des thiols. Les liaisons bore-soufre sont des réactifs utiles et sous-utilisés en synthèse organique. Leur utilité dans des réactions d’addition de Michael et de thioestérification est discutée. Il s’agit d’un bon exemple où l’utilisation d’un catalyseur sans métal permet de synthétiser un produit à valeur ajoutée.
iv
Abstract
The main interest of the Fontaine research group is in the development of metal-free catalysis with frustrated Lewis pairs (FLP). Dimer formation is known to cause loss of reactivity due to the energetic cost of dimer dissociation. By adding alkyl groups to the Lewis acid moiety of the 1-BH2-2-NR2-C6H4 type FLP can enhance the bulk of the abiphilic molecule, and thus disfavor the formation of the dimer. Those ambiphilic molecules (1-B(R)H-2-NR2-C6H4) are unstable and require the addition of a Lewis base in order to be isolated. The reactivity of these adducts towards catalytic borylation of N-methylpyrrole has been evaluated. The stability of the dimer seems to prevent degradation of such molecules.
The sensitivity of aminoboranes towards protic reagents is known. The investigation of the reactivity of [1-BH2-2-NMe2-C6H4]2 with thiols lead to the discovery of a novel metal-free catalyst for the borylation of thiols. Boron-sulfur bonds are useful yet underutilised reagents. Their ability to undergo Michael addition with enones and thioestérification with esters is discussed. It is a good example of the synthesis of a value-added product made possible by the use of a metal-free catalyst.
v
Table des matières
Résumé... iii
Abstract ... iv
Table des matières ... v
Liste des figures ... ix
Liste des tableaux ... xiv
Lite des abréviations ... xv
Remerciements ... xvi
1. Introduction ... 1
1.1. Problématique ... 1
1.1.3. Catalyse ... 3
1.1.4. Métaux et toxicité ... 5
1.1.5. Coûts associés à l’utilisation de métaux en catalyse ... 7
1.1.6. Impact environnemental des métaux ... 8
1.2. Catalyse sans métal ... 9
1.2.1. Acide de Brønsted ... 9
1.2.2. Acides et bases de Lewis ... 9
1.2.2.1. Catalyse par les acides de Lewis ... 10
1.2.2.2. Catalyse par les bases de Lewis ... 11
1.3. Paires de Lewis frustrées ... 13
1.4. Activation C-H ... 17
1.4.1. Intérêts de l’activation C-H ... 17
1.4.2. Borylation d’hétéroarènes par les FLPs ... 22
1.4.2.1. Bris du dimère ... 24
1.4.2.2. Bris du dimère et réactivité ... 25
1.5. Projet principal ... 26
2. Méthodes expérimentales ... 28
2.1. Synthèses sous atmosphère inerte ... 28
2.1.1. Rampe à vide et tubes de Schlenk ... 28
vi
2.2. Caractérisations... 31
2.2.1. Spectroscopie de résonnance magnétique nucléaire ... 31
2.2.2 Diffraction des rayons X de monocristaux ... 33
2.3. Chimie computationnelle ... 34
3. Effet des groupements R sur la réactivité des FLPs ... 36
3.1. Hypothèses ... 36
3.2. Synthèse de dérivés cibles ... 36
3.2.1. Synthèse des précurseurs ... 36
3.2.2.1. Substitution d’un groupement méthoxyde ... 38
3.2.2.1. Simple et double substitution... 41
3.2.3. Réduction ... 42
3.2.4. État fondamental par DFT ... 45
3.2.5. Lessivage de l’adduit LiH ... 47
3.3. Réactivité ... 50
3.3.1. Borylation du N-méthylpyrrole ... 50
3.3.2. Activation C-H du N-méthylpyrrole ... 51
3.4. Calculs DFT ... 52
3.5. Conclusion partielle ... 54
4. Borylation catalytique de thiols ... 55
4.2. Borylation des thiols par [1-BH2-2-NMe2-C6H4]2 ... 58
4.2.2. Réactivité avec nucléophiles protiques (Figure 3.3)... 59
4.3. Activité catalytique de [1-BH2-2-NMe2-C6H4]2 face à la borylation des thiols ... 61
4.3.1. Optimisation des conditions ... 61
4.3.2. Étendue de la réactivité ... 62
4.4. Mécanisme ... 64
4.5. Liens bore-soufre en synthèse ... 67
4.5.1. Addition de Michael « one-pot »... 67
4.5.2. Thioestérification ... 68
5. Conclusion ... 72
6. Section expérimentale... 75
6.1. Commentaires généraux ... 75
vii
6.2.1. Synthèse de 1-B(OMe)2-2-NMe2-C6H4 ... 75
6.2.2. Synthèse de 1-B(OMe)2-2-pip-C6H4 ... 76
6.3. Synthèse des composés du chapitre 3 ... 77
6.3.1. Synthèse du composé 1 ... 77
6.3.2. Synthèse du composé 2-Me ... 77
6.3.3. Synthèse du composé 2-iPr ... 78
6.3.4. Synthèse du composé 2-tBu ... 78
6.3.5. Synthèse du composé 2-Bn ... 79
6.3.6. Synthèse du composé 3-Me ... 79
6.3.7. Synthèse du composé 3-iPr ... 79
6.3.8. Synthèse du composé 3-tBu ... 80
6.3.9. Synthèse du composé 4-Me ... 80
6.3.10. Synthèse du composé 4-iPr... 81
6.3.11. Synthèse du composé 4-tBu ... 81
6.3.12. Synthèse du composé 4-Bn (à partir de 2-Bn) ... 82
6.3.13. Synthèse du composé 4-NMe ... 82
6.4. Tests à l’échelle RMN (Chapitre 3) ... 83
6.4.1. Test de lessivage de l’adduit LiH avec NEt3.HCl ... 83
6.4.2. Test de lessivage de l’adduit LiH avec PPh3.HBr ... 83
6.4.3. Test de l’activité catalytique de 4-Me ... 83
6.4.4. Test de l’activité catalytique de 4-iPr ... 83
6.4.5. Test de l’activité catalytique de 4-tBu ... 83
6.4.6. Test de l’activité catalytique de 4-Bn ... 84
6.4.7. Test de l’activité catalytique de 4-NMe ... 84
6.4.8. Test de l’activation C-H par 4-iPr ... 84
6.4.9. Test de l’activation C-H par 4-Bn ... 84
6.5. Test d’activation C-H en milieu ouvert (Chapitre 3) ... 84
6.6. Borylation des thiols ... 85
6.6.1. Méthode utilisée pour les composés 6a-p, 6u-w. ... 85
6.6.2. Méthode utilisée pour les composés 6q, 6s and 6t... 85
6.6.3 Méthode utilisée pour les composés 6r and 6x. ... 85
4.6.4. Données spectrales de la borylation de thiols ... 86
6.7. Thioestérification ... 92
viii
Annexes ... 101
Annexe A – Calculs DFT ... 101
Annexe B – Spectrométrie de résonnance magnétique nucléaire ... 127
ix
Liste des figures
Figure 1.1 Représentation des douze principes de la chimie verte constituant un procédé idéal. ... 3 Figure 1.2 Représentation d’une réaction hypothétique entre A et B pour former le produit C. L’énergie d’activation pour la réaction catalysée (en vert) est beaucoup plus faible que la réaction originale. ... 4 Figure 1.3 Cycle général pour les couplages catalytiques au palladium avec L = PR3. Suzuki-Myaura (R = aryle ou vinyle; R’ = aryle; M = B(OR)2, Stille (R = aryle ou vinyle; R’ = aryle; M = SnR’’3), Sonogashira (R = aryle; R’ = alcyne; M = Na).5 .. 5
Figure 1.4 Mécanisme du déréglage de l’équilibre oxydo-réducteur intracellulaire par des ions métalliques. La présence de métaux facilite la formation de radicaux hydroxyles. ... 6 Figure 1.5 Estérification et hydrolyse d’un ester catalysées par un acide de
Brønsted. ... 9 Figure 1.6 Schématisation de la formation d’un adduit de Lewis entre une base (L:) et un acide (Z) de Lewis. ... 10 Figure 1.7 Illustration de la réaction d’alkylation de Friedel-Crafts. ... 11 Figure 1.8 Réactions de formation de liens carbone-carbone catalysées par un cation fluorosulfonium. ... 11 Figure 1.9 Exemples d’amines courantes en chimie organique. La nucléophilicité et la basicité des amines peuvent être contrôlées par l’encombrement stérique et des groupements donneurs. ... 12 Figure 1.10 Réaction de formation d’un lien carbone-carbone catalysée par une base de Lewis. Le phosphazène (t-Bu-P4) permet la fonctionnalisation d’un
arylsilane avec un aldéhyde. ... 13 Figure 1.11 Représentation des paires de Lewis frustrées, ou un acide et une base de Lewis sont retenus de former un adduit par contraintes stériques ou
x
Figure 1.12 Activation réversible de H2 par une paire de Lewis frustrée 1-B(C6F5)2 -4-PMes2-C6H4. ... 14
Figure 1.13 Exemples de réactions d’hydrogénation catalysées par divers FLPs. 15 Figure 1.14 Structures d’ansa-aminoboranes rapportées par le groupe de Piers (A)31 et par le groupe de Repo (B et C).30 ... 16
Figure 1.15 Mécanisme d’hydrogénation d’alcynes par Repo faisant intervenir l’étape de protodéborylation.30 ... 17
Figure 1.16 Mécanisme de l’arylation directe faisant intervenir la métallation
déprotonation concertée. ... 21 Figure 1.17 Comparaison entre l’état de transition de la métallation déprotonation concertée et de l’activation C-H par un 1-BH2-2-TMP-C6H4. ... 21
Figure 1.18 Forme active d’un ansa-aminoboranes (A) et dimères les plus
courants (B = dimère trans, C = dimère H-pontant). ... 22 Figure 1.19 Cycle catalytique pour la borylation du N-méthylpyrrole par 1-BH2 -2-NR2-C6H4. ... 23
Figure 1.20 Équilibre atteint lorsque la température permet le bris du dimère. ... 25 Figure 1.21. Énergie libre des intermédiaires (∆G) et états de transition (∆G⧧) en kcal/mol impliqués dans l’activation C-H du N-méthylpyrrole par les
ansa-aminoboranes calculés au niveau de théorie wB97XD/6-31+G** SMD=chloroform. ... 26 Figure 1.22 Schématisation des objectifs du projet. Faciliter le bris du dimère permettrait une quantité plus grande de catalyseur sous sa forme active... 27 Figure 1.1 Rampe à vide (gauche) et bulleur à l’huile avec valve pressurisée
(droite)... 29 Figure 1.2 En ordre, bouchon et tube J-Young, tube de Schlenk et ballon Strauss avec valve en téflon. ... 29 Figure 1.3 Boîte à gants double possédant deux sas. ... 30
xi
Figure 1.4 Exemple de l’effet du couplage bore-proton. Le signal devient un triplet signifiant la présence de deux protons sur l’atome de bore. ... 32 Figure 1.5. Maille cristalline de l’adduit entre pyridine et 1-B(iPr)H-2-pip-C6H4
obtenue par diffraction des rayons X de monocristaux ... 34 Figure 2.1 Synthèse du 1-B(OMe)2-2-NMe2-C6H4 à partir de la
N,N-diméthylaniline. ... 37 Figure 2.2 Synthèse du 1-B(OMe)2-2-pip-C6H4 à partir de la 2-bromoaniline... 38
Figure 2.3 Synthèse du composé [1-BMe-2-NMe2-C6H4]2O (1-Me). ... 39
Figure 2.4 Représentation ORTEP du composé 1-Me par diffraction des rayons X de monocristaux. Ellipsoïde à 50% de probabilité; B2-O = 1,435 Å; N1-B2 = 1,732 Å; B2-O-B1 = 114,08°; R = 4,67%. Réseau cristallin monoclinique CC. ... 40
Figure 2.5. Mono et double alkylation selon l’utilisation d’un organolithien, d’un Grignard encombré ou d’un Grignard peu encombré. ... 42 Figure 2.6. Représentation ORTEP de 3-tBu obtenue par diffraction des rayons X de monocristaux. Ellipsoïde à 50% de probabilité; N = 2,09 Å; H = 1,94 Å; Li-O = 2,00Å; B-H = 1,28 Å; R = 12,77%. Réseau cristallin monoclinique P21. ... 43
Figure 2.7. Représentation ORTEP 1-B(Me)H-2-NMe2-C6H4B sous forme du dimère H-pontant obtenue par diffraction des rayons X de monocristaux. Ellipsoïde à 50% de probabilité; N1-B2 = 1,642 Å; B2-H = 1,192 Å, H-B1 = 1,374; B1-H-B2 = 128,4°, R = 5,31%. Réseau cristallin orthorhombique P c a 21. ... 45
Figure 2.8. Représentation des formes que peuvent prendre les
ansa-aminoboranes. ... 46 Figure 2.9. Structure de l’adduit entre pyridine et 4-iPr obtenue par diffraction des rayons X de monocristaux. Ellipsoïde à 50% de probabilité; N1-B = 2,943 Å; N1-H = 2,842 Å; N2-B = 1,625 Å; R = 4,59%. Réseau cristallin monoclinique P21/n. ... 49
Figure 2.10. Réaction d’activation C-H du N-Méthylpyrrole par les composés 4-iPr et 4-Bn ... 52
xii
Figure 2.11. Intermédiaires et états de transition impliqués dans la réaction
d’activation C-H par les composés 4. Dans le cas de 4-NMe, l’état fondamental est le dimère H-pontant plutôt que l’adduit avec la pyridine. ... 53 Figure 3.1. Réaction de thioboration entre d’un composé ayant un lien bore-soufre sur un accempteur de Michael, suivie de la méthanolyse du produit d’addition.65 . 56
Figure 3.2. Diverse réactions de fonctionnalisation rendues possibles après
l’insertion d’un composé diazo silylé dans le lien bore-soufre. ... 57 Figure 3.3. Réactivité de 5 avec des réactifs protiques. Les réactions mènent à des intermédiaires ayant des liens bore-hétéroatome. ... 60 Figure 3.4. Réactivité des intermédiaires 5-O, 5-N et 5-S avec le HBpin. La
métathèse avec pinacolborane est possible seulement dans le cas des liens bore-soufre. ... 61 Figure 3.5. Cycles catalytiques de la borylation de thiols par [1-BH2-2-NMe2-C6H4]2 (5). Le cycle A est favorisé pour tert-butylthiol en raison de son encombrement. Le cycle B est responsable de la borylation des substrats plus petits. La métathèse est plus lente dans le cycle B en raison de l’acidité plus faible du bore du
catalyseur... 65 Figure 3.6. Comparaison des spectres RMN 11B pendant la borylation catalysée par [1-BH2-2-NMe2-C6H4]2 à 20 mol% du thiophénol (A) et du tert-butylthiol (B). Le couplage avec les protons permet de montrer que l’état stationnaire du catalyseur porte un hydrure dans le cas du tert-butylthiol. Les autres signaux sont le produit borylé 33 ppm et le HBpin 28,2 ppm. ... 67 Figure 3.7. Réaction d’addition de Michael one-pot. L’addition d’un accepteur de Michael après la borylation catalytique du 4-méthylthiophénol par 5. Le produit a été isolé après une chromatographie flash. ... 68 Figure 3.8. Transthioestérification de l’acétate d’éthyle avec le p-tolylsulfur
pinacolborane. On observe l’apparition d’un nouveau produit seulement lorsqu’un équivalent de chlorure d’aluminium est ajouté. ... 70
xiii
Figure 3.9. Transthioestérification du 4-méthylbenzoate d’éthyle avec
2-méthoxyphénylsulfur pinacolborane avec le chlorure d’aluminium ... 70 Figure 3.10. Formation du thioester à partir de l’hémiacétal en conditions acides 71
xiv
Liste des tableaux
Tableau 1.1 Information pertinentes sur les noyaux actifs en RMN pour le projet effectué. ... 31 Tableau 1.2 Exemples de déplacements chimiques en RMN 11B ... 33
Tableau 2.1. Conditions de synthèse pour les dérivés 1-B(R)OMe-2-pip-C6H4 par monoalkylation du 1-B(OMe)2-2-pip-C6H4 ... 40
Tableau 2.2. Rendements et déplacements chimiques en RMN 11B{1H} des
composés [1-B(R)H2-2-pip-C6H4]Li (3). ... 44
Tableau 2.3. Stabilité de l’état fondamental des différentes formes des FLPs avec groupement alkyles par rapport à la forme ouverte en utilisant wB97XD/6-31+G** SMD=chloroform pour les dérivés de 1-B(R)H-2-NMe2-C6H4 ... 46
Tableau 2.4. Stabilité de l’état fondamental des différentes formes des FLPs avec groupements alkyles par rapport à la forme ouverte en utilisant wB97XD/6-31+G** SMD=chloroform pour les dérivés de1-B(R)H-2-pip-C6H4... 46
Tableau 2.5. Rendements et déplacements chimiques en RMN 11B{1H} des
adduits entre 1-B(R)H-2-pip-C6H4 et pyridine (4). ... 49
Tableau 2.6. Borylation catalytique du N-méthylpyrrole par les composés 4. ... 51 Tableau 2.7. Valeur énergétique des intermédiaires et états de transition pour l’activation C-H du N-méthylpyrrole par les composés 4 au niveau de théorie
wB97XD/6-31+G** SMD=chloroform. ... 54 Tableau 3.1. Optimisation des conditions pour la borylation du thiophénol
catalysée par [1-BH2-2-NMe2-C6H4]2 ... 62
Tableau 3.2. Étendue de la borylation des thiols catalysée par 1 et temps requis pour conversion complète par RMN 1Ha ... 63
xv
Liste des abréviations
{1H} Découplage en proton
6-31+G** Un ensemble de bases en DFT Ar Aryle
Å Angstrom (10-10 m) CO2 eq Équivalent en CO2 Cp Cyclopentadiényle Cp* 1,2,3,4,5-Pentaméthyl-cyclopentadiényle. d Doublet DFT Théorie de la fonctionnelle de la densité DIPEA Diisopropyléthylamine DMAP 4-Diméthylaminopyridine DME Diméthoxyéthane E Électrophile Et Éthyle
FLP Paire de Lewis frustrée HBpin Pinacolborane
HOMO Plus haute orbitale moléculaire occupée
HMB Hexaméthylbenzène
iPr isoproyle
J Constante de couplage (Hz) L Donneur d’électrons neutre LAH Aluminohydrure de lithium m Multiplet
M Métal
Me Méthyle
Mes Mésityle, 2,4,6-thiméthylphényle
m- Méta
n-Bu n-Butyle
NHC Carbène N-hétérocyclique
o- Ortho
ORTEP Oak Ridge Thermal Ellipsoid Plot
p- Para
Ph Phényle
ppm Partie par million Py Pyridine
q Quadruplet R Groupement alkyle
RMN Résonnance magnétique nucléaire s Singulet T Température THF Tétrahydrofurane TMEDA Tétraméthyléthylènediamine TMS Triméthylsilyle TMSBr Bromotriméthylsilane TMSCl Chlorotriméthylsilane Tol Toluene X Halogène
Z Accepteur d’électron neutre
d Déplacement chimique p Pi (orbitales moléculaires) s Sigma (orbitales moléculaires)
wB97XD Une fonctionnelle de DFT ∆G Énergie libre de Gibbs
∆H Enthalpie
xvi
Remerciements
Je tiens à remercier premièrement mon superviseur de maîtrise, le professeur Frédéric-Georges Fontaine. J’ai développé plusieurs compétences que je n’aurais pas aujourd’hui sans avoir bénéficié d’autant de liberté en laboratoire et d’aussi judicieux conseils.
Je veux également remercier les membres du groupe de recherche, spécialement Étienne Rochette, qui a été un mentor excellent. Il a été pour moi un immense bonheur de pouvoir discuter d’idées extravagantes et de chimie ésotérique avec vous tous.
Bien certainement, je tiens à remercier conformément, d’un merci sincère, les gens qui me sont chers, en l’occurrence ma famille et mes amis, qui ne survoleront jamais de leurs yeux ces paragraphes émouvants.
1
Introduction
Problématique
La protection de l’environnement s’est imposée au début du 21e siècle comme une problématique nécessitant l’action de tous et chacun, et bien sûr, des instances gouvernementales.1 Il est difficile, et peu réaliste, de compter uniquement sur les actions individuelles de la population pour préserver l’environnement, et ce, malgré les campagnes de sensibilisation. Il est également nécessaire de motiver les industries à diminuer leur impact environnemental. Celles-ci sont de grandes productrices de polluants et de gaz à effet de serre. Leur coopération pour la préservation de l’environnement est essentielle. Plusieurs moyens techniques peuvent être implémentés afin de minimiser la production de déchets. D’ailleurs, au niveau industriel, une minimisation des déchets et des besoins énergétiques peut représenter de grandes économies pour les compagnies, et est donc bénéfique pour les entreprises.2
La chimie joue un rôle, de près ou de loin, dans énormément de procédés industriels. Cela a permis aux industries chimiques de jouir d’un immense succès. Au Canada seulement, les exportations des industries chimiques génèrent plus de 30 milliards de dollars par année.3 Les industries chimiques œuvrent dans une foule de domaines incluant la pharmaceutique, l’agroalimentaire, l’énergie et les matériaux. Par exemple, on retrouve des procédés chimiques dans la production à grande échelle de médicaments et d’engrais. On doit également à la chimie la production des plastiques qui sont des matériaux extrêmement versatiles.
L’ubiquité des industries chimiques lie fortement la chimie et les industries. Les industries chimiques sont d’ailleurs souvent perçues comme source importante de pollution. Bien entendu, l’impact environnemental qu’on leur doit est important, que ce soit en termes de pollution atmosphérique, aquatique ou terrestre. Par exemple, dans les cas des émissions de gaz à effet de serre, les industries chimiques
2
canadiennes ont été responsables de l’émission d’environ 50 mégatonnes d’équivalents en CO2 pour l’année 2008.4
En conséquence, leur énorme succès et leur production importante de déchets ont certainement contribué à une mauvaise opinion du public à l’égard des industries chimiques. En effet, un sondage effectué en 1993 pour la Chemical Manufacturers Association montrait que moins du tiers des répondants croyaient que l’industrie se souciait de protéger l’environnement.5
Chimie verte
Pour pallier à ces problèmes environnementaux, et également pour redorer l’image de la chimie industrielle, douze principes ont été mis en avant afin de favoriser le développement de procédés moins polluants (Figure I.1). Ces principes forment les douze piliers de la chimie verte. Son but est de réduire l’impact environnemental de la chimie, que ce soit en industrie, en recherche ou au niveau académique.
La chimie verte vise à minimiser les effets négatifs de la pratique chimique sur l’environnement. Plus précisément, elle vise à prévoir et réduire la production de déchets, la consommation énergétique et la formation de produits de dégradation. Aussi, un chimiste doit considérer les risques reliés à la santé d’un procédé ou d’une réaction. Il doit minimiser l’utilisation de réactifs toxiques et la synthèse de produits toxiques. La chimie verte est un guide pour l’élaboration de procédés chimiques verts et économiquement viables.6
3
Figure I.1 Représentation des douze principes de la chimie verte constituant un procédé idéal.
Catalyse
La catalyse tient une place importante parmi les douze principes de la chimie verte. Plusieurs avantages justifient la place de la catalyse en industrie. Tout d’abord, un catalyseur est un additif sous-stoechiométrique à une réaction qui permet de diminuer la barrière énergétique globale de celle-ci, augmentant ainsi la vitesse de la réaction (Figure I.2). Le catalyseur se lie aux substrats pour la réaction désirée, puis libère le produit final et régénère le catalyseur. Cette capacité à se régénérer donne naissance aux cycles catalytiques, où il apparaît que le catalyseur peut réagir avec beaucoup plus qu’un équivalent du substrat. La catalyse permet souvent une meilleure sélectivité.7 Elle permet donc de minimiser la production de produits secondaires. Cela se traduit par une réduction des problèmes de séparation des produits et des sous-produits. Aussi, limiter la production de sous-produits limite du même coup la production de déchets. La catalyse intègre ainsi plusieurs principes de la chimie verte.
4
Figure I.2 Représentation d’une réaction hypothétique entre A et B pour former le produit C. L’énergie d’activation pour la réaction catalysée (en vert) est beaucoup plus faible que la réaction originale.
La catalyse est fortement associée à la chimie organométallique. En effet, les avancements en chimie organométallique ont souvent été destinés à la fabrication de catalyseurs à base de métaux de transition. Plusieurs caractéristiques justifient l’utilisation de métaux en catalyse, notamment la capacité d’un métal à changer de degré d’oxydation et la présence d’orbitales libres pouvant accepter les électrons et d’orbitales pleines pouvant donner des électrons sur une même espèce. Par exemple, plusieurs réactions de couplage au palladium débutent avec une addition oxydante, suivie par une transmétallation et d’une élimination réductrice (Figure I.3).8
5
Figure I.3 Cycle général pour les couplages catalytiques au palladium avec L = PR3. Suzuki-Myaura (R = aryle ou vinyle; R’ = aryle; M = B(OR’’)2; R’’ = alkyle ou aryle), Stille (R = aryle ou vinyle; R’ = aryle; M = SnR’’3; R’’ = alkyle), Sonogashira (R = aryle; R’ = alcyne; M = Na).5
Métaux et toxicité
Même si on associe souvent la toxicité aux métaux lourds, la rangée du tableau périodique n’est pas ce qui définit la toxicité d’un métal. Malgré l’absence de règles générales pouvant prédire la toxicité d’une espèce métallique, cette dernière dépend grandement de la voie d’exposition, de la bioaccessibilité, du degré d’oxydation du métal, de la solubilité de l’espèce, de la présence et de la nature des ligands, et de la dose. La solubilité et la voie d’exposition influencent la quantité de métal absorbé et comment il sera distribué et interagira à l’intérieur des cellules. Le degré d’oxydation déterminera la grosseur et la charge de l’ion métallique. C’est cette dernière qui interfère avec l’activité normale des cellules. Par exemple, les ions Ni2+ peuvent interférer avec les fonctions Mg2+ dans les enzymes de synthèse et de réparation d’acides nucléiques en raison d’un rayon ionique semblable (0,69 et 0,66 Å) et d’une préférence similaire pour les ligands.9 Des effets similaires peuvent être observés pour l’inhibition de la réparation de l’ADN par la protéine XPA, où le Cd(II)
6
et le Co(II) peuvent remplacer le Zn(II) de la protéine.10 Le doigt de zinc, la partie de la protéine pouvant se lier à l’ADN,11 a ainsi une affinité différente pour l’ADN et empêche la protéine de remplir sa fonction. Les métaux peuvent également dérégler l’équilibre oxydoréducteur des cellules. Le peroxyde d’hydrogène est un déchet cellulaire qui est rapidement éliminé par des enzymes (catalase et peroxydase). Cependant, la présence de métaux, comme Cr(VI), Fe(II), Co(II), Ni(II) et Cu(II), peut favoriser la formation de radicaux hydroxyles (•OH) à partir du peroxyde d’hydrogène (Figure I.4). Ces radicaux sont connus pour endommager les lipides, les protéines et l’ADN.12
Figure I.4 Mécanisme du déréglage de l’équilibre oxydoréducteur intracellulaire par des ions métalliques. La présence de métaux facilite la formation de radicaux hydroxyles.
En comparant la toxicité de différents chlorures de métaux pour leur toxicité chez le rat, il apparaît que la toxicité des métaux de la première rangée de transition peut
7
surpasser celle des métaux de la troisième rangée. En ordre de toxicité, on retrouve : NiCl2 > PtCl4 > CuCl2 » AuCl3 > FeCl3 > RhCl3 > PtCl2. Les chlorures de nickel et de cuivre, des métaux que l’on retrouve dans les organismes vivants, sont plus toxiques que ceux de rhodium(III) et de platine(II).13
Des instances gouvernementales imposent des limites à respecter en ce qui concerne les métaux à l’état de trace dans les produits destinés à la consommation.14 Cela entraine des coûts supplémentaires liés à la purification des produits, notamment pour les industries pharmaceutiques et agrochimiques.
Coûts associés à l’utilisation de métaux en catalyse
Outre les coûts associés à la purification des produits obtenus par catalyse avec des métaux de transition, plusieurs métaux sont reconnus pour être très dispendieux. L’or et le platine en sont des exemples bien connus. Par exemple, les sels métalliques de palladium et de rhodium utilisés comme précurseurs de catalyseurs coûtent entre 15,07 $/mmol pour le chlorure de palladium et 89,37 $/mmol pour le chlorure de rhodium (trace metals basis) selon le site web de Sigma Aldrich (en date du 25 avril 2018). Aussi, en catalyse homogène, les métaux sont complexés par des ligands qui, selon leur complexité, peuvent être responsables d’une grande part du coût du catalyseur.
Les phosphines (PR3) en sont un bon exemple. Elles sont une classe de ligands largement utilisée pour leur capacité à moduler les propriétés électroniques et stériques de complexes métalliques par le choix des groupements R. Leur capacité à jouer un rôle de ligand spectateur est un autre avantage. Les phosphines sont des donneurs s, mais peuvent être également des accepteurs p selon le choix du groupement R. Par exemple, on observe peu de rétroliaison dans le cas de la triméthylphosphine, mais pour la trifluorophosphine la rétroliaison est aussi forte que pour le ligand carbonyle.15 On retrouve une panoplie de phosphines commerciales qui permettent de choisir précisément les propriétés électroniques désirées.
8
Toutefois, le coût de ces phosphines peut être un sérieux désavantage pour des applications industrielles.
Impact environnemental des métaux
Les métaux de transition se retrouvent pour la plupart sous forme de minerais à l’intérieur de la croûte terrestre. Le cas du platine est un exemple flagrant de l’impact environnemental des métaux. La faible concentration de platine dans les minerais fait en sorte que l’extraction d’une énorme quantité de minerai est nécessaire à sa production. Effectivement, les minerais contiennent au plus 3,8 g de platine par tonne de minerai. 16
Pour la production annuelle de près de 161 kilotonnes, il faut extraire 4,23´1010 tonnes de minerais par année. 17
L’extraction et la purification du platine confient à ce métal précieux un impact environnemental très important. La production de gaz à effet de serre qui lui est associée s’élève à environ 33 kg en CO2 eq par gramme de platine, soit l’équivalent de 33 000 fois son poids en dioxyde de carbone. L’équivalent en CO2 (CO2 eq) est une unité utilisée pour décrire le potentiel d’un gaz à effet de serre à causer un réchauffement global. La consommation énergétique nécessaire à sa purification est d’environ 387 mégajoules par gramme de platine, assez pour conserver une ampoule écoénergétique de 10 W en fonction pendant plus d’un an.18
Il a été montré que le virage vers la chimie verte est en plusieurs points incohérents avec l’utilisation des métaux, principalement pour leur empreinte écologique et leur toxicité. Les aspects négatifs qui leurs sont liés sont source de motivation pour la recherche de systèmes sans métaux pouvant les remplacer. Plusieurs exemples de catalyseurs sans métaux sont bien connus et seront discutés dans la prochaine section. Sans être une revue exhaustive de la catalyse sans métal, la diversité des réactions pouvant être catalysées par dans systèmes sans métaux y sera explicitée.
9
Catalyse sans métal
Acide de Brønsted
Un exemple fort simple de catalyseur sans métal est le proton. La capacité d’un acide de Brønsted à catalyser certaines réactions est connue depuis longtemps.19 Une réaction très connue est la condensation d’un alcool et d’un acide carboxylique en ester. Pour que la réaction ait lieu, les conditions réactionnelles doivent être anhydres. D’un autre côté, en conditions aqueuses, l’acide de Brønsted va plutôt catalyser l’hydrolyse des esters pour reformer l’alcool et l’acide carboxylique correspondant. Le mécanisme d’action du catalyseur passe par la protonation du carbonyle.20 Le carbone du carbonyle sera alors beaucoup plus électrophile. Ainsi, un nucléophile faible, comme l’alcool et l’eau, peut attaquer le carbonyle et mener respectivement à l’ester et à l’acide carboxylique (Figure I.5).
Figure I.5 Estérification et hydrolyse d’un ester catalysées par un acide de Brønsted.
Acides et bases de Lewis
Selon le modèle développé par Gilbert Newton Lewis au début du 20e siècle, un acide de Lewis est une espèce ayant une déficience en électrons. Cette espèce possède généralement une orbitale vide pouvant accepter des électrons d’une autre espèce afin de satisfaire la règle de l’octet.21 Les éléments du groupe 13 du tableau périodique forment souvent des espèces acides. Par exemple, les espèces de bore et d’aluminium trivalents sont très répandues et utilisées comme acides de Lewis dans plusieurs réactions.22
Contrairement à l’acide de Lewis, une base de Lewis est une espèce qui possède une paire d’électrons libres, soit deux électrons qui ne participent pas à une liaison moléculaire. Cette paire d’électrons est donc disponible pour être partagée à une autre molécule. Des bases de Lewis basées sur les éléments du groupe 15 du
10
tableau périodique, comme les amines et les phosphines, sont très courantes.23 L’oxygène et le soufre peuvent également former des bases de Lewis, comme les éthers et les thioéthers, mais leur utilisation comme base est toutefois moins répandue que dans le cas des amines et des phosphines.
Les acides et les bases de Lewis sont généralement des espèces réactives. Lorsqu’on retrouve une base et un acide de Lewis dans le même milieu on obtient normalement la formation d’un adduit de Lewis. Cet adduit est le résultat du partage des électrons de la base avec l’orbitale inoccupée de l’acide (Figure I.6). Les deux espèces perdent leur caractère électrophile (acide) et nucléophile (base).
Figure I.6 Schématisation de la formation d’un adduit de Lewis entre une base (L:) et un acide (Z) de Lewis.
Acides de Lewis
L’utilisation d’acides de Lewis est très courante en chimie organique. Un exemple bien connu est le AlCl3, utilisé entre autres dans les réactions de Friedel-Crafts. Dans ces réactions, l’acide de Lewis permet l’attaque nucléophile des électrons d’un cycle aromatique sur un substrat électrophile (Figure I.7). Le groupe partant du substrat, généralement un halogénure, devient un meilleur groupe partant sous l’action de l’acide de Lewis.
11
Figure I.7 Illustration de la réaction d’alkylation de Friedel-Crafts.
Des acides de Lewis plus sophistiqués peuvent être développés et peuvent être des réactifs très utiles pour des réactions de formation de liens carbone-carbone. Par exemple, le groupe de Douglas Stephan a récemment montré que le cation fluorosulfonium permet d’effectuer l’hydroarylation d’un alcène avec le pyrrole. Le même acide de Lewis arrive également à catalyser l’hydrothiolation d’un alcène à partir du thiophénol (Figure I.8).24 Il s’agit d’un exemple d’un design habile avec pour but de créer un acide de Lewis très fort. L’idée générale est qu’une espèce de S(VI) devrait exhiber un caractère acide de Lewis fort. Les espèces de S(II) sont classifiées comme des donneurs s faibles. En augmentant le degré d’oxydation, le S(IV) devient un accepteur s.25 En suivant ce raisonnement, il apparaît que le soufre ayant un degré d’oxydation de +6 devrait avoir un caractère acide encore plus prononcé.26
Figure I.8 Réactions de formation de liens carbone-carbone catalysées par un cation fluorosulfonium.
Catalyse par les bases de Lewis
Les bases de Lewis azotées sont très utiles en chimie organique.27 On retrouve énormément d’exemples où il est possible de moduler la basicité et l’environnement stérique en vue d’obtenir la réactivité désirée (Figure I.9). Par exemple, la
12
triéthylamine et la N,N-diisopropylethylamine (DIPEA) sont deux bases très similaires ayant comme différence l’encombrement autour de l’azote. L’encombrement autour de l’azote de la DIPEA permet de rendre cette base moins nucléophile et donc plus sélective envers la déprotonation.28 La basicité peut être modulée entre autres par résonance. Un bon exemple est la différence de basicité entre la pyridine et la DMAP. Le pKa dans l’acétonitrile de l’acide conjugué à la pyridine est de 12,5 tandis que celui de la 4-diméthylaminopyridine (DMAP) est de 18,2.29 La forme protonée du DMAP (DMAPH) est très bien stabilisée par la forme de résonance où l’amine en position 4 sur la pyridine peut donner sa paire d’électrons au cycle.
Figure I.9 Exemples d’amines courantes en chimie organique. La nucléophilicité et la basicité des amines peuvent être contrôlées par l’encombrement stérique et des groupements donneurs.
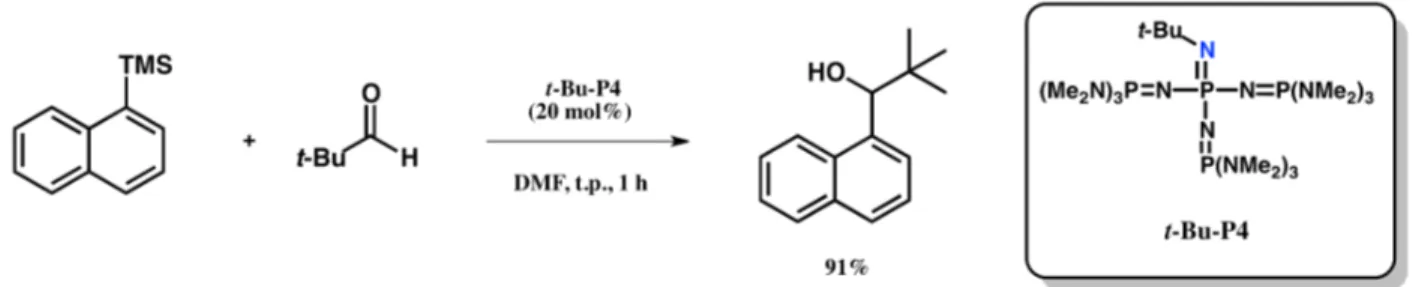
On retrouve des exemples où l’utilisation de bases permet de catalyser des réactions. Par exemple, des phosphazènes, qui sont principalement constitués d’atomes d’azote et de phosphore, sont rapportés pour catalyser des réactions de fonctionnalisation d’arylsilanes (Figure I.10)30 dans laquelle un lien carbone-carbone est formé. Le groupe de Kondo montre que le phosphazène peut activer le lien Si-Ar et ainsi former un phosphazénium et un carbanion. Ce dernier peut former un lien carbone-carbone en attaquant le carbonyle de l’aldéhyde, ce qui régénère le phosphazène.
13
Figure I.10 Réaction de formation d’un lien carbone-carbone catalysée par une base de Lewis. Le phosphazène (t-Bu-P4) permet la fonctionnalisation d’un arylsilane avec un aldéhyde.
Paires de Lewis frustrées
Comme mentionné précédemment, lorsqu’on retrouve dans un même milieu réactionnel une base et un acide de Lewis, on observe normalement la formation d’un adduit de Lewis. Il a récemment été démontré que, dans des cas particuliers, il est possible de conserver la réactivité de la paire de Lewis en prévenant la formation d’un adduit en modifiant l’encombrement stérique autour de la base et de l’acide de Lewis, ou encore en imposant des contraintes géométriques. Cela a comme effet de conserver le caractère nucléophile de la base de Lewis et le caractère électrophile de l’acide de Lewis.
Figure I.11 Représentations des paires de Lewis frustrées, où un acide et une base de Lewis sont retenus de former un adduit par contraintes stériques ou géométriques.
Il est possible de se servir de la réactivité de ces molécules ambiphiles pour activer des petites molécules, telles que le dihydrogène ou le dioxyde de carbone.31 On doit
14
cette nouvelle classe de composés, appelée paires de Lewis frustrées (FLPs), aux travaux pionniers de Douglas Stephan en 2006 lorsqu’il démontre que le composé 1-B(C6F5)2-4-PMes2-C6H4 active de manière réversible l’hydrogène (Figure I.12).32 Le domaine d’action des FLPs s’est rapidement étendu et est bien documenté.33 La versatilité de ces molécules ambiphiles a rapidement été comparée à celle des métaux de transition.34
La capacité à activer le dihydrogène ouvre la porte à des réactions d’hydrogénation catalytique. La capacité de libérer H2 est également cruciale à plusieurs réactions, comme il sera discuté plus loin. Le succès des FLPs dans les années suivantes confirmera qu’il s’agit d’un nouveau paradigme en catalyse sans métal.
Figure I.12 Activation réversible de H2 par une paire de Lewis frustrée 1-B(C6F5)2-4-PMes2-C6H4.
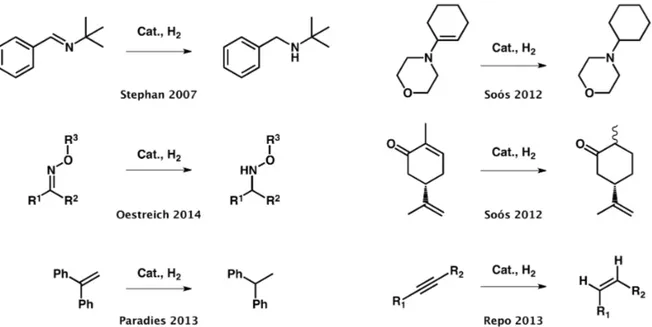
Effectivement, dans les quelques années suivant la découverte des FLPs, plusieurs réactions d’hydrogénation catalysées par des FLPs ont été rapportées. Parmi ces réactions, on retrouve d’abord l’hydrogénation d’imines, d’aziridines et de nitriles protégés.35 Ensuite, l’étendue de la réactivité pour l’hydrogénation par les FLPs s’est employée à l’hydrogénation d’énamines, d’éthers d‘énols silylés,36 d’énones,37 d’oxymes,38 d’oléfines39 et de systèmes poly-aromatiques.40
15
Figure I.13 Exemples de réactions d’hydrogénation catalysées par divers FLPs.
Alors que dans les 10 dernières années des centaines de systèmes catalytiques d’hydrogénation ont été rapportés, en 2013, le groupe de Timo Repo démontre un catalyseur de type FLP pouvant hydrogéner les alcynes en alcènes cis, qui est très pertinent pour le sujet traité dans ce mémoire.41 Plutôt qu’un FLP portant une phosphine et un borane, il rapporte la réactivité d’un FLP portant une amine et un borane en ortho sur un cycle benzénique. Cette classe de composé avait été rapportée par le groupe de Piers en 2003 avec l’ansa-aminoborane 1-B(C6F5)2 -2-NPh2-C6H4 (Figure I.14, A). Malgré qu’ils aient prédit une réactivité de cette structure avec des petites molécules comme H2, le composé rapporté ne permet pas d’activer l’hydrogène moléculaire.42 La structure proposée par le groupe de Repo diverge seulement par l’amine de la molécule. Les composés avec diméthylamine ou 2,2,6,6-tétraméthylpipéridine (TMP) en guise d’amine sont tous les deux capables de briser l’hydrogène moléculaire à température ambiante.43
16
Figure I.14 Structures d’ansa-aminoboranes rapportées par le groupe de Piers (A)42 et par le groupe de Repo (B et C).41
Peu après avoir publié ces composés dans la revue Dalton Transactions, le même groupe a montré dans la revue Nature Chemistry que le composé 1-B(C6F5)2 -2-NMe2-C6H4, sous deux atmosphères de H2 à 80 °C, est un catalyseur très efficace pour l’hydrogénation d’alcynes en alcènes cis. La forme active du catalyseur est obtenue après une première activation de l’hydrogène moléculaire qui permet de perdre un équivalent de pentafluorobenzène (C6F5H) pour doter le catalyseur d’un hydrure de bore. Ce dernier est nécessaire à l’hydroboration de l’alcyne par le catalyseur, qui est l’étape déterminant la conformation cis de l’alcène final. Après une deuxième activation de l’hydrogène, on observe l’élimination concertée de
l’alcène avec le proton acide de l’ammonium
(-NMe2H+), étape appelée protodéborylation, et le catalyseur est régénéré (Figure I.15, A).
17
Figure I.15 Mécanisme d’hydrogénation d’alcynes par Repo faisant intervenir l’étape de protodéborylation.
Activation C-H
La dernière étape du mécanisme suggéré par le groupe de Repo repose sur la protodéborylation de l’alcène. L’état de transition proposé est très semblable à celui rapporté par le groupe de Fagnou pour l’activation C-H d’arènes, qui sera discuté plus loin (Figure I.16).44 Dans le premier cas, un lien C-H est formé, tandis que dans le deuxième cas un lien C-H est brisé et permet la fonctionnalisation du lien. La possibilité de réversibilité de la protodéborylation du mécanisme du groupe de Repo permettrait l’activation C-H par des FLPs. Cette prise de conscience a marqué le début de l’intérêt de notre groupe de recherche à la réaction d’activation C-H par les FLPs.
Intérêts de l’activation C-H
La fonctionnalisation d’un lien C-H consiste à changer un lien carbone-hydrogène pour une autre fonctionnalité. La fonctionnalisation de tels liens résulte donc en produits à valeur ajoutée, ce qui est précisément le but de l’activation C-H. Lorsqu’on
18
tient compte que les liaisons carbone-hydrogène sont les liaisons les plus abondantes et les moins couteuses, la rentabilité de cette transformation parait indéniable.45
Cependant, l’abondance, en plus de sa robustesse, du lien C-H représente un défi pour réaliser des transformations sur un composé. Les défis de l’activation sélective peuvent être séparés en deux classes. Premièrement, l’activation d’un lien C-H particulier en présence de plusieurs liens C-H. Deuxièmement, le produit d’activation doit réagir moins rapidement que l’alcane dans les conditions de la réaction.46 Par exemple, un défi de l’oxydation du méthane au méthanol est de ne pas oxyder le méthanol en CO2. Également, le caractère inerte des alcanes face à la plupart des réactifs en chimie organique dû aux orbitales HOMO très stables et aux orbitales LUMO hautes en énergie, est également la source de plusieurs difficultés.
Les orbitales moléculaires des liaisons C-H sont non polaires et fortes. La stabilité de la HOMO rend le partage des électrons difficile et la haute énergie de la LUMO permet difficilement d’accepter des électrons. 47 Malgré qu’ils soient appelés paraffines (parum affinis ou sans affinités) les alcanes réagissent facilement avec l’oxygène de l’air. La combustion des alcanes est très favorable en raison des puits thermodynamiques que représentent le CO2 et l’eau. Malgré qu’elle soit inutile d’un point de vue synthétique, la combustion des alcanes peut révéler qu’ils sont réactifs face aux radicaux. Le bris homolytique du lien C-H non polaire des alcanes par des radicaux est d’ailleurs rapporté.48
Également, le caractère inerte des alcanes face à la plupart des réactifs en chimie organique est source de plusieurs difficultés. Ce caractère est souvent attribué à la force du lien (généralement entre 90 et 100 kcal/mol) et à la faible polarité du lien, conférant aux liens C-H d’alcanes une très faible acidité (pKa > 45).34 Une grande difficulté vient aussi de l’absence d’électrons π ou de doublets libres sur les alcanes.49 Plusieurs molécules peu réactives peuvent être rendues plus réactives par des métaux, notamment le monoxyde de carbone50 et l’azote moléculaire.51 La
19
capacité à se coordonner à un métal semble cruciale à cette nouvelle réactivité. Dans le même ordre d’idée, l’activation de liens C-H a d’abord été rapportée dans les cas d’hydrocarbures liés au métal par des interactions d’orbitales π de cycles aromatiques,52 ou d’hydrocarbure intramoléculaire faisant partie d’un ligand comme une phosphine.53 Le faible coût entropique du procédé intramoléculaire favorise la réaction d’activation C-H.
À défaut de posséder une paire d’électrons ou une orbitale π occupée, les alcanes peuvent interagir avec des métaux en partageant des électrons des liens C-H. Ce type d’interactions est appelé complexe s.47 Une fois le complexe formé, le bris du lien C-H est peu demandant de façon analogue au bris du lien H-H. Le métal affaibli la densité électronique de l’orbitale s du lien C-H, en même temps que les orbitales remplies du métal donnent par rétroliaison des électrons à l’orbitale s*.54 Dans le cas des arènes, les orbitales p favorisent la coordination au métal. Par exemple, le benzène peut former une liaison h6 avec un métal, où il est un ligand L3.55 Aussi, les liens C-H des arènes peuvent former des liaisons agostiques, qui favorisent grandement la fonctionnalisation du lien.56
La première observation directe de l’activation intermoléculaire d’un lien C-H d’un alcane a été rapportée par Janowicz et Bergman en 1982.57 L’irradiation à l’ultraviolet du complexe d’iridium à 18 électrons Cp*(Me3P)IrH2 entraine la perte d'hydrogène. Le complexe à 16 électrons résultant réagit rapidement avec le solvant pour former le produit d’activation C-H. Ils rapportent ainsi l’activation intermoléculaire C-H du benzène, du cyclohexane et du néopentane. D’autres complexes d’iridium, utilisant cette fois des ligands CO plutôt que des phosphines, ont permis d’activer le lien C-H du méthane.58 Plusieurs cas d’activation C-H d’alcanes par un complexe de rhodium ont aussi été rapportés.59
En 1997, Waltz et Hartwig rapportent des systèmes permettant la borylation sélective d’alcanes à la position terminale.60 Les systèmes utilisent des complexes de formule Cp*M(CO)nBR2 où le métal est le fer, le ruthénium ou le tungstène. Deux
20
années plus tard, Hartwig rapporte une version catalytique de ce système de borylation d’alcanes. Cette fois, des complexes de rhénium et de manganèse peuvent boryler de façon catalytique des alcanes terminaux lorsqu’en présence de B2pin2 (4,4,4’,4’,5,5,5’,5’-octamethyl-2,2’-bi-1,3,2-dioxaborolane), sous ultraviolet et sous deux atmosphères de CO.61
La fonctionnalisation de liens C-H activés par des réactifs pauvres en électrons est un choix qui s’est imposé. On peut expliquer ce choix à l’incompatibilité des complexes métalliques avec les bases de Lewis.62 Pour les raisons de compatibilité et pour leur versatilité en synthèse organique, la fonctionnalisation par borylation d’alcanes et d’arènes a rapidement été rapportée par plusieurs groupes.63 Le premier système de borylation catalytique du lien C-H d’arènes efficaces en conditions douces a été rapporté par les groupes de Miyaura et Hartwig.64
Une avancée majeure a été faite par le groupe de Fagnou avec l’étude mécanistique de l’arylation directe.65 La réaction d’arylation directe était déjà connue, cependant le manque de compréhension mécanistique minait son utilité en synthèse.66 Il a été rapporté que l’activation C-H d’un arène par le palladium procédait par un mécanisme de métallation déprotonation concertée.
En étudiant le mécanisme d’activation des liens C-H par un complexe de palladium, rapporté par le groupe de Fagnou, il est possible d’envisager l’activation de liens C-H par un catalyseur de type paires de Lewis frustrées. Pour l’étape de l’activation, le complexe de palladium forme un lien agostique avec les électrons du lien carbone-hydrogène, augmentant l’acidité du proton. Le ligand carboxylate peut déprotoner l’hydrogène, formant les liens carbone-palladium et hydrogène-oxygène (Figure I.16).67
21
Figure I.16 Mécanisme de l’arylation directe faisant intervenir la métallation déprotonation concertée.
Le palladium joue donc un rôle d’acide de Lewis, tandis que le ligand joue le rôle de base de Lewis. Notre groupe a montré qu’une paire de Lewis frustrée pouvait effectivement activer le lien C-H d’hétéroarènes par un mécanisme similaire.68 La similitude entre la métallation déprotonation concertée et l’activation C-H par un FLP est évidente lorsqu’on compare les états de transition des deux réactions (Figure I.17).
Figure I.17 Comparaison entre l’état de transition de la métallation déprotonation concertée et de l’activation C-H par un 1-BH2-2-TMP-C6H4.
22
Borylation d’hétéroarènes par les FLPs
L’étude des catalyseurs de type 1-BH2-2-NR2-C6H4 a montré que la forme active du catalyseur n’est pas son état fondamental. La forme active de l’aminohydroborane, dite «ouverte», est la forme où la paire d’électrons libres de l’azote n’est pas partagée à l’orbitale vide du bore (Figure I.18, A). Dans tous les cas, l’état fondamental de ces catalyseurs est une forme dimérique. Dans le cas d’une amine encombrée comme -TMP, la forme dimérique favorisée est semblable à la forme dimérique du BH3 avec des liaisons à trois centres deux électrons, que nous allons appeler forme trans (Figure I.18, B). Les amines moins encombrées comme avec – NMe2 et -pipéridine favorisent les dimères où un système ambiphile active le lien B-H d’une deuxième molécule ambiphile. Nous appelons ce dimère, qui était sans précédent avant le rapport de notre groupe,80 H-pontant (Figure I.18, C).
Figure I.18 Forme active d’un ansa-aminoboranes (A) et dimères les plus courants (B = dimère trans, C = dimère H-pontant).
Le mécanisme proposé commence donc par le bris du dimère (Figure I.19). Le catalyseur sous sa forme active peut ensuite procéder à l’activation C-H d’un hétéroarène. L’activation C-H, en passant par l’état de transition décrit précédemment, mène à un intermédiaire zwitterionique. Cet intermédiaire possédant un proton acide et un hydrure peut perdre un équivalent de H2 et mener à un intermédiaire où le bore porte un groupement aryle et un hydrure. La perte d’hydrogène procède par le même mécanisme concerté que l’activation de H2 qui est très connu dans la chimie des FLPs.33 Elle permet ici de favoriser entropiquement la réaction et est considérée irréversible si la réaction est en milieu ouvert. Le bore trivalent avec une géométrie planaire est acide en raison de son
23
orbitale vide. L’acidité du bore pour cette étape influence grandement la vitesse de l’étape. La métathèse permet donc de régénérer le catalyseur et de libérer l’hétéroarène borylé.
Figure I.19 Cycle catalytique pour la borylation du N-méthylpyrrole par 1-BH2 -2-NR2-C6H4.
Une deuxième réaction peut se produire à partir du dernier intermédiaire. La présence d’un hydrure à cette étape permet l’activation d’un deuxième équivalent d’un hétéroarène, suivi par une perte d’un deuxième équivalent de H2. Le nouvel intermédiaire obtenu après la double activation porte un bore beaucoup moins acide que le système ambiphile de départ. La métathèse est donc beaucoup plus lente pour l’intermédiaire portant deux groupements aromatiques. La métathèse peut même être irréalisable dans le cas du thiophène, où l’activation de deux équivalents du substrat est possible avec 1-BH2-2-TMP-C6H4, mais aucune activité catalytique n’est rapportée.69 La formation de cet intermédiaire en condition catalytique est source d’une perte d’activité.
24
Bris du dimère
Comme il a été mentionné brièvement précédemment, l’état fondamental des ansa-aminoboranes est dimérique, soit un dimère trans ou un dimère H-pontant. Il a été mentionné que l’encombrement de l’amine déterminait la nature du dimère favorable. Pour des amines plus encombrées comme TMP, le dimère formé est le dimère trans, tandis que pour des amines plus petites, le dimère H-pontant est favorisé. Un aspect qui n’avait pas été étudié est la difficulté à briser les dimères. Les dimères trans, bien qu’occasionnant une stabilisation autour de 8,7 kcal/mol par rapport à la forme ouverte, sont facilement brisés à des températures de -15 °C. En effet, le groupe de Repo a rapporté que le dimère trans du 1 BH2-2-TMP-C6H4 est en équilibre avec la forme monomérique fermée dans ces conditions.70 Comme on pourrait s’y attendre, l’équilibre avantage la forme monomérique en conditions diluées. La présence de cet équilibre montre que le bris du dimère est peu demandant et ne devrait pas influencer la réactivité du FLP dans les réactions subséquentes.
La structure asymétrique du dimère confère des signaux différents aux protons aromatiques des deux moitiés. À une température où le bris du dimère est accessible, les deux moitiés peuvent changer de « rôle », soit en monomère activé ou en monomère activant (Figure I.20). En utilisant la spectroscopie RMN de saturation de spin, il a été possible de déterminer la valeur de cette barrière énergétique.71 Le principe de base est que de saturer le spin d’un proton associé à une moitié affectera le proton correspondant de l’autre moitié. Donc, en irradiant le proton Ha, le proton Hb sera également affecté s’il y a un équilibre entre les deux formes du dimère. En comparant l’intégration des signaux à des températures différentes et des temps de saturation différents, il est possible d’estimer la barrière énergétique pour un bris du dimère. Ces études ont mené à une valeur de ∆G⧧ de 20,5 ± 3,1 kcal/mol pour le dimère H-pontant [1-BH2-2-pip-C6H4]2, alors que l’étude computationnelle du bris du dimère suggère que la réaction de dissociation peut
25
demander 20,7 kcal/mol (∆G⧧) pour ce même dimère, et jusqu’à 23,3 kcal/mol (∆G⧧) pour le dimère [1-BH2-2-NMe2-C6H4]2.
Figure I.20 Équilibre atteint lorsque la température permet le bris du dimère.
Bris du dimère et réactivité
Comme l’effet de l’encombrement a des répercussions sur la forme favorisée du dimère, on peut s’attendre qu’il y ait un effet sur l’accessibilité de la forme active du catalyseur et possiblement un effet sur la réactivité du catalyseur. Dans le cas où l’énergie d’activation du bris du dimère est supérieure à l’énergie d’activation globale de la réaction, le bris du dimère serait l’étape limitante de la réaction.
En comparant l’énergie de dissociation des dimères et l’énergie d’activation C-H du N-méthylpyrrole, il devient apparent que d’abaisser l’énergie de dissociation du dimère puisse avoir un effet bénéfique sur la réactivité des FLPs, surtout dans le cas d’amines moins encombrées (Figure I.21). Les calculs DFT suggèrent que, dans le cas d’amines peu encombrées comme diméthylamine et pipéridine, l’état de transition de la dissociation du dimère est supérieur en énergie à celui de l’activation C-H. Malgré que la DFT suggère que l’activation C-H par 1-BH2-2-NMe2-C6H4 est relativement facile, l’activité du catalyseur face à la borylation du N-méthylpyrrole est beaucoup plus faible que pour 1-BH2-2-Pip-C6H4. L’encombrement apparaît comme une composante importante déterminant la réactivité des FLPs. L’optimisation de celle-ci pourrait mener à un FLPs ayant un état de transition faible pour le bris du dimère ainsi que pour l’activation C-H.
26
Figure I.21. Énergie libre des intermédiaires (∆G) et états de transition (∆G⧧) en kcal/mol impliqué dans l’activation C-H du N-méthylpyrrole par les ansa-aminoboranes calculés au niveau de théorie wB97XD/6-31+G** SMD=chloroforme.
Projet principal
L’objectif principal de mes travaux de maitrise était de trouver des façons de défavoriser la formation de dimères tout en conservant la réactivité de type FLP. Deux possibilités ayant pour effet de faciliter le bris du dimère peuvent être schématisées. L’accessibilité de la forme active du catalyseur peut être augmentée en modifiant l’encombrement du FLP. Les deux types de dimères les plus fréquents sont les dimères trans et H-pontant. L’énergie d’activation pour le bris de ce dernier est beaucoup plus élevée que dans le cas du dimère trans. Un environnement légèrement plus encombré peut être suffisant pour favoriser le dimère trans. Le changement de dimère pourrait avoir un effet sur l’énergie d’activation globale d’une réaction, seulement si l’état de transition le plus élevé est celui du bris du dimère (Figure I.22, A). Ensuite, l’encombrement peut augmenter l’énergie fondamentale du FLP. Dans ce scénario, tous les états de transition sont plus accessibles, incluant l’état de transition pour le bris du dimère (Figure I.22, B).
27
Figure I.22 Schématisation des objectifs du projet. Faciliter le bris du dimère permettrait une quantité plus grande de catalyseur sous sa forme active.
Le deuxième chapitre portera sur les techniques expérimentales et instrumentales nécessaires à la réussite de ce projet. Le chapitre 3 portera sur la synthèse et la caractérisation de dérivés d’aminoboranes portant des groupements alkyles sur l’acide de Lewis du FLP. Le chapitre 4 décrira l’effet de groupements donneurs sur le bore du FLP et leurs effets sur l’étape de la métathèse en borylation. Cette étude mènera à la découverte d’un catalyseur efficace pour la borylation de thiols, qui y sera présenté. Le dernier chapitre sera essentiellement une discussion sur les résultats obtenus et les conclusions qu’ils ont apporté.
28
Chapitre 1. Méthodes expérimentales
1.1. Synthèses sous atmosphère inerte
La chimie des paires de Lewis frustrées est la plupart du temps sensible à l’air et/ou à l’eau. Bien que des versions de FLPs stables à l’air soient rapportées,72 les FLPs à base d’hydrures de bore sont très sensibles à l’eau. Les FLPs portant des phosphines sont très sensibles à l’oxygène de l’air. Pour ces raisons, les synthèses doivent être effectuées sous atmosphère inerte. La majorité des manipulations sont faites sous atmosphère d’azote en utilisant des techniques de Schlenk. Plusieurs produits sont également entreposés sous atmosphère inerte dans des ballons de type Strauss ou encore à l’intérieur d’une boîte à gants.
1.1.1. Rampe à vide et tubes de Schlenk
L’utilisation d’une rampe à vide, ou ligne de Schlenk, facilite grandement les synthèses sous atmosphère inerte (Figure 1.1). Elle est constituée de deux rampes et de plusieurs ports permettant d’y connecter plusieurs pièces de verrerie. L’une des rampes est reliée à une pompe à vide tandis que l’autre rampe est reliée à une source d’azote ou d’argon. Chaque port est doté d’une valve permettant de le connecter au gaz inerte ou au vide. L’alternance entre le vide et le gaz inerte permet de purger les pièces de verrerie de la majorité de l’oxygène et de l’humidité qu’elles peuvent contenir. Les manipulations sont faites sous une faible pression d’azote assurant que l’air ambiant ne puisse pas entrer dans les montages. La pression est générée par une valve pressurisée possédant un ressort, qui laisse passer l’azote à partir d’une pression déterminée. Cette valve permet l’utilisation d’un bulleur à l’huile, plutôt qu’un bulleur au mercure, tout en évitant les retours d’huile dans la ligne. L’évaporation des solvants sous vide est rendue possible par la présence de trappes à azote liquide. Celles-ci protègent la pompe en récoltant les solvants et autres produits volatils susceptibles de l’endommager.
29
Figure 1.1 Rampe à vide (gauche) et bulleur à l’huile avec valve pressurisée (droite). Les tubes et ballons de Schlenk possèdent un joint rodé ainsi qu’un bras possédant une valve en verre rodé. Le bras est la partie pouvant être raccordée à la ligne de Schlenk par un tuyau de caoutchouc. Les solvants et les réactifs qui sont purifiés et séchés sont généralement conservés dans des ballons Strauss munis de joints en téflon. Les transferts de solvants et de réactifs sont généralement faits en utilisant des seringues, des canules et des septums.
Figure 1.2 En ordre, bouchon et tube J-Young, tube de Schlenk et ballon Strauss avec valve en téflon.
30
1.1.2. Boîte à gants
Une boîte à gant permet de manipuler facilement des produits sous atmosphère inerte. Il est possible de peser des produits et de préparer des tubes RMN de produits sensibles à l’air ambiant. Le congélateur intégré permet également de faciliter la conservation et la recristallisation de plusieurs produits. Les produits et les équipements peuvent être entrés et sortis de la boîte à gants par l’un des deux sas. L’atmosphère contient normalement moins de 1 ppm d’oxygène et autour de 10 ppm d’eau.
Figure 1.3 Boîte à gants double possédant deux sas.
L’utilisation de tubes RMN de type J-Young permet de conserver le contenu du tube à l’abri de l’air ambiant. La valve en téflon permet de faire l’ajout de gaz et d’évaporer du solvant lorsque rattaché à une ligne de Schlenk. Il est possible de faire chauffer le tube à des températures légèrement supérieures à la température d’ébullition du solvant. Il s’agit d’un excellent outil pour réaliser des tests à très petite échelle et facilite grandement le suivi d’une réaction.
31
1.2. Caractérisations
1.2.1. Spectroscopie de résonance magnétique nucléaire
La RMN est la technique d’analyse la plus pertinente pour la réalisation de ce projet. Non seulement elle rend possible la caractérisation de la plupart des produits, elle permet aussi de suivre l’avancement d’une réaction. Au-delà de la résonance magnétique nucléaire du proton, la RMN peut s’appliquer à tous les noyaux ayant un nombre de protons impair et/ou un nombre de protons et de neutrons impair. Par exemple, l’hydrogène (1H), le carbone (13C) et le bore (11B) sont actifs par RMN.73 Le fait que le 13Csoit peu abondant rend les signaux sur les spectres du carbone plus faibles et demande un plus grand nombre d’acquisitions. Toutefois, les spectres en RMN 1H seraient beaucoup plus complexes à analyser si l’abondance du 13C était aussi grande que l’abondance de 1H en raison d’un plus grand nombre de pics dus au couplage des protons avec le carbone 13.
Tableau I.1 Informations sur les noyaux actifs en RMN pertinents pour le projet effectué.73
Noyaux Spin Abondance
(%) Sensibilité relative* 1H 1/2 99,98 1,000 11B 3/2 81,17 0,133 13C 1/2 1,11 0,016 31P 1/2 100 0,066
* Par rapport au proton et pour un nombre équivalent de noyaux dans un champ constant.
La spectroscopie en résonance magnétique du bore est particulièrement pratique au présent projet. En raison du petit nombre d’atomes de bore dans les molécules étudiées (1 à 2), le nombre de signaux est souvent faible et un bon indicateur du nombre de produits différents dans le tube RMN. Le moment quadripolaire du bore
32
a pour effet d’élargir les signaux. Les signaux ont généralement une largeur autour de 600 Hz. Dans le cas des espèces de bore tétracoordonnées les signaux sont souvent plus fins. Une optimisation adéquate de l’appareil permet d’observer le couplage entre le noyau du bore avec d’autres atomes.74 Le couplage bore-proton est un outil très pratique pour déterminer le nombre de protons présents sur un atome de bore (Figure 1.4).
Figure 1.4 Exemple de l’effet du couplage bore-proton. Le signal devient un triplet signifiant la présence de deux protons sur l’atome de bore.