En vue de l'obtention du
DOCTORAT DE L'UNIVERSITÉ DE TOULOUSE
Délivré par :
Institut National Polytechnique de Toulouse (INP Toulouse)
Discipline ou spécialité :
Chimie Organométallique et de Coordination
Présentée et soutenue par :
Mme GANNA GOGOLIEVA
le lundi 28 avril 2014
Titre :
Unité de recherche : Ecole doctorale :
CATALYSEURS GREFFES SUR SUPPORT ET LIBERES SOUS
STIMULUS EXTERNE
Sciences de la Matière (SM)
Laboratoire de Chimie de Coordination (L.C.C.)
Directeur(s) de Thèse :
M. EMMANUEL GRAS MME ODILE DECHY CABARET
Rapporteurs :
M. FLORIAN MONNIER, ECOLE NLE SUP DE CHIMIE DE MONTPELLIER Mme ELODIE ANXOLABEHERE-MALLART, UNIVERSITE PARIS 7
Membre(s) du jury :
1 Mme MONTSERRAT GOMEZ SIMON, UNIVERSITE TOULOUSE 3, Président
2 M. EMMANUEL GRAS, CNRS TOULOUSE, Membre
2 Mme CLOTILDE POLICAR, ENS PARIS, Membre
Remerciements
Je tiens à remercier en premier lieu les chefs d’équipe C et F : Philippe Serp et Peter Faller de m’avoir accueillie. Je remercie aussi la direction du LCC et de l’ENSIACET pour m’avoir permis de travailler entre leurs murs.
En deuxième lieu, je veux remercier mes directeurs de thèse : Emmanuel Gras et Odile Dechy-Cabaret. Emmanuel Gras pour son écoute attentive , la motivation et la persévérance qu’il a su me transmettre pendant ces trois dernières années , et Odile Dechy-Cabaret pour ses explications claires, son esprit de synthèse et la rigueur qu’elle a su m’enseigner. Je tiens également à les remercier pour leur accueil chaleureux, leur générosité et leur affection. Ils m’ont appris beaucoup de choses sur la chimie et sur moi-même qui me seront très utiles pour le reste de ma vie professionnelle et personnelle.
En troisième lieu, je souhaiterais remercier les équipes C et F avec notamment : Jérôme Volkman pour nos discutions scientifiques, Jérôme Durand pour ses connaissances pointues dans le domaine des nanotubes de carbone, Rosa Axet qui m’a enseigné de bonnes méthodes de travail et Christelle Hureau pour ses explications claires.
Ensuite mes remerciements vont aux étudiants qui sont devenus mes amis durant ces trois ans : Kévin Fourmy et Jamal El Karroumi qui m’ont permis de travailler dans une ambiance chaleureuse et conviviale, et avec lesquels j’ai partagé des moments de bonheur ; Bruno Machado qui a été l’un de mes principales soutiens avec ma meilleure amie Lisa Bratko, et Sofia De Leon qui est devenu le témoin de mon mariage.
Je voudrais également remercier tous les autres membres de mes équipes et en particulier : Nora Imlyhen, Sarah Cadet, Liping Zhang, Sabrina Noel, Hélène Eury, Gilles Caillot, Abdelouahd Oukhrib, Florence Gayet, Carole Leberre, Manon Genet, Idaline Chanteperdrix, Laurent Ropiquet, Anas Benyounes, Justine Harmel, Faqiang Leng.
J’aimerais aussi remercier les services d’analyses, et particulièrement Sonia Mallet-Ladeira pour les structures RX.
Enfin, j’adresse mes remerciements à ma famille et à ma belle-famille qui ont été d’un grand support. Mon mari, Alexandre Bastien, par son amour et ses attentions, m’a aidé à surmonter toutes les difficultés, c’est pourquoi je tiens à le remercier ici très sincèrement.
SOMMAIRE
Abréviations 1
Résumé 3
Introduction générale 5
Chapitre I. Elaboration de systèmes recyclables
1 Recyclage des catalyseurs 9
1.1 Pourquoi recycler ? 9
1.2 Greffage des complexes sur un support : catalyse « hétérogène » 9
1.2.1 Principe
1.2.2 Greffage covalent 1.2.3 Greffage non covalent
1.3 Immobilisation des complexes dans une phase : catalyse homogène 11 1.4 Immobilisation du catalyseur en fin de réaction : catalyse boomerang 11
1.4.2 Principe
1.4.2 Greffage en fin de réaction 1.4.3 Greffage réversible
1.4.4 Principe du projet Switchcat
2 Complexes commutables photochimiquement 17
2.1. Principe du switch photochimique sur les complexes
du ruthénium (II) polypyridine 17 2.2 Machines moléculaires commutables photochimiquement
à base de complexes du ruthénium. 21
2.2.1 Différents types de complexes [Ru(terpy)(N-N)(L)]2+
2.2.2 Exemples de machines moléculaires basées sur un cœur du type [Ru(terpy)(N-N)(L)]2+
2.3. Application à notre projet 23
3 Complexes commutables électrochimiquement 25
3.1 Principe du switch électrochimique sur les complexes du cuivre (I) polypyridines 25 3.2 Machines moléculaires commutables électrochimiquement
à base de complexes du cuivre. 27
3.2.1. Le complexe du cuivre caténane avec deux géométries distinctes 3.2.2. « Navette moléculaire » : rotaxane à deux stations
3.2.3. Les machines moléculaires à base de rotaxane : vers des mouvements rapides.
3.3 Application à notre projet. 36
Chapitre II. Complexes du palladium portant des ligands NHC
fonctionnalisés
1 Vers des complexes de palladium recyclables 45
1.1 Intérêt des complexes de palladium en catalyse 45 1.2 Système catalytique visé 45
1.2.1 Choix du ligand NHC 1.2.2 Choix du lien triazole
1.2.3 Diverses fonctionnalités pour le recyclage
2 Synthèse des imidazoliums précurseurs de NHC 56
2.1 Imidazoliums-triazoles portant un bras perfluoré 56 2.2 Ar-CN et Ar-NO2 : click direct et click inverse 57
2.2.1 Click direct 2.2.2 Click inverse
2.3 Terpyridine 62
3 Synthèse des complexes 65
3.1 Synthèse 65
3.1.1 Synthèse des complexes perfluorés porant un bras
3.1.2. Synthèse des complexes portant une fonctionalité CN et NO2
3.2 Caractérisation RMN 67 3.2.1 RMN 1H 3.2.2 RMN 13C 3.2.3 RMN 31P 3.3 Données RX 75 4 Evaluation en catalyse 79
4.1 Réaction modèle choisie 79
4.2 Résultats 79
4.2.1 Influence de la fonctionnalité sur l’activité 4.2.2 Variation des substrats et de la charge catalytique
4.3 Catalyse homogène ou hétérogène ? 83
5 Conclusion 83
6 Partie expérimentale 84
Chapitre III. Complexes du ruthénium et switch photochimique
1 Système catalytique envisagé 139
2 Synthèse de l’agrafe 141
2.1 Synthèse du précurseur 141
2.1.1 Synthèse de ligands terpyridines fonctionnalisés 2.1.2 Synthèses des complexes du ruthénium
2.2 Synthèse des complexes [RuCl(terpy’)(bipy)][PF6] 144
2.3 Synthèse de l’agrafe [Ru(terpy)(bipy)(H2O)]2+ 149
3 Faisablilité du switch photochimique 152
3.1 Test du switch photochimique sur le complexe modèle
[Ru(terpy)(bipy)(R-CN)]2+ 152
3.1.1 Synthèse du complexe modèle 3.1.2 Irradiation
3.1.3 Recomplexation par chauffage
3.2. Synthèse du complexe [Ru(NCPh)-Pd(NHC)]2+ 161
4 Vers le greffage de l’agrafe métallique sur les nanotubes de carbone :
résultats préliminaires 163
4.1 Greffage covalent à partir du sel de diazonium 164
4.1.1 Préparation des MWCNTs (nanotubes de carbone multi parois) 4.1.2 Greffage de la terpyridine sur les MWCNTs.
4.2 Greffage non covalent par π-stacking du pyrène 168
5 Conclusion et perspectives 170
6 Partie expérimentale 172
Chapitre IV. Complexes du cuivre et switch électrochimique
1 Système catalytique envisagé 207
2 Synthèse des complexes du cuivre 209
2.1 Complexes de Cu(I) 209
2.1.1 Le ligand bis-bipyridine. 2.1.2 Le ligand bis-bipyridine-NH2
2.1.3 Synthèse du Cu (I)-bisbipyridine
2.2 Complexes du Cu(II) 225
2.2.1 Complexes [Cu(II)-(bis-bipyridine)] et [Cu(II)(bipyridine)2]
2.2.2 Complexes hétéroleptiquesdu type [Cu(II)(bipyridine)(terpyridine)] 2.2.3 Synthèse du complexe [Cu(II)(terpy)2]
3 Tests électrochimiques sur système modèle 240
3.1 Etudes de la faisabilité du switch électrochimique 240 3.2 Voltamétrie cyclique à partir de complexes de Cu(II) 242
3.2.1 Analyse RPE
3.2.2 Ajout de terpy-PhNO2 dans une solution du complexe [Cu(II)(bis-bipyridine)]
3.3 Voltamétrie cyclique à partir de complexes de Cu(I) 249
3.3.1 Analyse RMN
3.3.2 Ajout de Terpy-PhNO2 dans une solution du complexe [Cu(I)(bisbipyridine)]
4 Conclusion 255
5 Perspectives 257
6 Partie expérimentale 258
7 Références bilbiographiques 275
1
Liste des abréviations utilisées dans ce manuscrit
NHC : carbène N-hétérocyclique CNTs : nanotubes de carbone L : ligand MLCT : metal-to-ligand charge-transfer PhCN : benzonitrile terpy : 2, 2’ ; 6’, 2’’-terpyridine bipy : 2, 2’-bipyridine phen : 1, 10–phénantroline dmp : 2,9-diméthyl-1,10-phénantroline dpp : 2,9-diphényl-1,10-phénantroline MeOBN : 2,6-diméthoxybenzonitrile
VSEPR : Valence Shell Electron Pairs Repulsion DCM : dichlorométhane
TA : température ambiante DIPA : diisopropylamine DMF : diméthyle formamide DMSO : diméthyle sulfoxyde
TsOH : acide paratoluène-sulfonique m-CPBA : acide métachloroperbenzoϊque THF : tétrahydrofurane
TON : turn-over number TOF : turn-over frequency Rdt : rendement
3
Résumé
La catalyse organométallique est à l'origine des avancées les plus significatives au cours des dernières années puisqu'elle a permis de découvrir de nouvelles voies d'accès à des molécules complexes; elle a ainsi révolutionné le monde de la synthèse organique. Bien que de nombreuses réactions s'appuyant sur des complexes organométalliques aient été développées, les catalyseurs organométalliques présentent encore deux inconvénients majeurs : leur toxicité, associée la plupart du temps à la présence de métaux lourds, et leur coût lié à la présence à la fois d'un centre métallique et de ligands élaborés. Dès lors de nombreuses recherches ont été lancées dans le but de mettre au point des techniques permettant de séparer simplement le catalyseur métallique des produits formés et de le recycler. C’est dans ce contexte que nous avons cherché à développer de nouveaux catalyseurs supportés pouvant être libérés pendant la réaction afin d’effectuer la catalyse en conditions homogènes et de nouveau greffés au support en fin de réaction afin de pouvoir séparer le catalyseur des produits de la réaction et le recycler ensuite via une nouvelle libération. Cette capacité de passage du catalyseur du support vers la solution et vice-versa est basée sur un complexe de coordination lié au support et appelé « agrafe contrôlable» dont la sphère de coordination peut être modulée via un stimulus externe : soit un complexe du ruthénium avec un stimulus photochimique, soit un complexe du cuivre avec un stimulus électrochimique, tels qu’exploités dans de nombreuses machines moléculaires. Les nanotubes de carbone sont choisis comme support, les ligands de type NHC sont choisis comme ligands de base du système catalytique et des complexes du cuivre ou du ruthénium portant des ligands de type bipyridine, phénantroline ou terpyridine constituent le cœur de l’agrafe contrôlable.
Des complexes de palladium basés sur des ligands NHC fonctionnalisés ont été synthétisés en utilisant un motif triazole comme lien entre le centre catalytique et la fonctionnalité impliquée dans la récupération du catalyseur. Les tests catalytiques dans des réactions de Suzuki-Miyaura ont montré que les complexes étaient actifs sur une large gamme de substrats et à des charges catalytiques faibles (jusqu’à 50 ppm) et ce, quelle que soit la fonctionnalité ajoutée. Des complexes du ruthénium portant des ligands terpyridine, bipyridine ou phénantroline ont été synthétisés et caractérisés. Le concept du switch photochimique a été validé sur un système modèle : nous avons pu montrer que sous irradiation à l’aide d’une lampe à vapeur de mercure, le complexe [Ru(II)(terpyridine)(bipyridine)(PhCN)][(PF6)2] était capable de libérer le ligand benzonitrile et que la réaction inverse se produisait par chauffage du complexe [Ru(II)(terpyridine)(bipyridine)(H2O)][(PF6)2] en présence de benzonitrile.
D’autre part, des complexes du cuivre portant des ligands de type bis-bipyridine et terpyridine ont été synthétisés et caractérisés. Le concept du switch électrochimique a été validé sur un système modèle : nous avons pu montrer par voltamétrie cyclique qu’en présence de terpyridine, le complexe [Cu(I)(bipyridine)2][BF4] pouvait s’oxyder et former un complexe de type [Cu(II)(bipyridine)(terpyridine)][(BF4)2] et que la réaction inverse se produisait par réduction.
Ces travaux démontrent le caractère contrôlable des agrafes choisies via un stimulus électrochimique ou photochimique, sur des systèmes modèles. L‘obtention de complexes de palladium comportant un ligand NHC-triazole fonctionnalisé, leur bonne activité et la voie de synthèse mise au point valident notre stratégie de récupération et ouvrent de nombreuses perspectives en catalyse.
4
Summary
Transition metal catalysed reaction is the source of the most significant advances in recent years since it revealed new access routes to complex molecules and as such has revolutionized the way of designing organic syntheses. Although many reactions based on complexes of transition metals have been developed, « organometallic » catalysts have still two major drawbacks: their toxicity, mostly related to the presence of heavy metals, and their cost mainly induced by the presence of both a metal center and sophisticated ligands. Therefore much research has been undertaken in order to develop techniques allowing a simple separation of the metal catalyst from reaction products and the recycling of the former. It is in this context that we sought to develop new supported catalysts that can be released during the reaction to perform catalysis under homogeneous conditions and being grafted back to the support (boomerang catalyst) at the end of the reaction in order to separate the catalyst from the products and then use the catalyst again, homogeneously in a new solution of substrate, through a new release. This ability to shift the catalyst from the support to the solution and vice versa is based on a coordination complex bound to the support and called the "controllable tether" which coordination sphere can be modulated via an external stimulus. Two type of tethers have been explored; one involving a ruthenium complex requiring a photochemical stimulus, and another using a copper complex and an electrochemical stimulus, similar to systems used in molecular machineries. Carbon nanotubes have been selected as solid support and NHC type ligands have been chosen for the robustness of their complexes in catalysis.
Complexes of palladium based on functionalized NHC ligands were synthesized using a triazole link between the catalytic center and the function involved in the recovery of the catalyst on the tether. Catalytic reactions of Suzuki - Miyaura showed that the complexes were active on a wide scope of substrates and at catalytic low loads (up to 50 ppm), and this, whatever the added recovery function is.
Ruthenium complexes bearing ligands such as terpyridine, bipyridine or phenanthroline have been synthesized and characterized. The concept of photochemical switch has been validated on a model system for which we have shown that under irradiation with a mercury vapor lamp, the complex [Ru(II)(terpyridine)(bipyridine)(PhCN)][(PF6)2] was able to release its benzonitrile ligand and the reverse reaction occurs by heating the complex [Ru(II)(terpyridine)(bipyridine)(H2O)][(PF6)2] in the presence of benzonitrile .
On the other hand, copper complexes bearing ligands of bis-bipyridine and terpyridine were synthesized and characterized. The concept of electrochemical switch has been validated on a model system for which we have shown by cyclic voltammetry in the presence of terpyridine that the complex [Cu(I)(bipyridine)2][BF4] could be oxidized and form a complex of the type [Cu(II)(bipyridine)(terpyridine)][(BF4)2]; the reverse reaction occuring upon reduction . This work demonstrates the controllability of the selected tethers via an electrochemical or photochemical stimulus on model systems. Obtaining functionalized palladium complexes with good activity through an efficient synthesis route validates our strategy of recovery and opens many opportunities in catalysis.
5
Introduction générale
Les dernières décennies ont connu un accroissement considérable du développement de méthodes de synthèse lié à l’essor formidable de la catalyse homogène exploitant les métaux de transition. Ce recours à des complexes organométalliques dans les synthèses organiques, conduit les chimistes à trouver des méthodes de récupération et de réutilisation des catalyseurs pour diminuer les taux de déchets de métal pour des raisons économiques et environnementales.
Deux approches sont classiquement envisagées pour la récupération des catalyseurs : soit l’ « hétérogénéisation » du catalyseur par son greffage sur un support solide, soit l’immobilisation du catalyseur dans une phase liquide. Si ces deux approches permettent une récupération aisée du catalyseur en fin de réaction, elles peuvent être limitées par une diminution de l’activité du catalyseur en conditions hétérogènes. Une stratégie alternative, appelée « catalyse boomerang » consiste à accrocher le catalyseur sur le support seulement une fois que la catalyse est terminée et à le relarguer dans le nouveau milieu réactionnel pour effectuer une autre réaction catalytique. Cette approche associe les avantages, a priori antagonistes, de la catalyse homogène (en termes d’activité et de sélectivité) et de la catalyse supportée (en termes de récupération du catalyseur).
Nous nous sommes intéressés à ce principe et nous avons proposé un nouveau modèle de liaison réversible du catalyseur sur un support via une agrafe contrôlable, tel que schématisé ci-dessous (Figure 1).
Figure 1. Catalyseur lié à un support via une agrafe contrôlable
L’agrafe envisagée peut exister selon deux états contrôlables : un état « on » (catalyseur attaché au support) et un état « off » (catalyseur en solution). La transformation d’un état à l’autre d’une agrafe peut être induite sous stimulus externe (photochimique ou électrochimique). La structure générale du système ciblé et son mode de fonctionnement est présenté sur le schéma suivant (Schéma 1). Sous stimulus externe, le système expulse le
6
catalyseur du support via le changement de la sphère de coordination de l’agrafe. Le catalyseur va réagir avec les réactifs dans la solution pour fournir les produits. A la fin de la réaction, sous le stimulus externe inverse, le catalyseur s’accroche au support via recoordination sur l’agrafe et peut ainsi être récupéré par simple filtration.
Schéma 1. Concept général de recyclage du catalyseur sous stimulus externe
Dans notre travail, les nanotubes de carbone sont choisis comme support afin d’exploiter leurs nombreuses propriétés, notamment leur surface spécifique élevée et le large éventail des réactions de fonctionnalisation possibles.
Des complexes de cuivre ou de ruthénium constituent le cœur de l’agrafe contrôlable ; leur sphère de coordination peut en effet être modifiée sous l’action d’un stimulus électrochimique ou photochimique.
Les catalyseurs choisis sont des complexes organométalliques basés sur les ligands carbènes N-hétérocycliques (NHC), connus pour leur stabilité, leur facilité de synthèse et leurs nombreuses applications en catalyse.
7
Dans une partie bibliographique, nous présenterons les diverses méthodes de recyclage de catalyseur envisageables et nous décrirons la structure et le principe de fonctionnement des complexes du ruthénium commutables photochimiquement et des complexes du cuivre commutables électrochimiquement tels qu’exploités dans de nombreuses machines moléculaires.
Le deuxième chapitre présentera la synthèse et l’évaluation en catalyse des complexes de palladium portant des ligands NHC fonctionnalisés sera décrite.
Le troisième chapitre présentera la synthèse et la caractérisation des complexes polypyridine du ruthénium choisis comme agrafe contrôlable photochimiquement et l’étude de la faisabilité du switch photochimique sur un système modèle.
Le quatrième chapitre présentera la synthèse et la caractérisation des complexes polypyridine du cuivre choisis comme agrafe contrôlable électrochimiquement et l’étude de la faisabilité du switch électrochimique sur un système modèle.
Chapitre I Elaboration de systèmes
catalytiques recyclables
Chapitre I. Elaboration de systèmes recyclables
9
1.
Recyclage des catalyseurs
1.1 Pourquoi recycler ?
Les dernières décennies ont connu un accroissement considérable du développement de méthodes de synthèse lié à l’essor formidable de la catalyse homogène exploitant les métaux de transition.
Ce recours à des complexes organométalliques dans les synthèses organiques, conduit les chimistes à trouver des méthodes de réutilisation des catalyseurs pour diminuer les taux de déchets de métal pour des raisons économiques et environnementales. Diverses approches « d’immobilisation » du catalyseur sont envisageables : (i) hétérogénéisation du catalyseur (greffage sur support solide) ; (ii) immobilisation du catalyseur dans une phase liquide (liquides ioniques, solvants perfluorés, glycérol) ; (iii) catalyseur boomerang (aptitude conférée au catalyseur de se décrocher de son support et de se raccrocher « à la demande ». Ces diverses méthodes de recyclage de catalyseurs sont présentées ci-dessous.
1.2 Greffage des complexes sur un support : catalyse « hétérogène »
1.2.1 Principe
L’immobilisation des complexes de métaux (les catalyseurs) sur les supports pour leur conférer une nature hétérogène est une méthode extrêmement attractive. La récupération des catalyseurs peut en effet s’effectuer par une simple filtration ou centrifugation. Selon la nature de l’interaction entre le support et les catalyseurs, le greffage de ces derniers peut être effectué (i) de façon covalente, (ii) de façon non covalente et (iii) par encapsulation. Ces trois types d’immobilisation des catalyseurs seront présentés ci-après.
1.2.2 Greffage covalent
Le greffage du catalyseur sur le support de façon covalente est la méthode la plus utilisée pour préparer le catalyseur immobilisé. L’immobilisation des complexes métalliques sur la surface peut être effectuée par liaison directe ou par l’intermédiaire d’un linker déjà greffé sur le support et réagissant avec le catalyseur. Les supports les plus utilisés sont des matériaux amorphes ou structurés (SiO2, Al2O3, zéolite, polymères) ou des supports carbonés
traditionnels tels que le charbon actif, le graphène ou les nanomatériaux émergents du carbone.1,2,3,4,5,6,7 L’utilisation de nanotubes de carbone (CNTs) comme support de
Chapitre I. Elaboration de systèmes recyclables
10
littérature.8,9,10,11,12 Les propriétés intéressantes des CNTs comme leur grande stabilité
mécanique et thermique ainsi que leur conductivité électrique élevée sont idéales pour qu’ils soient utilisés comme support de catalyseurs. De plus, il existe différentes façons de fonctionnaliser les nanotubes de carbone avec des entités chimiques qui permettent leur utilisation dans différents domaines incluant la catalyse.13
Le plus grand avantage de l’immobilisation des catalyseurs sur les supports de façon covalente repose sur la force des liaisons chimiques impliquées qui peut limiter le relargage (« leaching ») des espèces métalliques du support dans la solution pendant la catalyse. Néanmoins, cette méthode de synthèse des catalyseurs supportés demande beaucoup d’efforts préparatifs comprenant le greffage sur le support (fonctionnalisation) et la coordination du métal.
1.2.3 Greffage non covalent
Une méthode plus simple de fixation du catalyseur sur le support est l’immobilisation du catalyseur de façon non covalente via des interactions π-π, de Van der Waals, électrostatiques ou par encapsulation des catalyseurs dans des supports poreux.14,15,16,17 Les supports les plus
utilisés sont les tamis moléculaires microporeux (MCM), les zéolites, les polymères et les CNTs. Il a été montré que la modification non-covalente des nanotubes de carbone est une approche intéressante et différents structures moléculaires peuvent être immobilisées, tels que les molécules aromatiques, par exemple le pyrène. Ainsi, des complexes métalliques peuvent être greffés de façon non covalente sur les CNTs via π-π stacking entre le support et un motif pyrène porté par le catalyseur.18,19,20,21 Après la réaction catalytique, le catalyseur peut être
récupéré par filtration et réutilisé dans la réaction suivante.
L’inconvénient de cette méthode de greffage non covalent est le risque élevé de « leaching » des catalyseurs du support pendant la catalyse.
D’autre part, si les systèmes catalytiques hétérogènes sont faciles à recycler, ils sont souvent moins efficaces que les catalyseurs en solution homogène du fait de la limitation du transfert de masse.
Chapitre I. Elaboration de systèmes recyclables
11
1.3 Immobilisation des complexes dans une phase liquide : catalyse
homogène « supportée »
Une autre approche consiste à « immobiliser » le catalyseur dans une phase liquide à partir de laquelle le produit de réaction peut être facilement extrait. Ici nous parlons de catalyse homogène supportée. Cette approche est intéressante du point de vue industriel pour des raisons économiques et de sécurité. Divers milieux peuvent être envisagés : l’eau, les liquides ioniques, les solvants perfluorés et le glycérol ; de manière générale tout milieu liquide non miscible avec le solvant d’extraction des produits de réaction. Par exemple, en utilisant des catalyseurs solubles dans l’eau, on peut séparer les produits par simple filtration ou extraction.
22,23,24,25 La limite de cette approche est l’insolubilité des nombreux réactifs dans l’eau. Les
liquides ioniques sont beaucoup utilisés comme « support » pour le catalyseur mais leur capacité à dissoudre les substrats organiques peut devenir un problème quand il s'agit de récupérer le produit de la réaction dans un solvant classique.26 Une autre alternative est
d’immobiliser le catalyseur dans le glycérol, dont les produits peuvent être extraits sélectivement par extraction ou filtration.27,28 L’utilisation de systèmes catalytiques
comportant un rapport massique de fluor important dans les solvants perfluorés représente une autre approche basée sur la nature thermomorphique de ces solvants qui permettent à la réaction de s'effectuer dans des mélanges homogènes à température élevée tandis que la séparation des phases s’effectue à température ambiante.29
L'inconvénient principal de ces quatre approches est la difficulté de concevoir des ligands qui forment un système catalytique peu soluble dans le solvant organique utilisé pour la récupération du produit. L’efficacité de ce type de procédé repose sur le coefficient de partage de l’espèce catalytique et des réactifs et produits entre les deux solvants A et B. Le risque de pollution des produits par le catalyseur (et réciproquement du catalyseur par les substrats, produits ou sous-produits) est généralement difficile à contrôler.
1.4 Immobilisation du catalyseur en fin de réaction : catalyse boomerang
1.4.1 Principe
L’idée générale de cette approche est la combinaison de l’utilisation d’un support pour récupérer le catalyseur et de l’utilisation d’un catalyseur travaillant dans des conditions homogènes. Il devient ainsi possible d’associer les avantages, a priori antagonistes, de la catalyse homogène et de la catalyse supportée. Pour préserver la haute activité et la sélectivité du catalyseur dans les conditions homogènes, il convient d’accrocher le catalyseur sur le
Chapitre I. Elaboration de systèmes recyclables
12
support seulement quand la catalyse est terminée et le relarguer dans le nouveau milieu réactionnel pour effectuer une autre réaction catalytique.30 Cette méthode de recyclage des
catalyseurs a connu quelques exemples dans la littérature dont certains sont présentés ci-dessous.
1.4.2 Greffage en fin de réaction
Ce concept boomerang a été décrit par le Pr. A.G.M. Barrett dans la réaction de la métathèse d’oléfine catalysée par un complexe de ruthénium en 1999.31 Le principe est présenté sur le
schéma I.1. En utilisant le polystyrène comportant des sites vinyl 1 comme support, des catalyseurs de Grubbs de première génération ont pu être immobilisés via l’alkylidène pour conduire au catalyseur supporté 2. Pendant la réaction, le complexe de ruthénium se décroche du support, via une réaction de métathèse d’oléfine, effectue la réaction catalysée conduisant au produit de métathèse et au complexe intermédiaire 3 qui, lorsqu’il n’y a plus d’oléfine terminale en solution homogène, réagit avec le support pour régénérer 2. A la fin de la réaction, le produit est récupéré en solution dans le filtrat et le complexe de ruthénium sur le support. En utilisant de cette méthode, le catalyseur a été recyclé 3 fois.
Ru PCy3 PCy3 Cl Cl Ru PCy3 PCy3 Cl Cl 1 2 catalyseur supporté 3 en l'absence d'oléfine terminale Effet boomerang induit par le substrat
Schéma I.1. Premier exemple de catalyse boomerang.
Cette approche a depuis été revisitée en introduisant le concept de polymère métallique à bloc pour immobiliser des complexes de type Grubbs-Hoveyda (catalyseur auto-supporté, Schéma I.2).32 Pendant la réaction de métathèse d’oléfine cyclisante, le catalyseur supporté est
partiellement ou entièrement solubilisé et en fin de réaction il est récupéré par précipitation après reformation du polymère métallique par métathèse et expulsion d’éthylène. Ce type de système peut cependant présenter des variations de réactivités liées aux cinétiques de dissociation/association de la matrice oligomérique.
Chapitre I. Elaboration de systèmes recyclables 13 O O O O O O N N N N Ru Cl Cl Ph Cy3P Ru Ph PCy3 Cl Cl état de repos état actif N Ts N Ts N Ts en présence de sans diène
Schéma I.2. Catalyseur auto-supporté pour la métathèse d’oléfines cyclisante.
Une approche similaire a pu également être appliquée en catalyse asymétrique (Schéma I.3).33
Dans ce cas, des complexes de cuivre chiraux auto-supportés à base de ligands de type DiAzabisoXazoline (DAX) ont été employés dans une réaction de cyclopropanation énantiosélective.34 Dans ce cas, la force motrice favorisant le passage du complexe métallique
en solution homogène est l’interaction carbène-cuivre, plus important que l’interaction bisoxazoline-cuivre
Les deux catalyseurs du cuivre (tBuDAX-Cu(OTf)
2 et tBuDAX-Cu(OTf)) ont été synthétisés
sous la forme de polymères insolubles dans le dichlorométhane (schéma I.3). L’addition d’un dérivé diazoïque rend ces deux complexes solubles et en présence d’une oléfine telle que le styrène la réaction de cyclopropanation se déroule. En fin de réaction, un précipite observé qui correspond au retour à la forme polymérique des deux catalyseurs après consommation de tout le composé diazoïque. Les catalyseurs du cuivre se sont avérés stables et efficaces en catalyse avec seulement une érosion mineure de l’énantiosélectivité observée après 14 cycles.
Chapitre I. Elaboration de systèmes recyclables 14 CO2Et CO2Et N N N O O N O N N O tBu tBu tBu tBu Cu(OTf)n état de repos ... ... CO2Et N2 Ph Ph CO2Et Ph CO2Et Ph état actif après consommation du diazo
Schéma I.3. Cyclopropanation énantiosélective par un catalyseur auto-supporté.
1.4.3 Greffage réversible
Une autre approche de catalyseur boomerang a été développée par Wang et coll. Dans cet exemple, le complexe du ruthénium est lié à un pyrène qui est réversiblement immobilisé sur le support de type CNT via un stacking de type π-π. Une modification des conditions expérimentales permet le contrôle de l’interaction π-π principalement en fonction de la température (Schéma I.4).35
Chapitre I. Elaboration de systèmes recyclables 15 O O O Ru Cl Cl N N Mes Mes acétone 0°C O O O Ru Cl Cl N N Mes Mes acétone 35°C (decrochage) (acrochage) catalyseur RCM
Schéma I.4. Greffage réversible du complexe du ruthénium sur les CNTs proposé par Wang et al.
Le catalyseur de métathèse d’oléfines cyclisante, une fois encore un complexe de ruthénium Grubbs-Hoveyda, a été greffé sur des nanotubes de carbone mono-paroi (SWCNTs) par l’intermédiaire d’un noyau pyrène pour permettre la récupération du catalyseur après la réaction catalytique. Suite à une optimisation du solvant et de la température, la catalyse a été effectuée à 35°C dans l’acétone. A cette température, le complexe de ruthénium est en effet décroché de la surface du support et donc en solution homogène. Puis, à la fin de la catalyse, la ré-immobilisation du catalyseur a été effectuée à basse température (0°C). Les produits et le catalyseur se séparent par simple filtration ou centrifugation. Cette méthode accrochage-décrochage permet utiliser le même catalyseur dans 7 cycles. Anecdotiquement, les SWCNTs peuvent être rechargés avec le nouveau catalyseur de ruthénium lors d’une érosion de l’activité de celui-ci.
Chapitre I. Elaboration de systèmes recyclables
16
1.4.4 Principe du projet Switchcat
Nous avons montré ci-dessus quelques exemples de recyclage de catalyseurs qui sont re-immobilisés juste à la fin des réactions catalytiques, tout en conservant l’efficacité et la sélectivité de la catalyse homogène. Le projet Switchcat s’inscrit dans ce cadre mais vise à introduire un nouveau modèle de liaison réversible du catalyseur avec le support via une agrafe contrôlable (figure I.1).
Figure I.1 Greffage du catalyseur sur un support via une agrafe contrôlable.
Pour cela, nous pouvons définir deux états : un état « on » correspondant au complexe attaché au support via l’agrafe et un état « off» dans lequel le catalyseur n’est plus attaché au support et se trouve en solution homogène. Dans une première approche, nous sommes intéressés aux CNTs comme support pour leurs propriétés mécaniques et électroniques. L’agrafe envisagée peut avoir deux états stables qui se transforment l’un en l’autre sous un stimulus externe, photochimique ou électrochimique. Les catalyseurs envisagés sont des complexes portant des ligands NHC fonctionnalisés permettant l’emploi d’une large gamme de métaux en applications catalytiques, et présentant de très bonnes stabilités.
Une rapide analyse de la littérature permet d’identifier que les complexes de ruthénium et de cuivre comportant un fragment polypyridyl peuvent changer de sphère de coordination et/ou de géométrie sous stimulus externe (photochimique, électrochimique), conduisant ainsi à une deuxième structure de complexe. Une telle propriété permet de définir les deux états recherchés (« on » et « off »). De plus ces complexes peuvent revenir vers leur structure initiale sous l’effet d’un stimulus externe inverse ; la communication entre les deux formes est donc contrôlable et réversible. Afin de réaliser le projet Switchcat, nous nous sommes donc proposé d’utiliser le même type de complexes afin de pouvoir, par une modification de la sphère de coordination sous stimulus photochimique (pour les complexes polypyridyl du ruthénium) ou électrochimique (pour les complexes polypyridyl du cuivre), décrocher et accrocher le catalyseur au support via un linker adapté. Nous allons dans la suite de ce chapitre effectuer un rappel des données importantes relatives à ces complexes et à leurs applications, en particulier dans le domaine des machines moléculaires.
Chapitre I. Elaboration de systèmes recyclables
17
2.
Complexes commutables photochimiquement
L’étude photochimique des complexes de ruthénium (II) polypyridyl a été initialement décrite il y a 60 ans.36 Les complexe du ruthénium polypyridyl présentent de nombreux avantages :
(i) leur synthèse est relativement simple ; (ii) ils sont photochimiquement réversibles ; (iii) ils absorbent dans la région visible ; (iv) ils présentent des états excités à longues durées de vie ; (v) ils présentent une luminescence intense. Des complexes de cuivre avec le même type de ligands ont également été utilisés en photochimie supramoléculaire.37,38
2.1. Principe du switch photochimique sur les complexes du ruthénium (II) polypyridine
Les complexes de ruthénium (II) polypyridine ont montré des propriétés photophysiques et photochimiques intéressantes, qui ont conduit à leur utilisation dans de nombreuses applications médicales et technologiques.39,36
Le RuII polypyridine est un système octaédrique d6 dans lequel les ligands polypyridines ont
des orbitales donneuses de symétrie σ qui sont localisées sur les atomes d’azotes, et des orbitales donneuses et acceptrices, de symétrie π, qui sont délocalisées sous les noyaux aromatiques des pyridines. Les positions des orbitales dans le complexe du ruthénium (II) polypyridyl et les transitions électroniques entre ces orbitales sont présentées sur la figure I.2.40 L t2g(d 1, d 2, d 3) L1* L2* (d *,d *) e2g 1 2 3 4
Figure I.2 Représentation des orbitales et des différentes transitions électroniques dans un complexe RuII polypyridyl.
La géométrie octaédrique et le caractère fort du champ électrostatique engendré par le ligand polypyridine implique selon le modèle du champ cristallin que le centre métallique possède 6
Chapitre I. Elaboration de systèmes recyclables
18
électrons sur le groupe d’orbitales t2g dans l’état fondamental du complexe de ruthénium
polypyridyl. Les orbitales moléculaires du ligand sont des combinaisons linéaires d’orbitales 2s et 2p, elles sont donc plus basses en énergie que les orbitales du métal. Cette représentation orbitalaire est générale pour les complexes polypyridines dans la mesure où l’on se trouve avec ces ligands dans une géométrie octaédrique et en présence d’un champ fort, ce qui induit la levée de dégénérescence indiquée (t2g<e2g) et une valeur de ∆o supérieure à l’énergie
d’appariement qui conduit à la distribution électronique indiquée. L’exemple détaillé correspond classiquement au cas du [Ru(bipy)3].
Sous irradiation par la lumière, les électrons du métal peuvent effectuer des transitions entre les différents niveaux orbitalaires. Comme indiqué sur la figure I.2 il y a trois types de transitions électroniques possibles dans le complexe :
1) La transition (t2g-πL*) qui correspond à un transfert de charge du métal vers le ligand
(MLCT) ; dans ce cas le centre métallique est formellement oxydé et le ligand est réduit. Dans le spectre UV-visible, la bande d’absorption MLCT intense est caractéristique pour les complexes du ruthénium(II) polypyridyl. La bande d’absorption dans la région visible correspond à l’état singulet (1MLCT) et l'émission
se produit à partir de l’état triplet 3MLCT.
2) La transition (πL-π*) correspond aux transitions des ligands polypyridines et se trouve
dans la zone visible du spectre d’absorption du complexe dans la solution.
3) La transition entre des orbitales d du métal t2g-eg (dπ-dσ*) ou 3MC est interdite pour
les complexes du ruthénium (II) et est par conséquent généralement absente dans le spectre d’absorption du complexe du ruthénium(II) polypyridyl.
Après excitation des électrons du complexe, la relaxation vers l’état fondamental peut se faire soit par émission de lumière (désactivation radiative, observation d’une luminescence), soit par la désactivation non radiative conduisant globalement à la substitution photo-induite (ou photosubstitution) d’un des ligands du complexe de ruthénium (II) polypyridyl. Le mécanisme général de ces deux phénomènes est proposé dans la littérature et les explications seront présentées ci-dessous.39,41
Chapitre I. Elaboration de systèmes recyclables
19
Dans un état fondamental « ground state » (GS) le centre métallique possède 6 électrons sur les orbitales t2g (Figure I.3 i(a) et ii, iii (A)). L’énergie de chaque état du complexe et la
configuration des électrons qui correspond à ces états sont présentées sur la figure I.3 (i et ii,
iii). Sous l’effet de l’irradiation, un des six électrons des orbitales centrées sur le métal
acquiert une énergie suffisante pour effectuer la transition sur l’orbitale πL* centrée sur le
ligand qui correspond à l’état de transfert de charge du métal vers le ligand 1MLCT. (Figure
I.3 i(b) et ii, iii(B)) Pour la plupart des complexes du RuII polypyridyl, le premier état peuplé
est singulet. L’état triplet ne peut pas être peuplé directement (transition interdite) après absorption de photon mais peut être obtenu après désactivation de l’état excité supérieur.39 Il
s’agit du croisement inter-système, au cours duquel le système passe d’un état 1MLCT (Figure
I.3 i (b) ii, iii B) à 3MLCT (Figure I.3 i (c) et ii, iii (C)). En passant de l’état d’oxydation
formel du Ru(II) à Ru(III) après promotion, la rétrodonation des orbitales (t2g) du métal vers
les orbitales libres πL* des ligands diminue (t2g-πL*) du fait de la diminution de la densité
électronique lors du passage du Ru(II) au Ru(III). Le retour de l’état excité 3MLCT vers l’état
fondamental (GS) du système peut se faire selon deux chemins, (i) Désactivation radiative (luminescence) si l’énergie de l’état excité centré sur le métal (3MC) est significativement
plus haute que l’état excité du 3MLCT (figure 1.3 (ii)) ; (ii) Désactivation non radiative, si la
différence entre les deux états excités (3MLCT et 3MC) est faible ; dans ce cas l’électron peut
passer de l’orbitale πL*du ligand à une orbitale eg du métal. (Figure 1.3 i (d)). Dans cet état
excité 3MC (eg=dσ* figure I.2), l’augmentation de la densité électronique induit une
augmentation de la répulsion électronique le long de l’axe de la liaison σ métal-ligand ce qui provoque l’expulsion d’un des ligands du complexe du ruthénium polypyridyl (Figure I.3 iii). Le complexe du ruthénium (II), alors insaturé, réagit rapidement avec une molécule de solvant pour compléter sa sphère de coordination.42
Chapitre I. Elaboration de systèmes recyclables
20
Figure I.3 (ii) Energies des différents états de [Ru(bipy)3]2+, adapté des références 41,43 Globalement, l’ensemble de ce processus permet à ces complexes de ruthénium (II) de substituer photosélectivement un ligand donné lors de l'irradiation par de la lumière visible, d’où la notion de labilité photochimique. La différence énergétique entre les deux états excités
3MLCT et 3MC dans les complexes du ruthénium polypyridyl peut être contrôlée par les
propriétés électroniques et stériques des ligands chélatants, ce qui permet d’augmenter l’efficacité de la substitution photochimique. 44,45,46,47
Dans la littérature, les complexes de ruthénium (II) polypyridyl sont classifiés en quatre catégories principales en fonction de leurs ligands : (i) les complexes de type [Ru(bipy)2(L)2]2+, (ii) les complexes de type [Ru(terpy)2]2+, (iii) les complexes du type
[Ru(phen)2(L) ]2+ et [Ru(diimine)3]2+ et (iv) les complexes du type [Ru(terpy)(N-N)(L)]2. Les
structures des ligands sont présentées sur la figure I.4 ci-dessous.
N N N ligand tridente N N N N ligand bidente L 2,2'-bipyridine terpyridine 1,10-phénantroline N CN -CH3CN -H2O -Cl pyridine benzonitrile ligand monodente
Figure I.4 Structures des ligands tridentes, bidentes et monodentes.
Le cas des complexes qui possèdent plusieurs ligands monodentes est géométriquement plus compliqué. Cependant les complexes [Ru(bipy)2(L)2]n+ peuvent photoexpulser les ligands
Chapitre I. Elaboration de systèmes recyclables
21
le complexe [Ru(terpy)2]2+ non réactif vis à vis de la photoexpulsion.51 Les deux dernières
familles (iii) [Ru(phen)2(L) ]2+ où L est un ligand bidente et (iv) [Ru(terpy)(N-N)(L)]2+ (où
N-N est un ligand bidente comme la 1,10-phénantroline ou la 2,2’-bipyridine et L est un ligand monodente) sont plus récentes et ont des propriétés intéressantes. Sous irradiation, les complexes de type [Ru(phen)2(L)]2+ expulsent le ligand le plus encombré ou le plus faible
σ-donneur,52 le retour vers le complexe initial se faisant par simple chauffage en présence du
ligand expulsé.53,54 Ce type de complexe a été utilisé comme cœur dans la construction de
machines moléculaires commutables photochimiquement. Les complexes de type [Ru(terpy)(N-N)(L)]2+ présentent la même propriété photochimique consistant à expulser le
ligand monodente (L) sous irradiation en complétant sa sphère de coordination par une molécule de solvant coordonnée.55
Nous nous sommes plus particulièrement intéressés à ce type de complexes photoréactifs [Ru(terpy)(N-N)(L)2+] pour concevoir l’agrafe photochimiquement contrôlable liant le
support et le catalyseur. Quelques exemples de ce type de complexes et leurs applications dans la construction de machines moléculaire sont présentés ci-dessous.56
2.2 Machines moléculaires commutables photochimiquement à base de complexes du ruthénium.
2.2.1 Différents types de complexes [Ru(terpy)(N-N)(LLLL)]2+
Des complexes de type [Ru(terpy)(N-N)(L)]2+ avec différents ligands L (chlore, acétonitrile,
dérivés du benzonitrile, pyridine ou ligand soufré et eau) sont décrits dans la littérature.57,58,59,60 Il a ainsi été montré que l’efficacité de la photo-expulsion du ligand
monodente L du complexe [Ru(terpy)(phen)(L)]2+ dépendait fortement des facteurs stériques du ligand L. Collin et al. ont étudié la photosubstitution du 2,6-diméthoxybenzonitrile (MeOBN) par la pyridine pour des complexes de type [Ru(terpy’)(dmp)(L)]2+ (le ligand
terpy’ étant la 4’-(3,5-ditertiobutylphenyl)-2,2’ :6’,2’’-terpyridine et le ligand dmp étant la 2,9-diméthyl-1,10-phénantroline) et de type [Ru(terpy’)(phen)(L)]2+ (dans lesquels phen est la phénantroline) (schéma I.5). Ces travaux ont montré que pour le complexe du ruthénium (II) avec le ligand dmp encombré, le rendement quantique d’expulsion est 20 fois supérieur à celui du complexe avec la phénantroline non encombrée.61 Ceci s’explique simplement par les
Chapitre I. Elaboration de systèmes recyclables
22
contraintes stériques imposées par la substitution nettement plus importantes dans le cas du ligand dmp (Schéma I.5).
N N N Ru N N N MeO OMe 2+ N N N Ru N N N 2+ hν in C5H5N Φ = 0,0035 N N N Ru N N N MeO OMe 2+ N N N Ru N N N 2+ hν in C5H5N Φ = 0,079
Schéma I.5 Augmentation du rendement quantique de photosubstitution du complexe de ruthénium par encombrement de la sphère de coordination.
2.2.2 Exemples de machines moléculaires basées sur un cœur du type [Ru(terpy)(N-N)(LLLL)]2+
Une application de ce type de réactivité a été la construction d’un prototype de machine moléculaire basé sur un complexe scorpionate de type [Ru(terpy)(phen)(L)]2+.62 Dans ce
complexe, la terpyridine est liée de façon covalente avec le ligand benzonitrile via une chaine flexible de type polyéther. L’irradiation par de la lumière visible du complexe dans un mélange acétone-eau promeut la photoexpulsion du fragment benzonitrile et son remplacement par une molécule d’eau (Schéma I.6). La recoordination du fragment benzonitrile se produit à T.A pendant une journée où en chauffant pendant 2 heures dans l’acétone.
Chapitre I. Elaboration de systèmes recyclables 23 N N N Ru N N 2+ N O O O hν, H2O ∆, -H2O N N N Ru N N 2+ OH2 O O O N
Schéma I.6 Photolabilisation et recoordination thermique dans un complexe scorpionate.
La molécule scorpionate basée sur un complexe de type [Ru(terpy)(phen)(L)]2+ est une
première molécule photoréactive qui a montré un mouvement à grande échelle au niveau moléculaire.
D’autres exemples de machines moléculaires basées sur des complexes de type [Ru(terpy)(phen)(L)]2+ ont été développés par ce même groupe mais ne seront pas présentés ici.63
2.3. Application à notre projet
L’objectif de notre projet est le piégeage réversible d’un catalyseur métallique sur un support pour faciliter sa séparation des produits de réaction (comme un catalyseur hétérogène) tout en conservant les propriétés d’activité et de modulation de sélectivité classiquement associées aux catalyseurs homogènes. Nous cherchons donc à développer une méthode d’hétérogénéisation du catalyseur après un processus de catalyse homogène, en exploitant la mise en place d’une agrafe dont l’état « ouvert » ou « on » et « fermé » ou « off » puisse être contrôlé (nécessité de réversibilité). Il apparait que les complexes du ruthénium (II), sensibles à l’irradiation de la lumière et changeant de sphère coordination en expulsant le ligand, représentent des modèles d’agrafes potentiellement exploitables pour la réalisation de notre projet. Un complexe de type [Ru(terpy)(bipy)(L)]2+ pourra représenter l’agrafe
photochimiquement commutable nous permettant de lier réversiblement le catalyseur au support. Cette approche nécessite la fonctionnalisation d’un ligand du centre catalytique par un groupement nitrile qui permettrait d’attacher le catalyseur via l’agrafe sur son support en
Chapitre I. Elaboration de systèmes recyclables
24
chauffant après la catalyse et de le détacher sous irradiation dans la solution, pour effectuer la réaction catalytique. (Schéma I.7)
Schéma I.7 Présentation du système envisagé pour le switch photochimique.
Une autre approche peut être envisagée. Elle consiste à développer un système catalytique contrôlable via un stimulus électrochimique. Comme nous allons le décrire à la suite, les complexes de cuivre sont particulièrement adaptés à cette approche.
Chapitre I. Elaboration de systèmes recyclables
25
3.
Complexes commutables électrochimiquement
Comme nous l’avons vu précédemment, la commutation visée dans notre projet est basée sur une modification de la sphère de coordination du métal.
Une modification de l’environnement du métal peut être induite par un changement du degré d’oxydation du métal. Nous allons plus particulièrement nous intéresser au cas du cuivre, pour lequel le changement de degré d’oxydation peut induire un changement du nombre de coordination du métal.
3.1 Principe du switch électrochimique sur les complexes du cuivre (I) polypyridines
Le cuivre est stable en solution aux états d’oxydation (I) et (II). Au degré d’oxydation (I), le complexe du cuivre possède 10 électrons 3d et au degré d’oxydation (II) il possède 9 électrons 3d. Cela influe bien évidemment de manière très forte sur la géométrie et la coordination du centre métallique, le nombre de coordination d’ion du métal dépendant de sa configuration électronique.
Au degré d’oxydation (I) le cuivre est d10, et l’on peut dès lors le considérer, pour l’étude de
sa géométrie, comme un élément du groupe principal puisque seules les orbitales s et p sont impliquées dans des liaisons avec des « substituants donneurs » (Ligands L ou X). En effet, il n’y a pas de stabilisation à attendre d’une levée de dégénérescence des orbitales d puisque la sous couche est pleine (Σ∆E = 0). Sa géométrie peut donc être décrite en s’appuyant sur le modèle VSEPR (Valence Shell Electron Pairs Repulsion) : le cuivre (I) d10sera favorablement
entouré de quatre ligands et présentera une géométrie tétraédrique.
Le Cu(II) moins riche en électrons cherche par conséquent à s’entourer avec plus de ligands ou avec des ligands plus riches en électrons par rapport au cuivre (I). La sous-couche d n’étant pas complète, le Cu(II) entre dans la « catégorie des métaux de transition ». L’étude de sa géométrie fait appel, au modèle du champ cristallin. Si ce modèle est quantitativement mauvais, il reste extrêmement important dans la mesure où il prend en compte toutes les propriétés de symétrie du système, permettant ainsi d’obtenir le nombre et la dégénérescence des niveaux électroniques du complexe considéré et donc sa géométrie. Selon ce modèle, l’approche des ligands vers le métal central induit un champ électrostatique de symétrie non
Chapitre I. Elaboration de systèmes recyclables
26
sphérique engendrant l’ajout d’un potentiel dans l’hamiltonien dont découle une dégénérescence des orbitales du métal.
En symétrie octaédrique, les 5 orbitales d se subdivisent en deux blocs, dénommés selon les éléments de symétrie qui les caractérisent : (i) le bloc t2g qui contient les orbitales dxy, dxz, dyz ;
(ii) le boc eg comportant les orbitales dx2-y2 et dz2 (Figure I.5 a). L’ampleur du dédoublement
des orbitales eg et t2g dans un complexe octaédrique est notée ∆o. Pour une géométrie
tétraédrique, les orbitales triplements dégenérées (t2) sont énergétiquement plus hautes que les
orbitales doublements dégénérés (e) (Figure I.5 b).
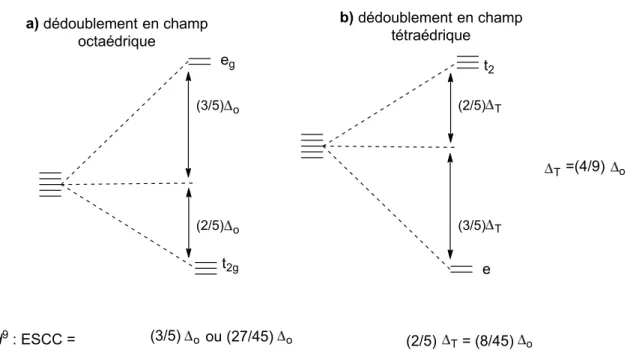
eg t2g (3/5) o (2/5) o (2/5) (3/5) t2 e T T a) dédoublement en champ octaédrique b) dédoublement en champ tétraédrique T=(4/9) o d9: ESCC = (3/5) o ou (27/45) o (2/5) T=(8/45) o
Figure I.5 Dédoublement a) octaédrique et b) tétraédrique dans le modèle du champ cristallin.
Le calcul de l’énergie électronique (ou plus précisément de l’énergie de stabilisation du champ cristallin, ESCC) correspondant à chaque géométrie pour un complexe d9 indique que
les complexes du cuivre (II) adoptent préférentiellement une géométrie dérivant d’un champ octaédrique (avec divers degrés de déformation tétragonale), et peuvent être pentacoordonné ou hexacoordonné.
Ces changements de géométrie et de nombre de coordination entre le cuivre(I) et le cuivre(II) peuvent être exploités pour induire sur un même composé un mouvement qui peut conduire à la libération d’un ligand. Ainsi, sous l’effet d’un stimulus électrochimique, un complexe de Cu(I)N4 tétracoordonné va s’oxyder en Cu(II)N4 instable qui cherchera à compléter sa couche
Chapitre I. Elaboration de systèmes recyclables
27
Cu(II)N6. La réduction du Cu(II) en Cu(I) engendrera l’effet inverse. Ce phénomène a été
exploité pour la construction de différentes machines moléculaires contrôlées électrochimiquement qui sont construites autour d’un centre cuivre pour les propriétés exposées ci-dessus.
3.2 Machines moléculaires commutables électrochimiquement à base de complexes du cuivre.
La plupart des machines moléculaires utilisent des structures caténanes (entrelacement de deux anneaux) et rotaxanes (anneau entourant un « fil »). Ces structures possèdent des contraintes stériques particulières qui maintiennent « l’unité » de l’édifice supramoléculaire tout en autorisant le déplacement d’un composant par rapport à un autre avec une amplitude significative. Les différentes machines moléculaires commutables électrochimiquement construites avec un cœur Cu-polypyridine seront succinctement présentées ci-dessous.
3.2.1. Le complexe du cuivre caténane avec deux géométries distinctes
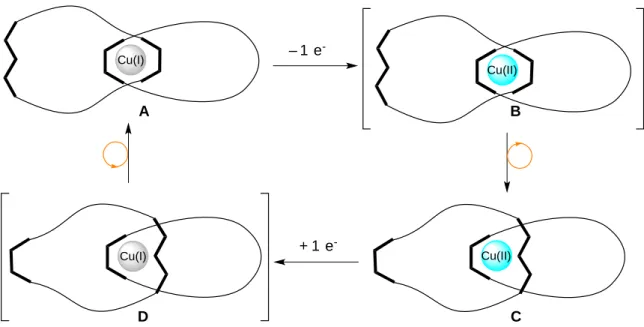
L’exemple du moteur moléculaire à base du caténane, dans lequel les mouvements sont contrôlés par oxydation ou réduction de l’ion central de cuivre, a été développé dans le groupe du Prof. Sauvage en 1994.64 Le caténane est composé de deux anneaux différents, entrelacés
et comportant entre eux un atome du cuivre ; ce dernier présente deux possibilités de coordination selon son degré d’oxydation. Le complexe du cuivre (I) préfère être entouré de quatre ligands (NC=4) alors que le complexe du cuivre (II) préfère avoir cinq ou six ligands (NC=5 ou 6). Sous stimulus électrochimique, le mouvement d’un anneau coulissant dans l’autre génère deux formes différentes qui sont commutables. Le principe du mouvement de ce type machine est présenté ci-dessous (Schéma I.8)
Chapitre I. Elaboration de systèmes recyclables 28 Cu(II) Cu(I) – 1 e -Cu(II) + 1 e -Cu(I) A B C D
Schéma I.8. Principe du mouvement moléculaire dans le complexe de cuivre caténane.
(Le symbole en U correspond au ligand bidente de type phénantroline et celui en W au ligand tridente de type terpyridine).
Un anneau du caténane contient un seul ligand bidente du type 2,9-diphényl-1,10-phénantroline (dpp) et est entrelacé avec le deuxième anneau contenant deux chélates différents : le ligand dpp et un ligand tridente de type terpyridine. Dans des conditions oxydantes, le complexe de cuivre (I) tétracoordonné A (deux ligands dpp), forme le cuivre (II) tétracoordonné B (espèce intermédiaire) qui se réarrange en cuivre (II) pentacoordonné C. A l’inverse, dans des conditions réductrices le cuivre (II) pentacoordonné C est transformé en cuivre (I) pentacoordonné D (espèce intermédiaire) qui se réarrange pour conduire à la forme favorisée A comportant quatre ligands. Les structures des complexes de cuivre caténanes A et
C sont représentés ci-dessous (Figure I.6).
O N O O O N N N N N O O N O O O Cu + O N O O O N N N N N O O N O O O 2+ Cu A C
Chapitre I. Elaboration de systèmes recyclables
29
3.2.2. « Navette moléculaire » : rotaxane à deux stations
Dans les caténanes, les mouvements de glissement d'un anneau par rapport à l'autre sont provoqués par la mise en mouvement soit d’un soit des deux anneaux. Dans le rotaxane présenté ici, l’anneau se déplace sous stimulus électrochimique le long de l’axe sur lequel il est enfilé (Schéma I.9).65
O O O O N N O N O O O N O O O O N N O N N N O Cu + O O O N N O N O 2+ N N O Cu – 1 e- + 1 e
-Schéma I.9 Navette moléculaire à base de cuivre.
La navette moléculaire est composée de deux parties : (i) un macrocycle possédant un ligand bidente de type phénantroline (dpp) et (ii) un composé formant l’axe avec deux chélates, un ligand bidente de phénantroline substitué et un ligand tridente de type terpyridine. Les deux chélates sont liés entre eux par une chaine alkyle et à chaque extrémité se trouve un groupement très encombrant qui joue un rôle de bouchon visant à éviter le désenfilage.
Chapitre I. Elaboration de systèmes recyclables
30
Le système fonctionne selon le même principe que les caténanes précédemment décrits. Le complexe de cuivre (I) se trouve dans un état tétracoordonné et le complexe du cuivre (II) est pentacoordonné. Le changement du degré d’oxydation induit le changement du nombre de coordination du cuivre et provoque le glissement du macrocycle relativement à l’axe comme schématisé sur le schéma I.10.
Cu(I) – 1 e Cu(II)
-Cu(II)
+ 1 e -Cu(I)
Schéma I.10 Principe du mouvement d’une navette moléculaire à base de cuivre
Ainsi au degré d’oxydation (II) du cuivre (donc juste après l’oxydation du Cu(I) tétracoordonné), la transition entre la forme tétracoordonnée moins stable et la forme
pentacoordonnée présente une constante de vitesse alors qu’au
degré d’oxydation (I) du cuivre (i.e. juste après la réduction du complexe pentacoordonné de Cu(II)), la transition entre la forme pentacoordonnée et la forme tétracoordonnée (plus stable) présente une constante de
Ces différences de constante de vitesse correspondent à un réarrangement du complexe Cu(I) pentacordonné en Cu(I) tétracoordonné plus rapide (environ 100s) que celui du complexe Cu(II) tétracoordonné en Cu(II) pentacoordonné (plus de 2 heures).
3.2.3. Les machines moléculaires à base de rotaxane : vers des mouvements rapides.
La vitesse du mouvement des machines moléculaires dépend de la nature des mouvements et des modes d’activation. Elle peut varier de quelques microsecondes pour les rotaxanes organiques commutables photochimiquement,66 à quelques secondes ou quelques minutes
Chapitre I. Elaboration de systèmes recyclables
31
pour les réactions de type « enfilage-défilage » et les processus d'oxydoréduction du centre métallique.67,68,64 Le saut du métal entre deux places définies est relativement lent quand il
s'agit de l'échange de ligand qui suit un processus d'oxydoréduction. Les machines moléculaires basées sur le Cu(II)/Cu(I) telle que celle présentée ci-dessus répondent lentement au stimulus électrique. Une façon d’accélérer leur mouvement est de changer la nature de celui-ci : en effet, en passant d’un mouvement de translation à un mouvement de rotation qui présente moins de restriction de mouvements, il est possible d’avoir un mouvement plus rapide et de déterminer les autres facteurs influençant la cinétique du mouvement. Ci-dessous sont présentés trois exemples de machines basées sur des rotaxanes rotatifs dans lesquels l’anneau peut tourner (pirouetter) entre deux positions autour de son axe. Nous illustrerons l’influence de la structure moléculaire sur la vitesse d’établissement de l’équilibre dans ce modèle (Schéma I.11). Ce facteur est effectivement important pour la récupération de catalyseurs rapide et efficace dans les systèmes que nous souhaitons développer.
Cu(I) – 1 e
-Cu(I)
Cu(II)
Cu(II)
+ 1 e
-Schéma I.11 Modèle général des rotaxanes rotatifs.
Le premier rotaxane (Figure I.7) de ce type contrôlé électrochimiquement a été développé par Raehm et al. en 1999.69 La force motrice de ce mouvement est basée sur les différences de
géométrie préférentielle des Cu (I) et Cu (II). L’anneau du rotaxane comporte deux chélates : un ligand bidente de type phénantroline et un ligand tridente de type terpyridine. L’axe possède exclusivement un ligand bidente, dpp que nous avons vu précédemment, et les
Chapitre I. Elaboration de systèmes recyclables
32
terminaisons de l’axe sont constituées des mêmes deux groupes encombrants sur ses extrémités (Schéma I.11).
N O N N O O O O O O N N N O N Cu 2+ (I.1)
Figure I.7 Structure du rotaxane rotatif à motif phénanthroline dans l’état Cu(II).
Dans ce premier cas, les valeurs cinétiques de mouvement dépendent fortement du sens dans lequel est effectuée la réaction redox. Ainsi au degré d’oxydation (II) du cuivre (donc juste après l’oxydation du Cu(I) tétracoordonné), la transition entre la forme tétracoordonnée moins stable et la forme pentacoordonnée présente une constante de vitesse
alors qu’au degré d’oxydation (I) du cuivre (i.e. juste après la réduction du complexe pentacoordonné de Cu(II)), la transition entre la forme pentacoordonnée et la forme
tétracoordonnée (plus stable) présente une constante de vitesse .
Ces différences de constante de vitesse correspondent à un réarrangement du complexe Cu(I) pentacordonné en Cu(I) tétracoordonné plus rapide (de l’ordre de d’une centaine de ms) que celui du complexe Cu(II) tétracoordonné en Cu(II) pentacoordonné (plus de 4 min).
Même si le mouvement de pirouette de l’anneau autour de l’axe est beaucoup plus rapide que la translation, la vitesse de cette machine reste faible pour une utilisation pratique telle que la construction des commutateurs. Afin de moduler cette cinétique, deux autres rotaxanes rotatifs ont été développés.70,71 La stratégie utilisée est basée sur la diminution de l’encombrement stérique autour du métal afin de faciliter les échanges de ligands. Afin de
Chapitre I. Elaboration de systèmes recyclables
33
pouvoir accéder à une géométrie plus linéaire (limitant les contraintes stériques à la rotation) et afin de limiter l’encombrement autour du métal, le motif phénantroline composant la pièce centrale du fil dans I.1 a été remplacé par un motif bipyridine. Ce ligand est en effet plus flexible que la phénantroline, du fait de la libre rotation entre les deux noyaux pyridines. Il a été supposé que cette flexibilité lui permettait de tourner plus facilement à l'intérieur de la cavité de l’anneau. Mais il est fort probable que la substitution en β plus qu’en α des azotes, conférant le caractère linéaire au fil, soit un paramètre plus important dans la mesure où il engendre moins de gêne autour du métal, comme cela apparaît clairement sur la figure I.8.
N O N N O N N N O N Cu O (I.2) 2+
Figure I.8 Structure d’un premier rotaxane rotatif à motif bipyridine dans l’état Cu(II).
Dans le cas de (I.2), les transitions entre les formes tétracoordonnées et les formes pentacoordonnées ont été significativement accélérées par rapport à (I.1). Le changement de degré d’oxydation induit le changement de nombre coordination du cuivre et provoque la rotation de l’anneau autour de l’axe comme présenté sur le schéma-carré I.12 ci-dessous.
CuIN4 CuIIN4 CuIIN5 CuIN5 e e e e -1 +1 +1 -1 1 E, 2C 3E, 4C