Modélisation gros grain de macromolécules végétales : champ de force paramétré par dynamique moléculaire et application à des assemblages cellulose-xylane
151
0
0
Texte intégral
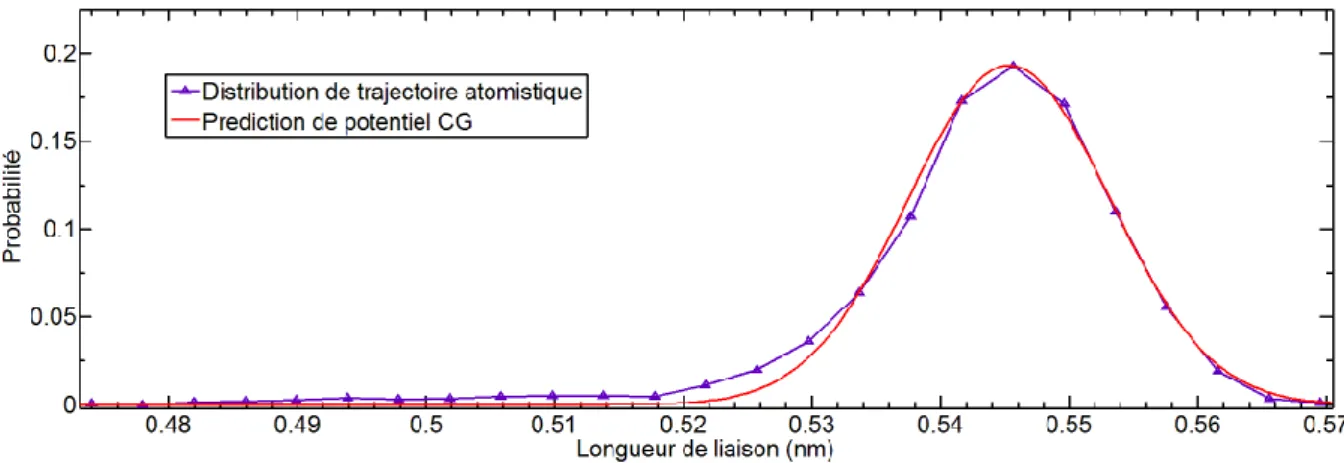
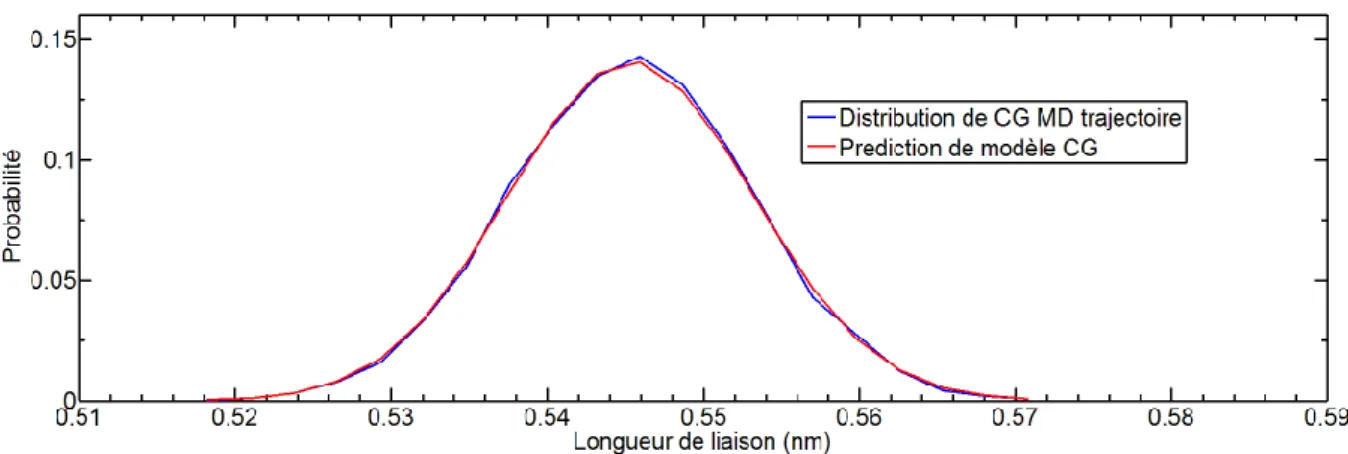
Figure
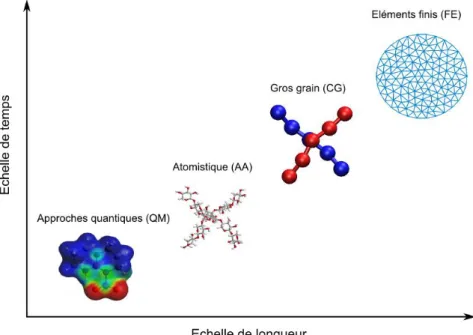
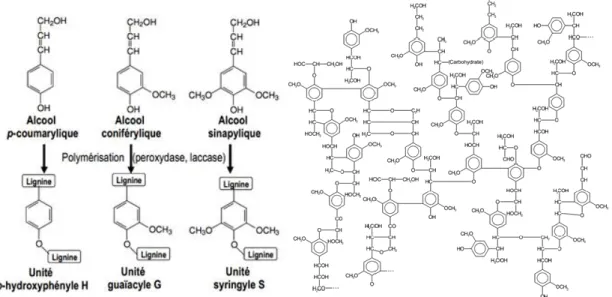
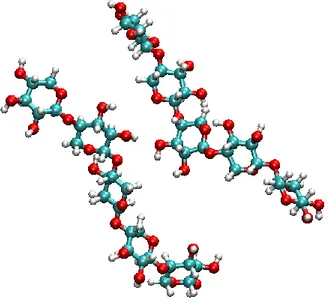

+7
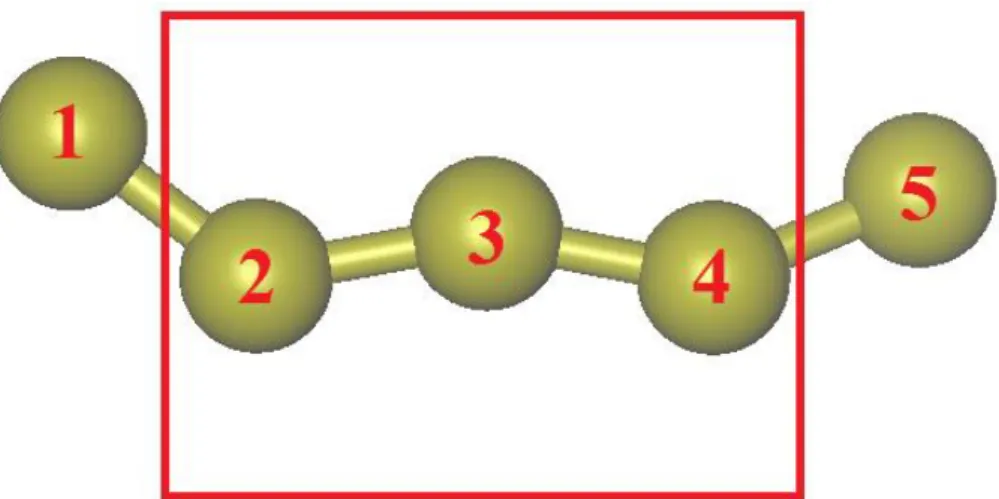



Documents relatifs