Faculté des Sciences, 4 Avenue Ibn Battouta B.P. 1014 RP, Rabat – Maroc Tel +212 (0) 37 77 18 34/35/38, Fax : +212 (0) 37 77 42 61, http://www.fsr.ac.ma
N° d’ordre 2886
THÈSE DE DOCTORAT D’ETAT
Présentée par
Saloua SEBBAHI
Discipline : Chimie
Spécialité : Chimie Physique
Option : Matériaux et Environnement
CARACTERISTIQUES PHYSICO-CHIMIQUES ET
THERMOMECANIQUES DE LA LIGNINE AU COURS DE LA
CARBONISATION ET DE L’ACTIVATION
APPLICATION DE LA LIGNINE BRUTE ET DE SES CHARBONS
DERIVES EN TANT QU’ADSORBANTS
Soutenue le 14 Juillet 2016
Devant le jury
Président :
Souad EL HAJJAJI PES, Faculté des Sciences, Rabat
Examinateurs :
Abdelillah HAKAM PES, Faculté des Sciences, Rabat
Fatima KIFANI-SAHBAN PES, Faculté des Sciences, Rabat
Mohamed RAHOUTI PES, Faculté des Sciences, Rabat
Lotfi RGHIOUI PES, Faculté des Sciences, Meknès
Mhamed TAIBI PES, ENS de Takaddoum, Rabat
Dédicace
Je dédie cette modeste thèse
A la mémoire de mes parents, de mon frère Abdel Ghani et de ma sœur Rokia,
A mon mari Saïd qui m’a constamment encouragé
Je lui rends ici, un vibrant hommage pour les sacrifices qu’il a consentis sur le plan familial afin que notre aspiration commune soit concrétisée
A mes enfants Charaf et Maha que j’aime beaucoup,
A mes frères et leurs petites familles,
A ma belle famille, frères et sœurs,
Remerciements
Le travail présenté dans ce mémoire de thèse a été réalisé au sein du Laboratoire de Spectroscopie, Modélisation Moléculaire, Matériaux et Environnement (LS3ME) et du Laboratoire de Thermodynamique et Energétique à la Faculté des Sciences de Rabat sous la direction conjointe de Mesdames les Professeurs Souad EL HAJJAJI et Fatima KIFANI-SAHBAN.
Je tiens tout d’abord à remercier Monsieur le Professeur Mohamed BOUKALOUCH, Directeur du Laboratoire de Thermodynamique et Energétique pour l’accueil qu’il m’a réservé, le respect qu’il m’a toujours témoigné, ainsi que Madame le Professeur Oum Kaltoum KABBAJ, Directrice du LS3ME. Je tiens tout particulièrement à remercier Madame le Professeur Souad ZAYDOUN, pour m’avoir admise en 2011 au sein de son équipe.
J’ai le grand plaisir d’exprimer mes vifs remerciements à mes deux encadrantes, les Professeurs Fatima KIFANI-SAHBAN et Souad EL HAJJAJI, qui ont bien voulu assurer un suivi régulier et efficace de mes travaux de recherche. J’apprécie à leur juste valeur la justesse des analyses et interprétations dont j’ai bénéficié de leur part et je tiens à leur témoigner ma profonde gratitude.
J’adresse mes remerciements et mes respects à Monsieur Abdelillah HAKAM, Professeur à la Faculté des Sciences de Rabat et à Monsieur Lotfi RGHIOUI, Professeur à la Faculté des Sciences de Meknès d’avoir accepté d’être rapporteurs et membres de jury de cette thèse. Je les remercie pour l’intérêt qu’ils ont porté à mon travail.
Monsieur Mhamed TAIBI, Professeur à l’ENS de Takaddoum et Monsieur Mohamed RAHOUTI, Professeur à la Faculté des Sciences de Rabat, m’honorent en acceptant de participer au jury de thèse, d’examiner mon rapport et d’en évaluer la teneur. Qu’ils me permettent de leur exprimer mes vifs remerciements.
Je tiens tout particulièrement à remercier Monsieur le Professeur Amar KIFANI, pour l’aide précieuse qu’il a bien voulu m’apporter, pour sa gentillesse et ses encouragements.
Ce travail a nécessité la contribution de plusieurs professeurs en France, en Espagne, à l’extérieur de la Faculté et au sein de la Faculté des Sciences de Rabat. Je tiens à exprimer mes plus vifs remerciements et toute ma gratitude au Pr. André ZOULALIAN de l’Université de Nancy I, au Pr. Jessous ARAUZO de l’Université de Saragosse, au Pr. Abdelmaleik DAHCHOUR, de l’Institut Agronomique et Vétérinaire Hassan II et au Pr. Abdelouhab ZERIOUH de l’Ecole Royale Navale de Casablanca. Je remercie aussi mes collègues et amis de la Faculté des Sciences de Rabat les Professeurs Mohamed LFERD, Directeur du CEDOC, Afaf EL HAJJI, Hassna ABOU EL MAKARIM, Faiza HAJJI, Fouzia GUEDIRA, Malika SERGHINI-IDRISSI, Aziz LAGHZIZEL, Said ARSALANE, Adnane EL HAMIDI, Kamal GUERAOUI et Abdelkrim BOUGOUZA.
J’adresse mes remerciements les plus sincères aux Docteurs Samir MEN LA YAKHAF et Adil EL YADINI, aux doctorants Layla EL FAKIR, Maryam EL MAROUANI et Mohammed BOULHAOUA ainsi qu’au technicien Abdelkader ABDENBI pour leur disponibilité et pour tous les services qu’ils m’ont rendus.
Résumé :
Les travaux de ce mémoire traitent des aspects physico-chimiques et thermomécaniques qui accompagnent les réactions de carbonisation et de gazéification de la lignine impliquées dans la préparation du charbon actif par la méthode physique.
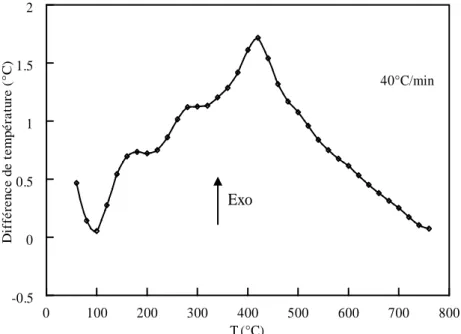
Le comportement thermique de la lignine a été suivi lors de sa carbonisation sous atmosphère inerte. La perte de masse de la lignine commence à partir de 180°C et se poursuit tant que la sollicitation thermique est maintenue. La perte de masse provoque une diminution de l’intensité des bandes des groupements fonctionnels initialement présents dans la lignine. La dégradation de la lignine est exothermique et s’accompagne d’une augmentation du volume qui provient de la formation d’une phase plastique par craquelures. La carbonisation de la lignine est contrôlée par la diffusion car la formation de la phase plastique empêche le dégagement des effluents gazeux et confère au matériau sa stabilité thermique.
Le carbonisât CL600 préparé pendant 2 heures à 600°C a été soumis et à la gazéification et à l’activation au CO2. La gazéification, suivie par thermogravimétrie en régime chimique, est
endothermique et s’effectue en une seule étape entre 640 et 800°C selon le mode de rétrécissement. Ce dernier est corroboré par les mesures des variations dimensionnelles. La température, la durée et la masse choisies pour effectuer l’activation de CL600 dans un lit fixe et dans des conditions favorables au développement de la micro, de la mésoporosité, de la surface spécifique et du maximum de groupements fonctionnels sont respectivement 700°C, 1 heure et 100 mg.
Pour améliorer le rendement de l’activation et réduire la résistance thermique des carbonisâts de la lignine, ceux-ci sont soumis à des traitements à l’air à des températures modérées. L’oxydation à l’air a un effet sur la perte de masse, sur la composition chimique, sur les variations dimensionnelles et sur la microstructure des carbonisâts de la lignine. La durée et la température retenues pour oxyder les carbonisâts sans les déstructurer sont respectivement 6 heures et 245°C. Dans ces conditions, l’analyse élémentaire indique que le contenu en oxygène de l’échantillon est augmenté et que sa perte de masse est inférieure à 8 %. L’apport en oxygène améliore la réactivité de l’échantillon vis-à-vis du CO2.
Le prétraitement à 245°C pendant 6 heures réduit de 50 % la durée d’activation. L’observation au MEB met en évidence la diminution des limitations physiques et l’évolution du matériau d’un comportement thermoplastique vers un comportement thermodurcissable. L’oxygénation à l’air des carbonisâts ne modifie pas le mode de déformation acquis par le matériau au cours de la carbonisation. Le prétraitement à l’air intensifie et améliore la résolution des bandes relatives aux différents groupements oxygénés sans pour autant atteindre l’intensité des bandes qui existaient initialement dans la lignine brute. Ces groupements, présents dans la lignine brute et dans son charbon actif (CAL) obtenu dans les conditions opératoires précitées, ont montré leur efficacité dans l’élimination du bleu de méthylène (BM) et de l’orange de méthyle (OM) en phase aqueuse. L’adsorption de BM sur CAL est comparable à celle sur CAC, alors que celle de OM est plus élevée. L’analyse IR a montré la fixation de BM et celle de OM sur le charbon actif de la lignine. Le processus d’adsorption est contrôlé par la chimisorption et les deux colorants sont adsorbés en multicouches sur des surfaces hétérogènes.
Mots clefs : Lignine, carbonisation, activation au dioxyde de carbone, pré-oxydation à l’air, charbon
Abstract :
This work addresses the physicochemical and thermomechanical aspects on the carbonization and gasification reactions of lignin during the preparation of activated carbon by physical method.
The thermal behavior of the lignin was followed during carbonization under an inert atmosphere. The mass loss of lignin starts from 180°C and continues as long as the thermal treatment is maintained. The mass loss causes a decrease in band intensity corresponding to the functional groups originally present in the lignin. The degradation of lignin is exothermic with an increase in volume that comes from the formation of a plastic phase by crazing. Carbonization of the lignin is diffusion controlled because of the plastic phase formation, which prevents the release of gases and gives the material its thermal stability.
The CL600 char prepared during 2 hours at 600°C and was subjected to the CO2 gasification and
activation. Gasification, followed by thermogravimetric in chemical system, is endothermic and is carried out in a single step between 640 and 800°C according to the restriction mode. This is corroborated by measurements of dimensional variations. The temperature, duration and the mass selected to perform CL600 activation in a fixed bed and in favorable conditions for the development of micro, mesoporosity, of the surface area and maximum functional groups are respectively 700°C, 1 hour and 100 mg.
To improve the yield of activation and reduce the thermal resistance of lignin char, they are subjected to treatment in air at moderate temperatures. The air oxidation has an effect on the mass loss, on the chemical composition, on the dimensional variation and the microstructure of lignin char. The time and temperature used to oxidize the lignin char without destruction are respectively 6 hours and 245°C. Under these conditions, the elemental analysis indicates that the oxygen content of the sample is increased and the mass loss is less than 8 %. The oxygen supply improves the reactivity of the sample towards CO2.
The pretreatment at 245°C for 6 hours reduces by 50 % the time of activation. The SEM observation highlights the decrease in physical limitations and evolution of material from a thermoplastic behavior to a thermosetting behavior. The air oxygenation of the lignin char does not alter the mode of deformation acquired by the material during the carbonization.
Pretreatment with the air increases and enhances the resolution of bands for the different oxygenated groups without reaching the intensity of the bands that originally existed in the raw lignin. These groups are present in the raw lignin and in its activated carbon obtained in the above operating conditions, give good efficiency in the elimination of methylene blue and methyl orange in aqueous solution. The adsorption of MB on CAL is comparable to that on CAC, while that of MO is higher. The IR analysis showed the adsorption of MB and MO on the activated carbon of lignin. The chemisorption process controls the adsorption and the two dyes are adsorbed in multilayers on heterogeneous surfaces.
Key words : Lignin, carbonization, activation by carbon dioxide, air pre-oxidation, activated carbon,
Introduction Générale ... 1
Références bibliographiques ... 4
Chapitre I La lignine Biosynthèse, Composition, Structure, Extraction et Propriétés I. Introduction ... 6
II. Description du bois ... 6
II.1. Matières extractibles ... 6
II.2. Matières minérales ... 6
II.3. Fraction aqueuse ... 7
II.4. Fraction organique ... 7
III. La lignine ... 8
III.1. Structure moléculaire de la lignine ... 8
III.2. Biosynthèse de la lignine ... 9
III.2.1. Structures hypothétiques de la lignine ... 14
III.2.2. Modèles moléculaires proposés pour la lignine ... 15
III.2.3. Géométrie de la lignine ... 16
III.2.4. Liaisons entre les unités de base de la lignine ... 17
III.2.5. Liaisons lignine-polysaccharides ... 17
III.3. Méthodes d’extraction de la lignine ... 17
III.3.1. Méthodes physiques ... 18
III.3.1.1. Extraction mécanique ... 18
III.3.1.2. Extraction par extrusion ... 18
III.3.2. Méthodes thermiques ... 18
III.3.2.1. Extraction à l’eau ou thermohydrolyse ... 18
III.3.2.2. Explosion à la vapeur ... 18
III.3.2.3. Explosion à l’ammoniac ... 18
III.3.2.4. Explosion au dioxyde de carbone ... 19
III.3.3. Méthodes chimiques ... 19
III.3.4. Extraction par des solvants organiques ... 19
III.3.4.1. Extraction par les alcools ... 20
III.3.4.2. Extraction par le 1,4-dioxane ... 20
III.3.4.3. Extraction par les acides ... 21
III.3.5. Méthodes biologiques ... 21
III.3.5.1. Extraction par hydrolyse enzymatique ... 21
III.3.5.2. Extraction par des agents biologiques ... 21
III.3.6. Méthodes d’extractions combinées ... 21
III.3.6.1. Extraction chimico-thermo-mécanique (CTMP) ... 21
III.3.6.2. Extraction en deux étapes ... 22
III.4. Propriétés de la lignine ... 22
III.5. Domaines d’applications de la lignine ... 22
IV. Conclusion ... 24
Références bibliographiques ... 25
Chapitre II Carbonisation de la lignine Analyses thermiques, Caractérisations et Cinétique I. Introduction ... 30
II. Etude bibliographique ... 30
II.1. Processus réactionnels proposés pour la pyrolyse de la lignine ... 31
II.2. Mécanismes de la dégradation thermique de la lignine ... 33
II.3. Produits de la dégradation thermique de la lignine ... 34
II.4. Effet de la méthode d’extraction sur la pyrolyse de la lignine ... 35
II.5. Pyrolyse des constituants de la lignine pris séparément ... 35
II.6. Cinétique de la pyrolyse de la lignine ... 37
III. Etude expérimentale ... 38
III.1. Matière première ... 39
III.2. Equipements expérimentaux ... 39
III.2.1. Appareil de mesure thermogravimétrique ... 39
III.2.2. Calorimétrie différentielle à balayage ... 40
III.2.3. Description du four ... 40
III.2.4. Procédure de carbonisation ... 41
III.2.5. Appareil d’analyse élémentaire ... 41
III.2.6. Méthode de mesure des variations dimensionnelles ... 41
III.2.7. Mesure des surfaces spécifiques ... 42
III.2.8. Caractérisation par diffraction des rayons X ... 42
III.2.9. Caractérisation par spectroscopie infrarouge ... 42
III.2.10. Caractérisation par microscopie électronique à balayage (MEB) ... 42
IV. Résultats et discussions ... 43
IV.1. Carbonisation de la lignine ... 43
IV.1.1. Analyse thermogravimétrique de la lignine ... 43
IV.1.2. Dérivée de la perte de masse de la lignine ... 45
IV.1.3. Analyse thermique différentielle de la lignine ... 46
IV.1.4. Superposition des tracés DTG et ATD ... 46
IV.1.5. Analyse par calorimétrie différentielle à balayage ... 47
IV.1.6. Comportement thermomécanique de la lignine ... 50
IV.1.7. Analyse par diffraction des rayons X ... 52
IV.1.8. Caractérisation par spectroscopie infrarouge ... 53
IV.1.8.1. Spectre infrarouge de la lignine brute ... 53
IV.1.8.2. Evolution des groupements fonctionnels au cours de la carbonisation ... 56
IV.2. Cinétique de la pyrolyse de la lignine ... 58
IV.2.1. Approche théorique ... 58
IV.2.2. Résultats ... 62
IV.2.2.1. Estimation des paramètres cinétiques par linéarisation de l’expression intégrale .... 62
IV.2.2.2. Intégration numérique de l’expression intégrale ... 63
V. Conclusion ... 68
Références bibliographiques ... 70
Chapitre III Activation au dioxyde de carbone des résidus de la carbonisation de la lignine Analyses thermiques, Caractérisations et Cinétique I. Introduction ... 77
II. Partie bibliographique ... 77
II.1. Production des charbons actifs ... 77
II.1.1. Matières premières utilisées pour la production des charbons actifs ... 78
II.1.2. Méthodes de préparation des charbons actifs ... 78
II.1.2.1. Activation chimique ... 78
II.1.2.2. Activation physique ... 79
II.1.2.2.1. Etape de carbonisation ... 79
II.1.2.2.2. Etape d’activation ... 80
II.1.2.3. Comparaison des deux méthodes ... 80
II.1.3. Types de charbon actif ... 81
II.2. Composition, structure et nature chimique de la surface des charbons actifs ... 81
II.2.1. Composition du charbon actif ... 81
II.2.3. Structure poreuse du charbon actif ... 83
II.2.4. Nature chimique de la surface des charbons actifs ... 84
II.3. Propriétés des charbons actifs ... 86
II.3.1. Tests physiques ... 86
II.3.2. Tests d’adsorption ... 87
II.3.3. Tests chimiques et physico-chimiques ... 87
II.4. Techniques pour tester la structure poreuse des charbons actifs ... 88
II.4.1. Volume total des pores, densité réelle et apparente ... 88
II.4.2. Porosimétrie de mercure ... 89
II.4.3. Spectroscopie infrarouge ... 89
II.4.4. Méthode de diffraction des rayons X ... 89
II.4.5. Microscope électronique ... 90
II.5. Applications des charbons actifs ... 90
II.6. Régénération des charbons actifs ... 92
II.7. Cinétique de la réaction de gazéification ... 92
II.7.1. Réactions de gazéification du charbon résidu solide de la carbonisation ... 92
II.7.1.1. Réactions char-oxygène ... 93
II.7.1.2. Réaction char-vapeur d’eau ... 93
II.7.1.3. Réaction char-dioxyde de carbone ... 93
II.7.1.4. Réaction char-hydrogène ... 94
II.7.2. Modèles cinétiques ... 94
II.7.2.1. Cas des particules initialement non poreuses ... 94
II.7.2.1.1. Modèle à cœur rétrécissant de taille invariable ... 95
II.7.2.1.2. Modèle à cœur rétrécissant de taille décroissante ... 95
II.7.2.2. Cas des particules initialement poreuses ... 96
II.7.2.2.1. Transformation complète des particules poreuses ... 96
II.7.2.2.2. Transformation des particules poreuses sans changement de volume ... 97
III. Etude expérimentale ... 98
III.1. Substance traitée ... 98
III.2. Appareil de mesure thermogravimétrique ... 98
III.3. Procédure d’activation ... 98
IV. Résultats et discussions ... 99
IV.1. Gazéification du carbonisât de la lignine ... 100
IV.2 Cinétique de la gazéification ... 100 IV.2.1. Estimation des paramètres cinétiques par la linéarisation de l’expression intégrale . 101
IV.2.2. Intégration numérique de l’expression intégrale ... 101
IV.3. Activation des carbonisâts de la lignine ... 102
IV.3.1. Effet de la température de carbonisation sur le rendement de l’activation ... 102
IV.3.2. Effet de la masse initiale sur le rendement de l’activation ... 103
IV.3.3. Evolution du rendement de l’activation avec la température et la durée de l’activation ... 104
IV.3.4. Variation des dimensions des carbonisâts de lignine avec la température et la durée de l’activation ... 105
IV.3.5. Caractérisation par microscopie électronique à balayage ... 106
IV.3.6. Caractérisation par spectroscopie infrarouge des carbonisâts activés ... 108
IV.3.6.1. Effet de la température de carbonisation sur les groupements fonctionnels des charbons activés ... 108
IV.2.6.2. Effet de la température et du temps d’activation sur les groupements fonctionnels des charbons activés ... 109
IV.3.6.3. Températures et durées de carbonisation et d’activation optimales ... 110
V. Conclusion ... 111
Références bibliographiques ... 112
Chapitre IV Pré-oxydation à l’air du charbon de la lignine suivie de l’activation au dioxyde de carbone I. Introduction ... 120
II. Etude bibliographique ... 120
II.1. Effet de la pré-oxydation sur la composition chimique ... 120
II.2. Effet de la pré-oxydation sur la structure poreuse ... 121
II.3. Effet de la pré-oxydation sur les propriétés plastiques ... 122
II.4. Mécanisme de l’oxydation du charbon ... 123
III. Etude expérimentale ... 124
III.1. Substance traitée ... 124
III.2. Procédure de la pré-oxydation ... 125
III.3. Méthodes de caractérisation ... 125
IV. Résultats et discussions ... 125
IV.1. Perte de masse au cours de la pré-oxydation ... 125
IV.2. Composition élémentaire au cours de la pré-oxydation ... 126
IV.3. Variation des dimensions des carbonisâts prétraités avec la température et la durée de la pré-oxydation ... 127
IV.4. Caractérisation par spectroscopie infrarouge des charbons pré-traités ... 128
IV.5. Caractérisation par microscopie électronique à balayage des charbons pré-traités ... 131
IV.6. Activation des carbonisâts pré-oxydés ... 133
IV.6.1. Evolution du rendement de l’activation avec la température et la durée de la pré-oxydation ... 133
IV.6.2. Variation des dimensions des charbons pré-traités et activés avec la température et la durée de la pré-oxydation ... 135
IV.6.3. Caractérisation infrarouge des charbons pré-traités puis activés ... 137
IV.6.3.1. Effet de la température et de la durée de pré-oxydation sur les groupements fonctionnels des charbons pré-traités puis activés ... 137
IV.6.3.2. Effet de la durée d'activation sur les groupements fonctionnels des charbons pré-traités ... 138
IV.6.4. Caractérisation par MEB des charbons pré-traités puis activés ... 140
IV.6.4.1 Effet du temps de pré-oxydation sur la morphologie des carbonisâts ... 140
IV.6.4.2 Effet du temps d’activation sur la morphologie des carbonisâts ... 142
V. Conclusion ... 144
Références bibliographiques ... 145
Chapitre V Élimination du bleu de méthylène et de l’orange de méthyle à partir de solution aqueuse par adsorption sur la lignine brute et sur ses charbons dérivés Caractérisations, Cinétiques et Isothermes d’adsorption I. Introduction ... 149
II. Etude bibliographique ... 149
II.1. Généralités sur la pollution ... 149
II.2. Colorants ... 149
II.3. Méthodes de dépollution ... 153
II.3.1. Phénomène d’adsorption ... 153
II.3.2. Mécanisme de l’adsorption ... 155
II.3.3. Principaux types d’adsorbants ... 155
II.3.4. Facteurs influençant le phénomène d’adsorption ... 156
II.3.5. Cinétique d’adsorption ... 157
II.3.6. Isothermes d’adsorption ... 158
II.3.7. Modèles d’isotherme d’adsorption ... 159
III. Etude expérimentale ... 161
III.1. Caractéristiques des adsorbats ... 161
III.1.1. Bleu de méthylène ... 162
III.1.2. Orange de méthyle ... 162
III.3. Mesure de la charge de surface de la lignine brute ... 163
III.4. Méthodologie de la Recherche Expérimentale ... 163
III.5. Protocole expérimental de la cinétique d’adsorption ... 164
III.6. Protocole expérimental des isothermes d’adsorption ... 165
IV. Résultats et discussions ... 165
IV.1. Etude expérimentale à l’aide de la MRE ... 165
IV.2. Cinétique d’adsorption ... 167
IV.2.1. Adsorption de BM et OM sur la lignine brute et sur ses charbons dérivés ... 167
IV.2.2. Caractérisation infrarouge des résidus solides de l’adsorption ... 170
IV.2.3. Modélisation de la cinétique d’adsorption ... 171
IV.3. Isothermes d’adsorption ... 173
V. Conclusion ... 176
Références bibliographiques ... 177
Conclusion Générale ... 180
Liste des abréviations H : p-hydroxy-phényle G : guaïacyle S : syringyle Coll. : collaborateurs C-C : liaison carbone-carbone C-O-C : liaison éther
O-Me : O-méthyle
CIMV : compagnie industrielle de la matière végétale DAEM : modèle de distribution de l’énergie d’activation FB : formule brute
PCS : pouvoir calorifique supérieur ATG : analyse thermogravimétrique
DTG : dérivée de l’analyse thermogravimétrique ATD : analyse thermique différentielle
DSC : calorimétrie différentielle à balayage
CL400 : carbonisât de la lignine obtenu à 400°C pendant 2 heures sous azote CL600 : carbonisât de la lignine obtenu à 600°C pendant 2 heures sous azote CL800 : carbonisât de la lignine obtenu à 800°C pendant 2 heures sous azote Act : activation
BO : rendement de l’activation ou taux d’activation appelé burn-off, 100 m m -m %BO c ca c
mc: masse du charbon avant l’activation
mca: masse après activation
Tact et tact : température et temps d’activation PO : pré-oxydation
Tpo et tpo : température et temps de pré-oxydation VD : variations dimensionnelles
J : jacobien de la transformation. J=V/V0
V : volume de l’échantillon à un instant t V0: volume initial
Jact : jacobien de la transformation au cours de l’activation Jpo : jacobien de la transformation au cours de la préoxydation RX : rayons X
IR : infrarouge
ATR : réflexion totale atténuée
FTIR : spectrométrie infrarouge par transformée de Fourier MEB : microscope électronique à balayage
MET : microscope électronique à transmission Tg : température de transition vitreuse
PEEK : poly-éther-éther-cétone
: vibration d’élongation ou de valence : vibration de déformation dans le plan : vibration de déformation hors du plan ass : vibration de valence associée as: vibrations de valence asymétrique
s: vibrations de valence symétrique
: degré d’avancement de la réaction,
f 0 t 0 m m m m α 0 m : masse initiale t m : masse à l’instant t
f() : mode de dégradation de la substance )
(
g : la forme intégrale de la fonction f(α)
K : constante de vitesse de la réaction, ) RT
E Aexp( k
E : énergie d’activation apparente (kJ/mol)
R : constante des gaz parfaits (8,314 Joules. K-1. mol-1)
A : facteur pré-exponentiel ou facteur de fréquence (min-1)
T : température absolue (K) T=T0+ t
T0: température initiale
: vitesse de montée en température dans le temps FWO : Flynn-Wall-Ozawa
KAS: Kissinger-Akahira-Sunose MD : mode de dégradation CC: coefficient de corrélation USB : unité structurale de base OM : orientation moléculaire CAP : charbon actif en poudre CAG : charbon actif en grain CAT : charbon actif en tissu CAE : charbon actif extrudé
IUPAC : Union Internationale de la Chimie Pure et Appliquée Ph-COOH : groupes carboxylique
Ph-OH : phénol O=Ph=O : quinone ph-C=OO-Ph’ : lactone Ph-(C=O-O-O=C) : anhydride Ph(-O-)Ph’ : éther Ph-C=O : carbonyle dHe: densité d’Hélium dHg: densité de mercure
R2 et R3 : modes de dégradation relatifs à la contraction respectivement de surface et de volume D3 et D8 : modes de dégradation relatifs à la diffusion ‘‘Jander’’ respectivement à trois et à deux dimensions
FSI : indexe de gonflement libre UV-Visible : ultra violet visible
1
k ,
k
2 etk
d: constantes de vitesse respectivement pour l’équation cinétique pseudo-premier ordre (min-1), pseudo-second ordre (g.mg-1.min-1) et diffusion intraparticulaire (mg/g min1/2),I : interception (mg/g).
Isotherme type S, dite ‘sigmoïdale’ Isotherme type L, dite de ‘Langmuir’ Isotherme type H, dite de ‘haute affinité’ Isotherme type C, dite de ‘partition constante’
qe: quantité adsorbée à l’équilibre par unité de masse de l’adsorbant (mg/g),
qm: quantité maximale adsorbée (mg/g),
t
q
: quantité adsorbée à un instant t par unité de masse de l’adsorbant (mg/g) Ce: concentration résiduelle à l’équilibre (mg/L)C0: concentration initiale (mg/L)
T : température absolue (K)
Q : variation d’énergie d’adsorption (J.mol-1)
Cs: solubilité du soluté
: constante reliée à l’énergie d’adsorption par E=1/0,5,
: exposant variable 1,5-3 pour les charbons actifs, n : paramètre de Freundlich
kL: constante de Langmuir (L.mg-1),
kF: constante de Freundlich relative à la capacité d’adsorption (mg(1-n)Ln.g-1),
kE: constante d’Elovich (L.mg-1),
kT: constante de Temkin (L.mg-1).
RL: constante adimensionnelle ou facteur de séparation ou paramètre d’équilibre
1/n : facteur d’hétérogénéité BM : Bleu de méthylène OM : orange de méthyle LB : lignine brute
CL : charbon de la lignine CAL : charbon actif de la lignine CAC : charbon actif commercial
MRE : méthodologie de la recherche expérimentale pHPCN: point de charge nulle
Numéro Titre de la figure page
Figure I.1 Structure moléculaire de la cellulose 8
Figure I.2 Structure moléculaire de l’hémicellulose 8 Figure I.3 Voies de biosynthèse des phényl-propanoïdes de la lignine 11 Figure I.4 Les trois principaux monolignols qui donnent naissance à la lignine :
alcool p-coumarylique; (2) alcool coniférylique; (3) alcool sinapylique 12 Figure I.5 Déshydrogénation de l’alcool coniférylique 12 Figure I.6 Représentation simplifiée des trois unités de base des lignines et de leurs précurseurs 12 Figure I.7 Délocalisation des radicaux obtenus par déshydrogénation enzymatique. Les radicaux
instables sont encadrés en pointillés 13
Figure I.8 Principales liaisons éther et carbone-carbone majoritairement présentes dans la lignine (R1= R2= H dans l’unité p-hydroxyphényl ; R1= OMe , R2= H dans l’unité
guaiacyl ; R1= R2= OMe dans l’unité syringyl)
13 Figure I.9 Structure de la lignine selon Freudenberg 14
Figure I.10 Structure de la lignine selon Adler 14
Figure I.11 Structure de la lignineissue du procédé papetier Kraft 15 Figure I.12 Modèle moléculaire de la lignine (Blanc = H; gris = C; noir = O) 16 Figure I.13 Structures chimiques des différents fragments de polymère de lignine de paille de blé
extraite par le procédé CIMV 16
Figure I.14 Les différentes liaisons lignine-carbohydrates 17 Figure I.15 Représentation schématique de la lignine kraft et Lignosulfonates 19 Figure I.16 Représentation schématique de la lignine organosolve 20 Figure II.1 Dégradation de la lignine par deux processus réactionnels 32
Figure II.2 Formation de radicaux 32
Figure II.3 Addition radicalaire 32
Figure II.4 Réactions de condensation de l’unité G 32 Figure II.5 Processus réactionnel pour la dégradation de la lignine à plusieurs étapes 33 Figure II.6 Energie de liaison (J/mol) des principales liaisons chimiques présentes dans la
structure de la lignine 34
Figure II.7 Exemple de molécules dimères type guaiacyl de la lignine 36 Figure II.8 Domaine de température des réactions de condensation, dépolymérisation et
carbonisation 37
Figure II.9 Schéma du four 40
Figure II.10 Tracé ATG de la lignine 44
Figure II.11 Tracés ATG et DTG de la lignine 45
Figure II.12 Tracé ATD de la lignine 46
Figure II.13 Tracés DTG et ATD de la lignine 47
Figure II.14 Thermogramsme DSC de la lignine, premier réchauffement 49 Figure II.15 Thermogramme DSC du carbonisât de la lignine, deuxième réchauffement 49 Figure II.16 Evolution du Jacobien de la transformation de la lignine en fonction de la
température 51
Figure II.17 Images prises par MEB de la lignine traitée à différentes températures 51 Figure II.18 Diffractogrammes des rayons X de la lignine et des carbonisâts de lignine 52 Figure II.19 Spectre infrarouge de la lignine brute 55 Figure II.20 Spectres infrarouges de la lignine brute et des résidus obtenus à 400, 600 et 800°C 57 Figure II.21 Taux de conversion expérimentaux et calculés pour les différents intervalles de
décomposition de la lignine 66
Figure III.1 Unité structurale de base (USB) 79
Figure III.3 Comparaison du réseau cristallin tridimensionnel
(a) graphite et (b) structure turbostratique 83 Figure III.4 Illustration schématique de la structure d’un charbon actif
(a) charbon qui a facilement subi la graphitisation, (b) charbon qui a subi la graphitisation à un degré faible
83 Figure III.5 Modèle de fragment de la surface du charbon actif oxydé proposé par Tarkovskya et
coll. 85
Figure III.6 Modèle de fragment de la surface du charbon actif proposé par Fanning et Vannice 86 Figure III.7 Evolution dans le temps d’une particule non poreuse, avec cendres adhérentes, sans
changement de volume 95
Figure III.8 Evolution dans le temps d’une particule non poreuse, sans cendres adhérentes 95 Figure III.9 Gazéification d’un solide poreux où la dimension externe de la particule rétrécit 97 Figure III.10 Réaction d'un réactif solide poreux avec un gaz pour former un produit solide poreux 97 Figure III.11 Evolution de la température du four et les gaz utilisés durant l’activation 99 Figure III.12 Analyses thermiques du carbonisât de la lignine : ATG (a), DTG (b) et ATD (c) 100 Figure III.13 Evolution du burn-off de CL400, CL600 et CL800 avec la durée de l’activation 103 Figure III.14 Evolution de BO de CL600 en fonction du temps d’activation pour trois masses
initiales différentes 104
Figure III.15 Effet de la température (a) et du temps (b) d’activation sur le rendement de
l’activation de CL600 105
Figure III.16 Evolution du Jacobien de transformation de CL600 en fonction de la température (a)
et du temps (b) d’activation 106
Figure III.17 Fractographies relatives à la face externe et interne de CL600 et de CL600 activé à
700°C pendant 30, 45 et 60 minutes 107
Figure III.18 Spectres infrarouges de CL400, CL600 et CL800 (a) et des mêmes carbonisâts activés à 700°C pendant 1h sous CO2(b)
108 Figure III.19 Spectres infrarouges de CL600 activé à différentes températures pendant 1h (a) et à
700°C pendant 30’, 45’ et 60’ (b) 109
Figure III.20 Images prises par MET de CL600 et de CL600 activé 110 Figure IV.1 Processus réactionnel schématique de la transformation des péroxydes en
groupements fonctionnels oxygénés 124
Figure IV.2 Effet de la température (a) et du temps (b) de pré-oxydation sur la masse de CL600 126 Figure IV.3 Analyse élémentaire des carbonisâts pré-oxydés 127 Figure IV.4 Jacobien de la transformation et perte de masse (a), rapports atomiques O/C et H/C
(b) de CL600 en fonction de la température de pré-oxydation 128 Figure IV.5 Spectres infrarouges de CL600 et de CL600 pré-traité à 140°C et 245°C pendant 6h 130 Figure IV.6 Spectres infrarouges de CL600 et de CL600 pré-traité à 140°C pendant 6h et 9h (a) et
à 245°C pendant 6h et 24h (b) 130
Figure IV.7 Fractographies de la face interne et externe de CL600 et des carbonisâts pré-oxydés 132 Figure IV.8 Rendements de l’activation en fonction de la température (a) et du temps (b) de
pré-oxydation 134
Figure IV.9 Evolution du burn-off de CL600 et de CL600 pré-oxydé en fonction du temps
d’activation 134
Figure IV.10 Jacobien de la transformation de CL600 en fonction de la température de
pré-oxydation 136
Figure IV.11 Jacobien de la transformation de CL600 en fonction du temps de pré-oxydation à
140°C (a) et à 245°C (b) 136
Figure IV.12 Evolution en fonction du temps d’activation du burn-off et du Jacobien de la transformation de CL600 et de CL600 pré-oxydé 137 Figure IV.13 Spectres infrarouges de CL600 pré-oxydé puis activé 138 Figure IV.14 Evolution des groupements fonctionnels avec la durée d’activation 139
CL600 pré-oxydé puis activé
Figure IV.17 Fractographies de la face interne et externe de CL600 activé à 700°C pendant 30, 45 et 60 minutes et de CL600 pré-oxydé à 245°C pendant 6 h puis activé dans les mêmes conditions
143 Figure V.1 Formules chimiques de certains colorants 152 Figure V.2 Domaines d’existence d’un soluté lors de l’adsorption sur un matériau microporeux
d'après Weber et Smith 155
Figure V.3 Classification des isothermes d’adsorption selon Giles et coll. 159 Figure V.4 Caractéristiques du bleu de méthylène 162 Figure V.5 Caractéristiques de l’orange de méthyle 163 Figure V.6 Diagramme Pareto de l’adsorption de BM (a) et de OM (b) sur LB 166 Figure V.7 Adsorption de BM (a) et OM (b) sur LB, CL, CAL et CAC 169 Figure V.8 Images prises par MET des charbons dérivés de la lignine 169 Figure V.9 Spectres infrarouges de la lignine brute et des charbons dérivés 170 Figure V.10 Spectres IR de BM (a), OM (a’) et de CAL avant (b) et après adsorption de BM (c) et
de OM (c’) 171
Figure V.11 Cinétique d’adsorption de pseudo premier-ordre de BM (a) et de OM (b) sur LB et
sur CAL 172
Figure V.12 Cinétique d’adsorption de pseudo second-ordre de BM (a) et de OM (b) sur LB et sur
CAL 172
Figure V.13 Tracés de la diffusion intraparticulaire pour l’adsorption de BM (a) et de OM (b) sur
LB et sur CAL 172
Figure V.14 Isothermes d’adsorption de BM (a) et OM (b) sur LB 173 Figure V.15 Isothermes d’adsorption de BM (a) et OM (b) sur CAL 174 Figure V.16 Isothermes de Langmuir (a) et de Freundlich (b) pour l’adsorption de BM sur LB et
sur CAL 175
Figure V.17 Isothermes de Langmuir (a) et de Freundlich (b) pour l’adsorption de OM sur LB et
sur CAL 175
Figure A.1 Thermogramme DSC de la lignine, 1erréchauffement 185
Figure A.2 Thermogramme DSC du carbonisât de la lignine, 2èmeréchauffement 185
Figure A.3 Principaux types de sites acides de groupes fonctionnels d’oxygène
(a) carboxyle, (b) phénolique, (c) quinonoïde, (d) lactone normale, (e) lactone type fluorescéine et (f) anhydride provenant de groupes carboxyles voisins
186 Figure A.4 Groupes fonctionnels de caractère basique, (a) chromène, (b) pyrone 186 Figure A.5 Jacobien de la transformation (Jpo) et perte de masse de CL600 en fonction de la
température de pré-oxydation 188
Liste des tableaux
N° Tableau Titre du tableau Page
Tableau I.1 Proportions des principaux constituants du bois dans les feuillus et dans les résineux
en % par masse de biomasse sèche 7
Tableau I.2 Composition élémentaire du bois, de la cellulose et de la lignine 8
Tableau I.3 Modes de couplage des radicaux phénoxy 10
Tableau I.4 Exemples de proportion des principales liaisons éther et carbone-carbone dans la
lignine du bois tendre et du bois dur 11
Tableau II.1 Pyrolyse des composés comparables à ceux qui existent dans la lignine 35 Tableau II.2 Pyrolyse des monomères ou dimères qui constituent la lignine 36 Tableau II.3 Paramètres cinétiques relatifs à la pyrolyse de la lignine 38 Tableau II.4 Analyse élémentaire, formule brute et pouvoir calorifique de la lignine avant et après
pyrolyse ( = 5°C/min)
39 Tableau II.5 Analyse globale de la lignine avant et après pyrolyse ( = 5°C/min) 39 Tableau II.6 Intervalle de dégradation de la cellulose, de l’hémicellulose et de la lignine 44 Tableau II.7 Attributions des vibrations fondamentales de la lignine et des carbonisâts de lignine 54 Tableau II.8 Modes de dégradation thermique des solides 61
Tableau II.9 Expressions intégrales 61
Tableau II.10 Paramètres cinétiques de la pyrolyse de la lignine 64 Tableau II.11 Paramètres cinétiques expérimentaux et calculés 67 Tableau III.1 Facteurs ayant une influence sur le processus de la carbonisation 80
Tableau III.2 Différents types de charbon actif 81
Tableau III.3 Principales caractéristiques des pores du charbon actif 84 Tableau III.4 Différentes applications du charbon actif 91 Tableau III.5 Réactions fondamentales de la gazéification du charbon 93
Tableau III.6 Analyses élémentaire de CL600 98
Tableau III.7 Paramètres cinétiques de la gazéification de CL600 101 Tableau III.8 Paramètres cinétiques de la gazéification de CL600 102 Tableau III.9 Valeurs des surfaces spécifiques de CL600 et de CL600 activé 110
Tableau IV.1 Analyse élémentaire de CL600 124
Tableau IV.2 Effet de la pré-oxydation sur le rendement de l’activation 134 Tableau V.1 Principaux groupes chromophores et auxochromes 150 Tableau V.2 Distinction entre l’adsorption physique et chimique 154 Tableau V.3 Modèles cinétique du pseudo-premier ordre, pseudo-second ordre et diffusion
intraparticulaire 158
Tableau V.4 Lois et modèles exprimant l’isotherme d’adsorption 160 Tableau V.5 Domaines expérimentaux des colorants BM et OM 164 Tableau V.6 Plan d’expérience des colorants BM et OM 164 Tableau V.7 Matrice d’expérience 23et la réponse étudiée (R%) 165
Tableau V.8 Coefficients des facteurs ayant une influence sur l’adsorption des colorants sur LB 166 Tableau V.9 Taux de rétention et temps d’équilibre des différents matériaux dans le cas du bleu de
méthylène et de l’orange de méthyle 169
Tableau V.10 Surfaces spécifiques de la lignine brute et des charbons dérivés 170
Tableau V.11 Paramètres des modèles cinétiques 173
Tableau V.12 Constantes des modèles de Langmuir et de Freundlich 175
Tableau A.1 Principales voies de pré-traitement 184
Tableau A.2 Attributions des vibrations fondamentales de la lignine brute, des carbonisâts de la lignine et des carbonisâts activés sous CO2à 700°C pendant 1h
189 Tableau A.3 Attributions des vibrations fondamentales de CL600, des carbonisâts activés à 700°C
pendant 30, 45 et 60 min et des mêmes activâts précédés d’une pré-oxydation 190 Tableau A.4 Attributions des vibrations fondamentales de CL600 et de CL600 pré-traité 191
1
Les travaux de recherche de notre laboratoire s’intéressent depuis plusieurs années à la valorisation en charbon actif des matières fossiles, des déchets lignocellulosiques [1-6] et synthétiques [7-14]. Ces travaux traitent essentiellement des aspects physico-chimiques et thermomécaniques qui accompagnent les réactions de carbonisation et de gazéification impliquées dans la préparation du charbon actif par la méthode physique. Cette orientation des travaux s’est imposée suite au lien mis justement en évidence entre les aspects physico-chimiques et thermomécaniques, lien qui contribue à mieux appréhender les mécanismes à l’origine de la formation du résidu de la carbonisation et de la stabilité thermique de celui-ci [1, 4, 5]. Les aspects précédemment cités n’ont pas fait l’objet de travaux dans la littérature et constituent, de ce fait, l’originalité des travaux de notre laboratoire. Nos recherches en cours et futures visent à améliorer le rendement en charbon, à réduire la stabilité thermique des carbonisâts lors de la gazéification et à enrichir la chimie de surface par des traitements sous air soit avant soit après carbonisation. Le suivi de l’évolution des caractéristiques physico-chimiques et thermomécaniques lors de tout traitement ultérieur à la carbonisation est primordial puisqu’il permet de déterminer les conditions optimales favorisant la formation des groupements fonctionnels nécessaires dans la dépollution par adsorption des eaux chargées en colorants.
Certains de ces objectifs ont déjà été atteints, puisque les travaux effectués sur le bois d’eucalyptus et sur son charbon actif montrent que la durée d’activation est réduite de 25 et de 50 % lorsque le bois est traité sous air respectivement avant et après carbonisation [15-17]. En outre, le bois et ses charbons dérivés de la méthode physique ont prouvé leur efficacité dans l’adsorption de colorants synthétiques, de substances aromatiques et de métaux en phase aqueuse [18, 19].
Le précurseur proposé pour préparer du charbon actif dans ce travail est le principal sous-produit de la délignification du bois. Il s’agit de la lignine qui se trouve dans la liqueur noire rejetée en grande quantité par les industries de pâte à papier. La production mondiale de liqueur noire est d’environ 50 millions de tonnes par an [20]. Seuls 10 % servent de combustible et le reste n’est pas valorisé [20]. Pourtant, la tendance actuelle de valorisation des déchets lignocellulosiques qui s’inscrit, entre autres, dans le cadre du développement durable et de préservation des écosystèmes suggère de tirer profit de la liqueur noire.
Précisons que la lignine est un polymère naturel et renouvelable. C’est l’un des constituants de la fraction organique du bois et le deuxième polymère carboné le plus abondant dans la nature après la cellulose. La lignine est source de produits chimiques, d’énergie et peut être valorisée, comme il est proposé dans ce travail, en adsorbant, en charbon et en charbon actif. A notre connaissance, l’élimination de plusieurs types de polluants en phase aqueuse par la lignine brute et par son charbon dérivé de
l’activation chimique a déjà fait l’objet de nombreux travaux [21-31], ce qui n’est pas le cas du charbon dérivé de l’activation physique qui est examiné dans ce travail. C’est encore une originalité et une contribution modeste aux recherches faites sur la dépollution des eaux. Au Maroc et à l’heure où une prise de conscience sur les problèmes de pollution de l’air, de l’eau et des sols se manifeste, la dépollution des eaux peut offrir la possibilité de réutilisation des eaux usées épurées notamment dans l’industrie et l’agriculture et peut par conséquent répondre à la problématique de l’eau potable qui devient de plus en plus rare et chère. Notons tout de même que l’intérêt du charbon actif n’est plus à démontrer dans la dépollution des eaux. En revanche, diversifier ses précurseurs, améliorer son rendement et ses caractéristiques sont d’un grand intérêt.
Les différents objectifs mentionnés précédemment nous ont conduits à subdiviser ce mémoire en cinq chapitres.
La biosynthèse, la structure, la géométrie et les méthodes d’extraction ainsi que les propriétés et les domaines d’application de la lignine sont décrits au premier chapitre. Le deuxième chapitre est relatif à la carbonisation de la lignine en atmosphère inerte. Les produits, les processus, les mécanismes et cinétique de la réaction précitée sont donnés dans la partie bibliographique de ce chapitre. Ensuite, dans la partie expérimentale et après la description des dispositifs et équipements expérimentaux utilisés, nous procédons à l’analyse des résultats de perte de masse, de thermicité, de variations dimensionnelles, de mode de dégradation et des caractérisations par IR, RX et MEB.
L’activation des résidus de la carbonisation est traitée au chapitre III. Les informations sur les différents précurseurs, les méthodes de préparation, de caractérisation, la structure, la porosité et la chimie de surface du charbon actif font l’objet de la synthèse bibliographique. La gazéification et l’activation du carbonisât de la lignine sous dioxyde de carbone par thermogravimétrie et en lit fixe respectivement sont traités dans la partie expérimentale. Les résultats analysés concernent les mesures de perte de masse, les variations dimensionnelles, le mode de dégradation, les surfaces spécifiques, la chimie de surface et la caractérisation aux microscopes électroniques et à balayage et à transmission.
Le quatrième chapitre est relatif au pré-traitement à l’air du carbonisât de la lignine suivi de l’activation au dioxyde de carbone. La synthèse bibliographique traite de l’effet de la pré-oxydation sur la composition chimique, la structure poreuse et les propriétés plastiques du charbon. Dans la partie expérimentale, nous examinons d’abord l’effet du pré-traitement à l’air sur la masse, la composition élémentaire, les dimensions, les groupements fonctionnels et la morphologie de surface des carbonisâts
3
de la lignine. Ensuite, nous comparons le comportement des carbonisâts non prétraités à celui des carbonisâts prétraités lors de l’activation. Les caractérisations des échantillons par IR, MEB et MET ont été faites pour les échantillons prétraités et prétraités puis activés.
L’élimination de deux colorants en solution aqueuse par la lignine et par ses charbons dérivés fait l’objet du chapitre V. La partie bibliographique présente les principales pollutions des eaux, les différentes méthodes de dépollution ainsi que le principe, le mécanisme et les isothermes d’adsorption. Dans la partie expérimentale, nous donnons les descriptions des caractéristiques des adsorbants et des adsorbats examinés et les protocoles expérimentaux de la cinétique et des isothermes d’adsorption. Les trois paramètres pris en considération pour effectuer les tests d’adsorption sont la masse de l’adsorbant, la concentration initiale de l’adsorbat et le temps de contact. Les conditions optimales des paramètres précités sont déterminées par une méthodologie de la recherche expérimentale basée sur les plans d’expérience. Les résultats sont présentés sous forme de comparaison entre les pouvoirs de rétention de la lignine brute et de ses charbons dérivés vis-à-vis du bleu de méthylène et de l’orange de méthyle. Une modélisation est proposée pour l’adsorption de ces deux colorants sur les adsorbants précités.
L’essentiel des résultats qui se dégagent des cinq chapitres de ce mémoire et les perspectives à venir sont présentés dans la conclusion.
Références bibliographiques
1- F. Kifani-Sahban, ‘Etude des aspects physiques et physico-chimiques de la pyrolyse lente de l’eucalyptus et des principaux constituants du bois’, Thèse de Doctorat d’Etat, Faculté des Sciences Rabat (1997).
2- F. Kifani-Sahban, L. Belkbir et A. Zoulalian, ‘Etude de la pyrolyse lente de l'eucalyptus marocain par analyse thermique’, Thermochimica Acta 284 (1996) 341-349.
3- F. Kifani-Sahban, L. Belkbir et A. Zoulalian, ‘Détermination des paramètres cinétiques de la pyrolyse lente de l’eucalyptus marocain’, Thermochimica Acta 289 (1996) 33-40.
4- F. Kifani-Sahban, A. Kifani, L. Belkbir, A. Zoulalian, J. Arauso and T. Cordero, ‘A physical approach in the understanding of the phenomena accompanying the thermal treatment of lignin’, Thermochimica Acta 298 (1997) 199-204.
5- F. Kifani-Sahban, A. Kifani, L. Belkbir, S. Bouhlassa, A. Zoulalian, J. Arauso et T. Cordero, ‘Variations dimensionnelles accompagnant le traitement thermique de la cellulose sous atmosphere inerte’, Thermochimica Acta 307 (1997) 135-141.
6- S. Sebbahi, M. Yaouiss, R. Rahho, F. Kifani-Sahban, M. Boukallouch et A. Kifani, ‘Gazéification du charbon de l’eucalyptus en présence de la vapeur d’eau’, Sciences-Letters 4(1) (2002).
7- M. Yaouiss, ‘Etude des mécanismes de la stabilité thermique du poly-éther-éther-cétone lors de sa pyrolyse et l’influence de cette caractéristique sur l’activation des résidus de la pyrolyse’, Thèse Nationale, Faculté des Sciences Rabat (2000).
8- M. Yaouiss, F. Kifani-Sahban, A. Zeriouh, L. Belkbir, T. Cordero et J. Arauzo, ‘Etude de l’influence des cycles de traitement thermique sur les mesures de perte de masse et des variations dimensionnelles du PEEK’, Journal de la Société Ouest Africaine de Chimie,
SOACHIM 007 (1999) 108-118.
9- M. Yaouiss, F. Kifani-Sahban, M. Boukallouch, A. Kifani, A. Zeriouh, L. Belkbir et J. Arauzo, ‘Mesure des variations de volume du PEEK soumis à un traitement thermique sous atmosphère inerte’, Thermochimica Acta 345 (2000) 47-52.
10- M. Yaouiss, F. Kifani-Sahban, A. Kifani, A. Zeriouh, L. Belkbir, T. Cordero et J. Arauzo, IIèmes Journées sur les Polymères Organiques et leurs Applications Industrielles, 27-28 Avril (1999) Kénitra Maroc.
11- M. Yaouiss, F. Kifani-Sahban, M. Boukallouch, A. Kifani, J. Arauzo et T. Cordero, ‘Evolution de la microstructure et des variations dimensionnelles du PEEK lors de son traitement thermique’, Journées Matériaux Sciences, Technologies et Applications (JMSTA) 1er et 2 octobre (1999) Faculté des Sciences, Université Moulay Ismaïl, Mekhnès.
12- M. Yaouiss, S. Sebbahi, R. Rahho, F. Sahban, M. Boukallouch, A. Kifani et J. Arauzo, ‘Etude des mécanismes de la stabilité du poly-éther- éther –cétone lors de sa carbonisation et l’influence de cette caractéristique sur l’activation des résidus de la carbonisation’, 5ème
Congrès de Mécanique 17-20 Avril 2001. Faculté des Sciences, Université My Ismail Meknès. 13- S. Sebbahi, M. Yaouiss, R. Rahho, F. Kifani-Sahban, M. Boukallouch et A. Kifani, ‘Comparaison entre le comportement physico-chimique et physique de la lignine et du PEEK sollicités thermiquement’, 5ème
Congrès de Mécanique 17-20 Avril 2001. Faculté des Sciences, Université My Ismail Meknès.
14- S. Sebbahi, M. Yaouiss, R. Rahho, F. Kifani-Sahban, M. Boukallouch et A. Kifani, ‘Comportements physico-chimique et physique d’une substance d’origine végétale comparés à ceux d’une substance d’origine synthétique’, Sciences-Letters 5(2) (2003) 1-6.
15- F. Saoud, ‘Contribution à l’étude de l’activation avec la vapeur d’eau du charbon de l’eucalyptus et du charbon du pin maritime’, Diplôme des Etudes Supérieures Approfondies, DESA Sciences du Bois, (2005) Faculté des Sciences, Rabat.
16- F. Saoud, S. Sebbahi, F. Kifani-Sahban, A. Sesbou et A. Hakam, ‘Contribution à l’étude de l’activation par la vapeur d’eau du charbon d’eucalyptus-effet de la préoxydation à l’air’, Proceeding, 3éme Ecole Science et Technologie du Bois, ESTB-III, Université Alakhawain, Ifrane Maroc (2005).
17- S. Sebbahi, F. Kifani-Sahban, S. El Hajjaji, M. Boukallouch, A. Kifani et A. Zoulalian, ‘Influence du pré-traitement à l’air sur l’activation du charbon obtenu à partir du bois
5
d’eucalyptus’, Proceeding, 3ème Colloque Maroco-Français en Chimie Moléculaire, Laboratoire International Associé (LIA), Université Mohamed V Rabat, Maroc (2012).
18- M. El Marouani, A. El Hajji, S. Sebbahi, L. El Fakir, S. El Hajjaji, A. Dahchour, N. El Hrech, M. Bouhaouss and F. Kifani-Sahban, ‘Removal of some pollutants in aqueous solution using eucalyptus sawdust-based activated carbon’, 4ème Colloque International Environnement et Développement Durable sur Les produits phytosanitaires : Impact sur la santé humaine et l’environnement : quelles solutions alternatives ? 08-10 Octobre (2015) Errachidia.
19- L. El Fakir, S. Sebbahi, M. El Marouani, M. Serghini-Idrissi, M. El M’Rabet, F. Kifani-Sahban, A. Dahchour and S. El Hajjaji, ‘Kinetic modelling and mechanism of dyes adsorption onto raw and activated eucalyptus sawdust samples’, soumis.
20- J. Van Dam, R. Gosselink and E. DE Jong, ‘Lignin Applications’, Wageningen UR, Agrotechnology & Food Innovations in :
http://www.biomassandbioenergy.nl/infoflyers/LigninApplications.pdf
21- N. Consolin Filho, E.C. Venancio, M.F. Barriquello, A.A.W. Hechenleitner and E.A.G. Pineda, ‘Methylene blue adsorption onto modified lignin from sugar cane bagasse’, Ecletica
quimica 32(4) (2007) 63-70.
22- Q. Feng, H. Cheng, J. Li, P. Wang and Y. Xie, ‘Adsorption behavior of basic dye from aqueous solution onto alkali extracted lignin’, BioResources 9(2) (2014) 3602-3612.
23- Y. Wu, S. Zhang, X. Guo and H. Huang, ‘Adsorption of chromium (III) on lignin’,
Bioresource Technology 99 (2008) 7709-7715.
24- G.C. Quintana, G.J.M. Rocha, A.R. Gonçalves and J.A. Velasquez. ‘Evaluation of heavy matal removal by oxidized lignins in acid media from various sources’, BioResources 3(4) (2008) 1092-1102.
25- A. Kriaa, N. Hamdi and E. Srasra ‘Adsorption studies of methylene blue dye on Tunisian activated lignin ’, Russian Journal of Physical Chemistry A 85(2) (2011) 279-287.
26- A. Kriaa, N. Hamdi and E. Srasra, ‘Removal of Cu (II) from water pollutant with Tunisian
activated lignin prepared by phosphoric acid activation’, Desalination 250 (2010) 179-187.
27- K. Mahmoudi, N. Hamdi, A. Kriaa and E. Srasra, ‘Adsorption of Methyl Orange Using Activated Carbon Prepared from Lignin by ZnCl2 Treatment’, Russian Journal of Physical
Chemistry A 86(8) (2012) 1294-1300.
28- B.O. Ogunsile and A.A. Anomo, ‘Adsorption of chromium (VI) and cadmium (II) from aqueous solution by soda lignin obtained from nypa palm leaves (nypafruitcans)’, Ozean
Journal of Applied Sciences 7(2) (2014) 43-56.
29- H. Palonen, F. Tjerneld, G. Zacchi and M. Tenkanen, ‘Adsorption of Trichoderma Reesei CBH I and EG II and their catalytic domains on steam pretreated softwood and isolated lignin’, Journal of Biotechnology 107 (2004) 65-72.
30- N. Tazrouti et M. Amrani, ‘Adsorption du Cr (VI) sur la lignine activée’, Revue des Sciences
de l’Eau 23(3) (2010) 233-245.
31- N. Tazrouti and M. Amrani, ‘Chromium (VI) adsorption onto activated kraft lignin produced from alpha grass (Stipa Tenacissima)’, BioResources 4(2) (2009) 740-755.
La lignine
6 I. Introduction
La lignine est l’un des principaux constituants du bois avec la cellulose et l’hémicellulose. Dans le bois, la cellulose représente le renfort, l’hémicellulose et la lignine la matrice. Les arbres sont les végétaux qui contiennent le plus de lignine. La lignine est un polymère tridimensionnel complexe et amorphe. Elle apporte aux végétaux la rigidité, une imperméabilité à l’eau et une grande résistance à de nombreuses substances chimiques et à la dégradation biologique. Elle possède des propriétés aromatiques en raison de la présence du phénol qui la compose.
Son pouvoir calorifique et son rendement en charbon après carbonisation sont plus élevés que ceux de la cellulose et de l’hémicellulose car elle contient plus de carbone que les deux autres constituants.
Avant de nous intéresser à la biosynthèse, à la structure, à l’extraction et à la valorisation de la lignine, il serait utile de commencer par une description succincte tant botanique que chimique du bois.
II. Description du bois
Le bois est une matière naturelle, renouvelable et très hétérogène. Les différentes espèces de bois peuvent être classées en deux groupes : les angiospermes et les gymnospermes, respectivement appelés les feuillus et les résineux [1, 2].
Le bois est constitué d’extractibles, de matières minérales, d’eau et de matières organiques.
II.1. Matières extractibles
Les produits d’extraction sont un mélange complexe de produits organiques qui peuvent être extraits du bois par des solvants appropriés. Leur présence et leur quantité varient avec l’espèce, les conditions écologiques et la saison [2].
II.2. Matières minérales
La fraction minérale est inerte et représente environ 1 %. Dans le cas du bois, la teneur en matières minérales est assimilable à la masse des cendres.
Les cendres se composent de métaux comme Ca, K, Mn, Na, P, …. avec une prédominance pour le Ca et le K. Dans le bois, ces métaux existent sous forme d’oxyde [2].
II.3. Fraction aqueuse
Le bois est un matériau hygroscopique qui échange l’eau de façon continue avec son environnement. Dans le bois, l’eau se trouve sous deux formes : eau libre et eau liée. Lorsque le bois sèche, l’eau libre s’évapore en premier. Si le bois continue à sécher et que l’eau liée s’évapore à son tour, le bois subit un retrait dimensionnel. Pour éviter les variations dimensionnelles, le bois doit être conservé à des conditions de température et d’humidité de l’air bien déterminées. La teneur en eau dépend des paramètres précités. Elle varie de 50 à 65 % lorsque le bois est fraîchement coupé. Cette quantité d’eau s’évapore lentement après stockage à l’air et se stabilise entre 10 et 15 % [2].
II.4. Fraction organique
La composition chimique de la fraction organique varie très peu d’une espèce à l’autre puisqu’elle est constituée de 50 % de carbone, 6 % d’hydrogène et 44 % d’oxygène. Ces pourcentages conduisent à la formule brute CH1.44O0.66 valable pour toutes les espèces de bois [2-4].
La fraction organique du bois contient trois constituants principaux : la cellulose, les hémicelluloses et la lignine. La teneur de ces constituants dans les feuillus et dans les résineux est indiquée dans le tableau I.1 [2-4] et leur composition élémentaire moyenne dans le tableau I.2 [5, 6].
La cellulose est un polysaccharide linéaire formée d’unités de -D glucose et le maillon de base est le cellobiose (figure I.1). La structure de la cellulose est une alternance de zones cristalline et amorphe.
Les hémicelluloses sont des polysaccharides constitués d’un mélange de différents hexoses (sucres en C6) et pentoses (sucres en C5) et parfois d’acides uroniques. Contrairement à la cellulose, les hémicelluloses sont amorphes (figure I.2).
La lignine est un polymère aromatique, c’est le constituant le plus complexe du bois. Sa description est donnée dans le paragraphe III.
Tableau I.1 : Proportions des principaux constituants du bois dans les feuillus et dans les résineux en % par masse de biomasse sèche [2-4]
Constituants Feuillus Résineux
Hémicellulose 29 ± 7 28 ± 3
Cellulose 47 ± 6 41 ± 4
8
Tableau I.2 : Composition élémentaire du bois, de la cellulose et de la lignine [5, 6]
% C % H % O % N
Bois 50 6 44 1
Cellulose 44 6 50 _
Lignine 63-67 5,5-6 27-34 _
Figure I.1 : Structure moléculaire de la cellulose
Figure I.2 : Structure moléculaire de l’hémicellulose III. La lignine
La lignine enveloppe les systèmes de conduction d’eau des racines jusqu’aux feuilles et protège le bois contre le vieillissement et la biodégradation. De plus, la lignine participe en grande partie à la rigidité du bois [7, 8]. La lignine et les hémicelluloses forment une matrice autour des fibres de cellulose [9].
III.1. Structure moléculaire de la lignine
La lignine est une macromolécule de nature phénolique, amorphe, tridimensionnelle et très complexe. Elle comporte un motif principal du type phényle-propane, substitué de façon variable et agencé dans la molécule selon divers modes de liaisons [3]. Le caractère aromatique rend la lignine hydrophobe.
Il n’y a pas une composition unique de la lignine car sa formation dépend de l’environnement physico-chimique où le végétal pousse. De plus la composition de la lignine varie au sein de la même espèce. C’est pour ces raisons qu’il est préférable de parler des lignines.
III.2. Biosynthèse de la lignine
Dans la plante, le glucose est obtenu par photosynthèse. Cette molécule va être transformée en acide shikimique qui va être transformé, à son tour, par voie métabolique en deux acides aminés : la phénylalanine et la tyrosine [7, 10-13] (figure I.3).
Ces deux acides aminés vont subir plusieurs réactions pour donner les monomères précurseurs de la lignine [10, 11, 14-16]. Les principales réactions sont indiquées ci-après :
* la désamination de la chaîne latérale avec formation d’une double liaison. Cela conduit à l’acide cinnamique ;
* l’hydroxylation du cycle en position para de la chaîne, avec formation d’acide p-coumarique et la thioestérification du groupe carboxyle par la Coenzyme A (formation du p-coumaroyl-CoA) ;
* la réduction de la fonction thioester en aldéhyde, puis en alcool ; * l’introduction du groupement méthoxyle.
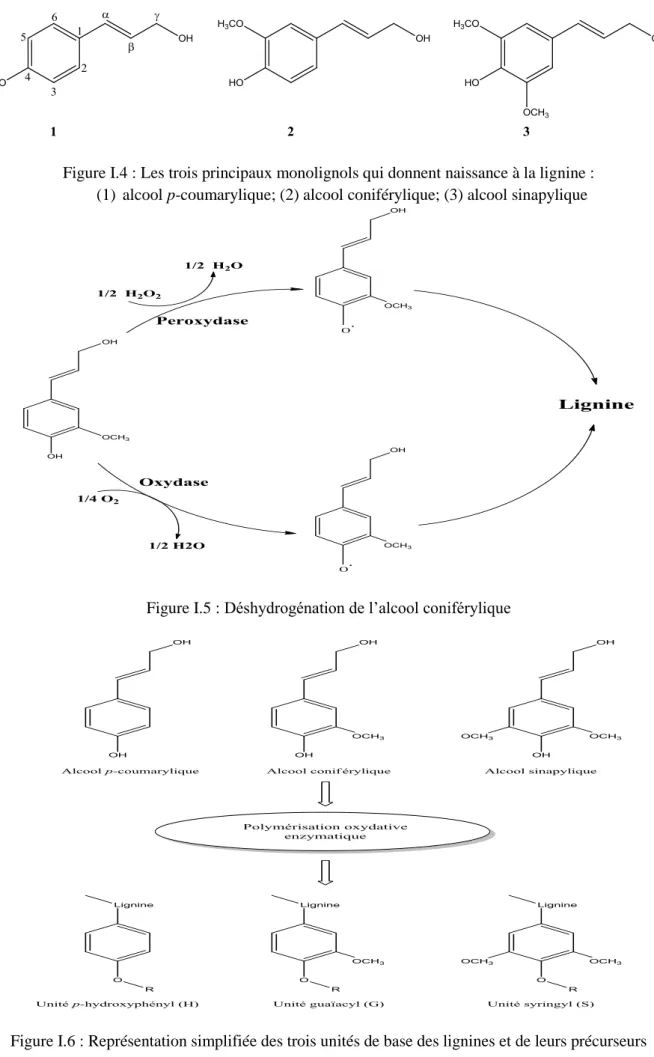
Les dénominations généralement attribuées aux précurseurs de la lignine sont monolignols, phénylpropanoïdes et alcools cinnamyles. Les principaux monolignols sont l’alcool p-coumarylique, l’alcool coniférylique et l’alcool sinapylique qui se distinguent par le degré de méthoxylation (figure I.4).
Les monolignols précités, biosynthétisés dans le cytosol, sont ensuite transportés vers la paroi cellulaire pour y subir une déshydrogénation par des peroxydases et/ou des laccases (figure I.5) [10, 17]. La déshydrogénation conduit à des radicaux qui vont s’incorporer dans le polymère de lignine [18]. Ces radicaux sont le p-hydroxy-phényle, le guaïacyle et le syringyle notés respectivement H, G et S (figure I.6).
La présence du noyau aromatique ainsi que de la chaîne carbonée entraîne la délocalisation du centre radicalaire [8, 19] (figure 1.7). La délocalisation donne ainsi lieu à de nombreux radicaux qui vont subir un couplage radicalaire combinatoire [8, 10, 20]. Les principaux modes de couplage des radicaux sont regroupés dans le tableau I.3 et présentés dans la figure I.8 [8, 21, 22].
Les radicaux relatifs à chacun des monomères H, G et S présentent des sites réactifs stables et d’autres thermodynamiquement et strériquement instables (figure I.7). En effet, l’alcool p-coumarylique contient cinq sites réactifs localisés sur C-1, C-3 et C-5
10
du noyau aromatique, HO-Phe et C- de la chaîne latérale. Toutes ces formes sont stables. Contrairement à l’alcool p-coumarylique, les alcools coniférilique et synapylique ne contiennent respectivement que quatre et trois sites réactifs. Pour l’alcool coniférilique, ces sites sont localisés sur C-1 et C-5 du noyau aromatique, HO-Phe et C- de la chaîne latérale. La forme V est instable. Pour ce qui est de l’alcool synapylique, les sites réactifs sont localisés uniquement sur C-1 du noyau aromatique, HO-Phe et C- de la chaîne latérale. Les formes III et V sont instables (figure I.7). La liaison -O-4 est la plus prépondérante au sein de la lignine issue aussi bien du bois tendre que du bois dur comme il ressort des valeurs du tableau I.4. Ainsi, le radical II constitue la forme de résonance la plus stabilisée et favorable à la liaison β-O-4 [8]. Des calculs en mécanique quantique sur la densité électronique de l’oxygène phénolique ont montré que la liaison β-O-4 est également la plus dominante [8].
La polymérisation des radicaux se fait d’une façon désordonnée et non répétitive, ce qui conduit à une multitude de lignines et par conséquent à la différence qui existe entre les lignines.
Les différentes lignines sont constituées de l’un, de deux ou des trois radicaux H, G et S phénylpropanols précités.
Les lignines des gymnospermes ou résineux sont constituées d’unités G avec de faibles quantités d'unités H. Cependant, les lignines des angiospermes dicotylédones ou feuillus sont composés d’unités G et S avec également de faibles quantités d’unités H. Les lignines de tiges des poacées (angiospermes monocotylédones) contiennent les trois unités H, G et S [4, 24].
Tableau I.3 : Modes de couplage des radicaux phénoxy [8]
I II III IV V
I Péroxyde instable -O-4 4-O-5 1-O-4 3-O-4
II -O-4 - -5 -1 -3
III 4-O-5 -5 5-5 1-5 3-5
IV 1-O-4 -1 1-5 1-1 1-3
Tableau I.4 : Exemples de proportion des principales liaisons éther et carbone-carbone dans la lignine du bois tendre et du bois dur [23]
Type de liaison
Structure dimère % de liaison Bois tendre
% de liaison Bois dur
-O-4 β−aryl éther phénylpropane 48 60
5-5 Biphényl et Dibenzodioxocyne 9,5 – 11 4,5
-5 Phénylcoumarane 9 – 12 6
-1 1,2-Diaryl propane 7 7
-O-4 α−aryl éther phénylpropane 6 – 8 6 – 8
4-0-5 Diaryl éther 3,5 – 4 6,5
- Structures à lien β-β de type résinol 2 3
12
Figure I.4 : Les trois principaux monolignols qui donnent naissance à la lignine : (1) alcool p-coumarylique; (2) alcool coniférylique; (3) alcool sinapylique
Figure I.5 : Déshydrogénation de l’alcool coniférylique
Figure I.7 : Délocalisation des radicaux obtenus par déshydrogénation enzymatique [19]. Les radicaux instables sont encadrés en pointillés
Figure I.8 : Principales liaisons éther et carbone-carbone majoritairement présentes dans la lignine (R1
= R2 = H dans l’unité hydroxyphényl ; R1 = OMe , R2 = H dans l’unité guaiacyl ; R1 = R2 = OMe dans
14 III.2.1. Structures hypothétiques de la lignine
La première structure de la lignine a été proposée par Freudenberg [15]. Ce dernier a obtenu la lignine par polymérisation enzymatique in vitro des alcools coumarylique, coniférylique et sinapylique (figure I.9). Ensuite, Adler [23] a extrait la lignine par oxydation de l'épinette, la structure comprend 16 unités (figure I.10). La structure de la lignine issue du procédé papetier Kraft [25] est donnée sur la figure I.11. Toutes les structures proposées pour la lignine diffèrent car celles-ci dépendent de l’origine et de la méthode d’extraction [26-30].
Figure I.9 : Structure de la lignine selon Freudenberg [15]
![Tableau I.2 : Composition élémentaire du bois, de la cellulose et de la lignine [5, 6]](https://thumb-eu.123doks.com/thumbv2/123doknet/2185476.10792/29.892.114.779.128.768/tableau-composition-elementaire-bois-cellulose-lignine.webp)
![Figure I.3 : Voies de biosynthèse des phényl-propanoïdes de la lignine [12, 13]](https://thumb-eu.123doks.com/thumbv2/123doknet/2185476.10792/32.892.99.862.194.1109/figure-voies-biosynthese-phenyl-propanoides-lignine.webp)
![Figure I.7 : Délocalisation des radicaux obtenus par déshydrogénation enzymatique [19]](https://thumb-eu.123doks.com/thumbv2/123doknet/2185476.10792/34.892.113.841.104.539/figure-delocalisation-radicaux-obtenus-deshydrogenation-enzymatique.webp)
![Figure I.13 : Structures chimiques des différents fragments de polymère de lignine de paille de blé extraite par le procédé CIMV [39]](https://thumb-eu.123doks.com/thumbv2/123doknet/2185476.10792/37.892.115.785.833.1082/figure-structures-chimiques-fragments-polymere-lignine-extraite-procede.webp)
![Figure I.15 : Représentation schématique de la lignine Kraft et Lignosulfonates [55] III.3.4](https://thumb-eu.123doks.com/thumbv2/123doknet/2185476.10792/40.892.104.791.697.961/figure-representation-schematique-lignine-kraft-lignosulfonates-iii.webp)
![Figure I.16 : Représentation schématique de la lignine organosolve [57] III.3.4.1. Extraction par les alcools](https://thumb-eu.123doks.com/thumbv2/123doknet/2185476.10792/41.892.107.786.174.381/figure-representation-schematique-lignine-organosolve-iii-extraction-alcools.webp)