N d’ordre:
UNIVERSITE TOULOUSE III – PAUL SABATIER Ecole Doctorale : Sciences de la matière
THESE
En cotutelle avecL’UNIVERSITE MOULOUD MAMMERI DE TIZI-OUZOU
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITE TOULOUSE III Spécialité : Physique des matériaux
Présentée par
E
l Hocine MEGCHICHE
Etude théorique de la migration de lacunes
et de l’oxygène interstitiel dans le nickel
solide soumis à un processus d’oxydation
Soutenue le Vendredi 16 Novembre 2007 devant le jury
C. Mijoule Professeur, ENSIACET, Toulouse Président
J.-C. Barthelat Professeur, UPS, Toulouse Directeur
M. Amarouche Professeur, UMMTO, Tizi-Ouzou Directeur
P. Sautet Directeur de recherche, ENS, Lyon Rapporteur
M. Zemirli Professeur, UMMTO, Tizi-Ouzou Rapporteur
A. Ziane Maître de conférences, UMMTO, Tizi-Ouzou Examinateur
UNIVERSITE TOULOUSE III – PAUL SABATIER Ecole Doctorale : Sciences de la matière
THESE
En cotutelle avecL’UNIVERSITE MOULOUD MAMMERI DE TIZI-OUZOU
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITE TOULOUSE III Spécialité : Physique des matériaux
Présentée par
E
l Hocine MEGCHICHE
Etude théorique de la migration de lacunes
et de l’oxygène interstitiel dans le nickel
solide soumis à un processus d’oxydation
Soutenue le Vendredi 16 Novembre 2007 devant le jury
C. Mijoule Professeur, ENSIACET, Toulouse Président
J.-C. Barthelat Professeur, UPS, Toulouse Directeur
M. Amarouche Professeur, UMMTO, Tizi-Ouzou Directeur
P. Sautet Directeur de recherche, ENS, Lyon Rapporteur
M. Zemirli Professeur, UMMTO, Tizi-Ouzou Rapporteur
A. Ziane Maître de conférences, UMMTO, Tizi-Ouzou Examinateur
Remerciements
Cette thèse est réalisée dans le cadre d’une coopération entre l’Université Paul Sabatier de Toulouse (France) et l’Université Mouloud Mammeri de Tizi-Ouzou (Algérie). Elle a été rendue possible grâce à une collaboration scientifique entre trois laboratoires de recherche : le laboratoire de Chimie et Physique Quantique (LCPQ) de l’IRSAMC, UMR 5626, UPS Toulouse (Professeur Jean Claude Barthelat), le laboratoire de Physique et Chimie Quantique (LPCQ), UMMTO Tizi-Ouzou (Professeur Mohand Amarouche) et le CIRIMAT, INP Toulouse, (Professeur Claude Mijoule),
Les calculs relatifs à ce travail ont été entamés initialement sur le cluster informatique que Monsieur F. Maury, Directeur du CIRIMAT et le Professeur E. Andrieu ont mis à notre disposition au LPCQ. Je les remercie vivement et sincèrement pour cette contribution. Merci à J. Trotard pour avoir installé le cluster au niveau de notre laboratoire LPCQ.
Une bonne partie de ce travail a été financée par le ministère algérien de l’enseignement et de la recherche scientifique que je remercie profondément. J’aimerais aussi remercier l’administration de L’UMMTO de m’avoir facilitée les procédures administratives durant la préparation de cette thèse.
Je tiens à remercier Messieurs Jean-Claude BARTHELAT et Mohand AMAROUCHE pour avoir accepté de codiriger cette thèse et pour m’avoir conseillé et soutenu tout au long de ce travail. Je tiens aussi à les remercier pour le temps qu’ils ont consacré à la correction de ce manuscrit.
Toute ma reconnaissance s’adresse à Claude MIJOULE pour la confiance qu’il m’a accordé en acceptant de superviser de près ce travail avec beaucoup de patience, de rigueur et de compétence. Sans lui, cette thèse n’aurait pas pris forme ; je le remercie pour ses inépuisables disponibilités, critiques, soutien ainsi que d’avoir accepté de présider le jury de cette thèse.
Je tiens à remercier chaleureusement les rapporteurs Monsieur Philippe SAUTET Directeur du Laboratoire de Chimie de l’Ecole Normale Supérieure de Lyon et Mourad ZEMIRLI Professeur à l’Université Mouloud Mammeri de Tizi-Ouzou pour m’avoir fait l’honneur de juger ce travail de thèse. Je remercie également Abdelhamid ZIANE et Daniel MONCEAU pour avoir accepté de faire partie du jury.
Je ne saurais oublier Mmes les Secrétaires des différents laboratoires, leurs aides et leurs disponibilités m’ont beaucoup touché.
Un grand merci à l’assistance technique apportée par les centres de calculs CALMIP et Grid5000 de l’Université de Toulouse.
Comment ne pas associer à ces remerciements mes compatriotes : Ammi Saïd, Aîmed, Hamza et Rachid pour leur formidable compagnie.
Enfin, j’ai une pensé toute particulière pour ma famille dont le soutien ne m’a jamais fait défaut durant toutes ces années d’études.
Résumé
L’objet de cette thèse est l’étude des défauts ponctuels dans le nickel solide. Le choix de cette thématique est élaboré pour apporter des éléments d’information sur les processus de formation et de migration des monolacunes, bilacunes et de l’oxygène interstitiel dans Ni ; d’autre part d’éventuelles interactions entre ces défauts, primordiales pour la compréhension des mécanismes d’oxydation de Ni et de ses alliages sont abordées. Nous avons effectué nos calculs avec la DFT et l’approche PAW (Chapitre 1). La méthode est validée après l’étude des différentes propriétés physiques de Ni massif (Chapitre 2). Dans le chapitre 3, nous avons déterminé les enthalpies de formation et de migration des monolacunes. Nous avons montré qu’il fallait tenir compte des effets d’expansion thermique pour bien reproduire les résultats expérimentaux. Il ressort de cette étude que l’énergie d’activation des monolacunes est indépendante des effets d’expansion thermique et que la courbure du graphe d’Arrhenius d’autodiffusion dans Ni est dûe à la contribution des bilacunes dans le processus de diffusion. Le chapitre 4 est dédié à l’étude de la formation et de la migration de celles ci. On montre que la bilacune est stable, que son énergie de migration est faible, mais que son énergie d’activation est élevée. Dans le dernier chapitre, nous étudions les processus d’insertion et de migration de l’oxygène interstitiel dans Ni ainsi que les interactions monolacune-oxygène. On montre que l’oxygène atomique s’insère préférentiellement dans les sites octaédriques et migre via les sites tétraédriques. Par ailleurs, les résultats obtenus indiquent une forte attraction entre l’atome d’oxygène et les mono- ou bilacunes. Il est donc possible que la diffusion de lacunes facilite celle de l’oxygène par un processus par paires.
MOTS CLES : DFT, Nickel, lacunes, Expansion thermique, Enthalpie de formation et
Abstract
The present work is concerned with the study of point defects in solid nickel. The goal of this work is to shed some light on the processes of formation and migration of monovacancies, divacancies and interstitial oxygen in Ni. Furthermore we study interactions between these defects which are of prime importance to understand the oxidation process in Ni and its alloys. The calculations are done within the DFT framework with PAW pseudopotential (Chapter 1). In the second chapter, we have validated our approach by calculating physical properties of Ni. Chapter 3 is dedicated to the study of monovacancies to determine their formation and migration enthalpies. We show that in order to reproduce the experimental results it is necessary to take into account the effects of thermal expansion. We find that the activation energy of a monovacancy does not depend on the thermal expansion effects and the curvature of the Arrhenius plot of the selfdiffusion in Ni may be due to the contribution of divacancies to the diffusion process. Chapter 4 is dedicated to the formation and diffusion of divacancies. We show that the divacancy is stable, its migration energy is weak but its activation energy is higher than that found for the monovacancy. In the last chapter, we study the process of the insertion and migration of interstitial oxygen in Ni and explore the vacancy-oxygen interactions. The oxygen is found to occupy preferentially octahedral sites and migrates through tetrahedral sites. Moreover there is a strong attraction between the oxygen atom and mono- and divacancy. Then, we may suppose that the diffusion of the point defects might enhance the diffusion of oxygen through pairwise process.
Key Words: DFT, Nickel, vacancies, Thermal expansion, formation and Migration
i
Table des matières
Introduction générale ...1
Chapitre 1 ...6
Méthodologie ... 6
1.1. Approche ab initio--- 6
1.2. Principes des calculs DFT --- 7
1.3. Echange et corrélation ---10
1.3.1. Position du problème---10
1.3.2. L’approximation de la densité locale ---14
1.3.3. L’approximation du gradient généralisé---15
1.3.3.1. Correction du terme d’échange ---16
1.3.3.2. Correction du terme de corrélation---17
1.4. Résolution numérique des équations de Kohn-Sham---17
1.5. Du pseudopotentiel au formalisme PAW ---21
1.5.1. Introduction---21
1.5.2. Pseudopotentiels à norme conservée---21
1.5.3. Pseudopotentiels à norme relaxée ---23
1.6. Le formalisme PAW ---24
1.7. Le code de calcul VASP ---26
1.7.1. Mise en œuvre du code VASP ---26
1.8. Conclusion---27
Bibliographie du chapitre 1 ...28
Chapitre 2 ...31
Validation de la méthode : Etude du nickel massif ... 31
2.1. Résultats ---32
2.1.1. Phase stable : cubique à faces centrées ---32
ii 2.1.1.2. Module de compressibilité ---36 2.1.1.3. Energie de cohésion ---36 2.1.2. Phases métastables CC et HCP ---37 2.2. Conclusion---40
Bibliographie du chapitre 2 ...42
Chapitre 3 ...44
Etude des monolacunes dans Ni ... 44
3.1. Rappels thermodynamiques : ---45
3.1.1. Concentration à l’équilibre des monolacunes ---45
3.1.2. Migration et diffusion des monolacunes ---47
3.2. Méthodes expérimentales d’étude des monolacunes ---49
3.2.1. Mesures de dilatation ---50
3.2.2. Annihilation des positrons---50
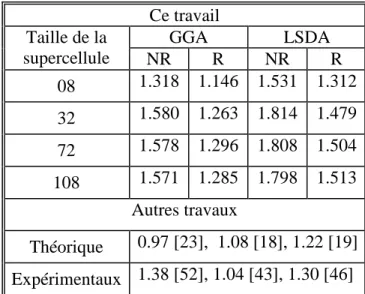
3.3. Enthalpie de formation d’une monolacune dans Ni ---51
3.3.1. Données théoriques et expérimentales---51
3.3.2. Modélisation de la monolacune : calculs en supercellule ---53
3.3.2.1. Détails techniques ---53
3.3.2.2. Taille de la supercellule---54
3.3.2.3. Maillage de la première zone de Brillouin ---55
3.3.3. Résultats et discussions ---57
3.4. Enthalpie de migration ---60
3.4.1. Modélisation---60
3.4.2. Résultats ---61
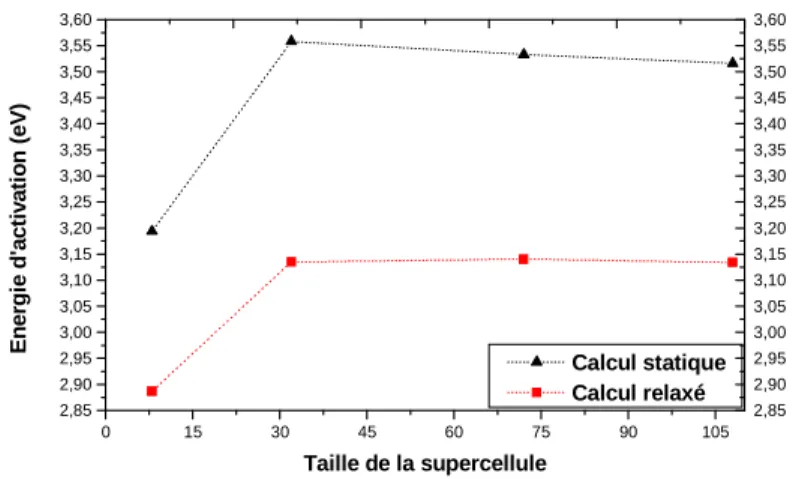
3.5. Energie d’activation ---63
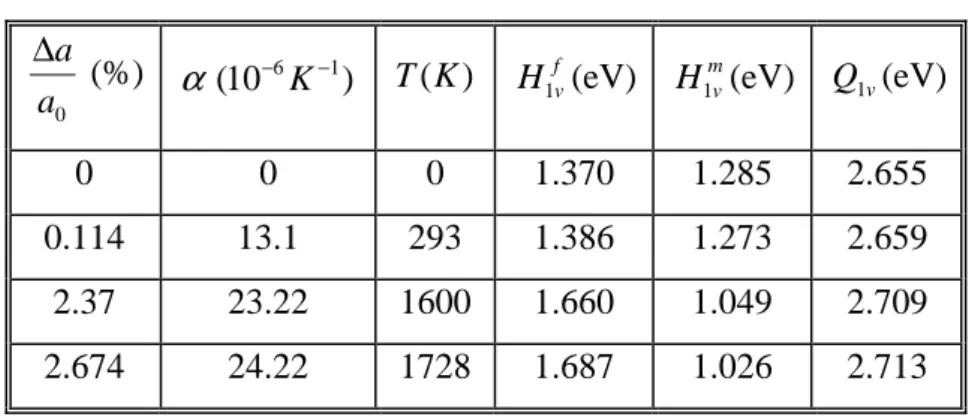
3.6. Contribution des effets thermiques: expansion thermique---66
3.6.1. Enthalpies de formation et de migration ---67
3.6.2. Effet de polarisation de spin ---68
3.6.3. Conséquences de l’invariabilité de l’énergie d’activation avec la température et écarts à la loi d’Arrhénius ---72
3.7. Conclusion---72
iii
Chapitre 4 ...78
Etude des bilacunes dans Ni ... 78
4.1. Introduction---78
4.2. Quelques données expérimentales et théoriques ---79
4.2.1. Travaux expérimentaux---80
4.2.2. Travaux théoriques ---83
4.3. Etude des bilacunes dans Ni ---84
4.3.1. Détails techniques ---84
4.3.2. Modélisation de la bilacune ---85
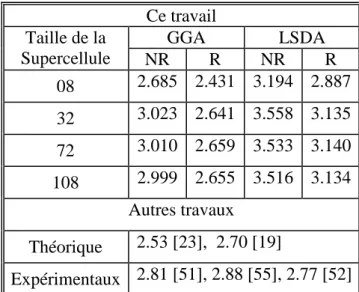
4.3.3. Résultats : Etude à 0 k ---86
4.3.3.1. Enthalpie de formation---86
4.3.3.2. Enthalpie de migration ---89
4.4. Effet d’expansion thermique ---92
4.4.1. Résultats ---92 4.4.1.1. Enthalpie de formation---92 4.4.1.2. Enthalpie de migration ---93 4.4.1.3. Energie d’activation ---94 4.5. Conclusion---95
Bibliographie du chapitre 4 ...96
Chapitre 5 ...99
Etude de l’insertion et de la diffusion de l’oxygène et de l’interaction
lacunes-oxygène dans Ni ... 99
5.1. Introduction---99
5.2. Revue bibliographique --- 100
5.2.1. Données expérimentales --- 100
5.2.1.1. Solubilité de l’oxygène dans le nickel --- 100
5.2.1.2. Diffusion de l’oxygène dans le nickel --- 101
5.2.2. Données théoriques--- 103
5.3. Insertion d’oxygène atomique et moléculaire dans Ni --- 104
5.3.1. Paramètres de calcul--- 104
iv
5.3.3. Résultats --- 107
5.3.3.1. Systèmes de référence : Oxygène atomique et moléculaire --- 107
Oxygène atomique--- 107
Oxygène moléculaire--- 107
5.3.3.2. Insertion et substitution de l’oxygène atomique dans Ni --- 108
Facteur structural --- 108
Facteur magnétique--- 110
Facteur électronique --- 111
5.3.3.3. Insertion de l’oxygène moléculaire --- 113
5.3.3.4. Solubilité de l’oxygène atomique dans les sites interstitiels et de substitution 115 5.3.4. Effets thermiques : dilatation du réseau --- 116
5.3.4.1. Energies d’insertion --- 116
5.3.4.2. Energies de solubilité --- 120
5.4. Diffusion de l’oxygène atomique dans Ni --- 121
5.4.1. Etude à 0 K --- 121
5.4.1.1. Principe --- 121
5.4.1.2. Résultats --- 124
5.4.2. Dilatation thermique --- 127
5.5. Interaction des monolacunes et des bilacunes avec l’oxygène dans Ni---- 129
5.5.1. Paramètres de calculs et modélisation --- 130
5.5.2. Résultats --- 132
5.5.2.1. Interaction de la monolacune avec l’oxygène --- 132
5.5.2.2. Interaction bilacune-oxygène --- 134
5.6. Conclusion--- 136
Bibliographie du chapitre 5 ...138
Conclusion générale...140
Annexe 1 : Liste des tables ...144
Annexe 2 : Liste des figures ...147
Annexe 3 : Publication 1...153
1
Introduction générale
Le comportement macroscopique d’un matériau peut dépendre souvent de la présence de défauts dans sa structure. C’est en effet la présence même de ces défauts dans tout cristal qui permet d’interpréter un grand nombre de propriétés parmi les plus intéressantes. Ainsi étudier les propriétés électriques des semi-conducteurs revient à étudier les propriétés des défauts cristallins dans les cristaux ioniques ou covalents. Toute la plasticité des cristaux repose sur l’existence de dislocations. Dans les métaux, la diffusion, le fluage, le frittage, pour ne citer que quelques exemples, sont étroitement liés à la présence et aux propriétés des défauts ponctuels. Par conséquent, augmenter les performances des matériaux revient toujours à trouver un compromis entre leur chimie, leur structure atomique et leur microstructure. Les métallurgistes savent parfaitement optimiser les propriétés de leurs matériaux en jouant sur ces trois composantes.
En service, les matériaux sont soumis à de nombreuses sollicitations qui en transforment les caractéristiques. L’exposition à un environnement oxydant induit une dégradation du métal par une croissance de la couche d’oxyde. Cette croissance est activée par des processus de diffusion, de transfert et également des réactions d’interface qui se font globalement en cinq étapes :
• adsorption et dissociation du dioxygène sur la surface du métal ;
•
réaction entre les atomes d’oxygène adsorbés et le métal pour former les germes d’oxyde ;•
croissance des germes et formation d’un film continu ;•
croissance du film d’oxyde en épaisseur par diffusion à travers le film d’oxyde ;•
rupture du film d’oxyde par les contraintes induites par sa croissance et la présence des défauts dans le substrat métallique.2 Dans le cas du nickel, ces mécanismes ont fait l’objet d’une étude expérimentale récente réalisée par Simon Perusin1. Au terme de cette étude, on a constaté une diffusion d’oxygène dans le nickel nettement supérieure à celle prévue en adoptant une approche classique de la diffusion. L’hypothèse d’une éventuelle assistance des lacunes présentes ou générées lors de la croissance de la couche d’oxyde dans le processus de diffusion d’oxygène a été proposée pour valider les constatations expérimentales.
Notre étude théorique a pour but de valider ou d’invalider ce dernier point. En toute rigueur, la simulation d’un tel processus ne peut se faire que dans le cadre de la méthode de dynamique moléculaire ou de type Monte Carlo Cinétique. Cependant la mise en œuvre de telles méthodes exige de connaître de façon précise les énergies de formation et de migration des défauts ponctuels (monolacune, bilacune) et de l’oxygène interstitiel dans le massif ainsi que la nature des interactions entre les lacunes et l’oxygène.
Le but que nous nous sommes fixé dans cette thèse est d’évaluer d’une part, les différentes énergies liées à la formation et la migration de lacunes ainsi qu’à l’insertion et à la migration de l’oxygène interstitiel et d’autre part, les énergies d’interaction lacune-oxygène. Cette étude est réalisée à l’aide de calculs à l’échelle atomique dans le cadre de la théorie de la fonctionnelle de densité (DFT) [P. Hohenberg et W. Kohn, Phys. Rev, 136 B864-B871 (1964)].
Le premier chapitre de ce travail est consacré au problème de la description des états électroniques dans un solide. En partant de l’approximation la plus simple, celle de Hartree [D.R. Hartree., Proc. Camb. Philo. Soc., 24, pp 89-110, 1928], nous présenterons les points fondamentaux de la théorie de la fonctionnelle de la densité. Comme nous le verrons, cette théorie est précise mais pour la rendre utilisable, plusieurs approximations ont dûe être introduites : l’approximation de la densité locale (LDA) qui est basée sur une description de la densité électronique à partir d’un gaz d’électrons libres ou l’approximation du gradient conjugué (GGA) qui améliore l’étude des systèmes à forte variation de densité électronique.
Nous donnerons ensuite un bref exposé de la méthode utilisée dans ce travail, à savoir la description des pseudopotentiels par la méthode PAW (Projected Augmented Waves) [P. Blochl, Phys. Rev., B50, 17953 (1994)] dans la formulation de Kresse et Joubert [G.
1 Thèse INPT : Conséquence de l’oxydation à haute température sur l’injection de défauts et le
3 raisons : d’une part, comme cette étude concerne la modélisation de défauts qui nécessite l’utilisation de mailles élémentaires de simulation assez larges, c’est-à-dire un nombre d’atomes à traiter important, nous nous sommes placés dans l’approximation des pseudopotentiels. La méthode des pseudopotentiels nous permet dans des temps de calcul raisonnables de prendre en compte la relaxation globale des paramètres structuraux dont la contribution aux différentes énergies liées à la formation et la migration des défauts ponctuels dans le métal est significative. D’autre part, vu la contrainte imposée par la forte localisation des électrons d dans le nickel et dans les métaux de transition en général, les pseudoptentiels de type PAW représentent une bonne alternative pour décrire correctement les interactions cœur-valence avec une base assez réduite associée aux pseudopotentiels « standards » [N. Troullier, et J.L. Martins, Phys. Rev., B 43, 8861 (1991), R. Hamman, M. Schluter, et C. Chiang, Phys. Rev. Lett., 43 1494 (1979) et G.B. Bachelet, D.R. Hamman, et M. Schluter, Phys. Rev., B 26, 4199(1982)].
Pour valider l’approche adoptée, nous présentons dans le deuxième Chapitre les résultats obtenus pour différentes grandeurs physiques du nickel massif comme le volume d’équilibre, la stabilité des différentes structures, le module de compressibilité et l’aimantation, à différents niveaux d’approximation. Nous présentons ensuite dans le Chapitre 3 les résultats obtenus pour les enthalpies de formation, de migration et d’activation d’une monolacune dans le nickel en suivant la méthodologie présentée au Chapitre 1. Ces calculs sont effectués avec polarisation de spin dans l’approximation locale ou celle du gradient conjugué. Afin de confronter nos résultats avec d’autres mesures expérimentales qui sont toutes effectuées à haute température, nous exposons dans la dernière partie de ce chapitre l’effet de la température à travers l’étude de la dilatation du réseau.
Compte tenu des résultats obtenus dans le Chapitre 3, le quatrième Chapitre est consacré à l’étude des propriétés énergétiques des bilacunes et à leur contribution au processus de diffusion dans le nickel. La stabilité relative des couples lacune-lacune est revue en détail dans les configurations de premier, second et troisième voisin ainsi que les différents mécanismes de migration. Ici aussi, nous explorerons l’effet d’expansion thermique sur les enthalpies de formation, de migration et d’activation des bilacunes.
4 Enfin dans le dernier chapitre, nous présentons les résultats concernant le système Ni-O. En premier lieu, nous nous intéresserons aux énergies d’insertion d’un atome d’oxygène dans le nickel massif par substitution (insertion dans une monolacune) et en position interstitielle (dans les sites octaédriques et tétraédriques). Par la suite, nous évaluerons les enthalpies de migration de l’oxygène correspondant soit à une transition directe entre deux sites octaédriques voisins (octa-octa) soit à une transition octa-octa en passant par un site tétraédrique intermédiaire. La seconde étape consistera à calculer les interactions lacune-oxygène dans le massif. L’objectif de ces calculs est double : d’une part, quantifier les énergies de liaisons de la paire lacune-oxygène et d’autre part, proposer un mécanisme de diffusion de l’oxygène couplé aux lacunes.
5
6
Chapitre 1
Méthodologie
Parmi les différentes méthodes ab initio disponibles, la théorie de la fonctionnelle de la densité (DFT) permet de traiter tous les éléments du tableau périodique, incluant les métaux de transition. Par expérience, elle s'est rapidement imposée comme étant la méthode quantique la mieux adaptée à l'objet de notre étude. La mise en œuvre est faite à travers le code de calcul VASP (Vienna Ab initio Simulation Package) développé dès 1991 à l'Institut de Physique Théorique de Vienne et dont nous exposerons succinctement les caractéristiques au fil des paragraphes. Commençons par un bref exposé des bases de la théorie employée.
Dans tout ce qui suit, nous considérons un système constitué de N atomes et a contenant N électrons en interaction. Toutes les équations sont écrites en unités atomiques e
(u.a.), c'est-à-dire avec
ħ
= me = e = 1, m étant la masse de l’électron et e la charge eélémentaire (un électron a donc une charge égale à -1).
1.1.
Approche ab initio
Lorsqu’on veut étudier la matière, on peut, en principe, calculer toutes les propriétés d’un ensemble d’atomes à l’aide de la mécanique quantique, en partant de l’équation de Schrödinger dépendant du temps :
) 2.1 (
{ } { }
(
)
(
{ } { }
)
t t r R i t r R H I i i I ∂ Ψ ∂ = Ψ , , , , avec :7
{ } { }
(
I i)
i i I I I r R V M H ˆ , 2 2 ˆ 2 2 + ∇ − + ∇ − =∑
∑
Le système étant décrit à l’aide d’une fonction d’onde à plusieurs particules
{ } { }
(
RI , ri ,t)
Ψ , où l’ensemble
{ }
RI contient les variables décrivant la position des noyaux, de masse MI, et{ }
r celles décrivant la position des électrons. i Vˆest l’opérateur associé àl’énergie potentielle et prend en compte les interactions noyau-noyau, électron-électron, et noyau-électron.
Il existe des solutions exactes de cette équation pour quelques systèmes très simples (atome d’hydrogène ou ion H2+ par exemple). Cependant, dans la plupart des cas, le recours à un certain nombre d’approximations s’avère inévitable. C’est pourquoi les nombreuses approches visant à obtenir des informations fiables sur tous ces systèmes sont en continuel développement [1]. Nous nous intéresserons ici à la théorie de la fonctionnelle de densité
(DFT, Density Functional Theory) qui est actuellement sans doute la plus efficace pour
traiter le genre de problèmes auxquels nous nous intéressons.
La fonction d’onde du système comporte un grand nombre de degrés de liberté, et son état fondamental peut être obtenu à partir de l’équation de Schrödinger indépendante du temps (état stationnaire) :
) 2.2 (
{ } { }
(
RI ri)
E(
{ } { }
RI ri)
H Ψ , = Ψ ,où E est l’énergie de l’état fondamental décrit par la fonction propreΨ.
On peut trouver une solution approchée de cette équation en introduisant plusieurs approximations (Born-Oppenheimer, méthode des électrons indépendants, introduction de la corrélation électronique par Interaction de Configurations,…). Une approche permettant de réduire la taille du problème tout en tenant compte de la corrélation est donc la DFT dont nous allons maintenant décrire le principe.
1.2.
Principes des calculs DFT
La DFT consiste à déterminer à partir de la densité électronique, les propriétés de l’état fondamental d’un système composé d’un nombre donné d’électrons en interaction coulombienne avec des noyaux fixes. Elle repose sur deux théorèmes fondamentaux démontrés par Hohenberg et Kohn [6] :
8 - L’énergie de l’état fondamental est une fonctionnelle unique de la densité
électronique
ρ
( )
r ;- Pour un potentiel Vˆ et un nombre donné d’électrons N , le minimum de e
l’énergie totale du système correspond à la densité exacte de l’état fondamental (principe variationnel).
Tout le problème consiste à déterminer cette fonctionnelle.
Peu de temps après la formulation des lois de la mécanique quantique, THOMAS et FERMI ont donné une expression approchée de l’énergie totale en fonction de la densité électronique ρ[7, 8]. Quarante ans plus tard, Kohn et Sham [9] introduisent une expression formelle de l’énergie totale du système en fonction de la densité électronique. Elle comporte deux étapes :
• Les orbitales sont réintroduites afin de traiter le terme d’énergie cinétique Tˆe de
manière exacte ;
• Le système étudié est redéfini par rapport à un système d’électrons sans interaction et de même densité
ρ
( )
r , de façon à considérer les termes d’interaction comme des corrections aux autres termes.Si l’on ne tient pas compte du spin des électrons , la densité s’écrit comme une somme sur les Nocc états occupés :
) 2.3 (
( )
( )
2 1 r f r i N i i occψ
ρ
∑
= = , avec e N i i N f occ =∑
=1où f , qui peut être fractionnaire [11], est le nombre d’occupation de l’orbitale i . Pour i traiter les systèmes avec polarisation de spin, il suffit de séparer la sommation de l’équation (2.3) en deux parties, l’occupation de chaque orbitale étant désormais fixée à 1 [12] :
) 2.4 (
( )
( )
( )
( )
( )
2 1 2 1∑
∑
↑ ↓ = ↑ = ↓ ↑ ↓ + = + = e Ne i i N i i r r r r rρ
ρ
ψ
ψ
ρ
où
ρ
↓ etρ
↑ désignent respectivement les densités associées aux états de spin down( )
↓ et up( )
↑ , avecNe +Ne =Ne↑ ↓
9 La prise en compte du spin permet de rendre compte des propriétés magnétiques. Elle modifie uniquement la forme analytique du terme d’échange- corrélation, car il est le seul à dépendre du spin.
L’énergie totale du système peut s’exprimer de la manière suivante :
( ) ( )
4 4 4 3 4 4 4 2 1 4 4 3 4 4 2 1 H e E T i i i KS tot r r r r r drd E ′ − ′ ′ + ∇ − =∑
ψ
ψ
∫
ρ
ρ
2 1 2 0 2 ) 2.5 (( )
[ ]
ρ
ρ
xc E J I i J J I N I I I E R R Z Z R r Z r dr ext a + − − − −∫
∑
∑
< = 4 4 4 4 4 4 3 4 4 4 4 4 4 2 1 1où Te0 est l’énergie cinétique du système sans interaction, EH le terme de Hartree, Eext inclut l’interaction coulombienne des électrons avec les noyaux et celle des noyaux entre eux, et le terme d’échange-correlation Exc
[ ]
ρ
comprend la déviation à l’énergie cinétique etles corrections au terme de Hartree, toutes deux dûes à la corrélation entre électrons. ZI et
J
Z désignent la charge des noyaux I et J.
Déterminer l’état fondamental du système revient alors à résoudre, de manière auto-cohérente, un ensemble d’équations aux valeurs propres appelées équations de Kohn-Sham : ) 2.6 (
( )
( )
( )
i i i e H xc ext H r V r V r i N V KS , , 1 , 2 2 K 4 4 4 4 4 3 4 4 4 4 4 2 1 = = + + + ∇ −ψ
ε
ψ
Avec :( )
( )
∫
−( )
′ ′ ′ = ∂ ∂ = r r r r d r E r V H Hρ
ρ
) 2.7 (( )
( )
∑
= − − = ∂ ∂ = Na I I I ext ext R r Z r E r V 1ρ
) 2.8 (( )
( )
r E r V xc xc ρ ∂ ∂ =10 Lorsque le spin est pris en compte, l’énergie d’échange-corrélation Exc
[ ]
ρdevientExc
[
ρ↓,ρ↑]
, et pour chaque valeur de σ ∈{ }
↑,↓ du spin, le potentiel correspondant s’écrit : ) 2.9 (( )
r E V xc xc ρ σ ∂ ∂ =Les équations de Kohn-Sham sont résolues en respectant l’orthonormalisation des fonctions d’onde : ) 2.10 (
( ) ( )
j ij i r r drψ ψ =δ∫
*La somme des trois termes VH +Vext +Vxc représente un potentiel effectif Veff local.. Le problème lié à la self-interaction qui résulte de l’utilisation de fonctions d’ondes indépendantes, doit être également pris en compte. Ce problème peut être résolu dans de très nombreux cas [13], mais pas toujours de manière satisfaisante pour les atomes et les molécules [14].
A ce stade, tous les termes de l’énergie, avec le potentiel associé, peuvent être évalués, excepté le terme d’échange-corrélation sur lequel nous allons maintenant porter notre attention.
1.3.
Echange et corrélation
1.3.1.
Position du problème
La densité électronique définie à partir d’un modèle de particules indépendantes, ne permet pas d’étudier en détail les effets d’échange-corrélation. Il faut également s’intéresser à la densité des paires, associée à la probabilité de trouver deux électrons en interaction et localisés dans deux éléments de volume donnés. On peut la définir de la manière suivante :
) 2.11 (
( ) ( ) ( )
r,r′ = r r′(
1+ f( )
r,r′)
2ρ
ρ
ρ
La fonction f est appelée facteur de corrélation et traduit le fait que les électrons interagissent. Quand f
( )
r,r′ =0 il n’y a pas d’interaction etρ
2 = Ne2 au lieu de(
e −1)
e N
N (qui correspond au nombre d’interactions entre électrons). Apparaît alors le problème de la self-interaction.
11 Dans le cas général, les effets dûs aux interactions entre électrons sont de trois sortes : 1- L’échange, encore appelé corrélation de Fermi, résulte de l’antisymétrie de la
fonction d’onde totaleΨ. Il correspond au fait que deux électrons de même spin ont une probabilité nulle de se trouver au même endroit et se traduit au niveau de la densité de paires par la relation
ρ
2( )
r,r =0. Cet effet est directement lié au principe de Pauli et ne dépend pas de la charge de l’électron.2- La corrélation de Coulomb est dûe à la charge de l’électron. Elle est liée à la répulsion des électrons en
r r− ′
1
. Contrairement à l’effet d’échange, elle est indépendante du spin. L’approximation de Hartree-Fock néglige cet effet de corrélation. Pour être correctement pris en compte, ce dernier nécessite l’utilisation d’un grand nombre de déterminants de Slater pour décrire la fonctionΨ, ce qui est fait à des degrés divers dans les méthodes de type CI.
3- Le troisième effet provient du fait que les fonctions d’onde électroniques sont formulées en termes de particules indépendantes. Il s’agit de la corrélation de self-interaction, qui doit conduire à un comptage correct du nombre de paires d’électrons.
De par sa définition même, l’approche de Kohn-Sham impose au terme d’échange corrélation de prendre en compte la correction du terme de l’énergie cinétique. En effet, même si la densité du système fictif considéré est la même que celle du système réel, l’énergie cinétique déterminée est différente de l’énergie cinétique réelle.
Afin d’inclure avec précision les effets d’échange-corrélation dans l’espace total et le potentiel effectif, la DFT s’est dotée d’un outil très pratique : le trou d’échange-corrélation.
Il sert à prendre en compte la diminution de la densité électronique dans tout l’espace dûe à la présence d’un électron en un point particulier. Il est défini par l’expression :
) 2.12 (
(
)
(
( )
)
( )
r r r r r r hxc − ′ ′ = ′ ρ ρ ρ , , 2où le premier terme représente la probabilité conditionnelle (notée P
(
r/r′)
dans la suite) de trouver un électron en r′ sachant qu’il y en a déjà un en r . Puisqu’il traduit une diminution de la densité électronique, le trou d’échange-corrélation est défini négatif.12 ) 2.13 (
(
, ′)
=−1 ′∫
dr hxc r rPuisqu’il est censé corriger le problème de self interaction, la distribution de charge du trou contient en effet exactement un électron. Cette règle montre par ailleurs que plus le trou est profond, plus il est localisé [15].
Pour déterminer de quelle manière l’énergie d’échange-correlation est reliée à ce trou, il faut revenir à la définition de l’énergie d’interaction électrostatique :
) 2.14 (
( )
∫
′ − ′′ = r r r r r drd Eel , 2 1 ρ2 soit en fonction de hxc : ) 2.15 (( ) ( )
∫
( ) ( )
∫
+ ′ − ′ ′ ′ − ′ ′ = r r r r h r r drd r r r r r drd E xc el , 2 1 2 1 ρ ρ ρOn reconnaît dans le premier terme l’énergie de Hartree. Le deuxième terme qui correspond exactement à l’énergie d’échange-corrélation, peut être vu comme l’interaction de chaque électron avec la distribution de charge de son trou d’échange-corrélation et prend en compte tous les effets énumérés précédemment. L’intérêt du trou d’échange-correlation apparaît dès lors évident : mieux on connaîtra ses caractéristiques et plus les modèles développés s’approcheront de la réalité.
L’approche conventionnelle consiste à traiter séparément l’échange et la corrélation. A cet effet, hxcest divisé en deux contributions :
) 2.16 (
( )
r r h h( )
r r hxc ′ = x + c ′ = , , σ1 σ2 σ1,σ2 xh désigne la partie échange, hc la partie corrélation et les σi correspondent aux
composantes de spin considérées. La raison d’être de cette séparation est purement pratique : le premier termehxpeut être obtenu dans l’approximation de Hartree-Fock. De plus, de par son origine (l’antisymétrie deΨ), ce terme est prédominant ; par la définition même de la probabilité conditionnelleP
(
r/r′)
, on a :) 2.17 (
( )
, ′ =−1 ′∫
dr hx r r13 ce qui signifie que la correction à la self-interaction est apportée par le trou d’échange. On peut aussi ajouter que la distribution de charge qui lui est associée possède à priori une symétrie relativement basse puisque sa forme dépend de la densité électronique.
Le trou de corrélation hc possède des caractéristiques différentes. A partir des équations (2.14) et (2.17), il vérifie : ) 2.18 (
( )
, ′ =0 ′∫
dr hc r rIl est important de noter que ni le trou d’échange, ni le trou de corrélation n’ont une signification physique. Seul le trou d’échange-corrélation total a un concept physique [16]. Dans l’approche de Kohn-Sham, la correction à l’énergie cinétique doit être intégrée au trou d’échange-corrélation. . Dans ce but, l’interaction coulombienne est paramétrée :
) 2.19 (
( )
( )
r r r dr r VH ′ ′ =∫
,λ
ρ
λet l’on fait varier progressivement le paramètre d’interaction λ de 0 jusqu’à 1.
Pour chaque valeur deλ, l’hamiltonien du système est adapté de manière à ce que la densité électronique demeure égale à celle du système réel afin de rendre celle-ci indépendante deλ. Ainsi les deux systèmes extrêmes sont-ils connectés par un continuum purement artificiel de systèmes dans lesquels les électrons interagissent partiellement : c’est la connexion adiabatique. On peut ensuite se servir de cette connexion pour déterminer l’énergie d’échange-corrélation : ) 2.20 (
( ) ( )
∫
( ) ( )
∫
∫
∫
= ′ − ′ ′ ′ − ′ ′ + = − = − = r r r r h r r d r r r r h r r d d dE E E xc xc xc xc xc , 2 1 , 2 1 1 0 1 0 0 1λ
ρ
ρ
λ λ λ λ avec : ) 2.21 (( )
r r d h( )
r r hxc ′ =∫
xc ′ − , , 1 0 λλ
Tout ceci donne un schéma de principe pour déterminer les termes d’échange-corrélation puisque la connaissance de hxc− nous mène directement à l’énergie, puis au potentiel d’échange-corrélation. La détermination ab initio du trou d’échange-corrélation n’est malheureusement possible que dans des cas triviaux. Le calcul de l’énergie et du potentiel d’échange-corrélation sont par conséquent basés sur des approximations [10].
14 En pratique, on pourra se servir de la sommation de l’équation (2.18) pour contrôler la qualité de l’approximation utilisée pour l’échange. Le terme de corrélation semble à priori beaucoup plus complexe à traiter, à cause de sa topologie à six dimensions (trois coordonnées par électrons). Néanmoins, en tenant compte de la symétrie sphérique de l’interaction coulombienne, une bonne approximation du trou de corrélation pourra se contenter, dans un premier temps, de reproduire les propriétés de la moyenne sphérique de ce trou, déjà moins complexe.
Nous allons voir dans la suite les deux principales approches utilisées : approximation de la densité locale et approximation du gradient généralisé.
1.3.2.
L’approximation de la densité locale
L’approximation de la densité locale (LDA, Local Density Approximation, ou LSDA,
Local Spin-Density Approximation) est basée sur le fait que, dans le cas d’un gaz d’électrons homogène, l’énergie d’échange-corrélation exacte par particule peut être déterminée à l’aide de calculs Monte-Carlo quantiques variationnels (VQMC, Variational Quantum Monte-Carlo) [17]. C’est une approximation qui repose sur l’hypothèse que les termes d’échange-corrélation ne dépendent que de la valeur locale de
ρ
( )
r . L’énergie d’échange-corrélation s’exprime alors de la manière suivante :) 2.22 (
( )
[
↓ ↑]
∫
= ρ εxc ρ , ρ LDA xc dr r Eoù εxc
[
ρ↓,ρ↑]
est l’énergie d’échange-corrélation par particule d’un gaz d’électrons libres, paramétrisée pour différentes valeurs de la densité électronique [18, 19].On pourrait s’attendre à ce qu’une telle approximation qui ne repose pas sur des critères physiques, ne donne des résultats corrects que dans les cas assez particuliers où la densité ρ varie peu. L’expérience a montré qu’au contraire, elle permet d’obtenir dans de très nombreux cas une précision équivalente, voire meilleure, à celle donnée par l’approximation Hartree-Fock [16].
Cette observation doit néanmoins être tempérée dans plusieurs cas. La LDA donne, par exemple une très mauvaise estimation du gap des isolants et des semi-conducteurs (environ 100% d’erreur), ce qui n’est pas très surprenant, car cette grandeur ne relève que partiellement de l’état fondamental. Plus gênant est le fait qu’elle ne permet pas de corriger le problème de self interaction.
15 Il a été également noté que :
- Les énergies de cohésion des solides sont systématiquement surestimées, et que l’erreur augmente au fur et à mesure que la taille ou la dimensionnalité du système diminue [19] ;
- Les longueurs de liaison à l’équilibre sont toujours sous-estimées, souvent faiblement, mais l’erreur peut atteindre 10% dans les petits systèmes.
- Les fréquences de vibration des petits systèmes sont généralement surestimées et l’erreur peut même avoisiner les 50% dans quelques cas pathologiques [20, 21]. Si l’on rentre un peu plus dans le détail, on s’aperçoit que pour les atomes, la LDA sous-estime d’environ 10% le terme d’échange et surestime d’à peu près 100% le terme de corrélation. Cette erreur de 100% a été attribuée au fait que, contrairement au gaz uniforme où il intervient pour moitié, le terme de corrélation dans les systèmes finis est beaucoup plus faible pour les électrons de même spin que pour les électrons de spin opposé [22]. Par conséquent, en retranchant ce terme, il est possible de diminuer notablement l’erreur sur la corrélation. En pratique, celle-ci diminue d’un ordre de grandeur [23].
Puisque, mis à part la mésestimation du gap, toutes ces insuffisances concernent les propriétés de l’état fondamental, il est tout à fait possible, en principe, d’améliorer la qualité des résultats sans perdre les avantages que procure la DFT.
1.3.3.
L’approximation du gradient généralisé
La plupart des corrections à la LDA utilisées aujourd’hui sont nées de l’idée consistant à tenir compte des variations locales de la densité
ρ
( )
r à travers son gradient( )
rρ
∇ . A cet effet, la LDA a été réinterprétée comme le premier terme d’un développement en série de Taylor en fonction de ce gradient. Cette approche, appelée approximation de développement du gradient (GEA, Gradient Expansion Approximation), aurait dû améliorer les résultats obtenus par la LDA. En réalité la mise en œuvre de cette approximation a abouti à des résultats désastreux, souvent moins bons que la LDA elle-même, la raison étant que ce développement a fait perdre toute signification physique au trou d’échange-corrélation : les règles de sommation n’étant plus vérifiées, le trou d’échange pouvait devenir positif [16, 24].
Ces problèmes ont été contournés, d’une part en annulant tous les termes issus de la GEA qui ne permettaient pas au trou d’échange de rester partout négatif, et d’autre part en imposant aux trous d’échange et de corrélation le respect des règles de sommation exposées
16 précédemment. Les fonctionnelles qui en ont résulté ont été appelées approximation du gradient généralisé (GGA, Generalized Gradient Approximation). En pratique, elles
traitent séparément la partie échange et la partie corrélation. Leur formulation est basée uniquement sur des principes mathématiques. On notera en particulier qu’elles ne peuvent apporter en elles-mêmes aucune aide à la compréhension des principes physiques sous-jacents [10, 16, 25].
1.3.3.1.
Correction du terme d’échange
Dans le cadre de la GGA, l’énergie d’échange peut être écrite de la manière suivante :
) 2.23 (
( )
( )
σ σ ρσ x F r dr E ExGGA = xLDA−∑∫
4/3 x avec : ) 2.24 ( 3 / 4 σ σ σρ
ρ
∇ = xLe terme xσreprésente, pour le spin
σ
, le gradient de densité réduit. Ce paramètre peut être vu comme une mesure locale de l’inhomogénéité du système et peut prendre des valeurs importantes soit pour un gradient important soit lorsque la densité est proche de zéro (e.g. dans la queue exponentielle loin des noyaux). D’un autre coté, les gradients et les densités élevés aux alentours des noyaux conduisent à des valeurs modérées de xσ[16]. Nous allons nous contenter d’expliciter la fonction Fx( )
xσ pour la GGA dûe à Perdew et Wang [27] et que nous noterons dans la suite PW91.) 2.25 (
[
σ↓ σ↑]
=(
↓[ ]
2σ↓ + ↑[ ]
2σ↑)
2 1 , , 91 , 91 91 PW x PW x PW x E E E ) 2.26 (( )
(
1(
)
(
)
4)
2 100 1 004 , 0 795 , 7 sinh 1645 , 0 1 1508 , 0 2743 , 0 795 , 7 sinh 1645 , 0 1 2 σ σ σ σ σ σ σ σ x x x x e x x x F x x + + + − + + + = − − − Avec : ) 2.27 (( )
2 1/3 4/3 3 2 σ σ σρ
π
ρ
∇ = x17
1.3.3.2.
Correction du terme de corrélation
Comme nous l’avons déjà évoqué, les corrections du terme de corrélation sont beaucoup plus complexes à formuler que celles du terme d’échange. De plus, même si leur influence sur les propriétés structurales et électroniques des systèmes étudiés est beaucoup moins significative que celle de l’échange, il n’en demeure pas moins que leur prise en compte se révèle absolument indispensable pour obtenir des résultats quantitativement satisfaisants. Nous ne détaillerons pas ici les expressions analytiques des différentes corrections car elles n’améliorent en rien la compréhension de la physique mise en jeu. Parmi les différentes corrections possibles, citons celle développée par Perdew en 1986 [26] dont le but est d’obtenir avec précision les énergies de corrélation dans le cas des atomes, des molécules et des surfaces métalliques. Une fonctionnelle différente mais également utilisée couramment est celle de Perdew et Wang [27] qui présente la particularité de corriger l’échange et la corrélation dans une même formulation. En particulier, elle a été construite afin de reproduire les propriétés du trou d’échange-correlation à la fois dans les limites de faibles et fortes densités, et donne aussi des résultats très satisfaisants pour les systèmes possédant des régions où la variation de la densité est grande comme c’est le cas pour les métaux de transition. C’est cette fonctionnelle que nous avons utilisés dans les calculs non locaux. Enfin signalons la fonctionnelle proposé par Perdew, Burke et Enrzerhof [28] qui est une reformulation de celle de Perdew et Wang.
1.4.
Résolution numérique des équations de Kohn-Sham
Nous ne prétendons pas expliquer en détail le fonctionnement d’un code DFT mais plutôt donner au lecteur une idée des paramètres mis en jeu pour aborder les chapitres qui suivent. Nous recommandons la lecture de l’article de revue [29] pour approfondir les points que nous allons aborder.
Pour résoudre numériquement les équations de Kohn-Sham, on développe les fonctions d’onde mono-électroniques
ψ
i sur des fonctions de base. La décompositionexacte des fonctions d’onde mono-électroniques implique que le nombre de fonctions de base doit être infini. Nous serons donc amenés à limiter le développement en utilisant certains critères pour pouvoir mener le calcul numérique. On peut choisir différentes bases de fonctions dont on retient généralement deux classes :
18 - les bases dites « localisées ». Ce sont souvent des bases construites sur des gaussiennes centrées sur les atomes du système ayant pour vocation de représenter les orbitales atomiques.
-
les bases dites « systématiques ». Ce sont des bases dont chaque fonction n’a pas nécessairement de réalité physique pour le problème étudié mais qui simplifient le traitement mathématique du problème. Elles ont par ailleurs l’avantage de pouvoir être facilement étendues afin d’améliorer la description de la fonction d’onde globale. Nous utiliserons par la suite des bases d’ondes planes. Suivant cette formulation, les fonctions d’onde mono-électroniques s’écrivent comme une somme continue d’ondes planes, ce qui revient à calculer la transformée de Fourier desψ
i.L’utilisation de bases continues n’est pas possible numériquement, il faut avoir une base discrète. Ceci peut être réalisé en travaillant sur des systèmes périodiques, en passant ainsi de la transformée de Fourier à des séries de Fourier. Par ailleurs pour les systèmes périodiques, le théorème de Bloch permet de décrire toute fonction d’onde comme le produit d’une onde plane de vecteur d’onde k (vecteur de la première zone de Brillouin) et
d’une fonction radiale F ayant la périodicité du système. Cette fonction F se décompose en série de Fourier, ce qui permet d’écrire :
) 2.28 (
( )
( )
ik G r G G k i r ik k i r e F r c e ). ( , , . , +∑
= =ψ
où les vecteurs G sont des vecteurs du réseau réciproque, c'est-à-dire tel que G.r =2πm avec r un vecteur du réseau direct et mun nombre entier quelconque.
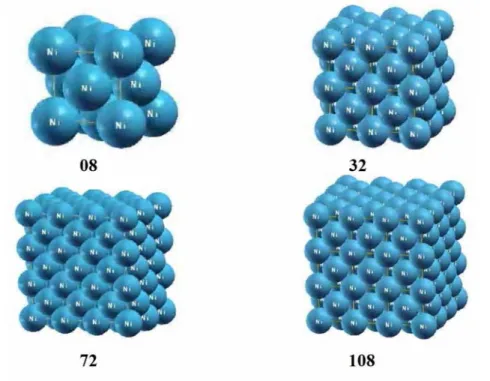
Le fait de rendre le système périodique permet d’exprimer exactement les fonctions d’onde de Kohn-Sham dans une base discrète. Ceci a pour conséquence de rajouter parfois dans le système à calculer des interactions qui n’existe pas dans le système réel, par exemple, la présence d’un défaut ( un défaut ponctuel qui nous intéresse dans cette présente étude) dans une boite reproduite par périodicité. La périodicité des répliques de ce défaut et l’énergie calculée n’est proche de l’énergie réelle de formation du défaut isolé que si les interactions entre répliques restent négligeables, comme illustré sur la Figure 1.1.
19
Figure 1-1 A gauche, le cas réel d'un défaut (lacune) dont on veut calculer l'énergie de formation.
On isole une supercellule autour de ce défaut. Lors du calcul ab initio, la supercellule est reproduite dans les trois directions de l'espace (à droite), ce qui amène à créer des répliques du défaut.
La boite élémentaire utilisée pour le calcul est appelée supercellule.
Lors d’un calcul, il faut vérifier que l’augmentation de la taille de la supercellule ne fait pas varier l’énergie du défaut, ce qui constitue un premier critère pour vérifier la convergence.
Les équations de Kohn-Sham exprimées dans la base des vecteurs G pour un vecteur k donné s’écrivent :
) 2.29 ( G k i i G k i G G G TF F G G c c G k , , , , 2 ) )( ( ) ( 2 1
δ
=ε
+ + − ′ ′ ′ ′∑
où
δ
GG′ représente le symbole de Kronecker et TF(F)(G) la transformée de Fourier de la fonction F prise en G.Pour résoudre numériquement ce problème, on coupe la base des vecteurs G au-delà d’un certain vecteur de coupureGc. On peut alors écrire l’équation (2.29) sous forme matricielle et associer aux valeurs propres
ε
i les vecteurs propres de composantes ci,k,G. Le choix de Gc est un autre paramètre important des calculs qui seront présentés par la suite. On associe à ce vecteur une énergie, que nous appellerons énergie de coupureEc définiepar : ) 2.30 ( 2 ) ( 2 2 c c G k E = h +
Plus cette énergie est grande, plus la base utilisée pour le calcul est grande. Les ondes planes de grande énergie correspondent aux variations rapides des fonctions d’ondes. Il
20 s’avère que les contributions des ondes planes de grande énergie deviennent négligeables quand G2devient grand (les ci,k,G tendent vers 0). Ceci justifie l’introduction d’une coupure. Il conviendra tout de même de vérifier la convergence en énergie de coupure en testant l’évolution de l’énergie avec l’augmentation deEc.
La diagonalisation donne les solutions des équations de Kohn-Sham pour un vecteur donné k de la zone de Brillouin. L’énergie totale du système s’obtient en intégrant sur la zone de Brillouin. Là encore, il faut discrétiser le problème pour pouvoir réaliser numériquement l’intégration. On notera que plus la supercellule est grande, plus la zone de Brillouin est petite, donc plus la discrétisation pourra être limitée (voire réduite au seul point Γ dans certain cas). Cette opération de discrétisation s’appelle le maillage au point
kde la zone de Brillouin.
Différentes méthodes ont été proposées ; la plus simple et la plus courante est celle proposée par Monkhorst et Pack dans leur article de 1976 [30]. C’est celle qui sera utilisée dans la suite de ce travail. Elle est constituée d’un maillage régulier dans les trois directions de l’espace de la zone de Brillouin. Nous utiliserons en plus un maillage uniforme nxnxn
qui amène à considérer n3 points k dont certains pourront être équivalents suivant la symétrie de la supercellule adoptée.
Nous venons de voir les différents paramètres utilisés par la suite permettant de résoudre numériquement les équations de Kohn-Sham :
- La taille de la supercellule;
- Le maillage aux points k (nécessaire pour intégrer dans la zone de Brillouin) ; - L’énergie de coupure, désignée par le terme anglais « cutoff » (qui limite la
taille de la base dans laquelle sont décrites les fonctions d’onde).
Ces différents paramètres nous servirons tout au long de ce travail. A ce stade, le calcul des fonctions d’onde Kohn-Sham tel qu’il vient d’être exposé n’est pas numériquement possible pour les atomes possédant de nombreux électrons. Les variations des fonctions d’onde proches des noyaux sont trop rapides pour être représentées avec une taille de base d’ondes planes raisonnable.
21 La notion de pseudopotentiel a été utilisée pour contourner cet obstacle. Elle consiste à modifier le potentiel au voisinage des noyaux de façon à omettre explicitement les électrons de cœur tout en conservant leur interaction avec les électrons de valence.
1.5.
Du pseudopotentiel au formalisme PAW
1.5.1.
Introduction
A l’aide des concepts développés jusqu’à présent, il est déjà possible de définir un schéma de principe afin de déterminer l’état fondamental électronique d’un système quelconque. Le problème étant que les calculs deviennent de plus en plus coûteux au fur et à mesure que la taille des atomes augmente, à cause d’une part, de l’augmentation du nombre d’électrons et d’autre part, du caractère localisé de certaines orbitales, comme par exemple les orbitales d des métaux de transition. En conséquence, certains éléments du tableau périodique vont pouvoir être modélisés avec beaucoup d’efficacité, tandis que d’autres, comme les éléments lourds ou les métaux de transition, vont nécessiter des moyens de calcul extrêmement puissants. Or dans la plupart des cas les électrons de valence sont les seuls à intervenir dans l’établissement des liaisons chimiques. Les électrons de cœur pourront donc être regroupés avec les noyaux pour constituer des ions rigides : c’est l’approximation du cœur gelé [31]. Afin de tenir compte des interactions qui
ont perdu leur caractère explicite, le potentiel effectif dans les équations de Kohn-Sham doit être remplacé, pour chaque espèce, par un pseudopotentiel. Celui-ci inclut :
- L’interaction du noyau avec les électrons du cœur ;
-
Le potentiel de Hartree provenant des électrons du cœur ;-
Une composante d’échange-corrélaton dûe à l’interaction entre électrons de cœur et de valence ;-
La prise en compte partielle, au besoin, des effets relativistes.1.5.2.
Pseudopotentiels à norme conservée
Par construction, un pseudopotentiel doit être additif : d’une part, il est obtenu à partir des calculs atomiques, et d’autre part, le potentiel effectif total doit être la somme des pseudopotentiels lorsque plusieurs atomes sont en présence. Il doit également être transférable, c'est-à-dire qu’on doit pouvoir utiliser le même pseudopotentiel dans des
22 environnements chimiques différents. Ces exigences constituent le fil directeur de la construction des pseudopotentiels, ainsi que leurs principaux critères de validation.
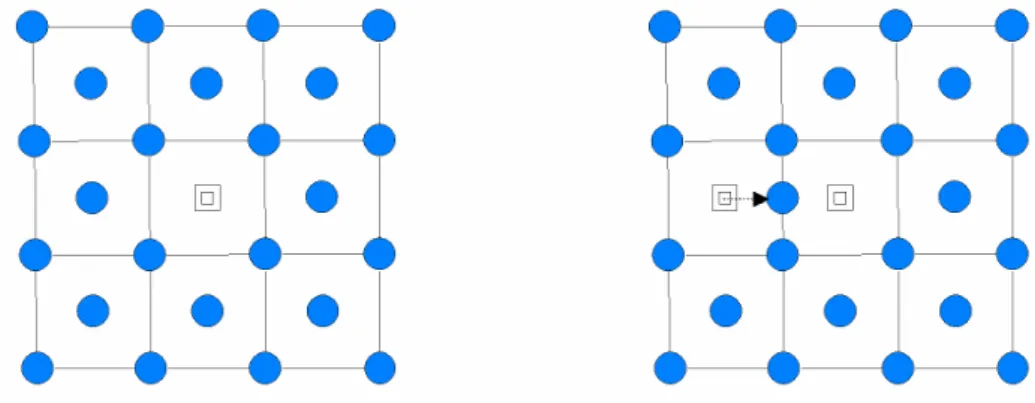
Figure 1.2 Pseudisation des fonctions d'onde de valence et du potentiel. Les noeuds et les
oscillations dûes aux conditions d'orthonormalisation sont supprimés, ce qui permet de décrire les pseudo-fonctions d'onde avec un nombre réduit d'ondes planes.
En pratique les fonctions d’ondes ψiv représentant les électrons de valence sont remplacées par des pseudo-fonctions d’onde ψips (Figure 1.2). Autour de l’atome, à l’extérieur d’une sphère de rayonrc, l’égalité ψips
( )
r =ψiv( )
r est imposée. A l’intérieur de cette sphère, la forme de ψips est choisie de manière à supprimer les nœuds et les oscillations dûs à l’orthonormalisation des fonctions d’onde [29]. Les pseudo-fonctions d’onde peuvent être ainsi obtenues avec beaucoup moins d’ondes planes que les fonctions d’ondes de Kohn-Sham.Le potentiel subit un traitement similaire. La singularité en r 1
− autour de l’atome est éliminée et la forme du pseudopotentiel est choisie de manière à ce que les pseudo-fonctions d’onde et les pseudo-fonctions d’onde de valence aient les mêmes énergies propres [31].
L’interaction entre les électrons de valence et les ions comprend l’interaction coulombienne écrantée par les électrons du cœur, celle des électrons de valence avec les noyaux, la répulsion cœur-valence dûe au principe de Pauli et les termes d’échange-corrélation. Celle-ci est prise en compte par l’introduction d’une dépendance par rapport au moment orbital du pseudopotentiel [29, 32].
La construction d’un pseudopotentiel est guidée par la volonté d’obtenir les mêmes énergies propres pour l’atome qu’avec un calcul incluant tous les électrons. Par ailleurs, le
23 pseudopotentiel ne doit pas diverger au voisinage du noyau afin de ne pas faire apparaître de terme de fréquence élevée dans l’espace réciproque [33].
Hamman, Schluter et Chiang [34], puis Bachelet, Hamman et Schluter [35] ou Troullier et Martins [36] ont mis au point, dans ce contexte, une méthode qui garantit une description de la densité de charge de valence. Celle-ci consiste à conserver la norme : les pseudo-fonctions d’onde de valence sont normées à l’unité. Les pseudopotentiels associés ont été appelés pseudopotentiels à norme conservée (norm-conserving pseudopotentials).
Mais la conservation de la norme trouve ses limites dans l’étude des systèmes ayant des orbitales de valence localisées (plusieurs ondes planes sont nécessaires pour les décrire au voisinage du noyau).
D’autres classes de pseudopotentiels ont étés proposées pour contourner la contrainte de conservation de la norme, ce sont les pseudopotentiels à norme non conservée ou relaxée.
1.5.3.
Pseudopotentiels à norme relaxée
Le rayon rc délimite la région du cœur. Plus ce rayon sera grand et plus les
pseudo-fonctions d’onde et le pseudopotentiel seront lisses, mais ils perdront en précision, puisqu’ils risqueront de dévier des grandeurs dont ils sont issus à des distances intervenant dans la liaison chimique, c'est-à-dire lorsque les orbitales de valence sont localisées, comme par exemple dans les métaux de transition ; dans ce cas le rayon de coupure rc doit rester relativement faible.
A cet effet, Vanderbilt a eu l’idée de relâcher la contrainte de la conservation de norme [37] qui était au centre de la génération des pseudopotentiels standard. En se débarrassant de la contrainte de la norme, Vanderbilt a construit une nouvelle classe de pseudopotentiels dans lesquels les pseudo-fonctions d’onde peuvent être arbitrairement lisses dans la région du coeur. C’est pourquoi ceux-ci sont qualifiés d’ultrasoft. Ce changement a pour effet de réduire l’énergie de coupure (Ecut) nécessaire pour décrire les orbitales localisées en autorisant l’utilisation d’un rayon de coupure plus grand que pour les pseudopotentiels à norme conservée. Néanmoins, ces fonctions d’onde ne permettent pas de retrouver toute la charge du système et il est nécessaire d’augmenter la densité électronique autour des atomes afin de récupérer la charge manquante. Pour plus de détails, voir la Réf. 37.
24 L’approche de Vanderbilt est actuellement largement utilisée dans les calculs ab initio, plus particulièrement pour les métaux de transition. Le grand avantage des pseudopotentiels Ultrasoft (USPP) est leur convergence extrêmement rapide en fonction de l’énergie de coupure. Les temps de calcul et la mémoire nécessaire pour effectuer un calcul sont extrêmement réduits.
Malgré cet avantage, les pseudopotentiels Ultrasoft présentent un inconvénient qui vient surtout des difficultés qu’il y a à les générer, et pour certaines gammes de matériaux, les pseudopotentiels ainsi générés n’assurent pas une meilleure transférabilité, ce qui est le cas des matériaux à fort moment magnétique.
Pour contourner ces inconvénients, nous avons adopté dans ce présent travail la méthode PAW développé par Blochl [38] avec une approche pseudopotentiels formulée par Kresse et Joubert [39]. Elle repose sur les mêmes idées que les pseudopotentiels ultrasoft mais considère une grille radiale autour de chaque atome pour reconstruire la densité. Les liens entre pseudopotentiels ultrasoft et la méthode PAW sont exposés dans la Réf. 39.
La méthode PAW version US-PP est implémentée dans le programme VASP incluant une base de pseudopotentiels de type PAW pour chaque élément.
1.6.
Le formalisme PAW
Le but de ce paragraphe n’est pas de présenter en détail la méthode PAW (la Réf. 38 est un bon point de départ pour cela), ni même de présenter spécifiquement l’implantation dans le VASP (voir plutôt les références [38, 39]) mais d’en résumer les grands principes .
L’idée de la méthode PAW est née du souci de restreindre le grand nombre de fonctions de base pour décrire une fonction d’onde typique dans un solide. L’approche PAW consiste à utiliser deux sortes de fonctions de base : l’une utilise des fonctions d’onde atomiques partielles à l’intérieur des cœurs, et l’autre un ensemble de fonctions d’onde planes dans la région interstitielle. Il y a donc une séparation de l’espace en deux zones. La raison de cette séparation est le comportement du potentiel effectif vu par les électrons de valence : dans la région interstitielle, la densité électronique totale est faible. Par conséquent, quelques ondes planes suffiront à décrire les états de valence de manière satisfaisante.