Mass spectrometry as a tool to dissect the
role of chromatin assembly factors in
regulating nucleosome assembly
par Marlène Gharib
Département de Biochimie, Université de Montréal Faculté de Médecine
Thèse présentée à la Faculté de Médecine
en vue de l’obtention du grade de Philosophiæ Doctor (Ph.D.) en biochimie
Décembre, 2010
Faculté des études supérieures et postdoctorales
Cette thèse intitulée:
Mass spectrometry as a tool to dissect the role of chromatin assembly factors in regulating nucleosome assembly
Présentée par : Marlène Gharib
a été évaluée par un jury composé des personnes suivantes :
Dr. Benoit Coulombe, président-rapporteur Dr. Pierre Thibault, directeur de recherche
Dr. Alain Verreault, co-directeur Dr. François Robert, membre du jury Dr. Momtchil Vodenitcharov, examinateur externe
Résumé
L'assemblage des nucléosomes est étroitement couplée à la synthèse des histones ainsi qu’à la réplication et la réparation de l’ADN durant la phase S. Ce processus implique un mécanisme de contrôle qui contribue soigneusement et de manière régulée à l’assemblage de l’ADN en chromatine. L'assemblage des nucléosomes durant la synthèse de l’ADN est crucial et contribue ainsi au maintien de la stabilité génomique. Cette thèse décrit la caractérisation par spectrométrie de masse (SM) des protéines jouant un rôle critique dans l’assemblage et le maintien de la structure chromatinienne. Plus précisément, la phosphorylation de deux facteurs d’assemblage des nucléosome, le facteur CAF-1, une chaperone d’histone qui participe à l'assemblage de la chromatine spécifiquement couplée à la réplication de l'ADN, ainsi que le complexe protéique Hir, jouant de plus un rôle important dans la régulation transcriptionelle des gènes d’histones lors de la progression normale du cycle cellulaire et en réponse aux dommages de l'ADN, a été examiné.
La caractérisation des sites de phosphorylation par SM nécéssite la séparation des protéines par éléctrophorèse suivi d’une coloration a l’argent. Dans le chapitre 2, nous demontrons que la coloration à l’argent induit un artéfact de sulfatation. Plus précisément, cet artéfact est causé par un réactif spécifiquement utilisé lors de la coloration. La sulfatation présente de fortes similitudes avec la phosphorylation. Ainsi, l’incrément de masse observé sur les peptides sulfatés et phosphorylés (+80 Da) nécéssite des instruments offrant une haute résolution et haute précision de masse pour différencier ces deux modifications.
Dans les chapitres 3 et 4, nous avons d’abord démontré par SM que Cac1, la plus grande sous-unité du facteur CAF-1, est cible de plusieurs sites de phosphorylation. Fait intéréssant, certains de ces sites contiennent des séquences consensus pour les kinases Cdc7-Dbf4 et CDKs. Ainsi, ces résultats fournissent les premières évidences que CAF-1 est potentiellement régulé par ces deux kinases in vivo. La fonction de tous les sites de
phosphorylation identifiés a ensuite été évaluée. Nous avons démontré que la phosphorylation de la Ser-503, un site consensus de la DDK, est essentielle à la répréssion transcriptionelle des gènes au niveau des télomères. Cependant, cette phosphorylation ne semble pas être nécéssaire pour d’autres fonctions connues de CAF-1, indiquant que le blocage de la phsophorylation de Cac1 Ser-503 affecte spécifiquement la fonction de CAF-1 aux structures hétérochromatiques des télomères. Ensuite, nous avons identifiés une intéraction physique entre CAF-1 et Cdc7-Dbf4. Des études in vitro ont également demontré que cette kinase phosphoryle spécifiquement Cac1 Ser-503, suggérant un rôle potential pour la kinase Cdc7-Dbf4 dans l’assemblage et la stabilité de la structure hétérochromatique aux télomères. Finalement, les analyses par SM nous ont également permi de montrer que la sous-unité Hpc2 du complexe Hir est phosphorylée sur plusieurs sites consensus des CDKs et de Cdc7-Dbf4. De plus, la quantification par SM d’un site spécifique de phosphorylation de Hpc2, la Ser-330, s’est révélée être fortement induite suite à l’activation du point de contrôle de réplication (le “checkpoint”) suite au dommage a l’ADN. Nous montrons que la Ser-330 de Hpc2 est phopshorylée par les kinases de point de contrôle de manière Mec1/Tel1- et Rad53-dépendante. Nos données préliminaires suggèrent ainsi que la capacité du complex Hir de réguler la répréssion transcriptionelle des gènes d'histones lors de la progression du cycle cellulaire normal et en réponse au dommage de l'ADN est médiée par la phosphorylation de Hpc2 par ces deux kinases.
Enfin, ces deux études mettent en évidence l'importance de la spectrométrie de masse dans la caractérisation des sites de phosphorylation des
protéines, nous permettant ainsi de comprendre plus précisement les mécanismes de régulation de l'assemblage de la chromatine et de la synthèse des histones.
Mots-clés: CAF-1, protéines Hir, spectrométrie de masse, coloration à l'argent, sulfatation, phosphorylation, assemblage de la chromatine, réplication de l'ADN,
Abstract
Nucleosome assembly entails a controlled mechanism that is tightly coupled to DNA and histone synthesis during DNA replication and repair in S-phase. Importantly, this contributes to the prompt and carefully orchestrated assembly of newly replicated DNA into chromatin, which is essential for the maintenance of genomic integrity. This thesis describes the mass spectrometric characterization of proteins critical in the regulation of nucleosome assembly behind the replication fork and chromatin structure. More specifically, the phosphorylation of Chromatin Assembly Factor 1 (CAF-1), a nucleosome assembly factor that uniquely functions during replication-coupled de novo nucleosome assembly in S-phase and the Hir protein complex, a second nucleosome assembly factor that also contributes to the transcriptional regulation of histone genes during normal cell cycle progression and in response to DNA damage, was examined.
We first demonstrated that characterization of protein phosphorylation by mass spectrometry (MS), which often relies on the separation of proteins by gel electrophoresis followed by silver staining for visualization, should be given careful considerations. In chapter 2, we report a potential pitfall in the interpretation of phosphorylation modifications due to the artifactual sulfation of serine, threonine and tyrosine residues caused by a specific reagent used during silver staining. Sulfation and phosphorylation both impart an 80 Da addition of these residues making them distinguishable only with MS systems offering high resolution and high mass accuracy capabilities.
Chapter 3 and 4 present the MS characterization of in vivo phosphorylation occurring on CAF-1 and Hir proteins, respectively. We first demonstrated that Cac1, the largest subunit of CAF-1, is phosphorylated on several novel residues containing the consensus sequences recognized by either Cdc7-Dbf4 (DDK) or cyclin-dependent kinases (CDKs). These results have provided the first evidence that CAF-1 is regulated by these two kinases in vivo. The function of all identified Cac1 phosphorylation sites was then assessed. In vivo phenotypic studies showed that the specific phosphorylation of Ser-503, a
Cac1 DDK-like site identified in our study, is essential for heterochromatin-mediated telomeric silencing. Cac1-Ser-503 did not appear to be required for other known functions of CAF-1, including DNA damage resistance and mitotic chromosome segregation, indicating that blocking Cac1 phosphorylation on Ser-503 sepcifically cripples CAF-1 function at telomeres. Next, biochemical purifications identified a physical interaction between CAF-1 and Cdc7-Dbf4. Consistent with this physical interaction data, in vitro kinase assay studies showed that Cdc7-Dbf4 specifically phosphorylates Cac1 Ser-503 thereby uncovering a novel role for Cdc7-Dbf4 in heterochromatin assembly and/or stability that is potentially mediated through CAF-1. Finally, MS analysis also showed that the Hpc2 subunit of the Hir protein complex is phosphorylated on several CDK- and DDK-like consensus sites. Furthermore, MS quantification of a specific phosphorylation site, Hpc2 Ser-330, was shown to be highly induced following the activation of the DNA damage checkpoint in response to DNA damage. We show that Hpc2 Ser-330 is phopshorylated by checkpoint kinases in a Mec1/Tel1- and Rad53-dependent manner. Our preliminary data suggest that the ability of the Hir protein complex to regulate the transcriptional repression of histone genes during normal cell cycle progression and in response to DNA damage is mediated through the regulated phosphorylation of Hpc2 by these kinases.
Finally, these two studies highlight the importance of mass spectrometry in characterizing protein phosphorylation events, which has yielded novel insights into the regulation of chromatin assembly by CAF-1 and histone synthesis mediated by Hir proteins.
Keywords: CAF-1, Hir proteins, mass spectrometry, silver staining, sulfation,
phosphorylation, chromatin assembly, DNA replication, telomeric silencing, histone gene regulation, DNA damage.
Table of contents
Résumé...i Abstract...iii Table of contents...v List of tables...ix List of figures...x List of abbreviations...xiii Aknowledgements...xviii 1. Introduction ... 11.1. Introduction to chromatin structure and organization ... 2
1.1.1. The nucleosome... 3
1.1.2. Histone post-translational modifications... 4
1.1.3. Euchromatin and heterochromatin ... 7
1.2. Genome duplication ... 10
1.2.1. DNA replication ... 10
1.2.1.1. Replication initiation ... 12
1.2.1.2. Replication elongation... 16
1.2.2. DNA repair ... 18
1.2.2.1. Cell cycle checkpoint activation... 19
1.2.2.2. DNA repair mechanisms ... 20
1.3. The role of PCNA in genome duplication and maintenance of chromatin structure ... 22
1.4. A general overview of chromatin assembly ... 29
1.4.1. Replication-coupled chromatin assembly ... 29
1.4.2. Histone chaperone ... 31
1.4.2.1. CAF-1 ... 31
1.5. Mass spectrometry as a tool to study protein phosphorylation ... 44
1.5.1. Mass spectrometry principles ... 46
1.5.1.1. Sample preparation... 46
1.5.1.2. Chromatographic separation of peptides ... 48
1.5.1.3. Mass spectrometry instrumentation... 49
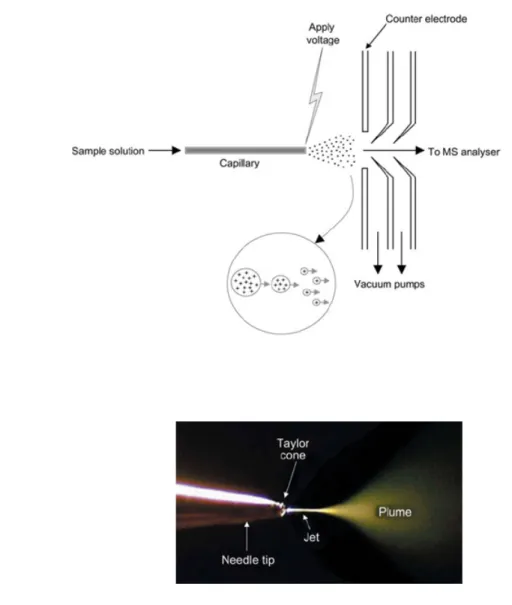
1.5.1.3.1. Electrospray ionization (ESI) source ... 50
1.5.1.3.2. Q-q-LIT and LTQ-Orbitrap mass analyzers ... 52
1.5.1.3.3. Peptide fragmentation under low energy collision ... 56
1.5.1.3.4. Database searching using MS/MS spectral data... 57
1.5.1.3.5. Relative quantification of peptide abundance... 59
1.5.2. Methods to detect protein phosphorylation ... 62
1.5.2.1. Enrichment techniques ... 63
1.5.2.2. Chemical derivatization... 65
1.5.2.3. Fragmentation methods for phosphopeptide identification... 67
1.5.2.4. Multiple reaction monitoring (MRM) ... 70
1.6. Research objectives ... 73
1.7. References ... 76
2. Artifactual sulfation of silver-stained proteins: implications for the assignment of phosphorylation and sulfation sites ... 110
2.1. Abstract ... 111
2.2. Introduction ... 112
2.3. Experimental procedures ... 115
2.3.1. Material and reagents ... 115
2.3.2. Protein separation by SDS-PAGE... 116
2.3.3. Destaining and in-gel digestion... 116
2.3.4. β-elimination ... 117
2.3.5. Mass spectrometry analysis... 117
2.3.6. Database searching ... 118
2.4. Results ... 120
2.4.1. Evaluation of staining protocols on the occurrence of protein sulfation artifact... 120
2.4.2. Sulfation artifacts on serine, threonine and tyrosine residues ... 124
2.4.3. Sodium thiosulfate gives rise to protein sulfation artifacts ... 127
2.4.4. Sulfation artifact caused by silver staining of phosphorylated ERK1... 129
2.4.5. Sulfation artifacts in silver-stained bacterial proteins ... 131
2.5. Discussion ... 136
2.6. References ... 143
3. Phosphorylation of CAF-1 by Cdc7/Dbf4 promotes telomeric silencing in Saccharomyces cerevisiae ... 148
3.1. Abstract ... 149
3.2. Introduction ... 150
3.3. Material and methods ... 154
3.3.1. Strains and plasmids... 154
3.3.2. Yeast strains ... 154
3.3.3. Purification of yeast CAF-1 ... 155
3.3.4. Recombinant proteins... 156
3.3.5. In-gel trypsin digestion... 156
3.3.6. In-solution trypsin digestion and phosphopeptide enrichment... 157
3.3.7. LC-MS/MS analysis ... 157
3.3.8. Proliferation, telomeric silencing and DNA damage sensitivity assays... 158
3.3.9. Co-immunoprecipitation of Cdc7-TAP and Cac1-FLAG3 ... 159
3.3.10. In vitro kinase assay ... 159
3.3.11. Chromatin binding assay ... 160
3.4. Results and discussion... 162
3.4.1. Purification of yeast CAF-1 and identification of in vivo phosphorylation sites by mass spectrometry ... 162
3.4.2. Cac1 Ser-503 phosphorylation contributes to telomeric silencing, but not
other functions of CAF-1 ... 169
3.4.3. DDK directly phosphorylates Cac1 in vitro ... 173
3.5. References ... 180
4. Preliminary phosphorylation analysis of the yeast Hir protein complex during normal cell cycle progression and in repsonse to DNA damage ... 184
4.1. Introduction ... 185
4.2. Material and methods ... 190
4.2.1. Purification of Hir3-TAP... 190
4.2.2. Purification of Hpc2-TAP from asynchronous, MMS and MMS + caffeine treated cells... 191
4.2.3. In-gel trypsin digestion... 192
4.2.4. LC-MS/MS analysis ... 192
4.2.5. MRM analysis ... 192
4.3. Results ... 193
4.3.1. Purification of the Hir protein complex from yeast and identification of in vivo phosphorylation sites by mass spectrometry ... 193
4.3.2. Phosphorylation of Hpc2 on Ser-330 is modulated in response to DNA damage and is a target of the DNA damage checkpoint. ... 198
4.4. Discussion ... 204
4.5. References ... 208
5. General conclusion and future perspectives ... 215
5.1. References ... 224
List of tables
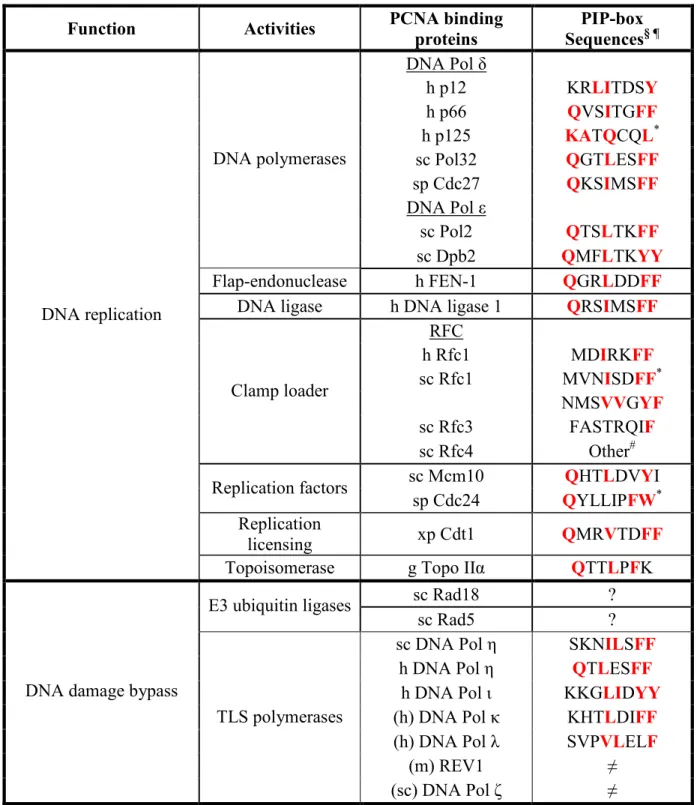
Table 1.1 PCNA binding proteins ... 26
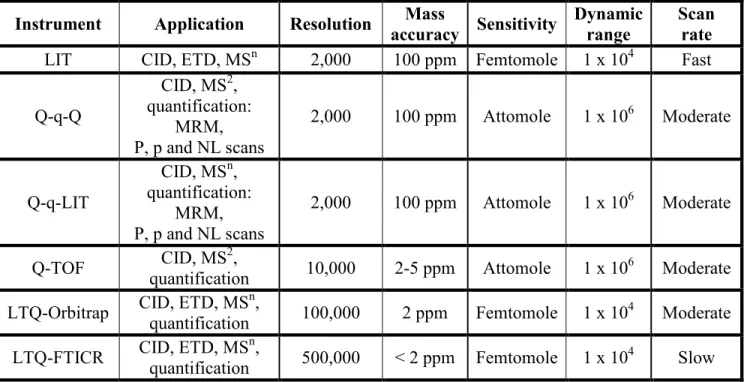
Table 1.2 Performance comparison of different instruments commonly used in mass spectrometry-based proteomics (Yates, 2009). ... 53
Table 2.1 Identification of sulfopeptides from yeast enolase... 123
Table 2.2 Identification of modified peptides from ERK1... 131
Table 4.1 Summary of Hpc2 phosphorylation sites ... 197
Table A1 Proteins identified from silver and Coomassie stained polyacrylamide gels…...228
Liste of figures
Figure 1.1 The crystal structure of the yeast nucleosome core particle ... 4
Figure 1.2 Initiation of DNA replication in eukaryotes.. ... 13
Figure 1.3 The process of DNA replication in S-phase. ... 14
Figure 1.4 Crystal structure of human PCNA.. ... 25
Figure 1.5 CAF-1 is composed of three evolutionary conserved subunits. ... 36
Figure 1.6 General workflow of a typical mass spectrometry experiment... 45
Figure 1.7 The electrospray ionization (ESI) process. ... 51
Figure 1.8 Main components of a triple quadrupole linear ion trap (Q-q-LIT) hybrid mass spectrometer. ... 54
Figure 1.9 Main components of a LTQ-Orbitrap hybrid mass spectrometer. ... 55
Figure 1.10 Nomenclature of peptide fragmentation patterns with collision induced dissociation (CID). ... 57
Figure 1.11 General approach of quantitative mass spectrometry.. ... 61
Figure 1.12 Overview of common approaches used for phosphopeptide analysis by mass spectrometry. ... 67
Figure 1.13 Example of a phosphopeptide analyzed by data-dependent neutral loss MS3.. 69
Figure 1.14 Schematic representation of the MRM technique using a Q-q-LIT instrument... 71
Figure 2.1 Intensity distribution of sulfopeptides and unmodified peptides from silver- and Coomassie Blue-stained yeast enolase. ... 122
Figure 2.2 Identification of modified peptides from yeast enolase. ... 125
Figure 2.3 Influence of sodium thiosulfate concentration on the abundance of silver stained-induced sulfopeptides.. ... 128
Figure 2.4 Identification of modified peptides derived from ERK1. ... 132
Figure 2.5 Identification of sulfopeptide artifacts from silver and Coomassie Blue stained gels of E. coli cell lysate (TCL)... 134
Figure 2.6 Identification of sulfopeptide artifacts from silver and Coomassie Blue-stained
gel band 2 (33 KDa identified in figure 2.5a) from E. coli cell lysates. ... 135
Figure 2.7 Proposed mechanism for sulfation of hydroxylated amino acids in silver-stained gels... 137
Figure 2.8 Distribution of 6 residues on either side of the sulfated amino acids identified in enolase and ERK1 using weblogo... 139
Figure 3.1 Cac1 is phosphorylated on Ser-94, Ser-238, Ser-501, Ser-503 and Ser-515 in vivo. ... 163
Figure 3.2 Cac3 Ser-14 is phosphorylated in vivo... 164
Figure 3.3 Cac1 is phosphorylated on DDK-like consensus sites... 166
Figure 3.4 Other Cac1 phosphorylation sites identified by mass spectrometry. ... 167
Figure 3.5 Conservation of Cac1 Ser-238 and Ser-503 throughout the Saccharomyces genus and other related fungi... 168
Figure 3.6 Phosphorylation of Cac1 Ser-503 is needed for telomeric silencing but not other functions of CAF-1... 172
Figure 3.7 DDK interacts with Cac1 in vivo and directly phosphorylates Cac1 Ser-503 in vitro.. ... 175
Figure 3.8 Mutation of Cac1 Ser-503 does not impair the association of CAF-1 with chromatin... 178
Figure 4.1 Purification and detection of the Hir protein complex using the tandem-affinity purification strategy... 194
Figure 4.2 Yeast Hpc2 is phosphorylated in vivo... 196
Figure 4.3 Hpc2 phosphorylation increases in response to the DNA damage checkpoint activation. ... 202
Figure 4.4 Hpc2 is phosphorylated on Ser-330 in response to DNA damage... 203
Figure A1 MS2 and MS3 spectra from modified peptides of yeast enolase………...229
Figure A2 MS2 and MS3 spectra from modified peptides of ERK1………...236 Figure A3 MS2 and MS3 spectra from modified peptides from E.coli protein extracts….244
Figure A4 MS2 spectra from sulfated yeast enolase peptide SIVPSGASTGVHEALEMR obtained from LTQ-Orbitrap (top) and Q-TOF (bottom)………...255
List of abbreviations
1D one dimensional
2D two dimensional
3`hExo 3`exoribonuclease 5-FOA 5-fluoroorotic acid 6-4 PP 6-4 photoproduct
ABF1 autonomously replicating sequence binding factor 1
AC alternating current
ACN acetonitrile
ACS ARS consensus sequence
Ade adenine
ADP adenosine diphosphate
Ag2S silver sulfide
AgNO3 silver nitrate
AP apyrimidinic/apurinic
ARS autonomously replicating sequence Asf1 Anti-silencing factor 1
ATM ataxia telangiectasia mutated ATP adenosine 5`-triphosphate ATR ataxia telangiectasa rad3-related Ba(OH)2 barium hydroxide
BER base excision repair
bp base pair
Cac chromatin assembly complex CaCl2 calcium chloride
CAF-1 chromatin assembly factor 1 CBB coomassie brilliant blue CBP calmodulin binding peptide CCR5 C-C chemokine receptor 5 Cdc cell division cycle
CDK cyclin dependent kinase
cDNA complementary DNA
ChIP chromatin immunoprecipitation Chk1/Chk2 checkpoint kinase 1/2
CID collision induced dissociation Clr4 cryptic loci regulator 4
Da daltons
Dbf4 dumbbell former 4
DDA data-dependent acquisition DDK Dbf4-dependent kinase DHB 2,5-dihydroxybenzoic acid DNA deoxyribonucleic acid
DNMT1 DNA (5-cytosine) methyltrasnferase 1
DSB double strand break
DTT dithiothreitol
ECD electron capture dissociation
EDT ethanedithiol
EDTA ethylene diamine tetra-acetic acid
EMS enhanced MS
EPI enhanced product ion
ERK1 extracellular signal-related kinase 1 ESI electrospray ionization
ETD electron transfer dissociation
FA formic acid
FAB fast atom bombardment
FACT facilitates chromatin transcription FEN-1 flap endonuclease 1
FT-ICR fourier transform ion cyclotron GCN5 general control non-derepressible 5 GCR gross chromosomal rearrangement GST gluthatione-S-transferase
H3PO4 phosphoric acid
HAT histone acetyltransferase HDAC histone deacetylase
HEPES N-(2-hydroxyethyl) piperazine-N`-(2-ethanosulfonic acid) HIR histone gene regulation
His histidine
HML homothallic mating type locus L HMR homothallic mating type locus R HP1 heterochromatin protein 1 Hpc2 histone promoter control 2
HPLC high performance liquid chromatography
HPO3 phosphate
IDA iminodiacetate
IDA information-dependent acquisition IDCL interdomain connecting loop
IDL insertion/deletion
IEF isoelectric focusing
IgG immunoglobulin G
IMAC immobilized metal affinity chromatography
IP immunoprecipitation
IPTG isopropyl-β-D-thiogalactopyranoside K3F2(CN)6 potassium hexacyanoferrate
Kb kilobase
KCl Potassium chloride
KOH potassium hydroxide
LB lysogeny broth
LC liquid chromatography
Leu leucine
LIT linear ion trap
m/z mass-to-charge
MCM minichromosome maintenance
MeOH methanol
MES 2-(N-morpholino)ethanesulfonic acid MgCl2 magnesium chloride
MMR mismatch repair
MMS methyl metanosulfonate
MOAC metal oxide affinity chromatography MRM multiple reaction monitoring
mRNA messenger RNA
MS mass spectrometry
MSA multistage activation MSH2/3/6 mutS homologue 2/3/6 Na2CO3 sodium carbonate
Na2S2O3 sodium thiosulfate
NaCl sodium chloride
NAD nicotinamide adenine dinucleotide
NaOH sodium hydroxide
NCBI national center for biotechnology information NER nucleotide excision repair
NH4OH ammonium hydroxide
NP-40 nonidet P-40
nt nucleotide
NTA nitrilotriacetate
NuRD nucleosome remodelling histone deacetylase complex NuRF nucleosome remodelling factor
OD optical density
ORC origin replication complex PBS phosphate buffered saline
PCNA proliferating cell nuclear antigen PCR polymerase chain reaction
PD plasma desorption
pI isoelectric point
PIP PCNA interacting peptide
PIPES 1,4-piperazine diethane sulfonic acid PMSF phenylmethylsulfonyl fluoride
Pol DNA polymerase
ppm parts per million
PRC2 polycomb repressor complex 2 pre-IC pre-initiation complex
pre-RC pre-replication complex Q-q-LIT quadrupole linear ion trap Q-q-Q triple quadrupole
Q-TOF quadrupole time of flight
Rb retinoblastoma
rDNA ribosomal DNA
RF radiofrequency
RFC replication factor C
RNA ribonucleic acid
Rnase H1 RNA nuclease H1
RP reverse phase
RPA replication protein A
RT retention time
S/N signal-to-noise
SAHF senescence associated heterochromatin foci Sas2 something about silencing 2
SCX strong cation exchange
Ser serine
Sir silent information regulator SLBP stem-loop binding protein snRNP small nuclear ribonucleoprotein SSB single strand break
ssDNA single stranded DNA
SUV39H1 suppressor of variegation 3-9 homologue 1
SV40 simian virus 40
T Ag T antigen
TAP tandem affinity purificatin TCEP tris (2-carboxyethyl) phosphine
TEL telomere
TFA trifluoroacetic acid
Thr threonine
TiO2 titanium dioxide
TLS translesion synthesis
TOF time of flight
TPST1/2 tyrosylprotein sulfotransferase 1/2
Tyr tyrosine
URA uracyl
UV ultraviolet
XIC extracted ion chromatogram
XPG xeroderma pigmentosum G
Aknowledgements
First of all, I would like to thank my research director, Dr. Pierre Thibault for giving me the opportunity to work in his lab and gain experience in the field of mass spectrometry as well as for his continual guidance, support and advice throughtout my Ph.D. I would also like to express my gratitude to my co-director, Dr. Alain Verreault for giving me the opportunity to work on exciting projects, for always having time for me, his advice and constant support, motivation through all of his helpful discussions and for providing me with a scientifically motivating environment. I will always be greatful for the opportunities they have given me to advance in their respective fields.
I would also like to thank Dr. Benoit Coulombe and Dr. Martine Raymond for being on my internal thesis committee and for their guidance of my research project throughout many committee meetings.
I am grateful to all past and present members of the Thibault and Verreault lab, for providing me help whenever needed, scientific discussions and constant moral support. I would also like to acknowledge Dr. Eric Bonneil for his help and advice in mass
spectrometry and Eun-Hye Lee for her assistance in molecular biology. Special thanks go to Louiza, Gaelle and Eun-Hye for their friendship.
I would also like to thank my parents and family who always encouraged me to pursue my goals in life. Special thanks go to my husband who had to experience first hand the life of a graduate student. He tolerated the long nights of research and encouraged me whenever I needed encouragement which promoted me to keep going through difficult times. I will always be grateful.
Finally, I would also like to thank MDS Sciex and the Faculté des Études Supérieures de l’Université de Montréal for their scholarships.
1.1. Introduction to chromatin structure and organization
The organization of DNA into chromatin fibres is a truly elegant mechanism that eukaryotic cells have evolved to regulate genome function. Although once widely thought to be mainly required for storing 2 meters of genomic DNA in a confined nucleus of just 10 µm in diameter, chromatin is also a fundamental regulator of many DNA-templated processes. In fact, changes in chromatin structure are at the basis of important functions of DNA such as gene transcription, DNA replication, DNA repair and silencing.
Histones are highly conserved proteins that play a central role in modulating the structure and the dynamics of chromatin. During S-phase of the cell cycle these highly conserved proteins associate with DNA to form the first level of packaging of the genetic material. Consequently, cells that replicate their DNA are also faced with the challenge of accurately duplicating their chromatin structures in an efficient and organized manner. This complex regulation is achieved partly through the combined actions of histone post-translational modifications (PTMs) and histone chaperones, whose function is to accurately escort histones with specific modification patterns from their point of synthesis to their delivery sites on newly replicated DNA.
This thesis will describe chromatin structure and the complexity of its maintenance during DNA replication. More specifically, the first part of the introductory chapter will focus on our current understanding of chromatin assembly and will highlight the key components that couple histone synthesis to their deposition on newly replicated DNA during S-phase.
1.1.1. The nucleosome
In the 1970s, the first insights into the nucleosomal organization of DNA were provided. A combination of physical and molecular biology studies revealed the bead-like structure of the nucleosome and suggested that it is mainly composed of DNA that is wrapped around proteins (Olins, 2003). Subsequent elucidation of the crystal structure of the nucleosome confirmed the molecular details of this nucleoprotein complex (Luger, 1997).
The nucleosome is the fundamental structural and repetitive unit of chromatin. Each nucleosome is composed of about 165 to 200 base pairs of DNA wrapped around an octameric histone protein core. This DNA length, which also comprises the linker region that separates adjacent nucleosomes, varies between different eukaryotic organisms and cell types of the same organism. The actual nucleosomal core consists of 147 base pairs of DNA wrapped in approximately 1.7 circular turns (~ 86 bases pairs per turn) around an octamer composed of two copies of each of the four core histones (Figure 1.1). The histone octamer, created by the direct association of a tetramer containing two copies each of histones H3 and H4 with two histones H2A and H2B dimers, is shaped like a cylindrical disc with a diameter of 13 nm and a height of 11 nm. Each of the histone proteins contain a globular domain known as the histone fold, composed of three alpha helices which are connected by two loops. Pairs of core histones use their globular domains to associate with each other through a handshake-like interaction surface. In addition, each histone contains unstructured N- and/or C-terminal globular domain extensions, comprising between 20-35 residues that are rich in basic amino acids and protrude outside of the nucleosomal surface. These exposed tails are critical for the folding of nucleosomes into higher-organization levels and the structure of chromatin as a whole. (Chakravarthy, 2005; Khorasanizadeh, 2004)
Figure 1.1 The crystal structure of the yeast nucleosome core particle. The DNA
molecule is wrapped around the histone octamer about 1.7 times to form an overall disk-like structure. The two strands of the DNA helix are coloured in dark and light blue. Histones are coloured as followed: H3: green, H4: yellow, H2A: red, H2B: pink (Khorasanizadeh, 2004)
1.1.2. Histone post-translational modifications
Years of research have documented a variety of post-translational modifications that are mainly clustered on the tails of histone proteins. The tails protrude outside of the nucleosome core particle where they are readily accessible to histone-modifying enzymes. These modifications include acetylation, methylation, phosphorylation, ubiquitylation and poly (ADP-ribosylation). It has been proposed that these covalent modifications constitute a « histone code » that is deciphered by various cellular machineries to influence chromatin structure and modulate a variety of nuclear processes (Jenuwein, 2001; Strahl, 2000).
One of the best characterized histone modifications is the acetylation of lysine residues (Kurdistani, 2003) which is associated with many functions. For example, mutational studies have provided evidence for the involvement of histone acetylation in transcription. Acetylation of lysine residues reduces the net positive charge of histone tails thereby weakening their affinity to DNA molecules. As a result, the genetic material is more accessible to transcription factors. Furthermore, acetylation can also regulate DNA replication and repair by specifically recruiting various replication and repair proteins that have modules that interact with acetylated residues. Such domains are termed bromodomains.
Experimental evidence also supports the notion that specific acetylation patterns can regulate the deposition of histones onto DNA and the formation of the nucleosome itself. In proliferating cells, the bulk of histone synthesis occurs during S-phase of the cell cycle, when new molecules of histones H2A, H2B, H3 and H4, the four main core histones, are deposited onto replicating DNA by histone chaperones to form nucleosomes. Immediately after their synthesis, histones H3 and H4 are deposited onto DNA in a pre-acetylated form. Newly synthesized histone H4 molecules were shown by pulse-labelling to be acetylated on 2 specific residues, lysines 5 and 12 (Chicoine, 1986; Sobel, 1995). This pattern of acetylation on new H4 molecules differs from those found in mature chromatin, in which H4 can be acetylated on one, two or three lysine residues, namely lysines 5, 8, 12 and 16. Even though acetylation of lysines 5 and 12 in new H4 molecules is evolutionary conserved, its function in chromatin assembly remains poorly understood. In yeast, acetylation of H4 lysines 5 and 12 is not essential for cell viability or nucleosome assembly. However, nucleosome assembly is severely hampered when H4 is simultaneously mutated at three residues, lysines 5, 8 and 12 in combination with mutations that prevent acetylation of the H3 N-terminal tail. This data suggests that a functional redundancy exists between H3 and H4 N-terminal tail acetylation sites (Ma, 1998). Finally, it has been proposed that acetylation of yeast histone H4 at lysine 91, within its core domain, plays an important role in the stability of the histone octamer and the proper
formation of chromatin structure (Ye, 2005). For histone H3, the acetylation pattern varies among different organisms. In budding yeast, new H3 molecules are preferentially acetylated on lysine 9 (Kuo, 1996) while in Tetrahymina, H3 is acetylated on lysines 9 and 14. In Drosophila melanogaster, lysines 14 and 27 are the preferred sites of acetylation (Sobel, 1995).
One recent and interesting finding is the highly abundant acetylation of newly synthesized H3 molecules on lysine 56, with levels that specifically peak during S-phase (Masumoto, 2005). In yeast, H3 lysine 56 acetylation plays a unique role in nucleosome assembly during DNA replication. Importantly, H3K56Ac acts through a mechanism that is non-redundant with other H3 and H4 acetylation sites and promotes replication-coupled nucleosome assembly by increasing the binding affinity of H3 molecules to histone chaperones such as CAF-1 (Li, 2008; Masumoto, 2005). Moreover, yeast cells that are defective in H3K56Ac are sensitive to DNA damaging agents that interfere with DNA replication (Driscoll, 2007; Han, 2007a; Han, 2007b; Masumoto, 2005). However, compared to the budding yeast Saccharomyces cerevisiae, H3K56Ac is much less abundant in human cells and whether H3K56Ac is involved in chromatin assembly in a mechanism that is similar to yeast remains an open question (Garcia, 2007).
Histone acetylation is performed by enzymes known as histone acetyltransferases (HATs), which catalyze the addition of an acetyl group from acetyl coenzyme A (acetyl-CoA) to the ε-amino group of lysine residues. Histone deacetylases (HDACs) catalyze the reverse reaction, namely the removal of acetyl groups from lysine residues. HATs can be divided into two groups: nuclear A-type HATs and cytoplasmic B-type HATs. A-type HAT enzymes have been implicated in gene transcription and DNA repair, whereas B-type HATs are involved in catalyzing the acetylation of newly synthesized histones (Brownell, 1996). One of the first identified B-type HAT is the evolutionary conserved Hat1, which forms a complex with Hat2 and catalyzes the acetylation of H4 on lysines 5 and 12 (Ai, 2004; Ruiz-Garcı]a, 1998). The HAT enzyme responsible for the deposition-related
acetylation of H3 molecules in Saccharomyces cerevisiae is unclear, although it has been suggested that a new GCN5-dependent complex termed HATB3.1, which shows a strong preference for free histone H3, might be responsible for lysine 9 acetylation (Sklenar, 2004). Finally, several groups have shown that Rtt109 is a member of a novel HAT family that acetylates H3K56 in a mechanism that requires both Asf1 and Vps75 chaperones (Driscoll, 2007; Fillingham, 2008; Han, 2007a; Schneider, 2006; Tsubota, 2007). In flies and humans, recent studies have shown that the Rtt109 homologue CBP/300 promotes H3K56 acetylation providing supporting evidence that acetylation of H3K56 by CBP/p300 promotes assembly of newly synthesized histones into chromatin in higher eukaryotes as well (Das, 2009; Tang, 2008).
1.1.3. Euchromatin and heterochromatin
During most of the cell cycle, chromatin from eukaryotic genomes is packaged into two varieties of structures named euchromatin and heterochromatin. Each type of chromatin structure is formed by the presence of distinct chromosomal proteins and histone post-translational modifications. Euchromatin is considered an open structure with loosely spaced nucleosomes and constitutes active genes that are proficient for transcription and replicate early during S-phase. On the other hand, heterochromatin is a highly condensed and relatively inaccessible structure that comprises late-replicating genes and contains either transcriptionally silenced genes or genomic regions that are devoid of genes (Richards, 2002). In the eukaryotic genome, several regions are packaged into specialized heterochromatin structures that contribute to various epigenetic phenomena. One classic example is the inactivation of the X-chromosome in female mammals, where one of the two X-chromosomes is packaged into silent chromatin while the active X remains transcriptionally active despite being exposed to the same nuclear environment (Richards, 2002). In yeast, regions near telomeres, mating type loci and ribosomal DNA repeats present many features of heterochromatin. For instance euchromatic reporter genes that are normally expressed in cells become strongly silenced when inserted within or near these
heterochromatic regions. This difference in gene expression reflects differences in chromatin structure (Rusche, 2003). Studies in fission yeast and metazoans have also established a role for heterochromatin in chromosome segregation, as mutations in factors that interfere with heterochromatin formation cause defects in cohesion and segregation of sister chromatids (Bernard, 2001).
In the budding yeast Saccharomyces cerevisiae, the establishment of silent chromatin at the silent mating type loci, known as HMLα and HMRa, is determined by cis-acting elements called silencers and trans-cis-acting proteins complexes such as Rap1, Abf1 and the Origin Recognition Complex (ORC). These bind to silencer DNA and help recruit Silent Information Regulator (Sir) proteins (Rusche, 2003). Sir2, Sir3 and Sir4 are essential for efficient silencing at both the HMLα and HMRa mating type loci and sub-telomeric regions, whereas Sir1 is specifically involved in silencing the mating type loci (Aparicio, 1991). While numerous histone acetylation sites are present in active euchromatin regions, nucleosomes in heterochromatin regions are hypoacetylated (relative to euchromatin) at all lysine residues of histones H3 and H4 and it has been demonstrated that Sir proteins are preferentially recruited to hypoacetylated histones (Carmen, 2002; Suka, 2001). Of particular importance for gene silencing is the Sir2-mediated deacetylation of histone H4 on lysine 16. Studies have suggested that deacetylation of H4K16 by Sir2, an evolutionary conserved NAD+-dependent histone deacetylase (HDAC), provides high-affinity binding sites for additional Sir proteins in adjacent nucleosomes and allows the spreading of the Sir complex along chromatin to form silent heterochromatin (Imai, 2000; Rusche, 2003; Tanny, 1999). Sas2 (Something about silencing 2), a histone acetyltransferase (HAT), counteracts the HDAC activity of Sir2 by acetylating H4K16. This creates a boundary that prevents heterochromatin from spreading into adjacent euchromatin regions (Kimura, 2002; Suka, 2002). In recent years, histone methylation has also emerged as an important mark that is associated with functionally specialized chromatin structures (Grewal, 2003). For instance, in fission yeast, flies and mammals, H3K9 methylation is associated with heterochromatin formation, whereas H3K4 and H3K79 di-methylation are excluded from
heterochromatin regions and are linked with transcriptionally active domains (Sims, 2003). Methylation of H4K9 by Clr4 in fission yeast and its orthologue SUV39H1 in flies and mammals directly binds to key structural components of heterochromatin known as HP1 proteins (Swi6 in fission yeast) (Bannister, 2001; Lachner, 2001; Nakayama, 2001; Rea, 2000). Once bound to K9-methylated H3 molecules, these proteins recruit histone modifying enzymes that create additional docking sites for Swi6/HP1 molecules, thus allowing the spreading of repressive chromatin (Hall, 2002) in a manner analogous to the spreading of Sir proteins in Saccharomyces cerevisiae heterochromatn. It has also been proposed that acetylation of H3K9 counteracts methylation of this site (the two modifications are mutually exclusive) and blocks the spreading of heterochromatin structures to adjacent regions (Litt, 2001). One striking observation is that, despite the divergence of the various proteins that are involved in heterochromatin formation and gene silencing from one organism to another, the molecular events that mediate the establishment and spreading of heterochromatin structures are highly conserved. In each case, the covalent modification of histones is necessary for recruitment of enzymes and structural proteins that polymerize and spread in a directional manner to form heterochromatin domains (Grewal, 2003; Moazed, 2001).
Once the silenced state of chromatin is established, the specific patterns of histone modifications must be preserved, such that silenced chromatin structures are faithfully maintained during DNA replication in S-phase and inherited after each cell cycle division, thereby ensuring perpetuation of the integrity and stability of heterochromatin structures (Grewal, 2002; McNairn, 2003). During DNA replication, the transient disruption of pre-existing heterochromatin structures raises the important question of how specialized chromatin states are reformed after replication. Several observations strongly suggest that some factors that function in DNA synthesis are also involved in epigenetic inheritance. For example, hypomorphic mutations in several different replication proteins reduce heterochromatin-mediated gene silencing in Saccharomyces cerevisiae and higher eukaryotes without affecting DNA replication processes (McNairn, 2003; Wallace, 2005).
One particularly important protein in which specific mutations affect heterochromatin-mediated silencing is the DNA polymerase processivity factor PCNA (Sharp, 2001; Zhang, 2000). Furthermore, studies have also demonstrated that chromatin assembly factors whose function is specifically coupled to DNA replication contribute to the maintenance and inheritance of silenced chromatin. Consistent with this, mutations in genes coding for chromatin assembly factors such as CAF-1 cause defects in gene silencing in sub-telomeric regions and the silent mating type loci (Enomoto, 1998; Monson, 1997). Interestingly, CAF-1 is targeted to replication forks during S-phase by direcly binding to PCNA (Shibahara, 1999). Precisely how these two proteins participate in the process of chromatin formation remains poorly understood. The following sections provide a detailed description of the process of DNA replication, followed by an overview of chromatin assembly events that occur behind the replication fork and how these processes work in conjunction to maintain chromatin structure in proliferating cells.
1.2. Genome duplication
The complete, accurate, and processive replication of DNA is vital for the maintenance of cell viability and genomic integrity in all organisms. As a result, eukaryotic cells have evolved molecular regulatory mechanisms to ensure that the duplication of the genetic material occurs with high accuracy and fidelity and, most importantly, takes place only once per cell cycle during S-phase (Bell, 2002; Waga, 1998).
1.2.1. DNA replication
DNA replication in eukaryotes is a complex process that involves the interaction of many evolutionary conserved proteins that act at chromosomal sites known as origins of replication. In Saccharomyces cerevisiae, each origin contains cis-acting “replicators”, which are specific DNA sequences known as autonomously replicating sequence (ARS)
elements. ARS contain multiple short functional elements, termed A1, B1, B2 and B3. The A1 element, which is highly conserved among all budding yeast origins, comprises an 11 bp ARS consensus sequence (ACS) that is essential for DNA replication. The A1 and B1 elements act together to create DNA binding sites for the origin recognition complex (ORC) and they are required for initiation of DNA replication (Rao, 1995). The remaining B elements are not essential but are collectively required as enhancers of origin efficiency (Rao, 1994; Theis, 1994). ARS are also present in other eukaryotes and humans; their sequences are however less well defined and can extend from 800 to over thousands of base pairs of DNA (Bell, 2002).
In prokaryotes, replication initiates at a single origin of a circular genome. In contrast, initiation of DNA replication in eukaryotes occurs at multiple origins distributed along linear chromosomes. The use of numerous origins of replication is an elegant mechanism that cells have evolved to efficiently replicate the entire eukaryotic genome within the relatively short time span of S-phase (Hyrien, 2003). Despite years of study, few origins have been mapped and functionally dissected in mammals and several other model
organisms. The only eukaryote where origins were mapped on a genomic scale is Saccharomyces cerevisiae. Using DNA microarray analysis, it was shown that the budding
yeast genome, which comprises 16 chromosomes, contains about 332 origins of replication (Raghuraman, 2001; Yabuki, 2002). Replication origins were also mapped by analyzing ORC and MCM2 (a component of the replicative DNA helicase that is recruited to ARS prior to origin firing) binding sites and these studies identified about 529 potential origins of replication (Wyrick, 2001; Xu, 2006).
Replication at multiple origins is controlled in a temporal order and individual origins are fired at different times throughout S-phase. Several observations have suggested a relationship between replication timing and the transcriptional potential of genes and/or the types of chromatin that flank the origins (Goren, 2003; McNairn, 2003). Most transcriptionally active euchromatin genes are replicated in the first half of S-phase whereas
most heterochromatin is replicated late in S-phase. In both yeast and mammalian cells, this timing is established in early G1 phase, where origins are repositioned and modified in specific nuclear compartments (Goren, 2003; Li, 2001; Raghuraman, 1997). This temporal program, which coordinates origin firing within tanscriptionally active and inactive regions, may be important for the propagation of specialized chromatin features to daughter cells (Dillon, 2002; Flickinger, 2001).
1.2.1.1. Replication initiation
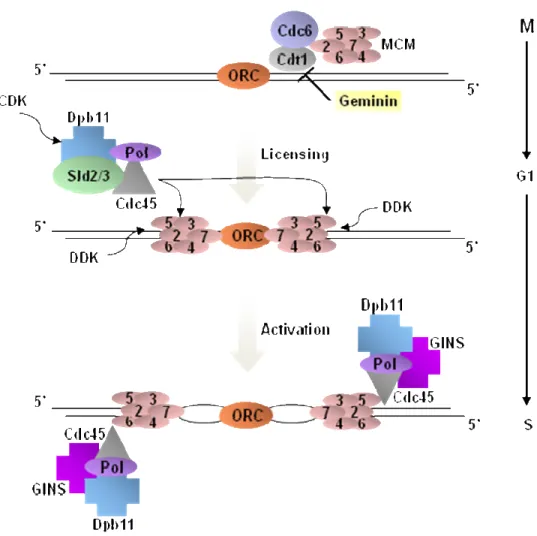
DNA replication from individual origins is generally bidirectional and tightly coordinated with cell cycle events. The basic mechanism of initiation of DNA replication in eukaryotic cells occurs in two steps. The origin licensing process, also known as the assembly of the pre-replicative multiprotein complex (pre-RC), is followed by the origin activation process (Figures 1.2, 1.3). Assembly of the pre-RC is initiated by DNA sequence specific binding of the origin recognition complex (ORC) to the replication origin (Bell, 1992). At the end of mitosis and early G1, Cdc6 and Cdt1 are independently recruited to ORC-bound replication origins (Figure 1.2). ORC, Cdc6 and Cdt1 form a complex, and are collectively required for stable association of the minichromosome maintenance 2-7 complex (MCM 2-7) helicase with the origin to form the pre-RC (Speck, 2007; Tanaka, 2002). At the G1/S phase transition and throughout S-phase, the conversion of the pre-RC into the pre-initiation complex (pre-IC) is necessary before DNA synthesis can begin (Figure 1.2). The pre-IC contains additional proteins that activate the DNA helicase activity of the MCM 2-7 complex which then unwinds the two DNA strands as a pre-requisite for origin firing. These include Cdc45, Sld2, Sld3 and Dpb11 as well as S-phase cyclin-dependent kinases (CDKs) and the Dbf4-cyclin-dependent kinase (DDK or Cdc7-Dbf4) which regulate distinct steps in the activation of replication origins (Zou, 2000).
Figure 1.2 Initiation of DNA replication in eukaryotes. Replication origins are
recognized by ORC. Cdt1, Cdc6 and the MCM helicase complex subsequently associate with origins in G1 to form pre-RCs. At this point, origins are “licensed” but not yet activated. In late G1, CDK and DDK become activated and in turn trigger origin activation by phosphorylating Sld2, Sld3 and the MCM proteins. This leads to the activation of the hexameric MCM helicase (MCM2-7), DNA unwinding and recruitment of other enzymes into the “replisome”. In order to prevent inappropriate pre-RC formation and re-replication in S, G2 and M phases, CDK also phosphorylate and inactivate certain components of pre-RC and inhibit further re-licensing after origin firing. In vertebrates, another level of regulation to block re-replication is mediated through geminin, a protein that inhibits Cdt1 and prevents the reloading of MCM proteins without interfering with ORC and Cdc6 association in G1 (Sclafani, 2007).
a) b) c) d) e) f)
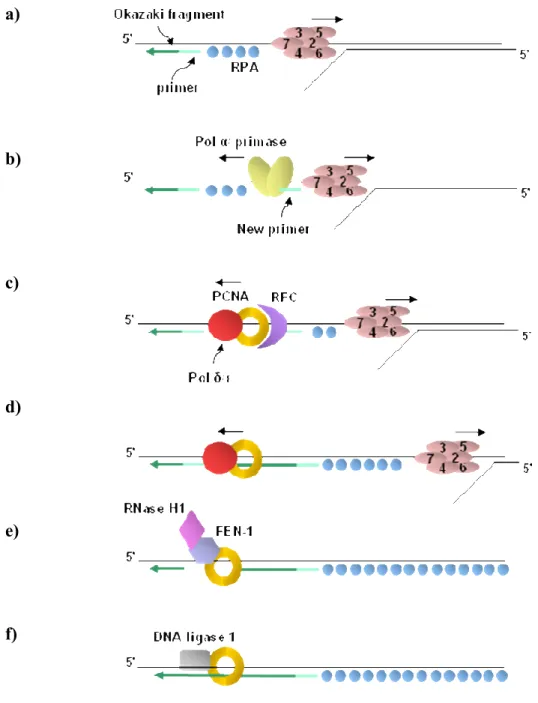
Figure 1.3 The process of DNA replication in S-phase. The events taking place during
DNA synthesis of the lagging strand are shown. Synthesis of the leading strand (not shown) requires only a subset of these events because it is synthesized in a continuous manner from a single primer, rather than in short fragments known as Okazaki fragments. These fragments need to be joined together by Okazaki fragment maturation enzymes whose action is only needed during lagging strand synthesis. a) The DNA helix is unwound by the MCM helicase and the resulting single stranded DNA (ssDNA) is coated with RPA proteins. b) Pol α/primase is recruited to the RPA coated ssDNA and synthesises a short
RNA/DNA hybrid primer, displacing the RPA molecules as it travels along the template.
c) The clamp loader RFC recognizes the completed primer-template junction, displaces Pol
α/primase and catalyses the loading of the PCNA sliding clamp and DNA polymerases δ or ε. d) Through their binding to PCNA, pol δ/ε processively synthesise DNA from the RNA/DNA primer until they reach the 5’ end of the next Okazaki fragment. Similar events take place at the leading strand, however since DNA synthesis occurs in 3’-5’ direction, leading strand synthesis is continuous. e) The RNA/DNA primer of the previous Okazaki fragment is removed by FEN-1 and RNase H1. f) The single-stranded gap generated by primer removal is filled in and the two DNA segments are joined by DNA ligase 1 (Morgan, 2007).
Some evidence indicates that phosphorylation of the MCM protein complex by Cdc7-Dbf4 is required for Cdc45 loading, which is an essential step for origin activation (Sclafani, 2002; Zou, 2000). It has been hypothesized that phosphorylation of MCM proteins induces a conformational change in the MCM2-7 complex that activates the helicase and is a signal for Cdc45 binding to origins (Fletcher, 2003; Sclafani, 2002). The essential role of CDKs in promoting origin activation has also been determined. CDKs phosphorylate Sld2 and Sld3 which enable them to interact with Dpb11, a subunit of the DNA polymerase ε holoenzyme (Tanaka, 2007; Yabuuchi, 2006; Zegerman, 2007). CDK phosphorylation of Sld2 is absolutely required for replication initiation and for Cdc45 loading onto origins (Masumoto, 2002). The GINS complex (consisting of Sld5, Psf1, Psf2 and Psf3) is also required for loading of Cdc45 onto origins and replication initiation (Gambus, 2006; Takayama, 2003; Yabuuchi, 2006). The combined action of CDKs and DDK is required to load other components of the replisome and activation of the DNA helicase, which unwinds DNA and allows DNA replication to proceed.
1.2.1.2. Replication elongation
Much of our knowledge of the steps taking place during DNA replication comes from the study of the simian virus 40 (SV40). In this system, DNA is replicated by the viral T antigen (T Ag) that acts in concert with other cellular proteins (Waga, 1998). After site-specific recognition of the origin of DNA replication by T antigen, its intrinsic helicase activity unwinds the DNA double helix near the origin. This step is necessary for binding the heterotrimeric single-stranded DNA (ssDNA) binding protein, known as replication protein A (RPA) (Figure 1.3a). RPA binds to stretches of ssDNA to protect and stabilize DNA strands exposed during DNA replication or repair (Wold, 1997). In a subsequent step called primosome assembly, the DNA polymerase α/primase complex (pol α/primase) is loaded on DNA through a direct interaction with RPA (Walter, 2000) (Figure 1.3b). The pol α/primase complex contains four evolutionary conserved subunits (p180, p70, p58 and p48 in human cells). The p180 and p48 subunits harbour the polymerase and primase activity, respectively. Once the replisome is assembled, pol α/primase synthesizes de novo a short hybrid RNA-DNA primer that contains about 30 nucleotides (nts) (Figure 1.3b). DNA pol α is the only eukaryotic enzyme that can initiate synthesis de novo, i.e. without the help of a primer (Hübscher, 2002). The two activities of DNA pol α/primase, DNA primase and DNA polymerase, act sequentially to generate the RNA-DNA primer. Key to this reaction is the primase activity, which can lay down a short RNA primer that acts as a seed for DNA polymerase α. Next, replication factor C (RFC), a heteropentameric ATPase protein complex, binds tightly to the primer template junction and locally promotes the assembly of the primer recognition complex (Mossi, 1998). This consists in loading the ring-shaped DNA sliding clamp Proliferating Cell Nuclear Antigen (PCNA) and its subsequent association with the pol δ holoenzyme (Figure 1.3c). The PCNA ring is able to slide freely along the DNA and, in conjunction with the replicative DNA polymerase, promotes processive DNA synthesis on the leading strand for at least 5-10 kilobases (kbs) (Matsumoto, 1990). Since DNA synthesis always occurs in the 5`to 3`direction, the lagging
strand needs to be synthesized in the direction opposite from that of the replication fork. As a result, lagging strand synthesis is discontinuous and divided into short, 180 nts Okazaki fragments (Hübscher, 2001) (Figure 1.3d). In vitro in the SV40 system, Pol δ seems to be the main replicative polymerase and is able to support DNA synthesis of both the leading and lagging strands (Fukui, 2004; Hübscher, 2002; Waga, 1998). Although pol ε is not essential for SV40 DNA replication in vitro, it is essential for cellular DNA replication (Karthikeyan, 2000; Waga, 2001; Zlotkin, 1996). Even though more studies are required to delineate the precise function of Pol ε in DNA replication, it has become clear that, in vivo, both Pol δ and Pol ε play essential roles as replicative DNA polymerases (Fuss, 2002). Recently, the first evidence for Pol ε to support DNA synthesis of the lagging strand was demonstrated (Pursell, 2007). In addition, Pol ε plays a unique role in a checkpoint that signals DNA damage during replication (Fuss, 2002).
Finally, the lagging strand is converted into a long continuous DNA product by a process called Okazaki fragment maturation. After the replicative DNA polymerase encounters an RNA primer at the 5` end of the previous Okazaki fragment, the primer is completely removed (Figure 1.3e) by two different nucleases, RNase HI and a 5`-3` endonuclease called Flap endonuclease 1 (FEN-1) (Hübscher, 2001; Waga, 1998). RNase HI cleaves the RNA primer at the 5`end of each Okazaki fragment leaving a single ribonucleotide which gets removed by FEN-1. PCNA also binds to FEN-1 and stimulates FEN-1 activity by at least 50-fold (Li., 1995). Recent biochemical and genetic studies strongly support the existence of a redundant pathway involving the participation of the helicase/endonuclease Dna2 with FEN-1 (Bambara, 1997; Hübscher, 2001). One hypothesis is that Dna2 displaces the RNA segment from the DNA template which then gets cleaved off by FEN-1. The gap created is then filled in by either pol δ or pol ε. The nicks left between adjacent Okazaki fragments are effectively ligated by another PCNA binding protein, DNA ligase 1 (Figure 1.3f). The superhelical strain that is caused by the unwinding of the DNA double helix is released by DNA topoisomerase I and then
replicated DNA molecules are decatenated by DNA topoisomerase II (Hübscher, 2001; Montecucco, 1998; Wang, 2002).
As mentioned above, “lagging” strand synthesis requires several reactions that must be tightly coordinated with synthesis of the “leading” strand. This coordination depends, at least in part, on PCNA and is required in order to avoid synthesis of the lagging strand from falling behind by hundred of nucleotides from leading strand synthesis events (Hübscher, 2001; Lee, 1998; Prelich, 1988). Furthermore, the DNA polymerase on the lagging strand is transferred from one Okazaki fragment to the other, rather than having new molecules being reloaded every 200bp, which also favours processivity of lagging strand DNA synthesis. Finally cells have evolved a looping back mechanism on the lagging strand that allows both strands to extend in the same direction and at similar rates. Studies using the bacteriophage T7 replication system have recently highlighted the importance of the primase-helicase T7-gp4 in limiting the speed of the leading strand polymerase. In fact, the looping of the lagging strand has been shown to allow the T7-gp4 primase-helicase to maitain contact with the nascent primer on the lagging strand as well as allowing the primer to be withing physical reach of the lagging strand replication complex. This mechanism of coordination provides efficient primer utilization and ensures adequate primer hand-off to the lagging strand T7 polymerase (Pandey, 2003).
1.2.2. DNA repair
The integrity of DNA is continuously challenged by DNA replication errors, metabolic products that damage DNA (e.g. oxidizing species) and genotoxic agents from the environment (e.g. chemical from tobacco smoke). Efficient detection of DNA damage and nucleotides misincorporated during DNA replication is particularly vital for dividing cells as replication and segregation of unrepaired DNA can seriously compromise genomic integrity. In order to ensure accurate transmission of genetic information, eukaryotic cells
have evolved elaborate surveillance and repair mechanisms that rapidly recognize and repair damaged DNA at the replication fork (Dinant, 2008).
1.2.2.1. Cell cycle checkpoint activation
In response to DNA damage, cells trigger an evolutionary conserved signalling cascade involving proteins of the DNA damage checkpoint response. These are designed to detect damaged DNA and coordinate cell cycle progression with DNA repair. More precisely, activation of the DNA damage checkpoint results in a number of downstream biological consequences whereby cell cycle progression through the G1, S or M-phase is transiently delayed and ongoing DNA replication is slowed down, providing cells more time to repair DNA lesions (Paulovich, 1995; Weinert, 1988). In addition, various genes and proteins involved in the repair of DNA lesions are activated by transcriptional and post-transcriptional mechanisms and become localized to sites of repair (Brush, 1996; D'Amours and Jackson, 2001; Gasch et al., 2001). Chromatin also undergoes structural changes to allow access of the repair machinery to damaged sites. These changes are mediated by histone post-translational modifications or recruitment of non-histone proteins, such as ATP-dependent chromatin remodelling factors and histone chaperones, to sites of DNA damage (Escargueil, 2008; Osley, 2006; Polo, 2006). At the top of the signalling cascade is a specific group of checkpoint proteins called the phospho-inositol kinase-related proteins. These include the protein kinases ATM/ATR in humans and Mec1/Tel in yeast (Lowndes, 2000; Rouse, 2002). These kinases play a central role in all checkpoint responses as they can respond to DNA damage generated by ultraviolet (UV) irradiation, gamma irradiation, DNA alkylating agents such as methyl methanosulfonate (MMS) and the replication inhibitor hydroxyurea (HU) that acts by depleting deoxyribonucleoside triphosphates (dNTP) (Abraham, 2001; Foiani, 2000; Rhind, 1998). Two classes of checkpoint effector kinases, CHK1 and CHK2 in mammals (Chk1 and Rad53 in Saccharomyces cerevisiae) function downstream of the phospho-inositol related kinases. These kinases become rapidly phosphorylated and activated in a Mec1/Tel1
(ATR/ATM)-dependent manner. Once activated, they modulate the activities of key effector proteins of the DNA damage response (Lowndes, 2000; Rouse, 2002). In budding yeast, an additional kinase named Dun1 is activated by Rad53 in response to DNA damage. Dun1 is involved in the control of transcriptional regulation and is needed to increase dNTP pools (Zhou, 1993).
1.2.2.2. DNA repair mechanisms
A number of highly sophisticated DNA repair pathways have evolved to cope with various types of DNA lesions. Single strand damages are repaired by three major damage repair pathways that include nucleotide excision repair (NER), base excision repair (BER), and mismatch repair (MMR). These processes function by excising a short stretch of the damaged DNA strand and then using the undamaged complementary strand to re-synthesize the correct DNA sequence (Ataian, 2006).
The NER pathway is specifically responsible for removing intra-strand helix distorting lesions such as UV-induced 6-4 photoproducts (6-4PPs) and cyclobutane pyrimidine dimers (CPDs). During NER, a large number of repair as well as replication proteins get sequentially deployed at the site of DNA damage to perform the incision as well as the gap-filling DNA synthesis and ligation steps. Incision is mediated by the Xeroderma pigmentosum G (XPG) endonuclease, a FEN-1 related nuclease that cleaves at the 3' side of the DNA lesion and is followed by an ERCC1-XPF mediated incision at the 5` side of the lesion (de Laat, 1999). Similarly to FEN-1, XPG also interacts with PCNA (Gary, 1997). The DNA repair synthesis step is carried out by DNA polymerase δ or ε, which fill in the gap with the correct nucleotides in a PCNA-dependent manner.
The BER pathway is mainly implicated in the repair of small non-distorting chemical alterations such as alkylation and oxidative damage to DNA bases (Zharkov, 2008). BER is initiated by the action of a DNA glycosylase that binds to the damaged DNA
base and hydrolyzes the N-glycosydic bond to release the damaged base from the deoxyribose. This cleavage generates an apyrymidinic/apurinic (AP) site that is subsequently processed by an AP-endonuclease. The synthesis step in BER is carried out by a short patch (single base) or a long patch (2-7 nts) pathway. In short patch BER, the missing nucleotide is filled in by DNA polymerase β, which also interacts directly with PCNA (Kedar, 2002). On the other hand, long patch BER utilizes the replication elongation apparatus, where FEN-1, PCNA, DNA polymerase β or δ and DNA ligase 1 are required for filling the gap and sealing the nicks (Klungland, 1997).
MMR targets mismatched bases and insertion-deletions mispairs (IDLs) that arise from replication errors (Iyer, 2005). Two distinct MutS-related heterodimeric complexes that have different mismatch recognition specificity can act to promote MMR. MSH2-MSH3 recognizes mispaired bases, while MSH2-MSH6 targets IDLs. The process also requires MLH1 which interact with MSH2 and MSH3 as well as other components of the replication machinery, including PCNA. Several studies have highlighted the important function of PCNA in MMR (Genschel, 2003; Gu, 1998; Umar, 1996). Importantly, PCNA is not only required for the DNA repair synthesis process, where it forms part of the DNA elongation apparatus, but appears to play a critical role in the initiation step as well. Recent observations revealed that PCNA enhances the mispair binding specificity of MSH2-MSH6 when a replication error is encountered (Flores-Rozas, 2000; Lau, 2003) This has led to the proposal that PCNA helps the MMR machinery mediate an efficient search for mismatches on newly replicated regions of DNA.
Finally, when the damaged DNA cannot be repaired, a fourth mechanism known as DNA damage bypass or DNA damage tolerance allows completion of DNA replication without removal of the damaged base. Although the DNA polymerase δ and ε replicate DNA in a highly processive and accurate manner, they are unable of copying DNA containing lesions such as CPDs, damaged bases and AP sites. When the replication machinery encounters such lesions, specialized polymerases termed translesion synthesis
(TLS) polymerases are recruited to stalled replication forks. Despite the conformational constraints imposed by the damaged bases, TLS polymerases can accommodate distorted template structures within their active site and replicate past these lesions. However, in contrast to replicative polymerases, TLS polymerases function with reduced fidelity on damaged DNA templates and are therefore mutagenic (Waters, 2009). Interestingly, several studies have suggested that stalling of the replication machinery at sites of DNA damage triggers post-translational modification of PCNA (Hoege, 2002). More specifically, PCNA ubiquitination promotes polymerase switching, where the replicative enzymes pol δ and ε are replaced by TLS polymerases, such as pol η which specialises in the bypass of UV-damaged CPDs (Kannouche, 2004; Stelter, 2003; Xiao, 2000).
1.3. The role of PCNA in genome duplication and maintenance
of chromatin structure
Proliferating cell nuclear antigen (PCNA) is a member of the DNA sliding clamp family, whose structure and function are evolutionary well conserved from yeast to humans (Moldovan, 2007). Alignment of amino acid sequences of PCNA from different organisms shows a relatively low level of sequence conservation. Human and yeast PCNA share only 35% amino acid sequence identity (Bauer, 1990). Nevertheless, crystallographic studies have shown that their three-dimensional structures are highly superimposable even with the homodimeric structure of the functional homologue of PCNA in bacteria, the β-subunit of DNA polymerase III (Krishna, 1994). The latter forms a homodimer and shows essentially no sequence similarity with PCNA in eukaryotes. In contrast to its functional equivalent in prokaryotes, eukaryotic PCNA forms a homotrimeric ring shaped complex with pseudo-six-fold symmetry. Because PCNA contains a central cavity, it is capable of encircling double-stranded DNA and sliding freely along it. Replication factor C (RFC) catalyzes the loading of PCNA by transiently disrupting the PCNA ring and rapidly re-closing it when it encircles DNA (Bowman, 2004; Majka, 2004). Its topological link to DNA allows PCNA