T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Physiopathologie Humaine
JURY Gilles Ponzio Franck Verrecchia Nicole Basset-Seguin Bernard Ducommun Guy Serre Michel Simon
Ecole doctorale : Biologie Santé Biotechnologies de Toulouse Unité de recherche : UMR5165 CNRS
Directeur de Thèse : Michel SIMON Rapporteurs : Gilles Ponzio et Franck Verrecchia Présentée et soutenue par Véronique Adoue
Le 08 décembre 2008
Titre : Le contrôle de l’expression des peptidyl-arginine désiminases dans les kératinocytes associe des éléments proximaux et une région intergénique distante, et implique les facteurs
2
-Remerciements
Je remercie Le Professeur Guy Serre de m’avoir accueillie dans l’Unité « Différenciation Epidermique et Autoimmunité Rhumatoïde » et de m’avoir permis de réaliser cette thèse.
Je remercie Messieurs Franck Verrecchia et Gilles Ponzio pour avoir accepté la lourde tâche d’être rapporteurs de ce manuscrit de thèse et de m’avoir fait l’honneur de juger ce travail.
Merci également à Mme Nicole Basset-Seguin et à Mr Bernard Ducommun d’avoir accepté de participer à mon jury de thèse malgré un emploi du temps chargé.
Merci à Michel Simon, mon directeur de thèse, qui a encadré ce travail et qui m’a témoigné sa confiance durant toutes ces années.
J’adresse également un grand merci à Stéphane Chavanas, sans qui ce travail n’aurait pas été possible. J’ai beaucoup appris à tes côtés, de la dissection de prépuces aux discussions plus « philosophiques » sur la régulation génique.
Je remercie particulièrement Catherine Bouchouata, Cathy, « ma technicienne préférée » pour avoir traversé ces années avec moi. J’ai beaucoup aimé travailler et discuter avec toi. Cette thèse est un peu la tienne.
Un énorme merci aux filles pour notre complicité et votre soutien inébranlable.
Merci à Anne-Aurélie Raymond, AA, d’avoir toujours été là. Quel plaisir de partager le même bureau et d’apprendre à te connaître, car tu n’es pas celle que tu laisses paraître. Ta présence rassurante me manquera beaucoup au Canada.
Merci à Fanny Coudane, Pafou, pour ton humour et ton sens de la dérision. Grâce à toi je remplissais tous les jours mon quota de fous rires. J’espère que tu ne pleureras pas en lisant ces mots, mais bon c’est vrai que tu es tellement sensible…
Merci à Julie Henry, youyou ou petite chine, pour ta bonté et ton (petit ?) grain de folie. Je peux t’annoncer que tu as remporté le concours de miss UDEAR 2008 ! Félicitations !
Merci à Cécile Caubet, Cissou, pour tes grands yeux, tes conseils, ta gentillesse et ta simplicité. Tu as su rester humble même après avoir découvert que tes fibres étaient bien amyloïdes !
Merci à Leila Gazeilles, tu es vraiment quelqu’un sur qui on peut compter.
Un grand merci à Mitou Ribouchon, ta bonne humeur est contagieuse, c’était un vrai plaisir de te croiser tous les jours.
Merci à Marie-claire Méchin pour ton implication. Merci à Rachida Nachat pour ta spontanéité.
Un grand merci à Nicolas Mattiuzzo et Laetitia Laurent, votre histoire d’amour secrète aura été le fil rouge de ma thèse.
3
-Merci à Émilie Leclerc pour ta patience et ton sens de l’organisation (ah…les séminaires…) !
Un grand merci à Mireille Sebbag et Céline Foulquier pour nos discussions aux allées (radio RLP j’écoute !) et pour vos conseils.
Merci à Carole Pons pour ton rire, ainsi qu’à ton fidèle binôme Romain Oger, les deux petits lutins du deuxième.
Régine Llobera parce que tu n’y vas jamais par 4 chemins et que c’est rafraîchissant !
Merci à Chiung-Yueh Hsu pour ta gentillesse et ta fraîcheur, Marina Weber-Vivat pour ton franc parlé, Cyril Clavel pour ton humour (œil du tigre).
Nathalie Jonca, Jacques Arnaud, Michel Thioulouse, Catherine Knodlseder, Christel Poisson, Hélène Brun et Claudie Offer pour votre gentillesse.
Merci également à Renan Destrade pour la « finesse » de tes blagues.
Et merci à tous ceux que je n’ai pas cités et que j’ai côtoyé tout au long de cette thèse.
Merci à mes amis pour leur soutien et leurs encouragements.
Merci à PJ pour ta présence, ton soutien et ta patience.
4
-Liste des abréviations
1,25(OH)2D, 1,25-dihydroxycholécalciférol AP-1, activating protein 1
Arg, arginine
ARNm, ARN messager ARNnc, ARN non-codants ARN Pol II, ARN polymérase II ARNr, ribosomique
ARNt, ARN de transfert CaR, récepteur au calcium
CaRE, éléments de réponse au calcium
CARM1, Coactivator-associated ARginine Methyltransferase 1 CDE, Complexe de Différenciation Epidermique
CDSN, cornéodesmosine
ChIP, « Chromatine ImmunoPrecipitation assay » CIT, complexe d’initiation de la transcription CNS, segments non-codants conservés CpG, Cytosine-phosphate-Guanine
∆Np63, isoforme de p63 sans domaine terminal transactivateur
DNMT, ADN méthyltransférases DP, desmoplakine
DSC, desmocollines DSG, desmogléine
EMSA, “Electrophoretic Mobility Shift Assays” ENV, envoplakine
ERK, “Extracellular signal-Regulated Kinases” FNH, facteur naturel d’hydratation
FT, facteur de transcription GAP, GTPase Activating Protein GEE, gaine épithéliale externe GEI, une gaine épithéliale interne GFAP, protéine fibrillaire acide gliale H2A, histone 2A
H2B, histone 2B H3, histone 3 H4, histone 4
HAT, histone acétyltransférase HDAC, histones désacétylase HMT, histones méthyltransférase HS, sites hypersensibles
K, kératine
LCR, « Locus Control Region »
MAPK, « Mitogen-Activated Protein Kinases » MBP, protéine basique de la myéline
MCF-7, lignée de cellules tumorales mammaires miARN, microARN
PAD, peptidyl-arginine désiminase PG, plakoglobine
PIP, “Percent identity plot” PKCα, protéine kinase Cα PKP, plakophilines PLC, phospholipases C PPL, périplakine
5 -PRMT1, PRotein arginine MethylTransferase 1 RXR, récepteur X aux rétinoïdes
S/MARs, Scaffold/matrix attachment regions SCCE, “Stratum Corneum Chymotryptic Enzyme” SCTE, “Stratum Corneum Tryptic enzyme” SCTP, “Stratum Corneum Thiol Protéase” SEP, sclérose en plaque
siARN, Small Interfering ARN snARN, small nuclear RNA
SPRR, petite protéine riche en proline SV40, Simian Virus
TAp63, isoforme de p63 avec un domaine terminal transactivateur TBP, TATA-binding protein
TGase, transglutaminase THH, trichohyaline
TPA, 12-O-tetradecanoylphorbol 13-acetate TSS, site d’initiation de la transcription
UTR, “untranslated region”
VDR, récepteur nucléaire de la vitamine D VDRE, élément de réponse à la vitamine D
6
-Résumé
Les 5 gènes PADI, regroupés en position 1p35-36 chez l’homme, codent pour les 5 peptidyl-arginine désiminases (PADs), des enzymes qui catalysent la désimination. Cette modification post-traductionnelle dépendante du calcium est impliquée dans plusieurs maladies humaines telles que la polyarthrite rhumatoïde et la sclérose en plaques. Les gènes PADI présentent des profils d’expression tissulaire et cellulaire spécifiques. En particulier, seules les PAD1, 2 et 3 sont exprimées dans l’épiderme, où elles sont plus abondantes dans les kératinocytes granuleux. Au cours de ma thèse, j’ai participé à la caractérisation du promoteur proximal de PADI1, et montré l’importance des facteurs MZF1 et Sp1 dans l’activation de la transcription de ce gène au cours de la différenciation des kératinocytes. L’action des promoteurs minimums des gènes PADI1, 2 et 3 et des facteurs de transcription s’y liant ne permet pas d’expliquer la spécificité d’expression de ces gènes. La régulation à distance étant un mécanisme déterminant dans le contrôle et la coordination de l’expression de gènes paralogues, notamment ceux regroupés au sein d’un locus, nous avons émis l’hypothèse de l’existence d’une telle régulation pour les gènes PADI. En effet, une région d’environ 8 kb conservée au cours de l’évolution et non-codante avait été identifiée entre PADI2 et PADI1, à 37 et 91 kb des sites d’initiation de la transcription de PADI1 et PADI3. Dans cette région, baptisée IG1, j’ai mis en évidence et caractérisé trois régulateurs distants (PIE, PIE-S1 et PIE-S2) qui présentent les propriétés des activateurs transcriptionnels : hypersensibilité à la DNase I, effet indépendant de l’orientation et dépendant du nombre de copies, spécificité d’action sur certains promoteurs et spécificité cellulaire. Dans les kératinocytes épidermiques cultivés in vitro et différenciés en présence d’une concentration élevée de calcium (1,5 mM), PIE est spécifique du promoteur de PADI3 et recrute les facteurs de transcription AP-1, c-Jun et c-Fos. PIE-S1 et PIE-S2 agissent en synergie pour activer les promoteurs des gènes PADI1 et PADI3. Leur action est dépendante d’un site de fixation putatif pour les facteurs MIBP1/RFX1 localisé dans PIE-S1 et d’un site AP-1 localisé dans PIE-S2. De surcroît, mon étude suggère que l’activité de cet activateur transcriptionnel bipartite est modulée, en fonction de l’état de différenciation des kératinocytes, par une compétition entre les facteurs c-Jun et JunD qui se lient au niveau du site AP-1 de PIE-S2.
Mon travail de thèse révèle les bases moléculaires de la spécificité d’expression de PADI1 et PADI3 au cours de la différenciation kératinocytaire : au-delà de leurs promoteurs proximaux, cette régulation met en jeu l’action à longue distance de deux activateurs transcriptionnels et implique les facteurs AP-1, c-Jun, c-Fos et JunD. C’est une première étape dans la compréhension des mécanismes du contrôle de l’expression des PADs et de la désimination des protéines. A l’image de mutations localisées dans d’autres régulateurs distants, PIE, PIE-S1 et PIE-S2 sont candidats à l’étude d’associations génétiques pour les maladies impliquant des troubles de la désimination.
7
-Summary
The five PADI genes are clustered in human 1p35-36 and encode peptidylarginine deiminases which catalyze deimination. This Ca2+-dependent post-translational modification has been implicated in the pathophysiology of severe human diseases as rheumatoid arthritis and multiple sclerosis. The PADI genes present different expression patterns. Only PADI1-3 are expressed in the epidermis, with increased expression levels in granular keratinocytes. During my PhD work, I was involved in the characterization of the PADI1 minimal promoter, and showed the crucial role of MZF1 and Sp1 factors in the transcriptional activation of this gene during keratinocyte differentiation. Activities of PADI1, 2 and 3 minimal promoters and binding of transcription factors on them do not explain the expression specificity of these genes. Long-range cis elements are critical regulators of transcription, particularly for clustered paralogous genes. We therefore hypothesized the existence of such a regulation for PADI genes. Indeed, we identified an 8 kb conserved intergenic sequence between PADI2 and PADI1, namely IG1, located 37 and 97 kb from the initiation start sites of PADI1 and PADI3 respectively. I highlighted and characterised 3 long-range regulators in IG1, PIE, PIE-S1 and PIE-S2, which presented all the hallmarks of transcriptional enhancers: DNase I hypersensitivity, orientation-independence, copy-number dependence and cell-type specificity. In normal human epidermal keratinocytes differentiated in vitro by a high-calcium containing medium (1.5 mM), PIE was shown to be specific for the PADI3 promoter and to require bound AP-1 factors, namely c-Jun and c-Fos. PIE-S1 and PIE-S2 acted in synergy to activate the PADI1 and PADI3 promoters. Their activity was dependent upon a putative binding site for the MIBP1/RFX1 complex and an AP-1 binding site located in PIE-S1 and PIE-S2, respectively. Furthermore, my work suggests a modulation of this bipartite enhancer during keratinocyte differentiation by a competition between c-Jun and JunD which bind on the latter AP-1 site.
My PhD study reveals the molecular bases of the specific expression of PADI1 and PADI3 during keratinocyte differentiation: beyond minimal promoters, this regulation involves two long-range enhancers and implicates AP-1 factors, c-Jun, c-Fos and JunD. This is the first step forward a better understanding of the regulation of both PAD expression and protein deimination. Since invalidation of distant regulators causes a variety of human diseases, PIE, PIE-S1 and PIE-S2 result to be plausible candidates in association studies on deimination-related disorders.
8
-Table des matières
INTRODUCTION INTRODUCTIONINTRODUCTION
INTRODUCTION... -...--- 10 10 10 - 10 --
-A. La régulation génique ... - 10 -
1. Les premières découvertes ... - 11 -
2. Organisation et régulation de la chromatine... - 12 -
a) Organisation ... - 12 -
b) Acétylation et méthylation des histones... - 15 -
c) Méthylation de l’ADN ... - 17 -
d) Modifications épigénétiques et cancer ... - 20 -
3. Structure et transcription d’un gène ... - 21 -
a) Structure et généralités ... - 21 -
b) Les régions régulatrices... - 22 -
c) L’initiation... - 23 -
4. La régulation de la transcription d’un gène... - 29 -
a) Le promoteur proximal... - 29 -
b) La régulation génique à distance ... - 29 -
5. Autres régulations ... - 35 -
6. Techniques d’analyse ... - 40 -
a) Essais luciférase ... - 41 -
b) Les gels retards... - 41 -
c) L’analyse de la sensibilité à la désoxyribonucléase I ou DNase I... - 42 -
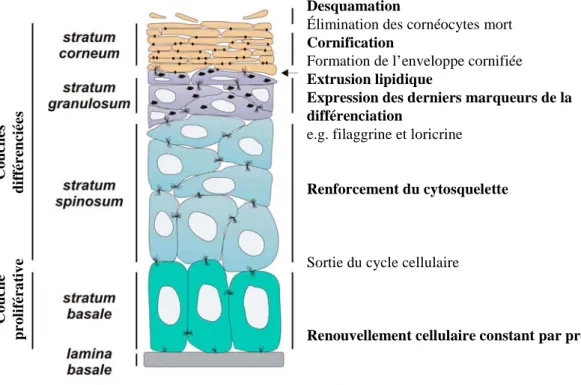
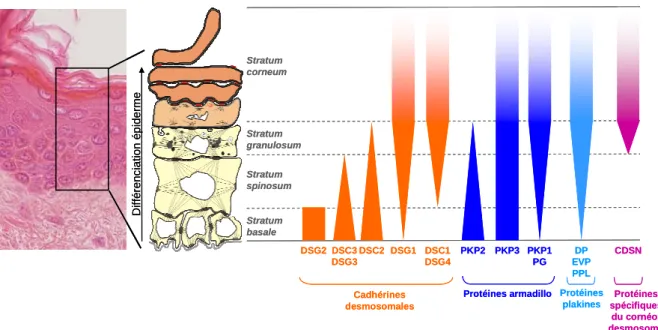
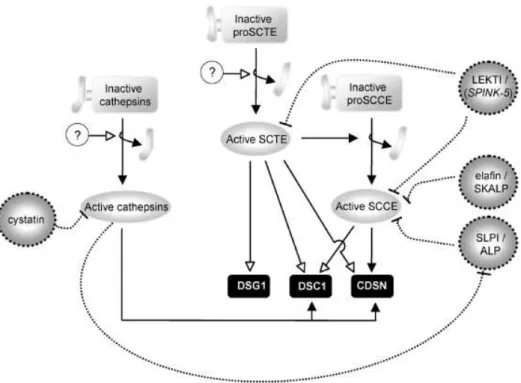
d) Immunoprécipitation de la chromatine... - 43 - B. La peau ... - 44 - 1. Généralités... - 44 - 2. L’épiderme ... - 45 - a) Généralités... - 45 - b) Structure ... - 46 - 3. La différenciation épidermique ... - 48 - a) Principales étapes ... - 49 - b) Desmosomes et cornéodesmosomes ... - 56 -
c) Les lipides intercornéocytaires... - 59 -
d) La desquamation ... - 61 -
4. Régulation du programme de différenciation... - 64 -
a) Rôle du calcium... - 64 -
b) Rôle de la vitamine D... - 66 -
C. La régulation génique dans l’épiderme ... - 68 -
1. Le complexe de différenciation épidermique... - 68 -
2. Les facteurs de transcription ... - 69 -
a) AP-1 ... - 69 -
b) Cas original de p63... - 74 -
c) FT spécifiques de la couche granuleuse – Modèles murins ... - 75 -
D. Les peptidyl-arginine désiminases ... - 77 -
1. Introduction ... - 77 -
2. Caractéristiques des PADs ... - 77 -
a) La désimination, une modification post-traductionnelle... - 77 -
b) Locus génétique, conservation et homologie ... - 78 -
c) Profils d’expressions ... - 80 -
9
-e) Inhibiteurs des PADs... - 84 -
3. Les PADs dans la peau : localisation et rôle ... - 85 -
4. Les PADs dans les follicules pileux ... - 87 -
5. Autres rôles physiologiques des PADs ... - 89 -
6. PADs et pathologies ... - 90 -
a) Polyarthrite rhumatoïde... - 90 -
b) Sclérose en plaque et autres maladies neurodégénératives ... - 91 -
c) Psoriasis et érythrodermie ichtyosiforme congénitale ... - 94 -
d) Cancers ... - 95 -
e) Néphropathie obstructive ... - 95 -
7. Régulation de l’expression des PADs ... - 95 -
RESULTATS EXPERIMENT RESULTATS EXPERIMENTRESULTATS EXPERIMENT RESULTATS EXPERIMENTAUXAUXAUX ...AUX... -...--- 99 99 99 - 99 -- -Publication N°1:... - 99 - Publication N°2:... - 110 - Publication N°3:... - 123 - DISCUSSION DISCUSSIONDISCUSSION DISCUSSION ... -...--- 137 137 137 137 --- -A. Caractérisation des promoteurs minimums des gènes PADI... - 137 -
B. Caractérisation d’éléments régulateurs distants ... - 139 -
1. IG1 peut-il être un gène ? ... - 139 -
2. Approche analytique : étude des CNS d’IG1 ... - 140 -
3. Approche globale : étude des 4 sous-fragments d’IG1 ... - 140 -
a) Identification d’un activateur bipartite : PIE-S1/S2 ... - 140 -
b) Implication du complexe MIBP1/RFX1 ... - 141 -
c) Implication des facteurs c-Jun et JunD ... - 142 -
4. Pourquoi PIE et PIE-S1/S2 n’agissent-ils pas sur PADI2 ? ... - 144 -
5. Interaction entre PIE-S1/S2 et les promoteurs de PADI1 et PADI3 : rôle de la boîte TATA ? ... - 145 -
6. Similitudes et différences entre PIE et PIE-S1/S2 ... - 146 -
a) Similitudes... - 146 -
b) Différences ... - 146 -
7. Perspectives ... - 147 -
a) Utilisation de siARN ... - 147 -
b) Réalisation de souris transgéniques... - 147 -
c) Utilisation d’oligonucléotides leurres ... - 148 -
d) Régulation à distance des gènes PADI dans d’autres types cellulaires ... - 148 -
e) Etude d’associations génétiques avec des pathologies épidermiques ... - 149 -
f) Régulation à distance dans l’épiderme : une généralité ? ... - 149 -
g) Caractérisation d’unités transcriptionnelles dans le locus des gènes PADI : étude des S/MARs... - 149 -
h) Etude de la régulation post-transcriptionnelle des gènes PADI ... - 150 -
C. Conclusion générale ... - 151 -
ANNEXE I ANNEXE IANNEXE I ANNEXE I ... -...--- 152 152 152 - 152 -- -Identification d’un nouveau gène dans le locus des gènes PADI ? ... - 152 -
ANNEXE II ANNEXE IIANNEXE II ANNEXE II ... -...--- 155 155 155 - 155 -- -Liste des publications ... - 155 -
BIBLIOGRAPHIE BIBLIOGRAPHIEBIBLIOGRAPHIE
10
-INTRODUCTION
A. La régulation génique
La découverte des lois de Mendel au début du vingtième siècle, constitue le début de la
recherche scientifique sur l’information génétique. Les progrès dans ce domaine sont passés
par quatre phases : la découverte des bases cellulaires de l’hérédité avec les chromosomes en
précéda la découverte des bases moléculaires, avec le rôle de l'ADN, qui fut initialement
démontré en 1944 par Avery et son équipe, puis la découverte de la double hélice d’ADN par
Franklin, Wilkins, Watson et Crick (prix Nobel de médecine en 1962) (Watson and Crick,
1953). Troisièmement le décryptage de l’information de base de l’hérédité, avec la découverte
des mécanismes biologiques par lesquels les cellules lisent l’information contenue dans les
gènes (Nirenberg en 1962) et l’invention des technologies de séquençage et de clonage
d’ADN recombinant. Enfin les 30 dernières années ont été marquées par le séquençage des
gènes et de génomes entiers, explorant le vaste domaine de la génomique. C’est ainsi qu’un
certain nombre de caractéristiques des génomes ont été annotées, comme les gènes, les
éléments transposables, la composition en GC et les « îlots CpG » ou encore les taux de
recombinaison, fournissant des indices sur leur fonction. Par exemple, les groupes (clusters)
de gènes HOX, très importants dans le développement, sont les régions du génome humain les
plus pauvres en éléments répétés, reflétant probablement une coordination complexe de la
régulation de ces gènes (Consortium, 2001).
Le séquençage du génome humain s’est achevé en 2004, aboutissant à une séquence finale de
2,85 Gb (2004). Le nombre total de gènes, bien que controversé, serait de 25 000 à 35 000
11
-répartis de façon non uniforme le long des 46 chromosomes humains (Consortium, 2001). Ces
chiffres correspondent seulement à deux fois plus de gènes pour l’homme que pour le ver (C.
elegans). Cependant, les gènes humains sont plus complexes, avec beaucoup d’épissages
alternatifs générant un plus grand nombre de protéines. Au-delà de ces chiffres, des
mécanismes de plus en plus complexes orchestrant dans l'espace et dans le temps l'expression
de tous ces gènes apparaissent au cours de l’évolution et sont fondamentaux. Cette régulation
intervient dans toutes les étapes du développement de l’embryon, au cours duquel les cellules,
contenant pourtant la même information génétique, se spécialisent et se multiplient pour
former divers organes et tissus, jusqu’à l’âge adulte dans le maintien de l’homéostasie du
corps tout entier. Comme certains gènes codent pour des protéines remplissant une fonction
très spécifique, il est impératif qu’ils soient exprimés aux moments opportuns et dans les
tissus appropriés. L’harmonisation de l'expression de ces gènes en réponse à des stimuli
internes et externes est primordiale, la moindre défaillance pouvant nuire à la survie de
l’individu.
1. Les premières découvertes
Les scientifiques ont entrepris d’étudier les mécanismes de régulation de l’expression des
gènes bien avant que la carte du génome humain ne soit disponible. Les premiers concepts de
régulation génique ont été évoqués par Jacob et Monod dès la fin des années 1950. Ils
proposèrent l'existence d'un « messager » servant d'intermédiaire entre les gènes et leurs
effecteurs biochimiques, les protéines. Ils firent également émerger les premières notions de
« promoteur », défini alors comme « opérateur », qui serait responsable du contrôle de
12
-2. Organisation et régulation de la chromatine a) Organisation
L’ADN, fractionné en chromosomes, est condensé sous forme de chromatine, une fibre de 30
nm de diamètre, la longueur totale de l’ADN humain étant estimée à 2 ou 3 mètres. Ainsi, une
molécule d’ADN linéaire hypothétique longue de 10 centimètres sera condensée en un
chromosome mesurant à peine 10 micromètres de long, ce qui implique une condensation
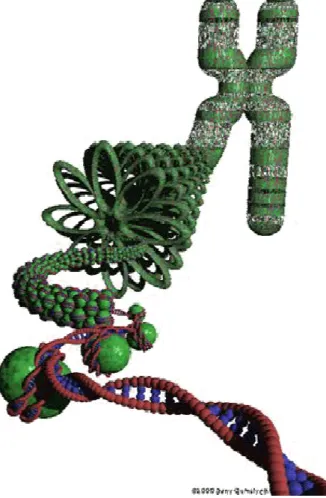
d’un facteur 10,000 (figure 1). La condensation de l’ADN en chromatine s’organise de
manière séquentielle et ordonnée. En premier lieu, 146 pb (paire de bases) d'ADN s'enroulent
autour d'un octamère d'histones (deux de chaque type : H2A, H2B, H3 et H4) riches en
résidus lysine et arginine, pour former un nucléosome condensant l’ADN par un facteur 7
(figure 2-a) (White et al., 2001). Deux nucléosomes sont séparés l'un de l'autre par une
cinquantaine de paires de base d'ADN et par l'histone internucléosomique H1.
13
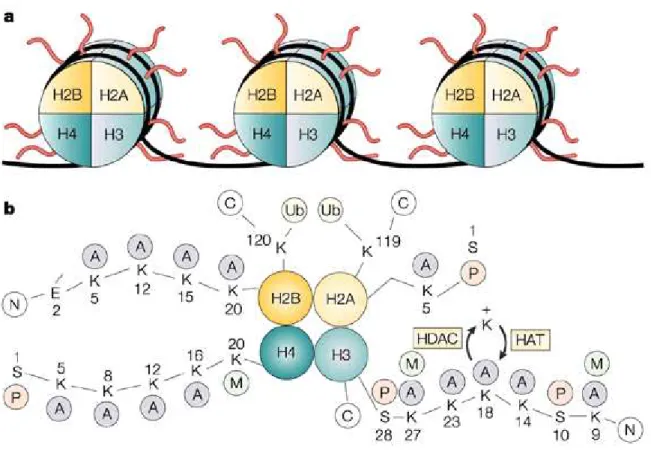
-Figure 2 : représentation de la structure des histones dans les nucléosomes.
a) Les protéines du cœur du nucléosome sont désignées comme H2A (histone 2A), H2B (histone 2B), H3 (histone 3) et H4 (histone 4). Chaque histone est présente en deux copies, ainsi l’ADN (noir) s’enroule autour d’un octamère d’histones – le « core » (cœur) du nucléosome.
b) Les queues amino-terminales des histones du coeur. Les lysines (K) des queues amino-terminales des histones H2A, H2B, H3 et H4 sont des sites potentiels d’acétylation/déacétylation pour les histones acétyltransférases (HATs) et les histones déacétylases (HDACs). L’acétylation neutralise la charge des lysines. A, acétyl; C, carboxyle terminal; E, acide glutamique; M, méthyl; N, amino-terminal; P, phosphate; S, sérine; Ub, ubiquitine (Marks et al., 2001).
Les nucléosomes vont ensuite être assemblés en des structures de plus en plus complexes, ils
se compactent, formant une hélice contenant 6 nucléosomes par tour, elle-même finalement
condensée en euchromatine (condensation légère, chromatine dite « ouverte ») ou en
hétérochromatine (condensation plus élevée) constituant un niveau d’organisation
supplémentaire (par exemple comme dans les centromères, les télomères et les régions
géniques inactives). La structure de la chromatine joue ainsi un rôle majeur dans la régulation
14
-Le niveau de compaction de la chromatine constitue le premier niveau de régulation des
gènes. Pour activer la transcription d’un gène dans une cellule, la chromatine située dans la
région de contrôle du gène doit être dans une conformation qui la rend accessible aux
différentes protéines régulant la transcription. Ainsi, les gènes localisés dans l’euchromatine
peuvent être transcrits. De la même façon, le gène dans sa totalité doit être accessible par la
polymérase pour être transcrit et donc localisé dans une zone décondensée de la chromatine.
La réorganisation locale de la chromatine et la modification des nucléosomes sont
indispensables à l’accessibilité des facteurs de transcription et de la machinerie générale de
transcription aux séquences régulatrices du gène (Davie and Chadee, 1998).
Les histones possèdent un domaine globulaire carboxy-terminal formant le cœur du
nucléosome et une queue amino-terminale présente à la surface du nucléosome (Uberbacher
and Bunick, 1985). Celle-ci va pouvoir être la cible de modifications post-traductionnelles
comme l’acétylation, la méthylation, la phosphorylation, la sumoylation et l’ubiquitination
engendrant des variations d’affinité pour l’ADN et les protéines régulatrices (figure 3) (Smith
and Denu, 2008). Ces modifications vont influencer l'état de compaction de la chromatine et
donc modifier les niveaux d'expression des gènes. Ce modèle est à la base de la notion de
« code des histones » (Jenuwein and Allis, 2001; Strahl and Allis, 2000), qui se superposerait
au code génétique proprement dit. L'acétylation des histones ainsi que la méthylation des
histones (ou de l'ADN) sont les modifications post-traductionnelles les plus directement liées
à l'état transcriptionnel d’une région génomique. Une nouvelle approche d’étude de la
régulation de la transcription consiste à analyser l’état des histones le long des chromosomes,
permettant l’identification de nouveaux éléments régulateurs, étape indispensable à
15 -Figure 3 : chromatine et nucléosomes.
Schéma du nucléosome et modifications post-traductionnelles de ses extensions. Le haut et le bas de la superhélice d’ADN sont colorés en bleu foncé et bleu clair respectivement; les histones H3 en jaune, H4 en magenta, H2A en cyan et H2B en vert, les extensions au-dessous du plan étant plus pâles que les extensions du dessus ; les résidus arginine et lysine sont en rouge. Les sites de modifications bien caractérisés sont indiqués : acétylation (*), méthylation (M), phosphorylation (P), ribosylation (R) et ubiquitination (U). D’après (Wolffe and Hayes, 1999).
b) Acétylation et méthylation des histones
L’acétylation et la méthylation des histones sont des formes épigénétiques de régulation des
gènes. Le terme « épigénétique » regroupe les modifications de l'expression des gènes qui ne
sont pas dues à des variations de la séquence d’ADN.
L’acétylation survient principalement sur les lysines des parties N-terminales des histones H3
et H4. C’est l’un des mécanismes impliqués dans la régulation de la chromatine les plus
étudiés (Eberharter and Becker, 2002). La réaction d’acétylation est le transfert d’un
groupement acétyl sur un résidu lysine localisé sur la queue N-terminale d’une histone
catalysé par une histone acétyltransférase (HAT). L’affinité des histones, chargées
16
-électrostatique. Lorsque les histones sont acétylées, leur charge positive diminue, entraînant
une modification de leur affinité pour l’ADN. Un niveau local d’acétylation élevé, ou
hyperacétylation, sur les queues des histones est souvent associé à une structure ouverte de la
chromatine permettant une meilleure accessibilité aux facteurs de transcription. A l’inverse,
une hypoacétylation locale, due à la suppression des groupements acétyls catalysée par les
histones désacétylases (HDACs), coïncide avec la répression de l’activité de transcription
(figure 2-b) (Felsenfeld et al., 1996).
La méthylation des histones sur des résidus lysine et arginine (Arg), catalysée par des
histones méthyltransférases (HMTs), est aussi reliée à la régulation de la transcription.
PRMT1 (PRotein arginine MethylTransferase 1) et CARM1 (Coactivator-associated
ARginine Méthyltransférase 1) en association avec des coactivateurs transcriptionnels des
récepteurs nucléaires (dont l’histone acétyltransferase p300) et avec p53 (An et al., 2004;
Wang et al., 2001) facilitent la transcription en méthylant spécifiquement l’Arg 3 de H4
(PRMT1) et l’Arg 17 de H3 (CARM1).
Les mécanismes permettant la déméthylation des histones sont moins connus (Schneider and
Shilatifard, 2006). Récemment, Chang et coll. ont découvert la première histone arginine
déméthylase JMJD6 capable de déméthyler l’Arg 2 de H3 et l’Arg 3 de H4 in vitro et in vivo
(Chang et al., 2007). D’autre part, la désimination (conversion d’un résidu arginyl en un
résidu citrulyl catalysée par une peptidyl-arginine désiminase (PAD) ; voir chapitre PAD) a
un effet négatif sur l’induction de la transcription médiée par la méthylation des Arg des
histones. L’enzyme PAD4 humaine pourrait convertir les méthyl-Arg en citrulline dans une
réaction de déméthylimination. Dans les granulocytes HL-60 (cellules myéloïdes humaines),
la désimination par PAD4 des Arg N-terminales des histones H3 et H4, incluant les sites
méthylés par les méthyltransferases, empêche leur méthylation par CARM1 (Hagiwara et al.,
2002) (Cuthbert et al., 2004; Wang et al., 2004). PAD4 peut également agir sur le cofacteur
17
-l’activité de PAD4 (Lee et al., 2005). Ces évènements de méthylation et de
déméthylimination agissent sur la fonction, la conformation et l’assemblage du complexe de
régulation de la transcription. Le recrutement et l’activité de PAD4 sont également liés à une
inhibition de la transcription des gènes de réponse aux œstrogènes dans les cellules MCF-7
(lignée de cellules tumorales mammaires). Inversement, la diméthylation des Arg empêche la
désimination de celles-ci par PAD4 (Cuthbert et al., 2004).
c) Méthylation de l’ADN
La méthylation sur cytosine de la molécule d’ADN est une modification épigénétique majeure
de la chromatine. Elle est catalysée par les ADN méthyltransférases (DNMTs), et consiste en
l’addition covalente d’un groupement méthyl, provenant du donneur S-adénosylméthionine,
sur le carbone 5 des cytosines des dinucléotides CG (ou CpG, Cytosine-phosphate-Guanine)
(figure 4). L’enzyme DNMT3 catalyse la méthylation de novo de l’ADN. Une grande
majorité (80 %) des dinucléotides CpG du génome sont méthylés : le génome est dit
globalement hyperméthylé. Les dinucléotides CpG peuvent être concentrés en « îlots CpG ».
Les îlots CpG sont des séquences d’au moins 200 pb avec un contenu en G/C d’au moins 50%
et un contenu en CpG supérieur à ce qui est statistiquement attendu (Gardiner-Garden and
Frommer, 1987). Ils sont majoritairement (70-80 %) localisés dans les régions situées en 5’
des gènes, en particulier des gènes dits de ménage, c'est-à-dire des gènes s’exprimant de la
même manière dans toutes les cellules d’un organisme et dont le produit est généralement
indispensable à la vie de la cellule. Dans les îlots CpG 80% des dinucléotides CpG ne sont pas
méthylés. A cause de l’existence des ces îlots, le génome est dit hyperméthylé globalement, et
hypométhylé localement. Les régions cibles de la méthylation sur CpG sont les gènes viraux
18
-inactif (13), et des régions spécifiques à chaque tissu. On appelle épigénome l'ensemble des
régions du génome spécifiquement méthylées dans une cellule ou un tissu particulier.
La méthylation des îlots CpG provoque l’apparition d’un contexte chromatinien répressif qui
bloque la transcription du gène cible (Bird, 2002; Jaenisch and Bird, 2003), alors que
l’absence de méthylation est associée à un contexte permissif (Razin, 1998). Certaines études
ont permis d’observer une répression stable de l’activité de la transcription de différents gènes
suite à leur méthylation (Schubeler et al., 2000).
La méthylation de l’ADN peut être modulée par des facteurs environnementaux (Jaenisch et
al., 2004) au cours du développement, mais aussi chez l’individu adulte. Elle peut être conservée au cours de la mitose par l’intervention de la DNMT1 dite de « maintenance ». Par
conséquent la méthylation de l’ADN sur cytosine est la seule variable connue qui modifie la
fonction du génome de façon pérenne sous l’action extérieure.
Des études in vitro dans des systèmes modèles suggèrent une association entre différenciation
cellulaire et la méthylation du génome. Notamment, dans un modèle de différenciation
kératinocytaire in vitro (voir chapitre épiderme), l’abondance en méthyl-cytosine est 3 fois
plus faible dans des kératinocytes normaux différenciés que dans des kératinocytes normaux
prolifératifs (Veres et al., 1989). De même une corrélation inverse a été mise en évidence
entre la méthylation et l’expression des gènes S100A2 et S100A6, analysée dans les fibroblastes, les kératinocytes humains normaux et la lignée HaCaT (Elder and Zhao, 2002). Plus récemment, la méthylation de l’ADN à été étudiée dans le processus de différenciation des cellules souches embryonnaires. Il a notamment été montré qu’après induction de la différenciation des cellules souches par différents stimuli, les gènes codant pour OCT4 et
NANOG, des marqueurs des cellules pluripotentes, sont méthylés et donc réprimés (Yeo et
al., 2007). Inversement, les régions promotrices d’OCT4 et NANOG sont déméthylés lors de la reprogrammation de fibroblastes dermiques en cellules souches pluripotentes induites
19
-Ainsi, la méthylation de l’ADN semblerait jouer un rôle très important dans la régulation des
gènes en inhibant leur transcription. Cette inhibition pourrait être la conséquence soit de la
réduction de l’affinité entre les facteurs de transcription activateurs et leur séquence d’ADN
cible, soit de la liaison de protéines ayant une grande affinité pour l’ADN méthylé et recrutant
in situ des facteurs répresseurs.
Figure 4 : méthylation de l’ADN par les DNMTs
Les mécanismes d'acétylation des histones et de méthylation de l'ADN sont liés. En effet,
l'ADN méthylé peut être lié par des protéines appelées Methyl-CpG-binding domain proteins
(MBD) (Meehan et al., 1989). Ces protéines peuvent ensuite recruter localement des histones
20
-d) Modifications épigénétiques et cancer
Des aberrations épigénétiques, notamment au niveau de l’acétylation des histones et de la
méthylation de l’ADN, sont souvent retrouvées associées à l’initiation et/ou à la progression
de tumeurs. Des études ont montré que le génome de tumeurs ou de tissus hyperplasiques
pré-tumoraux est hypométhylé globalement, et hyperméthylé localement. Ces mécanismes
pourraient contribuer à la carcinogénèse via l’inhibition des gènes suppresseurs de tumeurs
(hyperméthylation des îlots CpG des promoteurs de ces gènes), la surexpression des
oncogènes et/ou une diminution de la stabilité du génome (Counts and Goodman, 1995). On
peut se demander si ces modifications épigénétiques font partie des causes ou des
conséquences de la formation tumorale. Certaines données renforcent la première hypothèse.
L’équipe de Robertson a montré l’apparition de modifications du profil de méthylation avant
la formation de la tumeur (Robertson and Jones, 2000). En outre, une analyse statistique à
grande échelle concernant la distribution anormale des CpG méthylés dans 7 types de tumeurs
a montré qu’ils ne sont pas distribués aléatoirement mais selon un profil spécifique du type de
tumeur (Costello et al., 2000).
L’étude des mécanismes épigénétiques impliqués dans le développement des tumeurs s’est
intensifiée ces dernières années (Clark, 2007; Lopez-Serra and Esteller, 2008; Vucic et al.,
2008). De nouvelles stratégies thérapeutiques très prometteuses consistent à corriger ces
anomalies épigénétiques à l'aide de composés modulant l'acétylation des histones et la
méthylation de l'ADN. Des inhibiteurs de ces mécanismes constituent ainsi une classe
grandissante de composés anticancéreux (Allen, 2007; Kim et al., 2006; Palii and Robertson,
21
-3. Structure et transcription d’un gène a) Structure et généralités
La plupart des gènes de classe II, codant très majoritairement pour des ARNm (ARN
messager), sont constitués de deux séquences d'ADN distinctes : la séquence transcrite et la
séquence régulatrice.
La séquence transcrite va permettre la formation du transcrit primaire, une molécule d'ARN
directement synthétisée à partir du modèle ADN, composée d’exons (séquence codante) et
d’introns (séquence non codante), qui sera cappé et polyadénylé, et enfin « épissé ». L'ARN
peut alors être exporté dans le cytoplasme et devient un ARNm. L'ARNm sert alors de patron
pour sa traduction en protéine, à partir des acides aminés et par l’intermédiaire de ribosomes
et d’ARN de transfert (ARNt). La régulation de l’expression génique peut être réalisée à tous
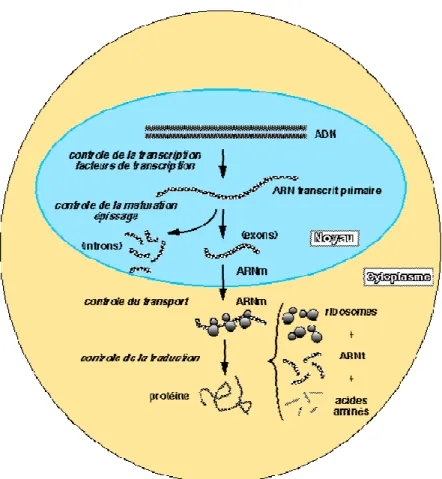
ces niveaux (figure 5).
Figure 5 : régulation de l'expression génique
22
-b) Les régions régulatrices
La séquence régulatrice, également appelée séquence promotrice, recrute l’ensemble des
acteurs indispensable à l’activation et à la régulation de la transcription d’un gène. Elle est
généralement située en amont de la séquence transcrite et contrôle la transcription du
messager. Des régions régulatrices peuvent également être situées dans les introns. Plus
particulièrement, de nombreux éléments régulateurs ont été retrouvés dans le premier intron
des gènes (Qiao et al., 2005; Suen and Goss, 2001). Il y a donc, en toute rigueur,
chevauchement entre séquences régulatrice et transcrite d’un gène.
Plusieurs séquences d’ADN hautement conservées au cours de l’évolution et agissant en cis
(car situés sur la même molécule d’ADN que le gène contrôlé) contiennent de nombreux sites
de fixation pour des facteurs de transcription, et permettent de réguler la transcription par
l’ARN polymérase II (ARN Pol II) :
- le promoteur proximal, au voisinage direct du site d’initiation de la transcription (TSS)
(généralement de -250 à +250 pb).
- une ou plusieurs régions régulatrices distales, localisées jusqu'à plusieurs centaines de kb
du TSS. Ces régions contiennent des séquences fixant des activateurs ou des inhibiteurs.
"L’enhancer" ou séquence activatrice a la propriété de stimuler l’expression génique
indépendamment de sa position (en 5’ ou en 3’), de son orientation et de sa distance vis à vis
du gène. Une première séquence de ce type a été identifiée dans le génome du virus SV40
(Simian Virus) (Benoist and Chambon, 1981). Des activateurs ou inhibiteurs intragéniques
(exoniques ou introniques) ou extragéniques, isolés ou regroupés, ont été identifiés.
- des éléments de délimitation ou insulateurs (insulator) divisés en deux classes : les
« enhancer-blocking » qui bloquent l’action d’activateurs ou de répresseurs transcriptionnels
distaux sur un promoteur, et les insulateurs barrières, inhibant la formation de
l’hétérochromatine et par conséquent l’inactivation transcriptionnelle d’un gène (Valenzuela
23
-Les MARs ou régions d’attachement à la matrice nucléaire (Scaffold/matrix attachment
regions ou S/MARs) font partie de la catégorie des insulateurs, et possèdent d’autres
propriétés. Les MARs jouent un rôle de régulateur transcriptionnel en réorganisant la
conformation de la chromatine. Ces régions de 300 pb à plusieurs kb sont présentes chez tous
les eucaryotes supérieurs. Les complexes MARs sont constitués de protéines qui se fixent à la
fois à la matrice nucléaire et à l’ADN, entraînant une compaction supérieure de la chromatine,
et la formation d’une « barrière » entre des domaines chromatiniens et transcriptionnels
indépendants (zones plus ou moins condensées de la chromatine). Les MARs peuvent
également servir à rapprocher des séquences distantes, donnant ainsi une nouvelle
« architecture » à la chromatine. Or certaines protéines se liant aux MARs ne sont présentes
que dans des types cellulaires spécifiques, d’où un rôle supplémentaire dans la régulation de
la transcription (Schubeler et al., 1996; Tetko et al., 2006).
c) L’initiation
L’activation de la transcription d’un gène requiert la coordination de nombreux facteurs. Des
protéines vont dans un premier temps interagir avec le core promoteur pour recruter l’ARN
polymérase et initier la transcription. Des facteurs de transcription, en réponse à des stimuli
internes et/ou externes (Felsenfeld et al., 1996), vont également participer à la modulation de
cette régulation, et peuvent dans certains cas activer la transcription de gènes en cascade.
(1) L'ARN polymérase
Cette enzyme catalyse la synthèse d’ARN à partir de molécules d’ADN ou d’ARN. Chez les
eucaryotes, trois sortes d'ARN polymérases sont responsables de la synthèse des quatre types
d'ARN (ARNm, ARNr (ribosomique) et ARNt, ainsi que les petits ARN : snARN (small
nuclear RNA), snoARN (Nucleolar) et miARN (Micro)). L’ARN pol II est un complexe
24
-protéines. L'ARN polymérase vient se fixer sur la région régulatrice du gène pour initier la
transcription. De nombreux facteurs protéiques vont être nécessaires à cette fixation. Parmi
ceux-ci on retrouve les facteurs de transcription.
(2) Les facteurs de transcription
On appelle facteur de transcription (FT) toute protéine nécessaire à l’initiation de la
transcription ou à son contrôle. Les mécanismes d’action des FT, qu’ils soient activateurs ou
répresseurs, sont variés. Ils peuvent agir au niveau de la condensation de la chromatine, de
l’initiation de la transcription et de l’élongation du transcrit. Ils se lient généralement à une
petite séquence « consensus » de l’ADN, conservée au cours de l’évolution et appelée site de
fixation. Mais cette liaison n’est pas toujours requise, puisqu’ils peuvent s’associer
physiquement à l’ARN polymérase ou à un autre FT qui, lui, est fixé directement à cette
dernière. Certains FT ont besoin d'un cofacteur ou d'une modification post-traductionnelle
pour être actifs, d’autres possèdent des domaines de liaison à des hormones (famille des
récepteurs nucléaires).
On distingue trois grands types de FT :
- Les facteurs généraux de la transcription, qui vont s’associer à l’ARN Pol II et
constituer la machinerie basale de transcription.
- Les FT ubiquitaires qui vont reconnaître des séquences du promoteur proximal et
augmenter le niveau basal de transcription. Par exemple, le facteur Sp1 se fixe sur de
nombreux promoteurs ayant une « boîte GC » (et en particulier ceux chevauchant un
« îlot CpG »).
- Les FT spécifiques d’un tissu donné et/ou d’un stade de développement. Ils font
souvent partie d’une cascade de signalisation intracellulaire, responsable de
25
-La séquence et/ou la structure du domaine de liaison à l’ADN sont utilisées pour classer les
FT en famille. Il existe 4 types principaux de domaine protéique de liaison :
hélice-tour-hélice, hélice-boucle-hélice-tour-hélice, glissière à leucine et doigt de zinc. Cependant plusieurs dizaines
d’autres ont été identifiés et la liste ne cesse de grandir. Ils sont classés de la façon suivante :
les superclasses (ex : la superclasse des domaines de base), elles-mêmes comportant plusieurs
classes (ex : la classe des glissières à leucine), sous-divisées en familles (ex : la famille AP-1)
et même en sous-familles (ex : sous-famille Jun), pour finalement atteindre les facteurs
eux-mêmes (ex : c-Jun). Si certains facteurs de transcription comme Sp1 et AP-1 sont associés à
l’activation de nombreux gènes, d’autres sont plus spécifiques (Bouwman and Philipsen,
2002; Shimano, 2001; Wisdom, 1999).
(3) Le « core » promoteur
Chez les eucaryotes, le « core » promoteur sert de plateforme d’assemblage du complexe
d’initiation de la transcription (CIT) (Butler and Kadonaga, 2002). Il correspond à une région
située autour du site d’initiation de la transcription de l’ARNm (nucléotide +1) et permet le
recrutement de l'ARN Pol II et des facteurs généraux de transcription. Il contient plusieurs
sites de fixation ou « boîtes » permettant le recrutement des protéines d’initiation. L’une de
ces régions, la boîte TATA ou « TATA box », est retrouvée dans moins de 25% des gènes
eucaryotes et correspond à une séquence régulatrice riche en adénine et thymine qu'on trouve
à environ 25 pb en amont du TSS (Yang et al., 2007). Parmi les autres motifs présents sur les
cores promoteurs, trois éléments ont été caractérisés : la séquence initiatrice (Initiator ou Inr)
présente dans 50 % des gènes et appelée ainsi car comprenant le site d’initiation de la
transcription (Smale et al., 1998), elle semble pouvoir recruter le facteur de transcription
général TFIID ; l’élément aval du promoteur (Downstream Promoter Element ou DPE)
26
-Kadonaga, 1996; -Kadonaga, 2002) ; l’élément de reconnaissance par le facteur TFIIB (BRE)
(Lagrange et al., 1998). Les éléments DPE et BRE sont chacun retrouvés dans 25 % des gènes
eucaryotes. Dans les promoteurs sont généralement associées une boîte TATA et une boîte
Inr, ou une boîte DPE et une boîte Inr, les boîtes TATA et DPE ayant la même fonction. La
boîte TATA est souvent présente sur les gènes dont l’expression est limitée à certains tissus.
Les gènes dont l’expression est ubiquitaire, par exemple les gènes de ménage, ne contiennent
généralement pas de boîte TATA ni de boîte Inr (Roeder, 1996). A la place, ces gènes
possèdent plusieurs sites d’initiation de la transcription ainsi que des promoteurs riches en
dinucléotides CG et/ou chevauchant un « ilot CpG ». Ainsi ils contiennent souvent des motifs
reconnus par le facteur de transcription Sp1, riches en CG (Bouwman and Philipsen, 2002).
Dans le cas des gènes de ménage, Sp1 permettrait le positionnement de la machinerie générale
de transcription au TSS.
(4) La machinerie basale de transcription
La transcription d’un gène implique l’interaction entre la chromatine, les protéines
régulatrices et la machinerie générale de transcription, également nommée holoenzyme. Elle
est constituée de 8 à 14 sous-unités dont l’ARN Pol II, des facteurs TFIIA, TFIIB, TFIID,
TFIIE, TFIIF et TFIIH et de coactivateurs variables (Thomas and Chiang, 2006). La plupart
des coactivateurs contiennent des activités enzymatiques de modification des histones, et en
particulier, une activité acétyltransférase. Ils facilitent ainsi l’accès à l’ADN en favorisant le
remodelage de la chromatine. C'est sur la boîte TATA, que se forme le complexe de protéines
qui va initier la transcription du gène. La formation du cœur du CIT commence par la liaison
de TFIID, lui-même contenant la protéine de liaison TBP (TATA-binding protein), sur la
boîte TATA. C’est l’évènement déclencheur de l’assemblage du complexe d’initiation,
27
-par TFIIF et l’ARN polymérase II et positionnera cette dernière au site d’initiation de la
transcription. TFIIF est le seul facteur qui reste associé à la polymérase pendant l’élongation.
Enfin TFIIE puis TFIIH sont recrutés. Les deux fonctions principales de TFIIE sont de réguler
les activités de TFIIH et de favoriser l’initiation de la transcription par son activité hélicase.
Aussi TFIIH catalyse l’ouverture de la double hélice d’ADN entre les positions -9 et +2 de
part et d’autre du TSS pour former la bulle transcriptionnelle. Enfin, TFIIH présente une
activité kinase impliquée dans la phosphorylation de l’ARN Pol II, déclenchant la libération
de l’ARN Pol II du CIT et le début de la transcription (figure 6). L’ARN Pol II s’associe à
différents facteurs pour l’étape d’élongation. La terminaison de la transcription est suivie du
recyclage de l’ARN Pol II sous sa forme non phosphorylée permettant à l’enzyme de
28
-Figure 6 : mise en place du complexe d'initiation.
29
-4. La régulation de la transcription d’un gène
En plus du « core » promoteur, d’autres séquences d’ADN agissant en cis régulent la
transcription de l’ARN Pol II : le promoteur proximal et les éléments distants contenant des
activateurs et/ou des inhibiteurs transcriptionnels.
a) Le promoteur proximal
Le promoteur proximal contient des boîtes ou des sites consensus de fixation de FT. Ces
boîtes sont situées plus en amont que la boîte TATA. On peut citer par exemple la "boîte
CAAT" située vers -100 pb environ du site d’initiation de la transcription, et la "boîte GC",
riche en guanine et cytosine, située entre les boîtes CAAT et TATA. Toute variation de
séquence ou de positionnement de ces éléments entraîne de fortes perturbations du niveau de
transcription. En effet, l’occupation du promoteur proximal par une combinaison de différents
FT est aussi importante pour le contrôle des gènes que la nature proprement dite des FT
intervenants. On appelle « module transcriptionnel » une suite ordonnée de sites de fixation de
FT dans un promoteur proximal.
D'autres participants existent encore. Il existe des promoteurs proximaux alternatifs (Ayoubi
and Van De Ven, 1996) (Andersen et al., 2006) et des promoteurs distaux, situés à des
milliers de nucléotides du promoteur proximal.
b) La régulation génique à distance
La régulation de l’expression d’un gène est très complexe, et ne se limite pas à l’étape
d’activation. Les gènes des eucaryotes supérieurs requièrent des séquences régulatrices
distantes pour coordonner leur expression spatio-temporelle et pour augmenter le niveau basal
d’expression du gène cible relié. Malgré la distance qui sépare les promoteurs distaux des
éléments proximaux, ces derniers agissent sur le promoteur proximal comme activateurs ou
répresseurs par un jeu de courbures de l'ADN ou d’autres mécanismes, par l’intermédiaire de
30
-Figure 7 : la régulation spécifique de la réplication.
http://www.snv.jussieu.fr/vie/dossiers/transcription/transcription.htm
(1) Modes d’interaction
Les mécanismes impliqués dans l’action d’un régulateur distant avec le promoteur d’un gène
cible situé jusqu'à plusieurs centaines de kb l’un de l’autre ne sont pas encore complètement
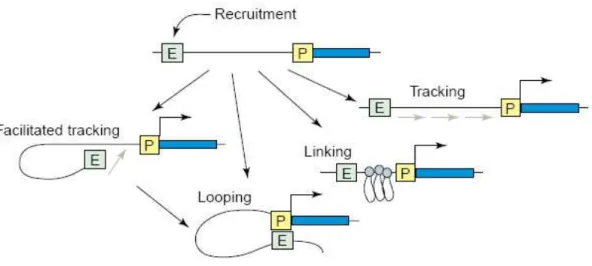
élucidés. Plusieurs modèles sont proposés pour expliquer cette interaction physique (figure
8):
- La formation d’une boucle chromatinienne (« looping »), permettant l’interaction
directe entre l’activateur distant et le promoteur. Ce modèle est retrouvé dans la
régulation des gènes du locus des cytokines Th2 (Spilianakis and Flavell, 2004).
- La formation de maillons le long de l’intervalle entre le régulateur et le point de départ
de la transcription (« linking »), générant une chaîne de complexes protéiques. Dans ce
cas la distance entre le régulateur distant et le promoteur ne peut pas être aussi grande
que lors de la formation d’une boucle chromatinienne unique.
- La poursuite (« tracking »). L’activateur distant propagerait son action le long de la
séquence qui le sépare du promoteur par l’intermédiaire d’activateurs et/ou de
coactivateurs qui vont par exemple modifier la chromatine de proche en proche. En
31
-au processus de poursuite. Egalement, la TnT associe « tracking » et transcription
(Zhu et al., 2007).
Figure 8 : modes d’interaction d’un régulateur distant avec le promoteur d’un gène cible. Quatre
principaux modèles sont proposés afin d’expliquer l’interaction entre « l’enhancer » (E) distant (après son recrutement) et le promoteur (P) : le « tracking » ou poursuite, le « linking » ou formation de maillons, le « looping » ou boucle chromatinienne et le « facilitated tracking » ou poursuite facilitée (Dean, 2006).
(2) Les LCRs
Des complexes de régulateurs transcriptionnels distants (également définis comme
« complexe d’activateurs ») sont appelés régions de contrôle du locus (Locus Control Region
ou LCRs) (Li et al., 2002). Les LCRs présentent trois propriétés majeures : ils influencent la
cinétique de la réplication et la déméthylation de l’ADN, activent la transcription, contrôlent
la tissu-spécificité et le niveau d’expression physiologique des gènes cibles. Ils présentent
généralement une activité dominante d’ouverture de la chromatine. Leur action est
indépendante de la position, et dépendante du nombre de copies, ils suppriment donc l’effet
de variégation dans la souris transgénique, et se distinguent ainsi des activateurs simples de
transcription (Simon et al., 2001a). Les LCRs sont distincts des promoteurs, des activateurs
32
-de tous ces régulateurs réunis, puisqu’ils contiennent un ou plusieurs -de ces éléments. Les
LCRs ont été décrits dans le contrôle de l’expression de nombreux gènes eucaryotes. On
retrouve plus particulièrement l’existence d’une régulation génique à distance dans la
coordination de l’expression de gènes homologues d’un même locus, comme c’est le cas pour
celui des bêta-globines qui est de loin le plus étudié (Li et al., 2002). C’est d’ailleurs le
premier à avoir été identifié dans les années 1980, à partir de l’étude de mutants naturels
(délétions) dans un contexte de β-thalassémie (Tuan et al., 1985; Van der Ploeg et al., 1980).
33
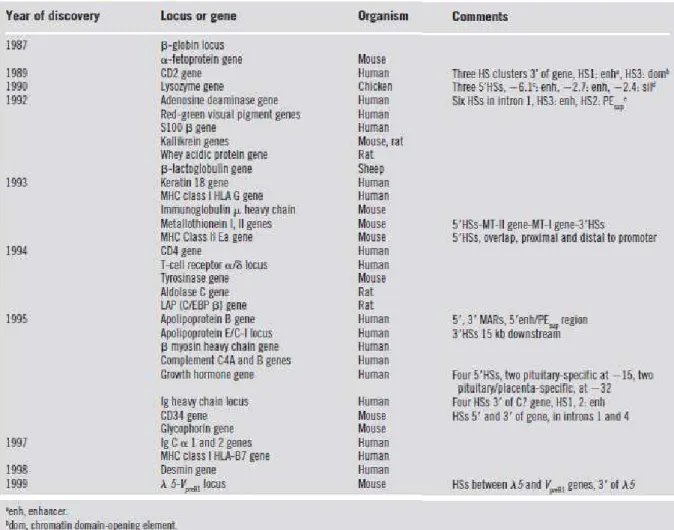
-Table 1 : LCR de mammifères et autres éléments « LCR-like » (Li et al., 1999)
(3) Exemple du locus des bêta-globines
Le complexe des bêta-globines comprend cinq gènes qui s’étendent sur 50 kb. On retrouve de
5’ en 3’ (centromérique vers telomérique) le gène embryonnaire ε, les deux gènes fœtaux Gγ, Aγ, et les gènes adultes δ et β. Ces gènes s’expriment uniquement dans les tissus érythroïdes, et de façon séquentielle au cours du développement. Cette spécificité est la conséquence de
multiples interactions entre les régions cis régulatrices du LCR situé en 5’ du locus (entre 6 et
25 kb en amont du gène ε) et les régions promotrices des gènes des bêta-globines. Cette région, dont l’importance a été soulignée par de nombreux travaux, intervient à différents
34
-niveaux de régulation et possède plusieurs propriétés dont l’activation de l’expression
tissu-spécifique des gènes de la famille des bêta-globines. Ce LCR comprend 5 sites hypersensibles
(HS) appelés ainsi en raison d’une sensibilité à la DNase I supérieure à celle des promoteurs
de gènes de bêta-globines (figure 9). L’utilisation de souris transgéniques a montré
l’importance fonctionnelle du LCRdes bêta-globines dans la coordination spatio-temporelle
des gènes du locus. C’est un puissant activateur transcriptionnel tissu-spécifique des gènes;
c’est également un élément insulateur : il abolit l’effet de position car il est capable de
conférer à un transgène une expression indépendante de son site d’intégration en l’isolant de
l’environnement chromatinien du génome hôte (Fraser et al., 1990) ; il agit sur l’origine et la
cinétique de la réplication d’ADN ; enfin il intervient dans l’ouverture de la chromatine. Des
études ont été menées afin de caractériser le rôle précis de chacun des 5 HS. Des séquences
minimales ou régions "core" de 200 à 500 pb hautement conservées durant l’évolution ont
ainsi été identifiées. L’activité majeure du LCR semble résider au niveau des sites HS2 et
HS3, et seuls les sites HS2, 3 et 4 présentent des activités d’activateurs transcriptionnels.
Cependant des spécificités fonctionnelles caractérisent chaque site. Le site HS1 confère au
gène de la β globine humaine une expression indépendante de sa position (Milot et al., 1996). Le site HS2 seul peut activer le gène de la β globine (Fraser et al., 1990). (Milot et al., 1996) ; il contient un enhancer puissant nécessitant la fixation des facteurs AP-1 (Activating Protein
1) et NFE2 en tandem (Ney et al., 1990a; Moi et Kan,1990; Ney et al., 1990b; Liu et al.,
1992; Bean et Ney, 1997, Forsberg et al., 2000). Il contient également deux séquences
inhibitrices (Labie et Elion, 1996)(Elnitski et al., 2001). Le site HS3 serait directement
impliqué dans la régulation de la transcription indépendamment du site d’intégration
(Philipsen et al., 1990) et dans l’ouverture de la structure chromatinienne. Le site HS4 a un
35
-MEL (érythroleucémique murine) (Pruzina et al., 1991). Le site HS5 est le seul dont la
fonction précise demeure encore inconnue.
Figure 9 : représentation schématique du locus des bêta-globines.
Les 5 gènesε, Gγ, Aγ,δ et β sont représentés par des rectangles colorés. Les 5 sites hypersensibles à la DNase I (HS) sont localisés par des flèches noires.
5. Autres régulations
Si les FT jouent un rôle majeur de régulateurs de la transcription, ils ne sont pas les seuls
acteurs de la régulation de l’expression ou de la fonction biologique d’une protéine puisque
celle-ci peut avoir lieu à d’autres niveaux :
1- La quantité des FT disponibles au CIT est limitante pour l’initiation de la transcription.
2- Le transcrit du gène, l’ARNm, peut lui-même être la cible d’une régulation lors de son épissage et de son exportation nucléaire. Par exemple, les ARE (AU-rich elements),
éléments riche en adénine et en uracile, situés en 3’ dans la région non-traduite
(untranslated region, UTR) de plusieurs ARNm, sont parmi les éléments responsables
de l'instabilité de ces ARNm et donc de leur courte demi-vie (Chen and Shyu, 1994).
3- La traduction de l’ARNm en protéine est également soumise à une régulation (Dever, 2002).
36
-5- Les ARN non-codants
Il existe de nombreux types différents d’ARN non-codants (ARNnc), présentant des fonctions
biologiques distinctes, parmi lesquelles on retrouve la régulation de l’expression des gènes
(figure 10).
Figure 10 : graphique représentants les différents types d’ARN possédant des fonctions biologiques distinctes.
Ils se répartissent principalement en 2 voies : les ARN codants et les ARN non-codants (http://www.snv.jussieu.fr/vie/dossiers/siRNA/index.htm).
Depuis le début du XXIème siècle, le nombre d’ARNnc connu a explosé, ils sont classés en
différentes catégories, selon leur fonction :
- Les ribozymes présentent la capacité de catalyser une réaction chimique.
- Les ARNt et ARNr sont impliqués dans la machinerie de synthèse des protéines.
- Les siARN (Small Interfering ARN) et miARN (microARN).
La technologie des siRNA (ou ARN interférence) a été découverte en 1990 par le
professeur Jorgensen qui, essayant de rendre des pétunias plus violets par introduction
d’un vecteur codant pour un pigment de cette plante, a obtenu des pétunias blancs (Napoli
et al., 1990). Le principe d’action des siARN est le suivant : des molécules d’ARN double-brin introduites dans une cellule, vont induire la dégradation spécifique de
l’ARNm homologue, inhibant ainsi l’expression des protéines correspondantes. La
découverte des siRNA a permis de dévoiler l’existence d’une voie naturelle de régulation
37
-En 1993, le groupe de V. Ambros décrivait lin-4, un gène de C. elegans ne codant pas
pour une protéine mais pour un ARN d’environ 21-23 nucléotides dont l’invalidation
affectait de façon très importante le développement du ver (Lee et al., 1993). Il s’agit du
premier miRNA découvert chez les eucaryotes.
Les principes d’action des siARN et des miARN sont très proches. Des molécules d’ARN,
précurseurs de grande taille (ou pri-miARN), sont transcrites à partir de leurs gènes
spécifiques localisés dans des introns et/ou exons (principalement) ou dans des régions
intergéniques. Leur maturation va impliquer au moins deux protéines apparentées à
l’ARNase III : Drosha et Dicer. Drosha convertit le pri-miARN en une structure en
épingle à cheveux irrégulière d’environ 70 nucléotides (ou pré-miARN). L’exportine 5
assure le transport du pré-miARN du nucléole vers le cytoplasme. Il est alors pris en
charge par Dicer qui permet la formation d’un duplex intermédiaire miARN:miARN, dont
un seul des deux brins formera le miARN mature (He and Hannon, 2004). Les miARN
simples brins vont ensuite interagir avec des protéines spécifiques pour former un
complexe ribonucléoprotéique stable nommé RISC-miRNP (RNA-induced silencing
complex-micro-ribonucleoprotein). Au sein de ce complexe, le miARN interagit avec des
ARNm par un jeu d’appariement de bases. Si la complémentarité d’appariement entre le
miARN et l’ARNm est parfaite, le complexe entraîne la dégradation de l’ARNm ciblé. Par
contre, si certains nucléotides restes non appariés dans le complexe miARN/ARNm
(complémentarité imparfaite), le RISC-miRNP entraîne alors l’inhibition de la traduction
de l’ARNm cible (figure 11). De tels défauts d’appariements ont généralement lieu dans
la région 3’UTR (He and Hannon, 2004).
Malgré leur petite taille, les miARN représentent un puissant système de contrôle de
l'expression des gènes. Les miARN sont impliqués dans de nombreux processus, dont le
développement (Farh et al., 2005), par exemple celui de l’épiderme (Yi et al., 2006), et
38
-2006). De plus, chaque tissu présente un profil d’expression unique de miARN, qui peut
être utilisé pour déterminer l’origine tissulaire d’une tumeur (Bloomston et al., 2007;
Zhang et al., 2008). On retrouve notamment la famille de miARN Let-7 dont les membres
sont présentés comme des inhibiteurs de la prolifération cellulaire, inhibant l'expression
du proto-oncogène RAS. Des dérégulations de l’expression de Let-7 ont été rapportées
dans de nombreux cancers (Jerome et al., 2007). Enfin, de nouvelles stratégies
thérapeutiques utilisant les miARN pourraient être utilisées dans le traitement des
infections virales dans les prochaines années (Sall et al., 2008). Les tables 2 et 3
répertorient des miARN connus dont la cible a été validée expérimentalement.
Figure 11: les miARN.
Petits ARN présentant un grand rôle dans la régulation génique. Principales étapes, de la synthèse du miARN jusqu’à son action sur l’ARNm cible (He and Hannon, 2004).
39 -Table 2
Table 3
Tables 2 et 3 : microARN et leurs cibles : exemples de microARN dont les fonctions/cibles
ont été validés expérimentalement.
Pour la Table 2 : AP2, APETALA2 ; Hid, head involution defective, Hoxb8, homeobox B8 ; PHB, PHABULOSA ; PHV, PHAVOLUA ; PTGS, post-transcriptional gene silencing ; REV, REVOLUTA; rld1 : rolled leaf1; SCL SCAPRCROW-LIKE; TCP, teosinte branched 1-cycloidea-PCF; d’après (He and Hannon, 2004).