UNIVERSITE TOULOUSE III - PAUL SABATIER
U.F.R. Sciences de la Vie et de la Terre
THESE
Pour obtenir le grade deDOCTEUR DE L’UNIVERSITE TOULOUSE III Discipline : Minéralogie expérimentale appliquée
Présentée et soutenue Par
Cédric LEBRE
Le 20 Décembre 2007
Elaboration et caractérisation de talcs
synthétiques pour l’amélioration des propriétés
physiques des matériaux composites industriels
(Revêtements de surface, plastique, peintures, …) incorporant du talc comme
charge minérale
_________
Directeur de thèse : Professeur François Martin
__________
Avant-propos
Ce mémoire ne serait pas ce qu’il est sans la collaboration, l’aide et le soutien de nombreuses personnes. Ces trois ans d’études ont été pour moi une expérience très enrichissante que ce soit au niveau du plan professionnel qu’au niveau des relations humaines.
Je tiens à remercier sincèrement François Martin, Professeur à l’Université Paul Sabatier de Toulouse, et Sabine Petit, chargée de recherche au laboratoire Hydr’Asa du CNRS à Poitiers, qui sont respectivement mon directeur et ma co-directrice de thèse. Ce sont deux personnes possédant de grandes connaissances dans le domaine des argiles qui ont bien voulu me transmettre une partie de leurs connaissances. Ils ont toujours été là pour me soutenir et m’aider dans ces travaux et ils m’ont permis d’intégrer le monde de la synthèse des minéraux argileux.
Je remercie également Jocelyne Ferret, Conseillé Spécial de la société Rio Tinto Minerals, qui m’a permis d’intégrer cette société et qui a tout fait pour que ça se fasse le mieux possible. Jocelyne m’a toujours soutenu et plus particulièrement lorsque le doute ou l’abattement s’installaient dans mon esprit. Ces conseils avisés et ses connaissances minéralogiques m’ont permis d’avancer dans les moments de stagnation.
Mes remerciements vont aussi à toutes les personnes de la société Rio Tinto Minerals qui ont contribué et soutenu le projet et plus particulièrement Didier Arseguel, Mike Greenhill-Hooper, Igan Hayati et Jean Sébastien Mas. Un grand merci aussi à tous les employés de la société qui ont facilité mon intégration dans la société.
Ces travaux n’auraient pas été ce qu’ils sont sans la présence de personnes très compétentes qui ont participé aussi à cette étude et plus particulièrement Eric Ferrage, Pierre Micoud, Emmanuel Gardes et Emmanuel Joussein.
Je n’oublierai pas de remercier toutes les personnes du laboratoire de Recherche et Développement de la société Rio Tinto Minerals : Sandrine Pejo Soucaille, Hélène Leroy, Nathalie Martinez, Catherine Maillard, Marie-Andrée Piquemal, Didier Galy, Gérard Martinez, Jean-Paul Massat, Yves Massat et Gilles Collard. Mais un grand merci à tous ceux qui ont quitté la société : Michèle Roudière, Nadine Lascassies, Lionel Truquet, Laurent Foucher et Laurent Cobigo. Une grande pensée pour une personne au grand cœur et courageuse qui nous a quitté : Serge Burato.
Sommaire
Avant-propos
5Introduction
11Chapitre I : Généralités sur les phyllosilicates
19I – Structure et chimie des phyllosilicates
211 - Le squelette de base des phyllosilicates 21
2 - La composition chimique 26
3 - Les structures réelles des phyllosilicates 27
A – Agencement des couches 27
B – Les ajustements structuraux 28
II – Le talc : phyllosilicate de type 2 : 1
311 - Structure du talc 31
2 - Les substitutions 34
3 - Solutions solides 35
A – Solutions solides tétraédriques 35
B – Solutions solides octaédriques 36
Chapitre II : Le protocole de synthèse hydrothermale
37I – Bibliographie de la synthèse hydrothermale
39Chapitre IV : Caractérisations des produits obtenus au cours
du protocole de synthèse hydrothermale
55
I – Le gel : précipité silicométallique
58II – Produit obtenu après le traitement hydrothermal
611 – Etude de l’influence du rapport solide/liquide introduit dans les réacteurs
61
A – Le protocole expérimental suivi 61
B – Etude de l’évolution de la cristallinité des produits synthétisés par diffraction des rayons X
62
C – Caractérisation des différents produits par analyse infrarouge
68
D – Discussion : essai d’interprétation 70
2 – Etude de l’influence de la durée de synthèse 71
A – Le protocole expérimental suivi 71
B – Etude de l’évolution de la cristallinité des produits synthétisés par diffraction des rayons X
72
C – Caractérisation des différents produits par analyse infrarouge
73
D – Discussion 78
3 – Etude de l’influence de la température de synthèse 79
A – Le protocole expérimental suivi 79
B – Etude de l’évolution de la cristallinité des produits synthétisés par diffraction des rayons X
80
C – Caractérisation des différents produits par analyse infrarouge
81
D – Caractérisation des différents produits par analyse thermogravimétrique
84
E – Discussion 86
III – Le talc synthétique nanométrique
871 – Analyses en diffraction des rayons X 87
2 – Analyses en proche infrarouge 88
3 – Analyses thermogravimétriques 90
4 – Observations au Microscope Electronique à Transmission 92
5 – Discussion 93
IV – Conclusion générale du chapitre
94Chapitre V : Synthèse des trois séries chimiques Ni,
Mg-Co et Ni-Mg-Co
I – Les trois séries chimiques : Mg-Ni, Mg-Co et Ni-Co
991 – Le protocole expérimental suivi pour la synthèse des trois séries 100
2 – Caractérisations de la série Mg-Ni 100
3 – Caractérisations de la série Mg-Co 119
4 – Caractérisations de la série Ni-Co 130
5 – Discussion 134
II – Un protocole de préparation du gel différent : influence sur
des produits synthétiques
136
1 - Si4Mg1.5Ni1.5 GM 136
2 - Si4Mg1.5Co1.5 GM 143
3 – Discussion 148
III – Conclusion générale du chapitre
149Chapitre VI : Le Nouveau Chapitre de Thèse : valorisation
des compétences
151
I – Cadre général, enjeux et présentation de la thèse
154II – Déroulement du projet
155III – Compétences techniques acquises
1561 – Méthodes analytiques 156
VI – Résultats et impacts de la thèse
161Chapitre VII : Conclusion générale : Apports des travaux et
perspectives
163
Bibliographie générale
171Annexes
179Liste des annexes 181
Annexe 1 :
PCT – ‘Préparation d’un interstratifié T.O.T. – T.O.T. gonflant’
183 Annexe 2 : PCT – ‘Procédé de préparation de composition talqueusecomprenant des particules minérales silico/germano-métalliques synthétiques’
231
Annexe 3 : PCT – ‘Procédé de préparation d’une composition de talc
synthétique à partir d’une composition de kérolites’
INTRODUCTION
Le talc est un minerai naturel utilisé dans le monde entier dans de nombreuses applications industrielles. Sa lamellarité est une des propriétés essentielles pour son utilisation comme charge minérale dans les domaines du papier, des polymères, des peintures, de l’alimentation animale et humaine, de la céramique, des engrais, des cosmétiques et produits pharmaceutiques et des caoutchoucs.
Une des dernières applications intégrant du talc comme charge minérale dans son process de fabrication se situe dans le secteur de l’aéronautique. Les revêtements de surface de certaines pièces aéronautiques étaient conçus avec du cadmium. Cet élément chimique étant cancérigène, une directive européenne a rendu son utilisation restreinte. Ces revêtements au Cd ont donc été remplacés par des revêtements de surface Zinc-Nickel (ZnNi) ou Nickel-Phosphore (NiP). Dans l’idée de rendre lubrifiant ces revêtements, des particules de talc ont été incorporées dans ces revêtements de surface, ce qui permet de diminuer l’usure lorsque deux pièces mécaniques sont en contact (figure 1). Cette étude, menée en commun par le LMTG et le Cirimat-Institut Carnot (Université Paul Sabatier, Toulouse III) en collaboration avec les sociétés Mécaprotec et Rio Tinto Minerals a fait l’objet de dépôts de brevets industriels (brevets industriels français, 2002, Martin et al et Brevets industriels internationaux, 2004, Martin et al.).
Il fallait donc que les particules de talc soient de tailles identiques et les plus petites possibles. De nombreuses études de broyage ont été réalisées par la société Luzenac Europe afin de diminuer la taille de ces particules. La meilleure taille des particules obtenue est voisine du µm. Une autre société, Nanomat, a essayé de broyer ce talc de manière intensive pour arriver à fabriquer des particules « nanométriques ». L’objectif n’a pas été atteint, une amorphisation du produit a été constatée, rendant ce produit sans grand intérêt, le talc perdant sa lamellarité. La société Nanomat ayant par la suite disparue.
L’idée était donc d’obtenir un talc de taille nanométrique avec une distribution granulométrique resserrée et pure pour plus de stabilité dans les bains électrolytiques (pas de minéraux associés). Les travaux ont débuté au cours de mon DESS Matériaux (UPS) avec trois objectifs majeurs : connaître la faisabilité du protocole de synthèse hydrothermale, protocole adapté des diverses études de synthèses de talc antérieures, connaître la qualité des talcs obtenus (lamellarité, cristallinité, …) et connaître la taille des particules obtenues. Les résultats ont montré que la synthèse hydrothermale de talc permet d’obtenir des tailles de particules de quelques nanomètres à quelques centaines de nanomètres, que les talcs synthétiques obtenus ont les mêmes caractéristiques physiques que les talcs naturels que le protocole de synthèse pouvait être étendu à un pilote semi-industriel.
C’est à la suite de ces travaux que j’ai réalisé une thèse (bourse CIFRE) ayant comme objectifs majeurs :
- l’optimisation un protocole de synthèse pour le rendre viable industriellement. Cette optimisation est passée par l’étude de l’influence des trois paramètres importants de la synthèse hydrothermale (température, durée et rapport solide/liquide introduit dans les réacteurs). Il fallait pouvoir disposer de talcs synthétiques de taille nanométrique, avec une granulométrie resserrée et en grande quantité (quelques centaines de g à quelques kg contrairement aux 300 mg obtenus au cours des travaux précédents.
- la mise au point d’un pilote semi-industriel de production de talc synthétique dans les conditions fixées par ce protocole,
- de juger les potentialités de ces talcs dans les matériaux composites : revêtement de surface, polymères, peintures, cosmétiques …
- d’étendre ce protocole de synthèse à des « talcs » ou le Mg des sites octaédriques est substitué par du Co, Ni, les talcs ainsi obtenus seront caractérisés et un intérêt sera porté aux propriétés physiques de ces talcs.
Ce mémoire contient les données et études menées au cours de ces trois années, Certains travaux effectués au cours de cette thèse ne sont pas décrits dans un souci de confidentialité.
Le premier chapitre de ce mémoire contient des rappels généraux sur la structure des phyllosilicates et en particulier du talc.
Le deuxième chapitre présente une brève bibliographie sur la synthèse hydrothermale suivie des premières pages des trois brevets déposés qui décrivent le protocole de synthèse établi dans ces travaux (cf. Annexes 1, 2 et 3).
Le troisième chapitre décrit les méthodes de caractérisation utilisées au cours de cette étude.
Le quatrième chapitre traite des différents produits synthétiques obtenus au cours du protocole, du gel silicomagnésien de départ jusqu’au talc synthétique, en étudiant les paramètres influants de la synthèse hydrothermale : rapport solide/liquide introduit dans les réacteurs, durée et température de synthèse.
Dans le cinquième chapitre, nous montrons l’existence de véritables solutions solides par substitution d’une partie ou de la totalité du Mg octaédrique par du Co et/ou du Ni et étudions les arrangements structuraux de ces « talcs » synthétiques.
Le sixième chapitre s’attache à décrire mes compétences personnelles et techniques acquises au cours de ces travaux de thèse en exposant le Nouveau Chapitre de Thèse : valorisation des compétences.
Pour terminer, une conclusion fait le bilan de ces travaux, les prospectives à venir seront présentées et sera suivie de la bibliographie et des annexes.
L’ARNT m’a accordé un financement pour réaliser cette étude au sein de la société Rio Tinto Minerals.
La société Rio Tinto Minerals appartient au groupe minier anglo-australien Rio Tinto. Le groupe possède et exploite des gisements de tout type pour couvrir une large gamme d’exploitation des ressources naturelles dont l’aluminium, le cuivre, l’argent, l’or, le nickel, le fer, le plomb, la bauxite, le zinc et l’uranium. Les minéraux exploités et commercialisés sont le talc, les borates, le sel, le zircon et les diamants. Les gisements sont répartis à travers le monde avec des usines en Europe, Amérique du Nord, Amérique du Sud, Asie, Australie et en Afrique. Multinationale employant 36000 salariés, le siège social est basé à Londres.
La société dans laquelle j’ai réalisé ces trois années de travaux a été restructurée en Février 2006 pour devenir une nouvelle entité du groupe Rio Tinto, Rio Tinto Minerals, anciennement Luzenac Europe (société d’exploitation du talc) regroupant les opérations talc, borate et sel. Cette entité du groupe emploie 3000 salariés, couvre les cinq continents et est le leader mondial dans l’exploitation et la commercialisation du talc (qualité, support technique, …).
Talc de Luzenac a célébré en 2005 les 100 ans d’exploitation de la carrière de talc de Trimouns dans les Pyrénées ariégeoises. Cette carrière de talc à ciel ouvert se situe à 1700 mètres d’altitude (figure 2) et est actuellement la plus grande carrière d’exploitation de talc au monde. Chaque année, 410 000 tonnes de talc sont extraites de ce gisement ce qui correspond à 7% de la production mondiale et 30% de la production de la société. Les réserves de ce gisement sont estimées à une centaine d’années d’exploitation.
Figure 2 : Photographie de la carrière de talc de Trimouns en Ariège
Rio Tinto Minerals possède d’autres carrières de talc en Europe (figure 3), sur le continent Nord Américain et en Australie. A l’heure actuelle, 11 gisements sont exploités à travers le monde et 19 usines de broyage en activité. On estime à environ 5.3 millions de tonnes la production mondiale de talc chaque année provenant de 250 gisements répartis à travers le monde, toutes sociétés confondues. Rio Tinto Minerals en vend près de 1.4 millions de tonnes par an. Cela représente 28% de la production totale, ce qui lui donne le rang de premier producteur mondial de talc.
J’ai donc était employé dans Rio Tinto Minerals au sein de son service Recherche et Développement d’Europe en tant que professionnel de l’innovation (dénomination de mon poste). Travaillant dans le domaine du talc, je m’attacherai à ne présenter uniquement que les applications industrielles de ce minéral.
Figure 3 : Rio Tinto Minerals en Europe
Les propriétés du talc, aussi bien structurales que chimiques, lui confèrent une utilisation dans de nombreux domaines industriels.
Les applications industrielles se servent en fait de ces nombreuses caractéristiques. En tout premier lieu, la première qualité du talc est sa grande inertie et stabilité chimique, ce qui permet de le mélanger à la plupart des produits organiques et minéraux. Le talc résiste à des attaques chimiques fortes et à des températures avoisinant les 900°C. Sa structure en feuillet, induisant une orientation préférentielle (plan (a, b)) lui confère une de ces propriétés les plus connue : l’onctuosité, la douceur. En effet, les feuillets sont reliés entre eux par des forces de Van der Walls, ce qui permet aux feuillets de glisser les uns sur les autres, conférant ainsi au talc ce
! ! "" # # $ $##!! $ $##!! % % &&''((
Figure 4 : Répartition des % de talc commercialisé par secteur industriel
Ces applications industrielles sont les suivantes : - Papier
L’utilisation du talc en papeterie est due à sa forme lamellaire, son affinité avec la cellulose et sa lipophilie. La charge minérale dans un papier oscille autour de 20%, et donne des qualités indispensables pour l’impression et l ‘écriture, évite les déformations du papier lors des variations d’humidité et surtout abaisse le prix de revient de la feuille. De plus le talc confère au papier une couleur blanchâtre avec des tendances bleu-vert, une excellente rétention sur machine, et une conservation des propriétés mécaniques de la cellulose. Enfin, une certaine catégorie de talc (granulométrie) permet d’empêcher les résines naturelles du bois, libérées lors de la fabrication de la pâte ou du papier, de s’agglomérer sous forme de masses poisseuses, occasionnant des défauts dans le papier et l’obturation des canalisations. N’oublions pas le rôle important du talc aussi pour les papiers couchés.
- Plastiques
Employé en raison de sa lipophilie et de sa lamellarité, le talc améliore les propriétés mécaniques et notamment la rigidité ainsi que la résistance mécanique des polypropylènes, permettant ainsi la réalisation de pièces techniques tels que les pare-chocs et tableaux de bord de voiture, les carcasses de machine à laver, les emballages alimentaires thermoformables. A faible taux, il résout des problèmes de blocking et facilite l’extrusion des films de PEhd et à forte teneur, il améliore les propriétés mécaniques du PVC ou des PEhd.
- Peintures
C’est son inertie chimique, sa lamellarité et sa lipophilie qui en font un élément essentiel des peintures (entre 1 et 5%). Il permet de modifier les propriétés d’écoulement et facilite
l’application, d’améliorer la résistance aux intempéries et à la corrosion, de renforcer le pouvoir couvrant en aidant à la dispersion des pigments, d’obtenir des effets satinés ou mats.
- Alimentation animale et humaine
Employé pour sa lamellarité, son onctuosité et son pouvoir lubrifiant ainsi que sa lipophilie, le talc joue le rôle d’anticollant dans l’alimentation humaine (confiserie, riz, etc…) ainsi que dans l’alimentation animale où il permet l’écoulement des tourteaux de soja, l’antimottage des granulés à forte teneur en mélasse, et un meilleur passage en filière.
- Céramiques
C’est la seule utilisation ou le minéral talc est détruit par cuisson et se transforme en proto-enstatite. Il est alors utilisé dans les faïences (entre 5 et 45%), leur conférant une forte dilatation thermique, supprimant la rupture de l’émail. Aussi usité dans des porcelaines électrotechniques (stéatites) très solides et isolantes, son taux d’incorporation peut avoisiner les 90%. Incorporé à haute température avec des argiles, il forme la cordiérite, composé à bas coefficient de dilatation, composant des pièces réfractaires résistantes aux brusques variations de températures (support de cuisson, résistances électriques, pots catalytiques). Une autre utilisation du talc est le renforcement de l’action fondante que ce dernier a sur les feldspaths dans les grès en permettant de baisser les températures de cuisson (avec 4% de talc).
- Engrais
Utilisé en raison de son hydrophobie, de son onctuosité, de sa forme lamellaire et de son inertie chimique, il permet l’enrobage des engrais après pulvérisation d’essence et d’amine. Ceci empêche la prise en masse et facilite la manutention et l’épandage. Il dilue en outre les ammonitrates ce qui écarte le danger d’explosion en abaissant la concentration en azote.
- Cosmétique et produits pharmaceutiques
Son onctuosité et son inertie chimique ont été les meilleurs supports publicitaires pour l’utilisation du talc (poudre de bébés, laits de toilette). Il permet pour les parfums une meilleure rémanence sur support poudre car il possède une bonne rétention d’odeur. En pharmacie, le talc est utilisé comme excipient, car étant inerte chimiquement, il n’altère pas les matières actives et ne perturbe pas les fonctions biologiques. Il limite l’usure des moules de fabrication lors du pressage des comprimés.
CHAPITRE I
Généralités sur les
phyllosilicates
I – Structure et chimie des phyllosilicates
1- Le squelette de base des phyllosilicates
La structure de base des silicates en feuillets, appelés aussi phyllosilicates, a été définie (Bailey et al. 1971b) comme étant "une couche continue bidimensionnelle de tétraèdres" ayant une composition de la forme Si2O5 dans laquelle chaque tétraèdre est relié à ses voisins en
partageant trois de ses sommets. Ces oxygènes partagés des tétraèdres forment un plan basal d'oxygènes de symétrie hexagonale (Figure 1).
Figure 1 : Agencement des tétraèdres SiO4 formant un hexagone représenté
proche de celui d'un tétraèdre isolé. Il en est de même de la longueur de la liaison Si-O qui dans les deux cas est proche de 1,62 Å (Figure 2).
Figure 2 : Distance Si-O et angle O-Si-O dans un tétraèdre SiO4.
Tous les oxygènes apicaux des anneaux de tétraèdres d'une même couche sont aussi coplanaires. Tout ce qui est compris entre le plan basal d'oxygènes et les oxygènes apicaux est nommé la couche tétraédrique.
L'autre élément structural principal des phyllosilicates est la couche octaédrique qui contient des cations en coordination YO6 entre deux plans d'atomes d'oxygènes. La couche
octaédrique est construite à partir d'octaèdres qui sont reliés les uns aux autres, c'est-à-dire que chaque cation Y partage deux de ses atomes d'oxygènes avec un cation Y d'un octaèdre voisin (Figure 3).
Il y a deux façons de réaliser cette liaison. Si tous les sites octaédriques sont occupés, chacun partage deux atomes d'oxygènes avec six cations voisins. Si deux sites octaédriques sur trois sont occupés, chaque cation Y partage deux atomes d'oxygènes avec seulement trois voisins Y. Lorsque tous les sites octaédriques sont occupés, la couche est dite trioctaédrique alors qu'elle est nommée dioctaédrique quand deux sites sur trois sont occupés (Figure 4).
Figure 4 : Représentation polyhédrale d’un feuillet trioctaédrique (a) et dioctaédrique (b)
Les groupes anioniques coordinateurs du cation Y sont soit (OH)6, soit (OH)4O2 ou soit
(OH)2O4. Ceci est fonction de la classe structurale des phyllosilicates et du nombre de cations
octaédriques 4 ou 6 :
-si tous les groupes anioniques sont des groupes hydroxyles OH, la couche est complète et apparaît seule, alternant avec les feuillets silicatés comme dans les chlorites ou bien comme une entité structurale unique dans les hydroxydes comme la brucite Mg(OH)2 ou la gibbsite Al(OH)3
Figure 5 : Représentation schématique de la forme des octaèdres en coordinence 6 pour la brucite et la gibbsite
- la couche octaédrique est de la forme (OH)4O2 et apparaît dans des minéraux tels que la
kaolinite ou les serpentines. Le plan sommital est entièrement composé d’OH, l'autre étant composé d' (OH)2O2. Chaque feuillet de ce groupe contient donc une couche tétraédrique et une
couche octaédrique, le tout étant appelé feuillet 1:1 (Figure 6).Les phyllosilicates de cette famille sont communément appelés TO (une couche tétraédrique et une couche octaédrique).
-la couche octaédrique est de la forme (OH)2O4 et apparaît dans les micas, vermiculites,
smectites, pyrophyllites et talcs. Deux couches tétraédriques encadrent une couche octaédrique. Les minéraux de cette forme appartiennent à la famille des phyllosilicates 2:1 (Figure 6).
En fait, les atomes d'oxygène du plan supérieur de la couche octaédrique sont déplacés de a/3 par rapport à ceux du plan inférieur. Les anneaux Si2O6 de la couche supérieure de tétraèdres sont
Figure 6 : Agencement des couches tétraédriques et octaédriques ; a : vue perpendiculaire à c* d’une couche idéale de tétraèdres ; b : couche octaédrique avec les groupes hydroxylés représentés en noir ; c : représentation schématique des feuillets 1 :1 et 2 :1 parallèle à c*.
Figure 7: Matérialisation du shift existant entre la couche tétraédrique et la couche octaédrique.
2- La composition chimique
A partir de ces considérations structurales, il est possible de représenter ces silicates en feuillet par la composition ionique de leurs tétraèdres et de leurs octaèdres. Les constitutions chimiques des phyllosilicates sont complexes mais peuvent être décrites de façon simple à partir des feuillets 1:1 et 2:1. Dans la famille des 2:1, le talc, minéral argileux de cette étude, est un phyllosilicate trioctaédrique, sa formule est Si8IVMg6VIO20(OH)4 et le nombre d'ions
dans chaque plan est :
Eléments constitutifs
Charges négatives
Charges positives
6 O 12 4 Si 16 4 O + 2 OH 10 6 Mg 12 4 O + 2 OH 10 4 Si 16 6 O 12 Total : 44 44
Le feuillet est électrostatiquement neutre. Tout remplacement d’un cation (ou d’un anion) par un élément de valence différente provoque une rupture de cet équilibre électrique, qui peut soit se pérenniser ou soit s’annihiler par d’autres substitutions ou lacunes. Pour des excès de charges négatives, la neutralisation est réalisée dans l'espace interfoliaire par des cations, des cations hydratés, ou des groupements hydroxydes de type brucitique par exemple.
Plusieurs possibilités de déficit de charge existent :
- substitution en site tétraédrique d’un atome tétravalent par un atome trivalent ou divalent.
- substitution en site octaédrique d'un cation trivalent par un cation divalent ou d'un cation divalent par un cation monovalent.
- présence d’une lacune octaédrique. - déshydroxylation.
Le déficit de charge peut être entièrement d'origine tétraédrique ou d'origine octaédrique mais il peut aussi provenir de substitutions hétérovalentes simultanées dans les deux couches. Ces déficits de charge sont compensés par des charges positives interfoliaires conférant ainsi à l'unité structurale une charge électrostatiquement neutre.
3- Les structures réelles des phyllosilicates
A – Agencement des couches
La description générale des classes structurales des minéraux silicatés en feuillets est basée sur une simplification géométrique qui mentionne une superposition exacte des couches tétraédriques et octaédriques au niveau des oxygènes apicaux.
Cependant, dans la plupart des minéraux, les atomes d'oxygène sont décalés de cet arrangement idéal (Figure 7). La raison de ces déplacements est due aux différences de dimensions latérales entre les couches tétraédriques et les couches octaédriques.
Les contraintes qu'elles doivent minimiser afin de s'agencer dépendent donc de la différence entre ces dimensions latérales. Ceci induit des effets sur la taille, la morphologie et la structure des minéraux argileux. Le degré relatif d'ajustement entre les deux couches peut être estimé en déterminant les dimensions latérales des couches octaédriques et tétraédriques lorsque
Ces ajustements structuraux sont de plusieurs types. B – Les ajustements structuraux
α) Rotations tétraédriques
Dans la grande majorité des argiles, les dimensions latérales de la couche tétraédrique sont plus importantes que celles de la couche octaédrique. Bradley (1940), Mathieson et Walker (1954), Newnham et Brindley (1956), Zvyagin (1957), et Radoslovitch (1961 et 1962) ont montré qu'il était relativement aisé pour la couche tétraédrique de réduire ses dimensions latérales par une rotation de tétraèdres adjacents dans le plan (001) (Figure 8). Ces auteurs ont proposé une relation (1) qui permet de quantifier la valeur (α) de l'angle de rotation nécessaire à l'ajustement.
cos
αααα = b
obs./ b
idéal(1)
Cette expression est une approximation car il est très difficile d'estimer le b idéal. Les rotations possibles sont comprises entre α = 10 ° et 30 °, angle limite de conservation des anneaux de six tétraèdres.
Figure 8 : Twist (α) des tétraèdres dans le plan (a, b)
Le sens de rotation des tétraèdres est gouverné par l'attraction des oxygènes basaux avec un ou plusieurs des trois groupes atomiques voisins :
- les hydrogènes liés aux OH adjacents ou les H2O de surface,
- les cations octaédriques à l'intérieur du même feuillet,
- les cations octaédriques d'autres feuillets ou des cations interfoliaires.
Dans les micas, les talcs et les pyrophyllites, seuls les cations octaédriques du même feuillet jouent un rôle alors que dans les autres feuillets 1: 1 et 2: 1, ces trois facteurs sont
importants. Ces rotations induisent donc une réduction de la symétrie de l'ensemble des anneaux de tétraèdres d'hexagonale à ditrigonale (Martin et al., 1996).
β) Autres ajustements
L'élasticité des couches tétraédriques et octaédriques permet par un épaississement de la couche tétraédrique, un amincissement de la couche octaédrique et ainsi de réduire l'écart des dimensions latérales entre ces deux couches. Au contraire, les couches octaédriques tendent à augmenter leurs dimensions latérales. Dans les feuillets dioctaédriques ou deux sites sur trois sont occupés, les cations trivalents repoussent les autres cations trivalents alors que les deux atomes d'oxygènes qui se partagent entre les cations adjacents, se rapprochent, raccourcissant donc le coté commun à deux octaèdres et allongeant les côtés non partagés. Ainsi deux types d'octaèdres apparaissent, un large inoccupé et deux petits possédant les cations. Ceci provoque un amincissement du feuillet et une légère augmentation des dimensions latérales. Ces ajustements ont un rôle mineur par rapport aux rotations tétraédriques. Dans les minéraux trioctaédriques, la répulsion des cations est identique dans les trois directions à 120° du réseau pseudo hexagonal. Mais les atomes d'oxygènes ne peuvent bouger qu'à l'intérieur des octaèdres perpendiculairement au plan basal et non le long des cotés communs. Par conséquent, l'aplatissement est plus petit et l'on obtient seulement 2% au contraire des 10% dans les dioctaédriques. En revanche, l'extension latérale est importante. Non seulement les variations de la symétrie idéale de la couche octaédrique permettent aussi bien des variations des dimensions latérales que des rotations des tétraèdres, mais aussi du paramètre b. Ce dernier est lui même influencé par la composition et la longueur des liaisons cation-oxygène de la couche octaédrique ainsi que de la composition de la couche tétraédrique (Figure 7).
Lorsque les substitutions en site tétraédrique se font par des cations de tailles différentes, les disparités de taille des couches ne peuvent pas s'estomper par le seul fait des rotations tétraédriques mais elles se font par un tilt des tétraèdres impliquant une non-coplanairité des oxygènes basaux (Figure 9).
La surface basale d'oxygènes prend un aspect « ondulé » dans laquelle deux oxygènes basaux sont légèrement redressés par rapport au plan basal et le troisième plonge vers la couche octaédrique (Martin et al., 1996)
.
II – Le talc : phyllosilicate de type 2 :1
1- Structure du talc
Le talc, silicate magnésien hydraté, de formule Mg3Si4O10(OH)2 est le pôle le plus simple
des phyllosilicates 2:1. Dans le cas idéal, les feuillets 2:1 sont neutres et donc aucun élément n'est présent dans l'espace interfoliaire. La figure 10 représente un feuillet « idéal » de talc.
Figure 10 : schéma d’un feuillet de talc (3D)
Ceci a engendré de nombreux travaux sur sa structure (Gruner, 1934 ; Hendricks, 1938; Stemple & Brindley, 1960 ; Ross et al., 1968 ; Russel et al., 1970 ; Daw et al., 1971 ; Bricker et
al., 1973 ; Rayner & Brown, 1973 ; Lindemann et al., 1975 ; Akizuki & Zussman, 1978 ; Giese,
1979 ; Ewans & Guggenheim, 1984 ; Wesolowski, 1984 ; Bleam, 1990). Gruner (1934) décrit pour la première fois le talc comme monoclinique et identique à la pyrophyllite. Après la confirmation de cette structure par Hendricks (1938) et Stemple & Brindley (1960), Ross et al. (1968) et Rayner & Brown (1973) montrent que le talc a une structure triclinique avec les paramètres suivant :
Plus récemment, Perdikatsis & Burzlaff (1981) par des affinements de structure ont montré que le talc avait une maille élémentaire pseudomonoclicique C-centrée avec un groupe d'espace P-1 (figures 12 et 13). Ils ont pu déterminer ainsi les paramètres de la maille qui sont :
a = 5,291Å b = 9,46Å c = 5,29Å α = 98,68° β = 119,9° γ = 85,27°
Une représentation en projection suivant le plan (010) pour un talc P-1 est mentionnée sur la figure 14.
Figure 14 : Représentation polyhédrale du talc P-1 suivant le plan (001)
2- Les substitutions
La plupart des talcs naturels sont proches de la composition idéale bien que des auteurs reportent des fortes teneurs en Fe, des teneurs faibles en Al et F, et des traces en Mn, Ti, Cr, Ni, Ca, Na et K (Chidester, 1962; Moine et al., 1962; Hemley et al., 1977; Noack et al., 1986 ; Martin et al., 1999 ; Petit et al., 2004 a et b). Des synthèses de talcs ont permis d'estimer les taux maxima de divers éléments pouvant être incorporésdans la structure : Al à 4% (Mc Kie, 1959; Yoder, 1952; Stemple & Brindley, 1960; Fawcett, 1962; Fawcett & Yoder, 1966) et F à 4,47% (Abercrombie et al., 1987). Le Fe3+ apparaît dans les talcs en substitutions tétraédriques en faible teneur (Forbes, 1969) et en site octaédrique pour Noack et al. (1986). Si le Fe3+ est très minoritaire, il en est différent pour le Fe2+ avec lequel on note un véritable équivalent du talc qui est la minnésotaite. De nombreux auteurs se sont intéressés à la possibilité d'existence d'une solution solide talc-minnésotaite (Gruner, 1934; Floran & Papike, 1978; Kager & Oen, 1983). Des études récentes (Guggenheim & Bailey, 1982; Gugenheim & Eggleton, 1986) montrent que la minnésotaite n'est pas l'équivalent ferreux du talc. Néanmoins Forbes (1969 et 1971) a obtenu une série chimique entre les talcs Mg3Si4O10(OH)2 et Fe3Si4O10(OH)2 mais ces produits ont été
réalisés à des températures de l'ordre de 600° C et des pressions d'environ 1000 bars. La willemseite est décrite comme un talc ayant du nickel en sites octaédriques (De Waal, 1970; Stubican & Roy, 1961; Wilkins & Ito, 1967). Des produits de synthèses (Stubican & Roy, 1961) sont mentionnés avec de fortes teneurs en Co (Mg Co ), Zn (Mg Zn ), Fe (Mg Fe ), Mn
(Mg92Mn8) et Cu (Mg55Cu45). Ces mêmes auteurs laissent apparaître la possibilité d'obtenir un
équivalent germanifère du talc Mg3Ge4O10(OH)2 mais sous des conditions de température et de
pression énorme (T > 500°C et P > 600 atm.). Des talcs Ni3Ge4O10(OH)2 ont été obtenus sous des
pressions supérieures à 1500 atm. Un talc Mg-Ge a été réalisé à une température de 670° C et une pression de 690 bars (Lyon & Ehlers, 1970). Récemment, Martin (1994), Martin et al., (1999) ; Petit et al., (2004 a et b) ont montré de très nombreuses substitutions dans des talcs naturels et les talcs synthétiques.
Ceci conduit à une formule structurale générale pour les argiles de type 2 :1 de la forme :
(R4+4-xR3+x)(R3+2-2a/3R2+a) O10 (OHy,F1-y)2 (A2+β A+α-2β)
avec R4+ = Si, Ge, … R3+ = Al, Fe, Ga, Cr, … R2+= Mg, Fe, Mn, Ni, Co, … A2+ = Mg, Ca, Cu, … A+ = K, Na, …
Il va de soit que la teneur en cations interfoliaires est par définition nulle dans le cas d’un talc. Néanmoins, il existe un nombre infini de combinaisons entre ces différents éléments et un nombre infini de teneurs possibles. Toutefois selon la règle de Goldschmidt, une différence de plus de 15% entre les rayons ioniques des cations interdit toute possibilité de solution solide étendue et donc de substitutions (dans le cas des substitutions homovalentes). Néanmoins, cette valeur peut être légèrement dépassée (quelques %) si les températures de formation sont importantes (effet Debye-Waller). Dans le cas des substitutions hétérovalentes, plusieurs processus peuvent se produire et notamment une compensation de charge afin que la neutralité du feuillet soit respectée, ceci permettant d’avoir une gamme relativement étendue de possibilités de substitution (Vedder, 1964 ; Hazen & Wones, 1972 ; Monier & Robert, 1986 a et b ; Robert et
al., 1993 ; Martin et al., 1999 ; Petit et al., 2004 a et b).
Ces notions de substitutions aussi bien tétraédriques qu’octaédriques qui peuvent apparemment aller d’un pôle à l’autre renvoient tout de suite à la notion de solution solide. Des exemples vont être pris pour matérialiser les deux types de solutions solides possibles dans les talcs : tétraédriques et octaédriques.
3- Solutions solides
A- Solution solide tétraédrique
Ce type de solution solide est invoqué pour expliquer la substitution de Si par Al, en couche tétraédrique. Le silicium n'est pas uniquement substituable par l'aluminium, mais il peut
B- Solutions solides octaédriques
Ce type de solution solide fait intervenir des substitutions cationiques en couche octaédrique.
En ce qui concerne les substitutions isomorphiques, les solutions solides octaédriques Mg-Métaux de transition (Ni, Co, Zn, Fe2+, Cu2+...), dans les minéraux T.O.T trioctaédriques ont fait l'objet de nombreuses études. Ainsi, pour des kérolites naturelles, la solution solide Mg-Ni est continue et complète (Manceau et al., 1985). Des travaux sur des phyllosilicates trioctaédriques de synthèse, confirment que la solution solide Mg-Ni est continue, quelle que soit la température de formation du minéral et sa nature, talc, kérolite ou stévensite (Wilkins & Ito, 1967 ; Mondésir, 1987). Enfin, la solution solide Ni-Co, dans des kérolites synthétisées à 150°C, est continue et complète (Grauby et al., 1991). Dans le cas de substitutions hétérovalentes, uniquement dans le cas des minéraux trioctaédriques, le taux maximal de substitution de R2+ par R3+ est de 33% (Foster, 1960 a, b et c). Pour ces taux de substitutions, le terme de minéraux di-trioctaédriques est employé. Pour les talcs, les taux de substitutions hétérovalentes sont très faibles (1 à 2%), et l’on peut considérer que la structure talc est conservée cristallographiquement (Martin et al., 1999 ; Petit et al., 2004 a et b).
CHAPITRE II
I – Bibliographie de la synthèse hydrothermale
De nombreux travaux ont eu pour sujet la synthèse hydrothermale de talc depuis plus d’un demi-siècle. Les premières synthèses de talc ont été réalisées par Bowen et Tuttle, en 1949, à une température de 490 °C. D’autres travaux ont conduit à la synthèse de talc par la suite avec les études de Yoder (1952), Greenwood (1963) et Whitney et Eberl (1982). L’ensemble de ces travaux a porté sur la synthèse hydrothermale de talc au sens strict du terme, avec du magnésium au sein des sites octaédriques. Mondésir (1987) met en évidence les transitions « stévensite en kérolite » et « kérolite en talc » en fonction de la température de synthèse. Le talc est synthétisé à partir de 220°C.
Une série chimique entre les « talcs » Mg3Si4O10(OH)2 et Fe3Si4O10(OH)2 aété obtenue
mais ces produits ont été synthétisés à des températures de l’ordre de 600°C et des pressions d’environ 1000 bars (Forbes, 1969). De même, des produits de synthèse ont été mentionnés (Stubican & Roy, 1961) avec des fortes teneurs en Co(Mg54Co46), Zn(Mg86Zn14), Fe(Mg52Fe48) et
Cu(Mg55Cu45). Ces mêmes auteurs laissent apparaître la possibilité d’obtenir un équivalent
germanifère du talc Ge4Mg3O10(OH)2 mais sous des conditions de température et de pression
élevées (T > 500 °C et P > 600 atm). Des talcs Ge4Ni3O10(OH)2 ont été synthétisés sous des
pressions supérieures à 1500 atm. Un « talc » Mg-Ge a été réalisé à une température de 670°C et une pression de 690 bars (Lyon & Ehlers, 1970). Wilkins & Ito (1967) ont réussi à synthétiser et à étudier des talcs avec divers cations octaédriques autre que Mg comme Ni2+ , Co2+ , Mn2+ et Zn2+.
Plus récemment, Martin et al. (1994, 1999, 2002, 2003 et 2004) ont montré l’existence de véritables solutions solides complètes et continues entre M2+3Ge4O10(OH)2 et M2+3Si4O10(OH)2
(M2+ = Co, Ni ou et/ou Mg) pour des températures de synthèse hydrothermale de 220°C et des pressions de vapeur d’eau de 16 bars, montrant ainsi qu’à basse température, la structure talc pouvait incorporer aussi bien dans la couche tétraédrique que dans la couche octaédrique des éléments autre que Si et Mg respectivement. Les synthèses qui ont été effectuées dans le cadre de cette thèse ont été faites en prenant pour base le protocole de synthèse mis au point par Martin (1994) pour la synthèse de talcs germaniféres.
II – Brevets internationaux déposés (PCT)
Cette étude a eu pour premier objectif d’adapter le protocole de synthèse mis au point par Martin, qui est un processus à basse température et basse pression, pour une future application
CHAPITRE III
Les différentes méthodes de caractérisation sont abordées dans les parties suivantes sans détailler chaque méthode de manière exhaustive. La description de ces différentes méthodes d’analyses est adaptée au minéral de cette étude : le talc synthétique.
I – La diffraction des rayons X appliquée aux phyllosilicates
La diffraction des rayons X est un phénomène de diffusion cohérente et élastique qui se produit lorsque les rayons X interagissent avec la matière cristallisée. Le rayonnement de la longueur d’onde des rayons X (0.1< < 10 nm) pénètre le cristal. Une partie de l’énergie est absorbée et l’autre partie excite les atomes de la structure. Les radiations émises partent dans toutes les directions de l’espace.
Les phyllosilicates, minéraux en feuillets, émettent des radiations par des plans atomiques qui sont parallèles. Ces plans émettent donc un faisceau cohérent diffracté dont la direction est définie par la loi de Bragg : n = 2dsin (hkl)
où
n : nombre entier correspondant à l’ordre de la diffraction : longueur d’onde du rayonnement utilisé
d : distance réticulaire en : angle de diffraction
Dans le cadre de cette étude, nous avons synthétisés des minéraux argileux dont la taille de chaque particule n’excède pas le µmètre (groupe talc-stévensite-kérolite). La méthode utilisée pour caractériser ces associations minéralogiques argileuses est la diffraction des rayons X. Chaque particule est un empilement de plusieurs feuillets. Ces feuillets émettent un ensemble de raies correspondant à des réflexions de Bragg (hkl) (figure 1).
Tableau 1 : tableau de diagnose (Eslinger & Peaver, 1988)
La détermination de la position d(001) de la raie de réflexion du plan (001) ainsi que la symétrie ou l’asymétrie de cette raie est la méthode classiquement utilisée pour exploiter les diffractogrammes sur lame orientée et déterminer la nature des argiles présentes dans l’échantillon.
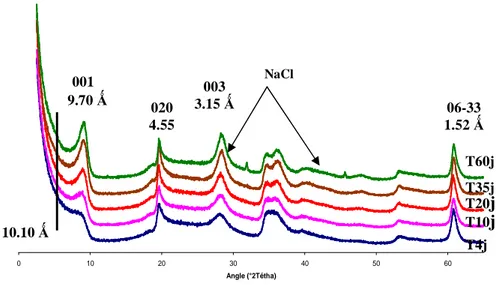
Cette raie (001), caractéristique des plans basaux des minéraux argileux, voit sa position et sa symétrie se modifier selon la nature des empilements des feuillets présents et l’état d’hydratation des produits. Le talc, objet de cette étude, est un empilement ordonné des feuillets. Les feuillets successifs sont translatés les uns par rapport aux autres d’une valeur constante. Le diffractogramme résultant est composé d’une série de raies symétriques correspondant aux différentes raies (hkl) (figure 2). La distance réticulaire pour le plan (001) du talc est 9.35 . Les trois autres raies du talc naturel prises en compte pour le caractériser sont les suivantes :
- Raie à 4.55 pour le plan (020) - Raie à 3.14 Å pour le plan (003)
- Raie à 1.52 Å pour les plans (060) et (330)
0 500 1000 1500 2000 2500 0 10 20 30 40 50 60 Angle (°2Tétha) In te n s it é ( c o u n ts /s ) CuK 1
Figure 2 : Diffractogramme de poudre d’un talc naturel
Lorsque l’empilement des feuillets est désordonné, le diffractogramme présente des raies de diffraction plus ou moins larges et asymétriques. C’est ce qui a été observé pour les kérolites (figure 3) et les stévensites (Brindley et al. , 1977). On parle aussi d’empilement turbostratique. Ces deux derniers minéraux se distinguent du talc par la position de la raie de diffraction du plan (001) qui est à 10 Å pour la kérolite et variable suivant l’état de l’interfeuillet pour la stévensite.
Plan (001) 9.35 Plan (020) 4.55 Plan (003) 3.14 Plan (06-33) 1.52
et voit sa distance réticulaire d(001) passer de 10 à des valeurs allant de 17 à 18 . Le gonflement de cette argile est un moyen de la détecter lorsqu’elle est associée à un autre minéral argileux qui n’a aucune capacité de gonflement (le talc par exemple). La kérolite présente un gonflement à l’éthylène glycol lent, partiel et difficile (Decarreau et al., 1989). Afin de mettre en évidence et de renforcer l’intensité des raies (001), les argiles sont déposées par sédimentation sur une lame de verre après saturation à l’éthylène glycol (Moore & Reynolds, 1989). Cette lame est ensuite analysée par diffraction des rayons X pour mettre en évidence les différentes argiles composant l’échantillon.
L’appareillage utilisé au cours de cette étude est un Philips X’Pert Pro. L’échantillon sous forme de poudre est déposé sur le porte échantillon. Chaque mesure est réalisée de 3°2 à 65°2 , avec un pas de 0.010° et 2 secondes par pas. Ce qui équivaut à 6201 points de mesure.
II – La spectroscopie infrarouge à transformée de Fourrier (IRTF)
La spectroscopie infrarouge à Transformée de Fourier est une méthode d’analyse simple à mettre en œuvre et non destructrice. Son principe est basé sur l’absorption d’un rayonnement infrarouge par le matériau analysé. Lorsque la longueur d’onde apportée par le faisceau infrarouge est voisine de l’énergie de vibration d’une molécule, cette dernière absorbe le rayonnement et nous enregistrons une diminution de l’intensité réfléchie ou transmise. Les analyses peuvent être effectuées en moyen infrarouge (domaine compris entre 4000 cm-1 et 400 cm-1) ou en proche infrarouge (domaine compris entre 11000 cm-1 et 4000 cm-1). Toutes les vibrations ne donnent pas lieu à une absorption, la géométrie de la molécule étant très importante. Pour une géométrie donnée, les modes de vibration actifs en infrarouge peuvent être déterminés par la Théorie des Groupes. La différence d’électronégativité des atomes et de leur masse influe aussi sur la position des bandes d’absorption d’un matériau donné.
Plusieurs modes de vibration des molécules en réponse à l’énergie absorbée existent en infrarouge :
- deux modes de déformation de la molécule (« streching »).
- deux modes de « courbure » ou « pliage » de la molécule (« bending »).
Des tables, établies par de nombreux auteurs, permettent d’attribuer les absorptions aux différents groupes chimiques présents dans le matériau analysé.
Cette méthode d’analyse permet donc de caractériser un matériau de composition chimique et de structure donnée par l’ensemble des bandes d’absorption composant son spectre.
L’objet de cette étude est le talc synthétique. De nombreux travaux ont traité du talc en spectroscopie infrarouge. Le talc a fait l’objet de nombreuses études en analyses en moyen infrarouge. Trois bandes de vibration sont caractéristiques du talc en moyen infrarouge (figure 4):
- 3675 cm-1 : vibration Mg3(OH) (« OH streching ») (Wilkins & Ito, 1967)
- 1016 cm-1 : vibration Si-O (« streching ») (Farmer, 1974) - 669 cm-1 : vibration OH (« OH bending ») (Farmer, 1974)
Figure 4 : spectre en moyen infrarouge d’un talc naturel
Le moyen infrarouge permet donc d’identifier grâce à la présence de ces bandes de vibration le
talc dans un échantillon.
Cette étude a utilisé cette méthode d’analyse en complémentarité avec la diffraction des rayons X pour étudier de manière précise la répartition des cations dans les sites octaédriques. C’est donc la région de vibration des groupements R3-OH la plus intéressante. Une étude récente de Petit et
al. (2004) a montré que l’analyse en proche infrarouge est plus rapide, complètement non
destructive et surtout plus sensible dans la région d’analyse de l’environnement cationique des OH. Ces travaux identifient la bande de vibration de Mg3(OH) à 7185 cm-1 (figure 5) la bande
pointée à 7153 cm-1 correspond à la vibration de 2 Fe3(OH). La position d’une bande de
vibration en proche infrarouge à l’aide de sa position en moyen infrarouge est déterminée par la relation suivante :
= W2 OH / (2 – W OH)
Avec :
: la constante d’inharmonicité (- 85.6 cm-1 dans l’étude de Petit et al., 2004)
W2 OH : position en cm-1 de la bande de vibration R3(OH) en proche infrarouge.
WOH : position en cm-1 de la bande de vibration R3(OH) en moyen infrarouge.
3675
1016
Figure 5 : spectre en proche infrarouge d’un talc naturel
Dans ces travaux, la spectroscopie en proche infrarouge amène des informations sur la répartition des différents cations au sein des sites octaédriques du talc synthétique et complète les informations obtenues par diffraction des rayons X. L’appareillage utilisé au cours de cette étude est un spectromètre Infra Rouge à Transformée de Fourier Nicolet Nexus. Les analyses dans le moyen infrarouge se font en transmission et en réflexion diffuse avec 64 scans entre 4000 cm-1 et 400 cm-1. La résolution est de 4 cm-1. Chaque pastille, pour une analyse en transmission, est préparée avec le mélange suivant :
- 1 mg d’échantillon à analyser - 150 mg de KBr
Avant d’être analysée, la pastille est séchée à l’étuve à 110°C pendant 12 heures.
Les mesures pour le proche infrarouge se font en réflexion diffuse entre11000 cm-1 à 4000 cm-1 avec 128 scans. La résolution est aussi de 4 cm-1. La préparation de l’échantillon est simple : la poudre est directement déposée sur un porte-échantillon pour être analysée. La totalité des analyses de ces travaux a été réalisée dans le proche infrarouge. Toutes les décompositions des spectres en proche infrarouge ont été effectuées à l’aide du logiciel « Peak Resolve ». Afin d’être le plus clair possible, le terme d’intensité relative intégrée d’une bande de vibration, employé au cours des décompositions des spectres, signifie que nous parlons de l’aire de la bande de vibration en question.
7185
III – Autres méthodes de caractérisation directes
Pour cette étude, nous avons utilisé d’autres méthodes de caractérisation. La première est une méthode d’observation directe des particules de talc synthétique à l’aide d’un Microscope Electronique à Balayage. L’échantillon à analyser est sous forme de poudre. Quelques milligrammes sont déposés sur un plot recouvert d’un revêtement en carbone qui est déposé dans la chambre du microscope. Cette analyse rapide permet de vérifier la nature de la particule observée par une analyse chimique ponctuelle ainsi que d’observer la forme des particules. Cette technique nous a permis aussi d’estimer la taille des particules synthétiques et mais surtout l’état d’agglomération de ces particules. L’appareil utilisé pour cette étude est un microscope environnemental FEI Quanta 200. De l’imagerie a également été réalisée au MEB FEG du service TEM Scan de l’Université Paul Sabatier.
A l’état désaggloméré, les particules de talc synthétique sont nanométriques. Afin de pouvoir les observer, des analyses ont été effectuées au Microscope électronique à Transmission (MET). Cette technique d’analyse a permis de visualiser de très fines particules et l’empilement de quelques feuillets de talc synthétique. Afin de désagglomérer au maximum les particules à observer, quelques mg de poudre sont dispersés dans de l’alcool (dispersion 1 pour 20). Le mélange est ensuite ultrasonné pendant 2 minutes à une puissance de 70W. La grille MET est immergée dans le mélange ultrasonné rapidement et laissée sécher pendant 15 minutes. L’observation se fait directement sur cette préparation.
Les produits synthétisés ont été aussi caractérisés par analyse thermogravimétrique (ATG). Cette technique d’analyse consiste en la mesure de la variation de masse, sous atmosphère inerte (Argon ou Azote) d’un échantillon en fonction de la température. Il est donc possible de déterminer la température de déshydratation et de déshydroxylation d’un minéral. Le talc naturel présente une température de décomposition à 942°C (Figure 6) et présente plusieurs températures de déshydroxylation caractéristiques des minéraux associés.
Figure 6 : analyse thermogravimétrique d’un talc naturel
L’appareillage utilisé dans cette étude est une thermobalance Netzsch STA-409. 20 mg de poudre sèche sont utilisés pour chaque mesure. L’acquisition se fait sous balayage d’Azote, pour une montée en température de 17°C par minute, de l’ambiante à 1150°C.
900°C
talc chlorite
CHAPITRE IV
Caractérisations des produits
obtenus
Le protocole de synthèse hydrothermale établi et décrit dans le chapitre II renferme plusieurs étapes dont chacune d’entre elle correspond à un produit de synthèse. Chacun de ces produits intermédiaires a fait l’objet d’étude.
Le protocole est divisé en trois étapes correspondant à trois produits obtenus au cours de ce protocole de synthèse :
- première étape : préparation du gel silicométallique.
- deuxième étape : obtention d’un produit synthétique en sortie de réacteur après le traitement hydrothermal pendant une durée et à une température de synthèse déterminées.
- troisième étape éventuelle: traitement thermique à 550°C pendant 5 heures.
La synthèse hydrothermale renferme trois paramètres importants et influant sur les caractéristiques des produits obtenus : le rapport solide/liquide introduit dans les réacteurs, la durée de synthèse et la température de synthèse. Ce chapitre décrit les produits intermédiaires et le produit final obtenu par le protocole de synthèse établi tout en étudiant chaque paramètre de cette synthèse et leur influence sur les caractéristiques du produit obtenu après le traitement hydrothermal.
I - Le gel : précipité silicométallique
Ce précipité silicométallique est obtenu par mélange de trois réactifs de départ : solutions de métasilicate de sodium, de chlorure de magnésium et d’acide chlorhydrique tel que le rapport Mg/Si = 0.75 (stœchiométrie talc).
Ce précipité est aussi communément décrit dans la littérature comme un gel de silicate de magnésium hydraté (Brew & Glasser, 2004). La figure 1, observation réalisé au microscope électronique à balayage, montre un produit sans ordre et structure apparente. Les particules présentent un aspect moutonneux et sont agglomérées.
Figure 1 : Observation au microscope électronique à balayage du gel silicomagnésien
L’analyse en diffraction des rayons X (figure 2) ne montre aucune raie nette de diffraction de ce produit hormis trois pics de diffraction intenses attribués au chlorure de sodium, produit formé au cours de la coprécipitation et résultant d’un mauvais lavage du gel. Quatre amorces très larges de raies de diffraction apparaissent : 4.3 – 9.3 , 3.6- 3.0 , 2.6 – 2.3 et 1.5 – 1.6 . Ces quatre larges raies de diffraction ont été attribuées à un gel de silicate de magnésium hydraté de très basse cristallinité, précurseur du talc synthétique, au cours de travaux antérieurs : Temuujin et
CuK 1
0 10 20 30 40 50 60
Angle (°2Tétha)
Figure 2 : Diffractogrammes de rayons X du gel silicate de magnésium hydraté
La figure 3 représente le spectre en moyen infrarouge de ce coprécipité et le tableau 1 regroupe les différentes bandes de vibration de ce spectre et leurs origines.
Figure 3 : spectre en moyen infrarouge du gel de silicate de magnésium hydraté
3678 9.3-4.3 3.6-3.0 2.6-2.3 1.5-1.6 NaCl 3430 1636 1017 901
La bande à 3678 cm-1 est attribuée au streching Mg3-OH des phyllosilicates de type 2 :1 (Farmer,
1974 ;Takahashi et al., 1994). Les deux bandes de vibration à 3430 cm-1 et 1636 cm-1 sont attribuées à H2O et traduisent le fort degré d’hydratation de ce gel. La forte bande de vibration à
1017 cm-1 avec son épaulement à 901 cm-1 est assignée au « streching » Si-O des sites Q². La bande de vibration à 788 cm-1 est caractéristique du « streching » Si-O des sites Q1. Ces analyses en moyen infrarouge sont confirmées par les analyses du 29Si par résonance magnétique nucléaire (figure 4). Deux résonnance sont détectées : une à -84.7 ppm caractéristique des sites Q1 et l’autre à -91.0 ppm caractéristique des sites Q².
Les sites Q3 n’apparaissent pas dans cette analyse. Comparé à l’analyse 29Si en résonance magnétique nucléaire d’un talc naturel qui ne présente qu’une seule résonnance à -95.4 ppm (Q3) et à la vue des analyses précédentes, il est possible de conclure que ce gel de silicate de magnésium hydraté n’a pas une structure identique au talc mais est une phase contenant une couche de type brucite (complète ou non) et des tétraèdres de silicium isolés ou associés par paire.
II – Produit obtenu après le traitement hydrothermal
Le précipité silicomagnésien, gel de silicate de magnésium hydraté, décrit dans la partie précédente est soumis au traitement hydrothermal.
1- Influence du rapport solide/liquide introduit dans les réacteurs
Les premières synthèses hydrothermales ont suivi le protocole consistant à préparer un gel silicomagnésien avec les proportions stœchiométriques du talc (Mg/Si = 0.75), à le laver et à le sécher à l’étuve à 110°C pendant 12 heures. La poudre obtenue était ensuite dispersée dans l’eau et ce mélange mis dans les réacteurs de synthèse (50ml de volume). Il s’agit dans cette partie d’étudier l’influence de la variation de la quantité de poudre de gel silicométallique et d’eau introduites dans le réacteur sur les caractéristiques des produits obtenus avec le traitement hydrothermal (rapport solide / liquide).
A – Le protocole expérimental suivi
Après formation du coprécipité silicométallique à stœchiométrie talc, 4 atomes de silicium pour 3 atomes de magnésium, ce coprécipité a été séché à l’étuve à 110°C pendant 12 heures. Le produit séché a été broyé au mortier en agate afin de le rendre sous forme de poudre. Cette poudre a été dispersée dans l’eau déminéralisée et placé dans plusieurs réacteurs, chaque réacteur correspondant à un rapport solide/liquide (tableau 2) pour une durée de synthèse de 21 jours à 220°C et à une pression de 16 bars.
Coprécipité originel
Nomenclature des produits obtenus pour
les conditions de synthèse fixées Quantité d’eau introduite (ml) Quantité de poudre introduite (mg) Si4 Mg3 O11, nH2O Si4 Mg3 O11, nH2O Si4 Mg3 O11, nH2O Si4 Mg3 O11, nH2O R=10 R=20 R=40 R=100 30 20 20 10 300 400 800 1000
B – Etude de l’évolution de la cristallinité des produits synthétisés par diffraction des rayons X
La dissymétrie des raies de diffraction pour les plans (001) des réseaux cristallins pour chaque produit obtenu traduit une hétérogénéité cristalline due à l’interstratification de feuillets avec différentes épaisseurs (figure 5) et d’un état d’hydratation différent de l’ensemble de l’échantillon.
0 10 20 30 40 50 60
Angle (°2Tétha)
CuK 1
Figure 5 : diffractogrammes de rayons X sur poudre des échantillons étudiés à 220°C
Les échantillons R100, R40 et R20 montrent une amorce de raie de diffraction à des angles compris entre 4 et 7 °2 (figure 5). La présence de cette raie est caractéristique de la présence de feuillets gonflants. Cette argile ne peut être qu’une stévensite (smectite magnésienne), la chimie des produits étant strictement contrôlée lors de la préparation du gel silicomagnésien. L’échange des sites interfoliaires des feuillets gonflants avec du calcium est une méthode de préparation permettant d’homogénéiser leur épaisseur aux environs de 14 (2 couches d’eau) et de préparer les conditions optimales pour une solvatation à l’éthylène glycol. Par le contraste d’épaisseur ainsi obtenu, après une saturation au calcium. Il est possible de mettre en évidence la présence de feuillets gonflants. Les analyses en diffraction des rayons X sur lame orientée des échantillons saturés calcium confirment la présence de la stévensite associé au talc. La raie de diffraction du plan (001) du talc est présente mais une autre raie de diffraction apparait aux faibles angles d’analyse (figure 6). Pointée à 15 , cette raie caractérise la stévensite et augmente en intensité lorsque le rapport solide/liquide augmente. La composition des produits obtenus évolue donc avec le rapport solide/liquide. Lorsque ce rapport augmente, la teneur relative de la stévensite augmente et celle du talc diminue.
R100 R40 R20 R10 9.71 4.55 3.17 1.52
0 5 10 15 20 25 30 35 40
CuK 1
Figure 6 : diffractogrammes de rayons X sur lame orientée des différents échantillons saturés en calcium
Une modélisation des diffractogrammes de rayons X expérimentaux des échantillons saturés en Ca et éthylène glycol a été réalisée par Eric Ferrage afin de déterminer les proportions des feuillets de type talc et stévensite et leur empilement.
Afin de pouvoir interpréter et discuter les résultats issus de cette approche, la description rapide des principaux principes de cette méthode basée sur l’approche probabiliste markovienne est nécessaire.
Si nous avons un interstratifié composé de deux types de feuillets A et B dans un empilement à M feuillets, il est possible de décrire les motifs des différentes sous entités (ou séquences d’empilements) constituées de n+1 feuillets (avec 1<n<M-1). Par exemple pour n=1 et 2 :
n=1 : 2 feuillets dans la sous entité : AA, AB, BA, BB.
n=2: 3 feuillets dans la sous entité:
AAA, AAB, ABA, BAA, BBA, BAB, ABB, BBB.
Ainsi, pour un interstratifié composé de X types de feuillets différents, dans chacune de ces sous entités constituées d’un nombre y de feuillets il y aura Xy motifs de succession différents.
Si nous pouvons admettre que les différents motifs de succession pour chacune de ces sous 001 (9.6 - talc) 001 (15 - stévensite) R10 R40 R20 R100