Structural properties of cation exchange membranes: characterization,electrolyte effect and solute transfer
10
0
0
Texte intégral
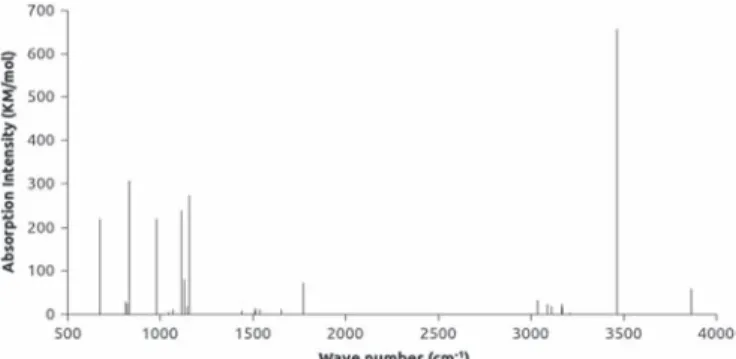
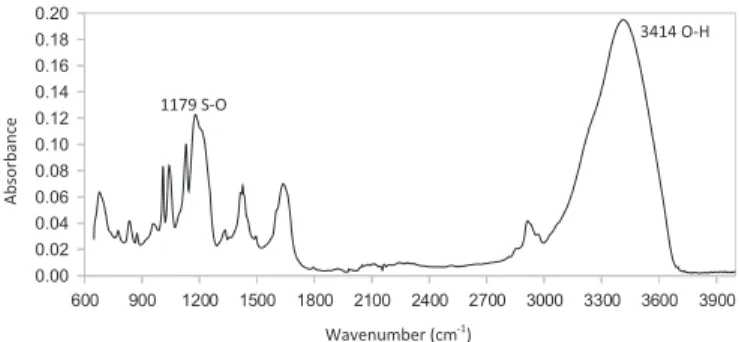
Figure


+2

Documents relatifs
