Interventions pharmacologiques affectants
spécifiquement la signalisation du récepteur D2 de la
dopamine médiée par la beta arrestine 2
Thèse
Gohar Fakhfouri
Doctorat en neurosciences
Philosophiæ doctor (Ph. D.)
Interventions pharmacologiques affectants
spécifiquement la signalisation du récepteur
D2 de la dopamine médiée par la beta
arrestine 2
Thèse
Gohar Fakhfouri
Sous la direction de :
Jean Pierre Julien, directeur de recherche
Jean Martin Beaulieu, codirecteur de recherche
Résumé
Le système dopaminergique joue un rôle essentiel dans plusieurs processus physiologiques : au niveau de l’activité locomotrice, des émotions, de l’humeur, des performances cognitives et des fonctions neuroendocrines. Le récepteur D2 de la dopamine (D2R) fait partie de la famille des récepteurs couplés aux protéines G (RCPG). D2R est couplé à la sous-unité Gαi/o qui inhibe l'adénylyl cyclase, réduisant ainsi la synthèse du messager secondaire cAMP. En parallèle, D2R signalise aussi via un mécanisme dépendant de βArr2 conduisant à une déphosphorylation/inactivation d’Akt et à une déphosphorylation/activation concomitante de son substrat, la glycogène synthase kinase 3β (GSK3β). GSK3β est inhibée en réponse aux antipsychotiques, aux stabilisateurs de l'humeur et aux antidépresseurs. L'inhibition pharmacologique ou génétique de GSK3β imite les effets de ces agents sur le comportement des rongeurs. Alors que les antipsychotiques sont en grande partie des antagonistes de D2R, le lithium perturbe un complexe de signalisation composé de Akt:βArr2:PP2A qui se forme en aval de l'activation de D2R. La lamotrigine et le valproate ne sont cependant pas des ligands de D2R. Ces derniers bloquent plutôt les canaux sodiques dépendant du voltage (Nav).
Au chapitre 2, la caractérisation de l'effet de la lamotrigine sur la réponse locomotrice aux agents dopaminergiques révèle une modulation post-synaptique de l’activité locomotrice médiée par D2R et de la signalisation βArr2, mais pas par celle du récepteur D2 de la dopamine. Nous démontrons que dans le striatum D2R forme un complexe avec Nav1.6, une cible de la lamotrigine, et que l’extrémité C-ter du canal est responsable de cette interaction. L'association de D2R/Nav1.6 ne modifie pas les propriétés du canal, n'a aucun effet sur l'inhibition de cAMP médiée par la protéine G mais conduit plutôt à une modulation sélective de βArr2, mais pas de βArr1, ni de l'interaction βArr2/AP2. Fait intéressant, l’association de D2R/Nav1.6 nui à l’internalisation, qui est potentialisée par la lamotrigine. La surexpression de l’extrémité C-ter de D2R dans les neurones D2R striataux aboli la signalisation de D2R médiée par βArr2 ainsi que la réponse locomotrice à l'activation du récepteur, reproduisant les effets de la lamotrigine. La surexpression de l’extrémité C-ter de D2R dans les neurones du cortex préfrontal médian est associée à une altération de la mémoire de travail. Ces observations indiquent l'existence d'interactions fonctionnelles et physiques entre Nav1.6 et D2R entraînant une modulation négative de la signalisation de D2R médiée par βArr2. Nous proposons que l'interaction D2R/Nav1.6 altère les mécanismes d'internalisation en raison d'un encombrement stérique et/ou d'une conformation non permissive, qui est ensuite stabilisée par la lamotrigine via une liaison à Nav1.6. Il est concevable que l’internalisation altérée empêche βArr2 de former le complexe de signalisation avec Akt après l'activation de D2R, ce qui correspond aux effets du traitement avec la lamotrigine et à la surexpression de l’extrémité C-ter in vivo. Dans le chapitre 3, suite aux conclusions du chapitre 2, du rôle nouvellement apprécié de D2R en tant que facteur de risque de dépression, et de notre étude précédente révélant la capacité de la fluoxétine à bloquer le canal cardiaque Nav1.5, nous avons examiné le rôle du D2R dans les effets de cet antidépresseur chez la souris. Nous démontrons que l'efficacité des traitements chroniques avec des antidépresseurs et leurs effets sur la neurogénèse associés à la fluoxétine nécessitent l'expression et la fonctionnalité du D2R dans le cerveau. Cette dépendance à l'action de D2R est spécifique à la fluoxétine et n'est pas partagée par la tranylcypromine, un inhibiteur de la monoamine oxydase. Néanmoins, la fluoxétine a
conservé son effet comportemental aigu chez les souris knock-out Drd2. Cela signifie que le rôle de D2R dans les voies associées à la neurogenèse et à un comportement de type antidépresseur dépendant de la neurogenèse est sélectif et jusque-là inconnu. Fait intéressant, une évaluation électrophysiologique a révélé que la fluoxétine, mais non la tranylcypromine, est un bloqueur de Nav1.6 au niveau neuronal fournissant ainsi un mécanisme par lequel la fluoxétine peut exercer ses effets dépendant du D2R. Enfin, notre étude montre que le BDNF est un médiateur de l’effet associé à D2R dans l’hippocampe lors de l’administration chronique de la fluoxétine.
Prises dans leur ensemble, nos données suggèrent un nouveau mécanisme selon lequel les bloqueurs de Nav peuvent agir en tant que modulateurs sélectifs de la voie βArr2 de la signalisation D2R, conférant ainsi des effets antidépresseurs et stabilisateurs de l'humeur. Ensemble, nos résultats montrent comment il est possible d'obtenir une signalisation biaisée d'un RCPG donné en ciblant un partenaire non RCPG avec lequel il interagit.
Abstract
The dopaminergic system plays an essential role in a multitude of physiological processes including motor behavior, emotion, mood, cognitive performance and neuroendocrine function. The dopamine D2 receptor (D2R) is a prototypical G protein coupled receptor (GPCR) that couples to Gαi/o to inhibit adenylyl cyclase, thereby reducing the synthesis of the secondary messenger cAMP. In addition, D2R signals through a beta-arrestin2 (βArr2)-dependent mechanism leading to dephosphorylation/inactivation of Akt and concomitant dephosphorylation/activation of its substrate glycogen synthase kinase 3β (GSK3β). GSK3β is inhibited in response to antipsychotics, mood stabilizers, and antidepressants. Moreover, pharmacological or genetic inhibition of GSK3β mimics the effects of these agents on rodent behavior. While antipsychotics are largely D2R antagonists, lithium disrupts a signaling complex comprised of Akt:βArr2:PP2A that forms downstream of D2R activation. However, lamotrigine and valproate are not known ligands at D2R. Instead, they both block neuronal voltage gated sodium channels (Nav). In chapter 2, characterization of lamotrigine impact on the locomotor response to dopaminergic agents revealed a post-synaptic modulation of D2R-mediated locomotion and βArr2 signaling, but not that of dopamine D1 receptor. We demonstrated that in the striatum, D2R forms a complex with Nav1.6, a target of lamotrigine, and that the Ctail domain of the channel is responsible for this interaction. D2R/Nav1.6 association did not alter channel properties, had no effect on G protein-mediated cAMP inhibition but led to a selective modulation of βArr2, but not βArr1 recruitment, and of βArr2/AP2 interaction. Interestingly, D2R/Nav1.6 association hindered internalization, which was further potentiated by lamotrigine. Overexpression of Ctail in striatal D2R neurons abolished βArr2-mediated D2R signaling and locomotor response to D2R activation, replicating lamotrigine effects. Overexpression of Ctail in the medial prefrontal cortex D2R neurons was associated with working memory impairment. These observations indicate the existence of a functional and physical Nav1.6/D2R interaction resulting in negative modulation of βArr2-mediated signaling of D2R. We propose that D2R/Nav1.6 interaction impairs internalization machinery due to induction of steric hindrance and/or a non-permissive conformation, which is further stabilized by lamotrigine via binding to Nav1.6. It is conceivable that impaired internalization incapacitate βArr2 from forming the signaling complex with Akt following D2R activation, consistent with the effects of lamotrigine treatment and Ctail overexpression in vivo. In chapter 2, in light of our findings in chapter 1, the newly appreciated role of D2R as a risk factor for depression, and our previous study revealing fluoxetine’s ability to block cardiac Nav1.5, we examined the role of D2R in the effects of this antidepressant in mice. We demonstrated that chronic antidepressant efficacy and the associated neurogenic effects of fluoxetine require the expression and functionality of brain D2R. Such D2R-dependency of action was specific to fluoxetine and not shared by the monoamine oxidase inhibitor tranylcypromine. Nonetheless, fluoxetine retained its acute behavioral effect in Drd2 knockout mice. This signifies a selective and previously unappreciated role for D2R in pathways associated with neurogenesis and neurogenesis-dependent antidepressive-like behavior. Interestingly, electrophysiological assessment revealed that fluoxetine, but not tranylcypromine, is a blocker of neuronal Nav1.6, therefore providing a mechanism by which fluoxetine can exert its D2R-dependent effects. Lastly, our investigation pointed to BDNF as a mediator of D2R-associated effect of chronic fluoxetine in the hippocampus.
Collectively, our data suggest a novel principle by which Nav blockers can act as βArr2 pathway-selective modulators of D2R signaling, thus conferring antidepressant and mood stabilizing effects. Together, our findings showcase how biased signaling of a given GPCR can be achieved by targeting a non-GPCR interacting partner.
Table of contents
Résumé ... ii Abstract ... iv Table of contents ... vi List of Figures ... ix List of abbreviations ... x Acknowledgement ... xii Foreword ... xiii Introduction ... 1G protein coupled receptors (GPCRs) ... 2
G protein subunits ... 2
G protein-coupled receptor kinases (GRKs) ... 6
Regulators of G-protein signaling (RGS) ... 7
Arrestins... 7 GPCR trafficking ... 9 Effect of cations on GPCRs ... 11 Ligand bias ... 11 Dopaminergic system ... 14 Dopmaine... 14
Dopamine pathways and circuits ... 15
Dopamine receptors ... 16
Dopamine receptor expression ... 17
Dopamine receptor signaling ... 18
GSK3 ... 25
GSK3 regulation by dopaminergic system ... 26
GSK3 regulation by other mechanisms ... 27
GSK3 in mood disorders and its importance as a therapeutic target ... 28
GSK3 substrates ... 30
GSK3 substrates likely to be involved in mood disorders and schizophrenia... 30
D2R oligomerization ... 36 A2AR-D2R ... 38 NMDAR-D2R ... 38 5-HT2AR-D2R ... 39 OXTR-D2R ... 39 NTS1-D2R ... 39 Other D2R heterodimers ... 40
Voltage gated sodium channels ... 40
Allosteric modulation of voltage gated sodium channel ... 42
Bipolar disorder ... 43
Pharmacotherapy ... 44
Antidepressant medications and neurogenesis ... 46
Neurogenesis ... 46
Antidepressant-induced neurogenesis ... 47
1 CHAPTER: TRANS-MOLECULAR REGULATION OF BETA-ARRESTIN-MEDIATED DOPAMINE D2 RECEPTOR SIGNALING BY VOLTAGE GATED
SODIUM CHANNELS. ... 52
1.1 RÉSUMÉ ... 53
1.2 ABSTRACT ... 54
1.3 INTRODUCTION ... 55
1.4 MATERIALS AND METHODS ... 57
1.5 RESULTS ... 66
1.5.1 Lamotrigine modulates dopamine-mediated locomotion in a D2R-dependent fashion 66 1.5.2 Lamotrigine modulates beta-arrestin2-mediated biochemical response to amphetamine in D2R neurons ... 69
1.5.3 Lamotrigine does not affect D2R coupling to G protein-dependent cAMP inhibition or β-arrestin2 recruitment in a heterologous expression system ... 74
1.5.4 Voltage-gated sodium channel, the target of lamotrigine, forms a complex with D2R 74 1.5.5 The C-terminal region within Nav1.6 is responsible for Nav1.6/D2R interaction ... 78
1.5.6 D2R activation does not affect Nav1.6 channel currents ... 81
1.5.7 D2R/Nav1.6 interaction differentially modulates D2R coupling to signaling effectors ... 84
1.5.8 D2R/Nav1.6 interaction hinders D2R internalization ... 87
1.5.9 Lamotrigine potentiates Nav1.6-driven hindrance of D2R internalization ... 87
1.5.10 Viral-mediated overexpression of the D2R-interacting region of Nav1.6 in the striatal D2R neurons replicates lamotrigine behavioral and signaling effects ... 88
1.5.11 Viral-mediated overexpression of the D2R-interacting region of Nav1.6 in the mPFC D2R neurons impairs working memory ... 93
1.6 DISCUSSION ... 95
2 CHAPTER: ROLE OF DOPAMINE D2 RECEPTOR IN FLUOXETINE-INDUCED NEUROGENESIS ... 99
2.1 RÉSUMÉ ... 100
2.2 ABSTRACT ... 101
2.3 INTRODUCTION ... 102
2.4 MATERIAL AND METHODS ... 103
2.5 RESULTS ... 108
2.5.1 Dopamine D2 receptor is not required for antidepressant effects of acute Fluoxetine ... 108
2.5.2 Dopamine D2 receptor is indispensable for antidepressant effects of chronic Fluoxetine in the novelty-suppressed feeding test ... 109
2.5.3 Dopamine D2 receptor is necessary for neurogenic effects of chronic Fluoxetine ... 111
2.5.4 Dopamine D2 receptor mediation of neurogenic effect is selective for fluoxetine and not a common property of antidepressant treatment ... 112
2.5.6 Chronic fluoxetine relies on beta-arrestin2-mediated dopamine D2 receptor
signalling ... 115
2.5.7 Fluoxetine is a blocker of neuronal voltage gated sodium channels ... 118
2.5.8 Dopamine D2 receptor contributes to chronic fluoxetine effects by enhancing brain-derived neurotrophic factor in the hippocampus... 119
2.6 DISCUSSION ... 120
Conclusion ... 126
List of Figures
Figure 1-1 Paradigms of GPCR-mediated signaling and multiple roles of βarrestins ... 9
Figure 1-2 Mechanisms of ligand-induced biased agonism ... 13
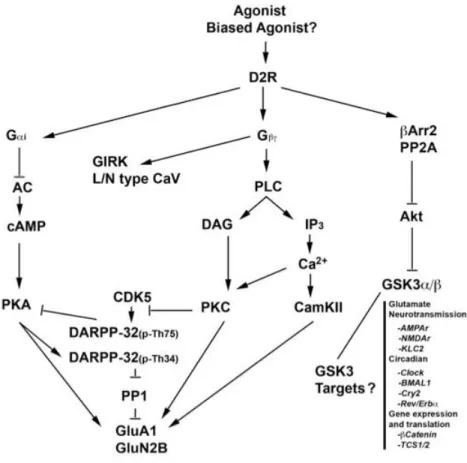
Figure 1-3 Schematic diagram representing the signalling cascades activated by the D2 dopamine receptor (D2R). ... 25
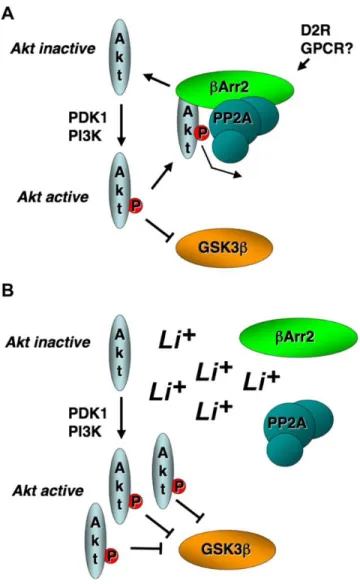
Figure 1-4 Model for Akt-mediated indirect inhibition of GSK3β by lithium ... 29
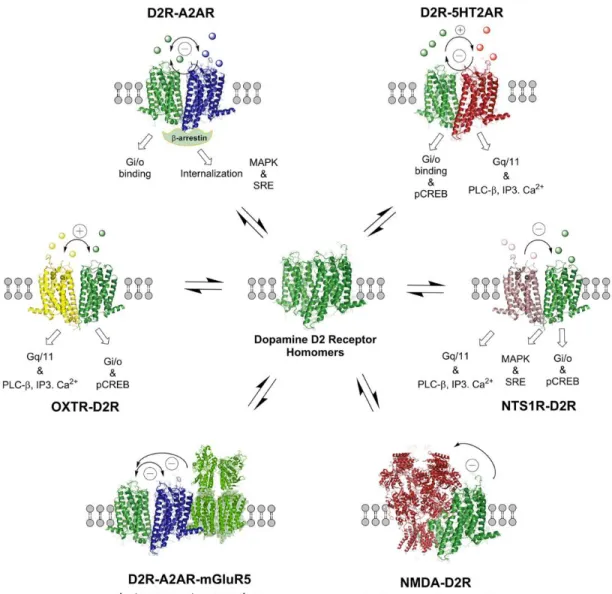
Figure 1-5 The D2R heteroreceptor complexes mainly found in the striatum ... 37
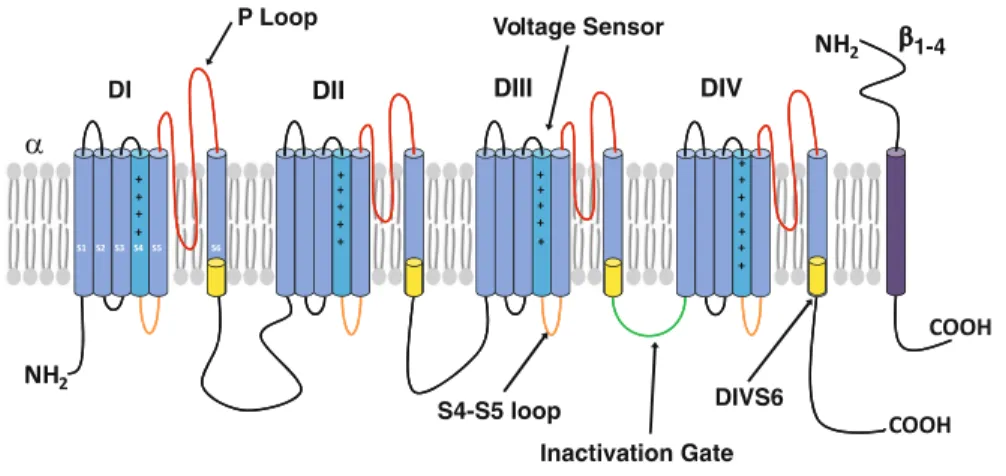
Figure 1-6 Organization of voltage-gated sodium channels ... 42
Figure 1-1: Lamotrigine inhibits locomotor response to dopaminergic stimulation in a D2R-dependent manner ... 69
Figure 1-2: Lamotrigine reverses modulation of Akt/GSK3β by amphetamine in the striatum but has no effect on D2R signaling pathways in vitro... 71
Figure 1-3: Lamotrigine reverses amphetamine-induced GSK3β dephosphorylation selectively in striatal D2R neurons ... 73
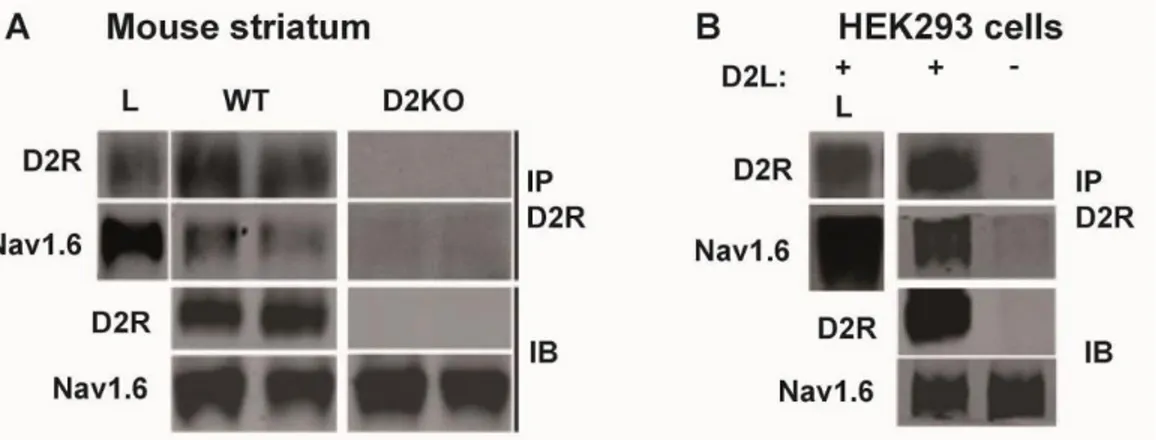
Figure 1-4: D2R forms a complex with neuronal voltage gated sodium channels, the target of lamotrigine, in the striatum ... 76
Figure 1-5: Voltage-gated sodium channel Nav1.6 forms a complex with D2R in vitro and in vivo ... 77
Figure 1-6: The C-terminal region within Nav1.6 is responsible for interaction with D2R 80 Figure 1-7: D2R does not affect Nav1.6 channel currents but Nav1.6 modulates signaling events following D2R activation ... 83
Figure 1-8: Nav1.6 and its D2R interacting Ctail differentially modulate D2R coupling to signaling events ... 86
Figure 1-9: Viral-mediated overexpression of the D2R-interacting region of Nav1.6 in accumbal D2R neurons replicates lamotrigine effects ... 90
Figure 1-10: Viral-mediated overexpression of the D2R-interacting region of Nav1.6 in dorsal striatum D2R neurons differentially modulates locomotor response to amphetamine and quimpirole ... 92
Figure 1-11: Viral-mediated overexpression of the D2R-interacting region of Nav1.6 in the mPFC D2R neurons impairs working memory ... 94
Figure 2-1: Drd2 genotype and fluoxetine differentially affect performance in forced swim test and novelty suppressed feeding ... 110
Figure 2-2: D2R is essential for fluoxetine-induced hippocampal cell proliferation ... 113
Figure 2-3: Fluoxetine relies on βArr2-mediated D2R signalling to induce hippocampal effects... 117
List of abbreviations
Akt : Protein kinase B Βarr : Beta-arrestin
BDNF : Brain-derived neurotrophic factor
BRET : Bioluminescence resonance energy transfer CaMKII : Calcium/calmodulin-dependent PK II cAMP : Cyclic adenosine monophosphate CCP : Clathrin-coated pit
COMT : Catechol-O-methyltransferase
CREB : cAMP-responsive element-binding protein CRMP2 : Collapsin response mediator protein 2 D1R : Dopamine D1 receptor
D2L : Long dopamine D2 receptor D2R : Dopamine D2 receptor D2S : Short dopamine D2 receptor DAG : Diacylglycerol
DARPP-32 : Dopamine- and cAMP-regulated phosphoprotein of 32 kDa DISC1 : Disrupted in schizophrenia 1
DOPAC :3,4-Dihydroxyphenyl-acetic acid
EPAC : Exchange protein directly activated by cAMP ERK : Extracellular-signal regulated kinase
Fxr : Fragile X mental retardation-related protein1 G protein : Heterotrimeric guanine-nucleotide binding protein GAP : GTPase activating protein
GIRK : G-protein-coupled inwardly rectifying potassium channels GPCR : G-protein-coupled receptors
GRK : G-protein coupled receptor kinase GSK3 : Glycogen synthase kinase3 HVA : Homovanillic acid
IP3 : Inositol trisphosphate KO : Knockout
MAO : Monoamine oxidases
MAOIs : Monoamine oxidase inhibitors MAP : Mitogen-activated protein mPFC : Medial PFC
MSNs : Medium spiny neurons NAc : Nucleus accumbens
Nav : Voltage gated sodium channels NET : Norepinephrine
NPCs : Neural progenitor cells PFC : Prefrontal cortex PKA : Protein kinase A PKC : Protein kinase C
PIP2 : Phosphatidylinositol- 2-phosphate PIP3 : Phosphatidylinositol-3-phosphate
PLCβ : Phospholipase Cβ PP2A : Protein phosphatase 2A PPI : Pre-pulse inhibition RTKs : Receptor tyrosine kinases SERT : Transporters for serotonin SGZ : Subgranular zone
SN : Substantia nigra
SNRIs : Serotonin and norepinephrine reuptake inhibitors SSRIs : Selective serotonin reuptake inhibitors
SVZ : Subventricular zone
TAAR1 : Trace amine-associated receptor 1 TCAs : Tricyclic antidepressants
TH : Tyrosine hydroxylase
VMAT2 : Vesicular monoamine transporter 2 VTA : Ventral tegmental area
Acknowledgement
I would like to thank my supervisor Dr. Martin Beaulieu for giving me an opportunity to do my Ph. D in his lab. This work would not have been possible without his continued support. Every scientific discussion with him enhanced my knowledge in the field of my research and helped me think and act independently.
I would like to thank Prof. Jean Pierre Julien for doing me the honor of accepting my co-supervision. I am grateful for his insightful comments at my committee presentation and professional guidance throughout my studies.
I would like to express my gratitude to each of the members of my Dissertation Committee: Prof. Stephen Ferguson, Prof. Claude Gravel, and Dr. Benoit Labonté for accepting to evaluate my thesis.
I am grateful to all of those with whom I have had the pleasure to work during this project, Dr. Annie Barbeau, Pavel Powlowski, Jivan Khlghatyan and all my previous and current colleagues, whose warm personality and positive attitude made the lab a pleasant place to work.
I would like to extend my special thanks to Dr. Sai Sampath, Louis-Charles Béland and Geneviève Soucy for their kind help and support.
Nobody has been more important to me in the pursuit of this project than my family. Most importantly, I wish to thank my loving and supportive husband Reza, who provides unending inspiration. I dedicate this dissertation to my parents; whose love and guidance are with me in whatever I pursue.
Thank you Gohar Fakhfouri
Foreword
This dissertation includes a literature review that outlines the current knowledge about the role of non-canonical signaling pathway of dopamine D2 receptor in mood disorders. The first part of the introduction describes G-protein coupled receptors, their trafficking and modes of signaling. The second part of the introduction reviews the dopaminergic system, classification of receptors, their distribution and signal transduction mechanisms, with focus on D2 receptor (D2R) and its effector GSK3β. In this context, D2R oligomerization is explained and the importance of GSK3β in mood disorders discussed. The third part deals with pharmacotherapy of mood disorders and the last part describes antidepressant-induced neurogenesis.
Chapter one of the dissertation is presented in the form of a research manuscript where I am the first author. The manuscript will be submitted to a valid journal in near future.
“Trans-molecular regulation of beta-arrestin-mediated dopamine D2 receptor signaling by voltage gated sodium channels”
Gohar Fakhfouri, Pavel Powlowski, Clementine Quintana, Thomas Del' Guidice, Hugo Poulin, Jean Martin Beaulieu
Authors contributions: Gohar Fakhfouri performed more than 90% of the experiments, analyzed the results and wrote the manuscript. Pavel Powlowski performed RNA profiling and helped with plasmid generation and BRET experiments, Clementine Quintana performed stereotaxic injections, Thomas Del' Guidice performed immunostaining, Hugo Poulin did patch clamp recordings, Jean Martin Beaulieu designed the study and wrote the manuscript.
Chapter two of the dissertation is presented in the form of a research manuscript where I am the first author. The manuscript will be submitted to a valid journal in near future.
“Role of dopamine D2 receptor in fluoxetine-induced neurogenesis”
Gohar Fakhfouri, Quentin Rainer, Stella Manta, Morgane Lemasson, Olivier Thériault, Jean Martin Beaulieu
Authors contributions: Gohar Fakhfouri performed BRET assays and western blot, analyzed the results and wrote the manuscript. Quentin Rainer performed behavioral tests
and immunohistochemistry, Stella Manta performed ELISA and western blot, Morgane Lemasson performed immunohistochemistry, Olivier Thériault did patch clamp recordings, Jean Martin Beaulieu designed the study
G protein coupled receptors (GPCRs)
GPCRs are known to be a superfamily of membrane proteins that bind hormones, neurotransmitters, and other extracellular cues to transmit information about the external world to the interior machinery of cells (Lefkowitz, 2004).
For a cell to adapt to its environment, it must be able to receive extracellular cues and then elicit an appropriate intracellular response to those cues. Although there are multiple receptor families (i.e., receptor tyrosine kinases, ion channels, nuclear receptors), GPCRs represent the largest and most pharmacologically important family. Approximately 1% of the human genome is dedicated to these receptors, and nearly a third of the pharmaceuticals currently on the market target one or more of these receptors (Wootten et al., 2018). In addition to being the largest component of the “druggable” proteome, GPCRs are also responsible for our ability to perceive the visual, olfactory, and gustatory cues in our environment. Missense or truncation mutations to individual codons in genes encoding GPCRs result in a myriad of pathological conditions, including color blindness, retinitis pigmentosa, pseudohermaphroditism, and Hirschsprung’s disease (Violin et al., 2014). At the most basic level, GPCRs consist of seven α-helical transmembrane stretches with an extracellular N terminus and an intracellular C terminus. Although the precise mechanism of activation of the heterotrimeric G-protein probably varies from family to family, in simplest terms upon binding of a hormone, neurotransmitter, ion, or other stimuli, the GPCR undergoes conformational changes that allow the activation of the G α-GDP/Gβγ complex. Upon the binding of an activating ligand, the GPCR catalyzes the release of GDP and subsequent binding of GTP on the Gα subunit (Kimple et al., 2011).
G protein subunits
The heterotrimeric guanine-nucleotide binding protein (G protein) family consists of numerous, subunit isoforms, with 20 different G protein α-subunits, 5 different β-subunits and 12 different γ-subunits that can associate in various combinations to produce a broad array of potential G protein heterotrimers. GPCR coupling specificity and downstream
target regulation are driven largely by the identity of the Gα subunit, and these can be classified into four different families (Campbell and Smrcka, 2018).
Gαs
The Gα subunits of the Gαs family were the first G proteins discovered and were purified on the basis of their ability to stimulate adenylyl cyclase. Gαs-GTP directly binds to adenylyl cyclase, resulting in increased catalytic activity and cyclic adenosine monophosphate (cAMP) production. There are three Gαs isoforms including two splice variants, Gαs short and Gαs long, and the distinct gene product Gαolf (Campbell and Smrcka, 2018).
Gαi
Gαi family of G proteins bind directly to and inhibit adenylyl cyclase isoforms, thereby leading to a decreased cAMP concentration in cells. The Gαi family consists of Gαo, Gαi1, Gαi2, Gαi3, Gαz and Gαt. All three Gαi isoforms (1, 2 and 3) inhibit adenylyl cyclase in biochemical experiments with no clearly distinguishable isoform-specific functions (Linder
et al., 1990). Gαo also weakly inhibits adenylyl cyclase and has no other clearly defined
function, although recent work suggests a role for Gαo in the Golgi apparatus regulating neurite outgrowth. All of the Gαi family members, except for Gαz, are inhibited by pertussis toxin (PTX) through ADP-ribose modification of a unique cysteine at the C-terminus of Gαi subunits, which inhibits the interaction of Gαi with receptors, presumably by steric occlusion (Murayama and Ui, 1983; Campbell and Smrcka, 2018).
Gαq and Gα11
This family of G proteins activates phospholipase Cβ (PLCβ) downstream of GPCR activation. Gαq and Gα11 share 90% homology (Lee et al., 1992). PLC hydrolyses the membrane lipid inositol trisphosphate (IP3) and diacylglycerol (DAG), each of which initi-ates a signal transduction cascade. IP3 leads to the release of calcium into the cytoplasm and DAG activates protein kinase C (PKC); both of these pathways are ubiquitous regulators of cell physiology (Singer et al., 1997). Gαq or Gα11 regulation of PLCβ and
subsequent Ca2+ release are major drivers of cell function throughout the body. These
include platelet aggregation, smooth muscle contraction and exocytosis, as well as many others. Gαq family members in humans are Gαq, Gα11, Gα14 and Gα15, all of which can activate PLCβ (Wilkie et al., 1991). These subunits have distinct tissue distributions, but no biochemical specificity for target interactions has yet been defined and most physiological data suggest that Gαq and Gα11 have overlapping functions (Campbell and Smrcka, 2018). However, one study has shown that Gαq and Gα11 have different roles in GPCR-mediated sensitization of mechanical and thermal nociception in mice that were often distinct and non-compensatory. Another alternative Gαq/11-regulated pathway involves a direct interaction between Gαq and PKCζ that is important for extracellular signal regulated kinase 5 (ERK5) activation in cardiac myocytes and fibroblasts in response to angiotensin II (Sanchez-Fernandez et al., 2014; Campbell and Smrcka, 2018).
Gα12 and Gα13
Gα12 and Gα13 were originally identified in a homology screen for novel G protein subunits (Kelly et al., 2006). Later, these subunits were found to directly interact with RhoGEFs and increase their ability to activate the GTPase Rho. In fact, RH-RhoGEFs combine the GTPase activating protein (GAP) activity and effector activity into a single molecule (Suzuki et al., 2009).
(A well-established downstream effector of Gα12/13-mediated signaling is the monomeric GTPase RhoA, which is a regulator of a variety of intracellular processes including formation of actin stress fibers and assembly of focal adhesions, gene transcription, and control of cell growth (Diverse-Pierluissi et al., 2000; Witherow et al., 2000). The Gα12 or Gα13 pathway is the major pathway through which GPCRs activate Rho. However, it is noteworthy that Gαq/11 can also bind to p63RhoGEF to initiate the activation of Rho by converting Rho–GDP to Rho–GTP and depending on the cell type and the receptor, Gαq/11-dependent Rho activation may predominate (Campbell and Smrcka, 2018).
Gβγ subunits
The seminal work by Logothetis and colleagues showing regulation of inwardly rectifying K+ channels (GIRKs) by Gβγ subunits disproved the then prevailing notion of Gα subunits being the only transducers of G protein signals (Logothetis et al., 1987). It is now well accepted that the Gβγ subunit heterodimers are bona fide signal transducers and can activate a wide range of effector targets. One group of the most important targets for Gβγ comprises N-type and P/Q-type Ca2+ channels, which are localized in presynaptic terminals
and participate in neurotransmitter release. Activation of Gαi heterotrimeric proteins inhibits these channels through their Gβγ subunits (Herlitze et al., 1996).
There are 5 different Gβ isoforms and 12 different Gγ isoforms in humans. Gβ1-4 are highly homologous, sharing ~80–90% sequence identity, and Gβ5 shares about 50% identity with those four. Gβ5 is unusual in that it is not thought to interact with Gγ subunits in vivo but rather forms dimers with regulator of G protein signaling (RGS) proteins (Snow
et al., 1998). No clear specific differences have been noted between Gβ1-4 in biochemical
reconstitution or cell transfection experiments, although some subtle preferences have been noted. Co-transfected Gβ5γ is often less effective at activating downstream targets, but because Gβ5 subunits are not thought to interact with Gγ or Gα subunits in the traditional sense, this apparent biochemical selectivity does not likely reflect physiological specificity (Lindorfer et al., 1998).
Gγ subunits are more diverse, with intrafamily identities as low as 25%. Similar to Gβ subunits, activation of most effectors by Gβγx combinations does not reveal any particular specificity either with purified proteins or upon transfection. Gγ1-containing Gβγ complexes are consistently less potent that other Gβγ subunits with respect to effector activation, but because Gγ1 is almost exclusively expressed in the retina, this cannot be taken as evidence for physiological Gγ subunit target specificity (Wickman et al., 1994). Most effectors bind to a highly conserved surface on Gβ subunits that roughly corresponds to the Gα-Gβγ interface. In GPCR-G protein crystal structures, interactions between receptors and Gγ subunits have not been observed. Gγ subunits are not directly involved in target interactions in cases for which this has been defined. These data likely explain why
no clear, specific biochemical roles have been attributed to individual isoforms of Gβγ. On the other hand, a number of Gγ and Gβ subunit knockout or knockdown studies indicate exquisite specificity in vivo (Campbell and Smrcka, 2018). This finding strongly suggests specific roles for Gβ and Gγ subunits in physiological signaling, but the mechanistic nature of this specificity is not understood. An emerging concept is that different Gγ subunits may impart specific Gβγ plasma membrane affinities, but the physiological relevance of this is not yet clear (Cho et al., 2011; Ajith Karunarathne et al., 2012).
Interestingly, Gβγ subunits do not undergo major conformational changes upon G protein activation and their activities are regulated by the activity of the Gα subunits. Activation of Gα and subsequent dissociation from Gβγ exposes a protein–protein interaction surface on Gβ that can bind to downstream targets (Campbell and Smrcka, 2018). Despite lack of a direct action on Gβγ, PTX has been shown to inhibit the activation of most Gβγ subunit signaling pathways in cells. The reason is that PTX modifies Gαi in the Gαiβγ heterotrimer, preventing Gαi activation, thereby inhibiting release of Gβγ. This observation indicates that despite the requirement for Gβγ assembly with all Gα subunits for efficient GPCR coupling, and the presumed Gβγ release upon activation of all GPCRs, Gβγ signaling downstream seems to preferentially involve Gαi-coupled receptors (Campbell and Smrcka, 2018). One of the targets downstream of Gβγ signaling is GRK2, which upon activation selectively inhibits Gβγ signaling, functioning as a negative feedback. This observation has been used to develop small molecules that bind directly to Gβγ and inhibit Gβγ signaling based on the C-terminal Gβγ binding fragment of GRK2 (Bonacci et al., 2006; Campbell and Smrcka, 2018).
G protein-coupled receptor kinases (GRKs)
G protein-coupled receptor kinases (GRK) initiate homologous desensitization by phosphorylating the third cytoplasmic loop or tail of activated GPCRs (Pitcher et al., 1998), thereby allowing cells to adapt to changing extracellular signals. The seven mammalian GRKs are grouped into three subfamilies based on sequence identity and gene structure. The GRK1 subfamily consists of rhodopsin kinase (GRK1) and GRK7; the GRK2 subfamily consists of adrenergic receptor kinase-1 and -2 (GRK2 and GRK3); and the
GRK4 subfamily consists of GRK4-6. All GRKs have a 500-amino acid structural core consisting of a regulator of G protein signaling (RGS) homology (RH) domain and a kinase domain closely related to those of other AGC family members, including protein kinase A (PKA) and protein kinase B (Akt). However, each GRK subfamily has distinct N and C termini containing elements known to regulate kinase activity and to mediate membrane targeting (Lodowski et al., 2006).
Regulators of G-protein signaling (RGS)
The GTP hydrolysis activity intrinsic to the Gα subunit was initially thought to control the lifetime of G-protein α subunits in their GTP-bound state and the in vitro kinetics of GTP hydrolysis observed by Gαs supported this hypothesis (Cassel et al., 1979); however, intrinsic rates of GTP hydrolysis measured in vitro could not account for the fast deactivation kinetics seen with other G-proteins in the cellular context (Vuong and Chabre, 1991). Today we know that RGS proteins are responsible for the rapid turnoff of G protein coupling. The first evidence that the cycle of nucleotide binding and hydrolysis by Gα subunits could be modulated by binding partners came from the report of Berstein et al demonstrating that the Gαq/11 effector phospholipase C (PLC) β1 could also increase the rate of GTP hydrolysis by Gαq/11 (Berstein et al., 1992). Later, another group of RGS were discovered, which are called GTPase-accelerating proteins (GAPs). Examples include RGS4 and G alpha interacting protein (GAIP) that attenuate bradykinin (Gαq-mediated) and somatostatin (Gαi-mediated) signaling (Huang et al., 1997). As a third mechanism, it has been suggested that some RGS proteins can alter the number of free Gβγ subunits available to interact with their effectors by enhancing the affinity of Gα subunits for Gβγ after GTP hydrolysis, thus accelerating reformation of the heterotrimer (De Vries et al., 2000).
Arrestins
While arrestins were named based on their initially discovered ability to arrest (turn off) GPCR signaling, it is now evident that arrestins regulate GPCR trafficking as well as G protein-independent signaling (Rankovic et al., 2016). The arrestin family, consisting of
two retinal isoforms, visual arrestin (arrestin1) and cone arrestin (arrestin4), and two nonvisual arrestins, β-arrestin1 (arrestin2) and β-arrestin2 (arrestin3), belong to a superfamily of structurally and functionally related scaffolding proteins that trace their origins to prokaryotes and occur in all eukaryotes except plants (Ferguson, 2001). Depending on the type of cell and its metabolic state, arrestin family proteins can be found distributed diffusely in the cytosol, bound to the cytoskeleton, concentrated at the centrosome, coating internalizing endosomes, and inside the nucleus. These many pools of arrestin are integral to the control of cell metabolism, division, motility, and crosstalk and are adapted to provide diverse but highly specific signal integration (Peterson and Luttrell, 2017).
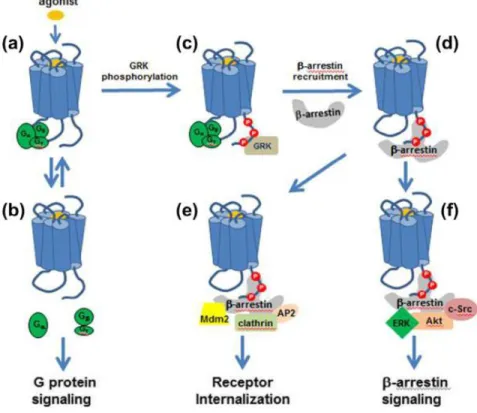
In addition to their canonical function of GPCR desensitization and internalization (see GPCR trafficking), β-arrestins have been appreciated as independent signaling units by virtue of their crucial role as both adaptors and scaffolds for an increasing number of kinases, phosphatases, E3 ubiquitin ligases, deubiquitinases, cAMP phosphodiesterases, elements of the nuclear factor kB signaling pathway, and regulators of small GTPase activity (Perry et al., 2002; Shenoy and Lefkowitz, 2005; Cahill et al., 2017; Peterson and Luttrell, 2017). A general scheme for the β-arrestin-mediated regulation of GPCR function is depicted in figure 1. It is the interaction with this diverse set of partners that enables arrestins to perform their diverse roles and positions them as critical regulators of GPCR signal transduction and permits them to integrate GPCR-mediated signals with other inputs (Peterson and Luttrell, 2017).
Figure 1-1 Paradigms of GPCR-mediated signaling and multiple roles of βarrestins
Binding of an agonist (a) results in activation of signaling pathways by G proteins (b), as well as βarrestins (f), in addition to desensitization and internalization by βarrestins (d and e) (Rankovic et al., 2016).
GPCR trafficking
Following agonist binding, GPCRs undergo conformational changes that regulate the activity of downstream effector systems to mediate various cellular responses. The extent and duration of GPCR signaling is tightly regulated by mechanisms that terminate the initial signaling and later re-establish the capacity of the receptors to respond to new agonist exposure. The removal of GPCRs from the cell surface, also known as internalization or endocytosis, plays an important role in these processes (Claing et al., 2002; Marchese et al., 2003). For most GPCRs, rapid feedback desensitization is initiated by GRKs that phosphorylate agonist-occupied GPCRs to create high affinity binding sites for β-arrestins, which in turn uncouple the receptor from its cognate G protein (Lefkowitz and Shenoy, 2005). β-arrestins also target receptors for endocytosis by linking them to the
endocytic machinery, including clathrin and the clathrin adaptor AP-2 (Laporte et al., 1999). By controlling receptor recycling following endocytosis, β-arrestins have also been shown to regulate the rate of receptor resensitization (Prossnitz, 2004).
Based on their interaction with β-arrestins, GPCRs are divided into three main classes. Class A, which includes receptors such as the β2-adrenergic (β2AR), endothelin A and dopamine receptors, interacts transiently with β-arrestins and can rapidly recycle back from the endosomes to the cell surface. Class B receptors, such as vasopressin V2, CC-chemokine 5 and prostaglandin EP4, interact more stably with β-arrestins, leading to a complex that resides for extended periods of time into endosomes. Receptors tightly associated with β-arrestins in endosomes are only poorly recycled to the cell surface and eventually targeted for lysosomal degradation. The two classes of receptors also differ by their binding preference toward β-arrestin1 and β-arrestin2. Indeed, whereas class A receptors binds with greater affinity to β-arrestin2, class B receptors do not show preference between the two β-arrestins (Hamdan et al., 2007). More recently, class C receptors were described (Simaan et al., 2005). Class C GPCRs are defined by two unique structural features: first, they possess a large extracellular domain that is distal to the 7TM and contains the orthosteric sites; second, they form constitutive dimers with unique activation modes compared with other classes of GPCRs. Class C GPCRs are composed of metabotropic glutamate receptors (mGlu receptors), GABAB receptors, Ca2+-sensing
receptors (CaS receptors), sweet and amino acid taste receptors, pheromone receptors, odorant receptors in fish and several orphan receptors (Chun et al., 2012; Wootten et al., 2018).
Although the roles of β-arrestins and clathrin-coated vesicles in GPCR endocytosis have been well characterized, alternative pathways involving non-coated vesicles, such as caveolae, or other non-clathrin and non-caveolae mediated routes, have also been described for several receptors (Chini and Parenti, 2004). Also, the requirement for β-arrestin does not seem to be universal, and endocytosis of some GPCRs through either clathrin-coated vesicles or caveolae was proposed to be β-arrestin-independent (Okamoto et al., 2000). To characterize the various endocytic routes used by different receptors, several pharmacological and biochemical tools have been used. These include blockers that do not
discriminate between clathrin-dependent or caveolae-mediated endocytosis (e.g. hypertonic sucrose, low temperature, concanavalin-A, and dominant negative mutants of dynamin) as well as inhibitors that are believed to selectively inhibit clathrin-coated vesicle (e.g. monodansylcadaverine, chlorpromazine, as well as dominant negative mutants of β-arrestin and Eps-15) or caveolae-mediated endocytosis (e.g. filpin and nystatin) (Hamdan et al., 2007). Whereas it was initially believed that β-arrestins were specific regulators for GPCRs, growing evidence demonstrates its role in the regulation of non-GPCRs membrane proteins endocytosis (Chen et al., 2003; Hamdan et al., 2007).
Effect of cations on GPCRs
Physiological cations can allosterically influence GPCRs. Studies carried out in rat brain homogenate in the early 70s clearly demonstrates a differential impact of Na on agonist versus antagonist binding to opiate receptors, suggesting a Na-driven conformational change in receptor. Using Molecular Dynamics simulations, high-resolution crystallography and NMR studies, researchers show that Na exerts negative allostery on many class A GPCRs by stabilizing a functionally inactive conformation of the receptor. Examples include A2AR. In contrast, divalent Mg2+, Ca2+, and Mn2+ enhanced binding of
μ-, δ-, and κ-opioid receptor agonists in brain homogenates. Allosteric effects of Mg2+,
Ca2+, and Na+ have been observed with A2AR and the muscarinic M2 receptor, while Ca2+
is known to allosterically regulate many family C GPCRs (Ye et al., 2018)
Ligand bias
Classically, agonists were thought to deploy the entire signal repertoire of a receptor, and thus pharmacological selectivity was defined as selectivity of a drug, hormone, or neurotransmitter for a particular receptor type. In fact, in the classic two-state model of receptor function, receptors were either ‘on’ or ‘off’. The discovery of partial agonists and inverse agonists revealed new levels of pharmacological properties, often differentiating these agonists from full agonists and neutral antagonists, but still consistent with the two-state model. Thus, it was envisioned that receptors adopt two two-states, and ligand binding preferentially stabilizes the inactive state (for antagonists and inverse agonists) or the active
state (weakly for partial agonists, more strongly for full agonists). This paradigm dominated receptor theory and drug discovery for several decades and afforded a level of simplicity for investigators. However, several examples were found that did not fit this paradigm: compound sets yielded different relative potencies in different assays, which could not be explained by off-target actions. These findings challenged the notion that receptors are only capable of a single spectrum of responses. In light of these observations, the classical pharmacological theory has been revised to incorporate the possibility that receptors engage distinct subsets of their full signaling repertoire. Receptors are now envisioned as occupying an array of discrete conformations, and non-overlapping sets of these conformations are associated with different signaling mechanisms such as G protein coupling, β-arrestin recruitment, receptor trafficking, and other signals. When conformations linked to different signals are distinct, ligands that selectively stabilize these conformations will give rise to pharmacological responses that differ not just quantitatively but also qualitatively from responses to traditional agonists. These compounds are thus biased towards subsets of receptor function relative to a reference ligand. By convention, the reference ligand is defined as unbiased or balanced and is usually an endogenous agonist or clinically validated drug, although examples of endogenous biased ligands also exist (Violin et al., 2014). Figure 2 summarizes mechanisms of ligand-induced biased agonism. Ligands, for example, can show bias for either G protein- (G protein-biased) or β-arrestin-mediated (β-arrestin-biased) signaling. Using this framework, we could hypothesize that such ligands would more selectively target beneficial signaling and even block or negate detrimental or unwanted actions of receptor activation (e.g. side effects, toxicity or tolerance). Indeed, over the last decade a diversity of biased ligands for GPCR have been identified that selectively activate G proteins or β-arrestins, and several of these seem to have distinct and advantages functional consequences when compared to traditional ligands (Whalen et al., 2011). It is noteworthy that signaling bias is not merely governed by ligands, and interactions of a given GPCR with other GPCR or non-GPCR partners may regulate the nature, kinetics, strength, and fine-tuning of GPCR signaling (Wootten et al., 2018).
Figure 1-2 Mechanisms of ligand-induced biased agonism
a A schematic illustrating conformational dynamics occurring in G protein-coupled receptors (GPCRs). GPCRs can move between various inactive-like (R, R′ and R′′) and active-like (R* and R**) states. This can occur in the absence of ligand (Apo, black line); however, the energy barrier to achieving these states makes their occurrence a low probability. Addition of agonist or G protein (blue line) can decrease the energy required to reach active states, but full conformational change is favored by the addition of both agonist and G protein (green line). b–d Biased agonism can arise via multiple mechanisms.
Distinct ligands induce different conformations within the receptor, resulting in different recruitment profiles for transducer proteins such as G proteins and arrestins (b). Ligand-induced receptor conformations can promote different conformational changes within scaffolding proteins such as arrestins, which in turn promotes activation of different downstream signaling pathways (for example, different MAPKs) (c). Different ligands can induce distinct conformational rearrangements within G proteins that result in differences in the rate of GTP–GDP exchange. Ligands that induce a faster rate of GTP association (and hydrolysis) (top panel) allow quantitatively more G protein and downstream signaling events per unit of time than ligands that induce a slow rate of exchange (bottom panel) (d). Pi, inorganic phosphate (Wootten et al., 2018).
Dopaminergic system
Dopmaine
During dopamine synthesis, the amino acid tyrosine is hydroxylated to levodopa by the enzyme tyrosine hydroxylase (TH). Levodopa then undergoes decarboxylation by dopa-decarboxylase to produce dopamine (Carlsson, 1987). In adult neurons, dopamine is loaded into synaptic vesicles by the vesicular monoamine transporter 2 (VMAT2). Following synaptic release, dopamine can either be preferentially up-taken or degraded, depending upon neuronal circuit (Rampino et al., 2018). Dopamine transporter (DAT) is expressed in dopamine neurons and is in charge of rapid reuptake of released dopamine form the synaptic cleft, thereby playing an essential role in controlling extracellular levels of dopamine and therefore dopamine signaling, especially in the striatum (Jones et al., 1998). DAT belongs to the family of neurotransmitter: sodium symporters, which also include the clinically relevant transporters for serotonin (SERT), norepinephrine (NET) and GABA and glycine (SLC6) (Adhikary et al., 2017). DAT harnesses pre-existing sodium gradients to actively translocate dopamine across the membrane of presynaptic neurons against its concentration gradient. Importantly, DAT constitutes the prime target for the stimulatory action of psychostimulants such as cocaine and amphetamine. Whereas cocaine acts as a high affinity, competitive inhibitor, amphetamine is a substrate that upon entry through the transporter promotes the release of dopamine via DAT. DAT is also a target for commonly
used mediation used against ADHD such as methylphenidate and modafinil (Runegaard et
al., 2019). Other transporters such as NET have also been implicated in Dopamine reuptake
in brain regions expressing low levels of DAT (Smith and Greene, 2012).
Catechol-O-methyltransferase (COMT) constitutes the main enzyme responsible for dopamine degradation, which methylates dopamine into 3-metoxytyramine. Degradation by COMT is the principal mechanism of dopamine clearance in the prefrontal cortex (PFC). Dopamine can also be converted to 3,4-Dihydroxyphenyl-acetic acid (DOPAC) by the monoamine oxidases (MAOA and MAOB). The final product of dopamine catabolism is homovanillic acid (HVA) either from 3-metoxytyramine by MAOs or from DOPAC by COMT (Rampino et al., 2018).
Dopamine pathways and circuits
The brain’s dopamine system is mainly classified into four neural pathways: 1. The
nigrostriatal pathway projects from substantia nigra (SN) pars compacta in the midbrain to
the dorsal striatum. This pathway mediates habitual and compulsive responses and exhibits both facilitatory and inhibitory regulation of movements. 2. The mesolimbic pathway projects from the ventral tegmental area (VTA) to limbic brain regions, including the amygdala, the nucleus accumbens (NAc) of the ventral striatum and hippocampus. This pathway is central for reward-related processes and governs motivation, goal-directed behaviors and attribution of incentive salience to reward-related cues. 3. The mesocortical
pathway projects from the VTA to the PFC and is mainly involved in executive function
such as selective and sustained attention, flexible responding to stimuli, planning and goal-directed behavior, and monitoring and inhibitory control. 4. The tuberoinfundibular
pathway projects from the hypothalamus to the pituitary gland, where dopamine inhibits the
secretion of prolactin (Runegaard et al., 2019).
Although the above-mentioned classification of the dopaminergic transmission is still relevant, recent advancements gives rise to a remarkably more complicated picture of this system, with heterogeneous populations of neurons releasing dopamine via different mechanisms in different brain regions (Morales and Margolis, 2017). For example, in
labeling studies the midbrain dopamine system displays a highly heterogeneous projection patterns, firing and release probability, and intracellular molecular properties (Lammel et
al., 2008). Moreover, some midbrain dopamine neurons have been shown to release other
neurotransmitters including glutamate, GABA and serotonin (Runegaard et al., 2019). Lastly, complex input and output network projections onto and from dopamine neurons, picture dopamine mainly as a modulator rather than a regulator of neuronal excitability in target regions. Once released from presynaptic terminals, dopamine activates its receptors, which transduce and determine the further processing of the dopaminergic signal (Beier et
al., 2015; Runegaard et al., 2019). Dopamine receptors are well-established targets in the
clinical pharmacology of numerous disorders and conditions such as schizophrenia, Parkinson's disease, bipolar disorder, depression, restless leg syndrome, hyperprolactinaemia, pituitary tumours, hypertension, gastroparesis, nausea and erectile dysfunction (Beaulieu et al., 2015).
Dopamine receptors
Dopamine exerts its effects via the stimulation of five different G-protein coupled dopamine receptors, which are organized in two groups based on their coupling and structures of the encoding genes. 1. D1-like dopamine receptors, including D1R and D5R, are generally coupled to Gαs G-proteins and are encoded by genes that do not contain introns. 2. D2-like dopamine receptors, including D2R, D3R and D4R, are mainly coupled to Gαi/o G-proteins and are encoded by genes that contain several introns (Runegaard et
al., 2019), with six introns found in the gene that encodes the D2R, five in the gene for
D3R, and three in the gene for D4R (Gingrich and Caron, 1993). D1-like receptors are found exclusively postsynaptically on dopamine-receptive cells, such as GABA-ergic medium spiny neurons (MSNs) in the striatum. D2R and D3R, on the other hand, are expressed both pre and postsynaptically (Beaulieu and Gainetdinov, 2011). Alternative mRNA splicing generates two isoforms of D2R, named long (D2L) and short (D2S) isoforms, which differ by an insertion of 29 amino acids in the third intracellular loop of the D2L receptor. D2L is preferentially involved in postsynaptic responses, such as the synergistic stimulation of motor activity produced by D1/D2 receptors, haloperidol-induced catalepsy and act as heteroreceptor, regulating the release of heterogeneous
neurotransmitters. In contrast, the D2S receptor seems to be preferentially expressed by presynaptic dopaminergic neurons, where it acts as an autoreceptor, inhibiting dopamine synthesis and release (Lindgren et al., 2003).
Dopamine receptor expression
Dopamine receptor subtypes are broadly distributed in the brain and in the periphery. In the brain, D1R display high expression levels in the nigrostriatal, mesolimbic, and mesocortical areas, such as the caudate-putamen (striatum), NAc, substantia nigra, olfactory bulb, amygdala, and PFC, and lower levels in the hippocampus, hypothalamus, cerebellum, retina, and thalamus. Regarding D5R, a low level of expression is found in various brain regions, such as cortical areas including the PFC, premotor cortex, cingulated cortex, entorhinal cortex, as well as substantia nigra, hypothalamus, hippocampus, and dentate gyrus. D5R has been detected, albeit at quite low levels, in the caudate nucleus and NAc (Missale et al., 1998; Beaulieu and Gainetdinov, 2011).
The major regions expressing high levels of D2R are the striatum, the NAc, and the olfactory tubercle. D2R also occur at substantial levels in the SN, VTA, hippocampus amygdala hypothalamus, cortical areas, septum and the pituitary gland (Seeman, 2006; Beaulieu and Gainetdinov, 2011; Khlghatyan et al., 2018b).
It is important to note that MSNs of the dorsal striatum and NAc can be clearly separated into two principal subgroups that are defined by their projection sites and by expression pattern. In particular, the MSNs that project to the medial globus pallidus and the substantia nigra pars reticulata comprise a direct striatonigral pathway that selectively expresses D1R. The second group of MSNs projects to the lateral globus pallidus and forms the indirect striatopallidal pathway. The MSNs in the latter pathway selectively expresses D2R and indirectly reach the substantia nigra pars reticulata through synaptic relays in the lateral globus pallidus and the subthalamic nucleus. In addition to these main groups, there is a low-abundance subpopulation of MSNs in the dorsal striatum that express both D1R and D2R (Valjent et al., 2009). Likewise, coexpression of D1R and D2R was also observed in the pyramidal neurons in the PFC, albeit at a higher density (Zhang et al., 2010b).
D3R shows more limited expression pattern, with highest levels detected in the limbic areas including the shell of the NAc and the olfactory tubercle and lower levels in the striatum, the substantia nigra pars compacta, VTA, hippocampus, septum, and various cortical regions. D4R displays the lowest level of expression in the brain, with documented expression in the PFC, amygdala, hippocampus, substantia nigra pars reticulate, hypothalamus, globus pallidus, thalamus and retina. In the periphery, the kidney, adrenal glands, sympathetic ganglia, gastrointestinal tract, blood vessels, and heart show the expression of all subtypes of dopamine receptors with varying proportions (Beaulieu and Gainetdinov, 2011).
Dopamine receptor signaling
Dopamine receptor coupling to cAMP production
Dopamine receptor functions have typically been associated with the regulation of cAMP and PKA via G protein-mediated signaling. Through coupling to Gαs/olf, the D1-like receptors stimulate the production of the second messenger cAMP and activation of PKA. In contrast, D2-like dopamine receptors negatively regulate the production of cAMP, resulting in a decrease in PKA activity (Beaulieu and Gainetdinov, 2011). Several PKA substrates, such as cAMP response element-binding protein (CREB), ionotropic glutamate receptors (AMPA and NMDA), and certain ion channels have been shown to be affected by dopamine receptor stimulation (Beaulieu and Gainetdinov, 2011). Among them, the dopamine and cAMP-regulated phosphoprotein of 32kDa (DARPP-32) is a multifunctional phosphoprotein that is predominantly expressed in MSNs and acts as an integrator of cell signaling in response to multiple neurotransmitters, including dopamine. Phosphorylation of this molecule at threonine 34 (T34) by PKA or protein kinase G activates the protein phosphatase 1 (PP1) inhibitory function of DARPP-32 (Greengard et al., 1999). Therefore, activation of D1R increases whereas stimulation of D2R reduces the phosphorylation of DARPP-32 at T34, the latter being presumably a consequence of a reduction in PKA activation and/or the dephosphorylation of threonine 34 by the protein phosphatase calcineurin that is activated by increased intracellular Ca2+ downstream of D2R stimulation
Phosphorylated DARPP-32 at T34 has been shown to amplify PKA signaling. In contrast, phosphorylation of DARPP-32 at T75 by cyclin-dependent kinase 5 in response to sustained D1R activation abolishes its inhibitory activity on PP1 and converts it to an inhibitor of PKA (Svenningsson et al., 2004). A thorough ethological characterization of DARPP-32 KO mice showed that several dopamine-associated behaviors, including basal locomotion, are not overtly disrupted in these mice. Moreover, behavioral responses to apomorphine and cocaine are only partially affected in DARPP-32 knockout mice (Beaulieu and Gainetdinov, 2011).
In addition to their effects on PKA, other cAMP-regulated molecules may also mediate physiological actions downstream of dopamine receptors. Exchange proteins directly activated by cAMP (Epac1 and Epac2) are a group of such signaling proteins that are highly enriched in the striatum (Beaulieu and Gainetdinov, 2011). Epac2 has been implicated in D1R-mediated synapse remodeling and depression in cultured rat cortical neurons (Woolfrey et al., 2009).
Non-canonical signaling of D1-like receptors via Gαq coupling
For the first time in 1989, it was reported that the D1-like receptor agonist SKF 82526 stimulates PLC activity independently of cAMP in renal tubular membranes (Felder et al., 1989). Activation of PLC leads to the production of IP3 and diacylglycerol (DAG). This results in the activation of PKC by DAG and an increased mobilization of intracellular calcium in response to IP3 (Berridge, 2009). The increase of intracellular calcium in the cytoplasm leads to the activation of calcium-dependent PKC variants as well as other calcium-regulated enzymes, such as the calcium/calmodulin-dependent PK II (CaMKII) and calcineurin (PP2B). The most common way for a GPCR to regulate PLC activity is by coupling to Gαq. However, the molecular determinants regulating this non-canonical coupling of D1-like receptors are still not completely understood and it is possible that such coupling occurs in certain neuronal populations and/or following D1-like receptor interaction with other proteins. More recent in vivo observations also support the regulation of PLC by D1-like receptors in the striatum. Acute systemic administration of cocaine, amphetamine, apomorphine or the D1-like receptor agonist SKF 81297 to WT mice
increases striatal IP3 synthesis (Medvedev et al., 2013). Using selective antagonists as well as KO animals revealed that the observed production of IP3 requires D1R, but not D2 receptor activation. Importantly, PLCβ inhibition antagonized the effects of these dopaminergic drugs on forward locomotion, suppressed spontaneous locomotor hyperactivity in DAT KO mice and reduced the restoration of locomotion by L-DOPA in dopamine-depleted mice (Medvedev et al., 2013). These data strongly support a contribution of PLC in mediating the effects of dopamine on forward locomotion. However, expression of D5R not D1R in HEK293 cells induces calcium mobilization after stimulation (So et al., 2009). Moreover, dopamine as well as D1-like receptor agonists SKF 83959 and SKF 38393 failed to increase IP3 levels in brain slices prepared only from D5R KO not D1R KO mice (Friedman et al., 1997), suggesting that activation of D5R is sufficient to activate Gαq/11 in response to selected doses of certain D1-like receptor agonists. In the hippocampus, formation of ghrelin receptor (apo-GHSR1a):D1R:Gαq heteromeric complexes leads to signal transduction via Gαq-PLC-IP3-Ca following D1R activation and expression of early markers of hippocampal synaptic plasticity. Interestingly, activation of this pathway occurs at the expense of canonical DRD1 Gαs cAMP signalling (Kern et al., 2015). Thus, several independent studies support the regulation of PLC-mediated signaling through dopamine receptors, however, these studies are in disagreement with regard to the detailed mechanism of this regulation (Beaulieu et al., 2015).
Regulation of Gβγ signaling and ion channels by D2-like receptors
D2-like receptors also modulate intracellular calcium levels by acting on ion channels or by triggering the release of intracellular calcium stores. This modulation appears to stem from signaling responses mediated by the Gβγ subunits of heterotrimeric G proteins, which dissociate from Gα subunits following D2R activation. D2R-regulated Gβγ subunits are involved in the negative regulation of N-type calcium channels in striatal cholinergic interneurons (Yan et al., 1997). It was shown that D2R stimulation suppresses L-type Ca2+
currents and excitability in enkephalin-expressing MSNs. But the suppression was not mediated by inhibition of adenylyl cyclase. Rather, D2R stimulation mobilizes intracellular Ca2+ stores via Gβγ activation of a PLCβ1 pathway, leading to a calcineurin-dependent
suppress spike activity and Ca2+-dependent gene transcription but activate Ca2+-dependent
intracellular enzymes (Hernandez-Lopez et al., 2000). Lastly, Gβγ subunits are also involved in the association/regulation by D2R of G protein-coupled inwardly rectifying potassium channels (GIRKs) (Kuzhikandathil et al., 1998). The activation of GIRKs downstream of D2R and other GPCRs has an inhibitory effect in neurons and could potentially mediate some functions of dopamine in vivo (Beaulieu and Gainetdinov, 2011).
Regulation of ERK1/2 by dopamine receptors
Mitogen-activated protein (MAP) kinases have been shown to be signaling intermediates involved in the regulation of dopamine-associated behaviors (Berhow et al., 1996). Results obtained from heterologous cell culture systems suggest that both D1- and D2-like dopamine receptors can regulate the MAP kinases extracellular-signal regulated kinases 1 and 2 (ERK1 and ERK2) (Beom et al., 2004). In vivo, the administration of amphetamine, cocaine, or the D2-like receptor antagonist haloperidol has been shown to enhance striatal ERK1 and ERK2 phosphorylation (Beaulieu and Gainetdinov, 2011). Moreover, ERK2 phosphorylation has also been shown to increase in response to elevated dopamine levels in the striatum of DAT-KO mice (Beaulieu et al., 2006). D1R has been demonstrated to be essential for the activation of ERK in MSNs, while, D3R has been associated to inhibition of ERK-mediated signaling in the striatum (Zhang et al., 2004). In contrast, in the lactotrophs of the pituitary gland, D2R activation drives the activation of the ERK 1/2 pathway and isoform specific overexpression and KO studies revealed that D2S but not D2L is the only isoform required for ERK 1/2 activation (Iaccarino et al., 2002; Radl et al., 2013). Using a heterologous expression system, Quan et al showed that Gαi2 protein is involved in D2R-mediated ERK activation but β-arrestins are either not involved or play minor role (Quan et al., 2008).
Coupling of D2-like receptors to βArr2, Akt and glycogen synthase kinase
It is now well established that in addition to G-protein dependent pathway, D2R signals through βArr2-mediated mechanisms. Apart from the roles in GPCR desensitization and internalization (Lohse et al., 1990; Ferguson et al., 1996a), the two ubiquitous arrestins, βArr1 and βArr2, have also been shown to act as molecular scaffolds for signaling