Pour l’obtention du grade de
DOCTEUR DE L’UNIVERSITÉ DE POITIERS
(Faculté de Médecine et Pharmacie)(Diplôme National – Arrêté du 7 août 2006)
Ecole Doctorale : Biosanté N°524
Champ disciplinaire : Biologie, Médecine, Santé
Secteur de Recherche : Aspects Moléculaires et Cellulaires de la Biologie
Présentée par
Sylvain NORMAND
***********************Étude des profils cytokiniques et des mécanismes moléculaires
induisant l’inflammation dans un modèle mimant les déficits en
mévalonate kinase : un rôle central pour l’IL-1 et la caspase-1
dans les fièvres périodiques héréditaires.
***********************
Directeur de thèse : Dr. Jean-Claude LECRON
***********************
Soutenue le 16 Décembre 2009 Devant la Commission d’Examen
JURY
M. CHAMAILLARD Chargé de Recherche, INSERM, Institut Pasteur de Lille Rapporteur H. YSSEL Directeur de Recherche, INSERM, Montpellier Rapporteur L. CUISSET MCU-PH, INSERM, Hôpital Cochin, Paris Examinateur
E. SOLAU PU-PH, Université de Poitiers Examinateur
T.BERGÈS Professeur des Universités, Université de Poitiers Examinateur
Ce travail de thèse a été effectué au sein du laboratoire inflammation, tissus épithéliaux et cytokines (LITEC, EA4331) dirigé par le Dr. Jean-Claude Lecron.
J’adresse tous mes remerciements au Dr. Mathias Chamaillard et au Dr. Hans Yssel pour avoir accepté d’être rapporteurs de ce travail. Qu’ils trouvent ici l’expression de ma gratitude.
Je remercie également très chaleureusement le Dr. Laurence Cuisset et le Pr. Thierry Bergès d’avoir accepté d’évaluer mon travail.
Je remercie infiniment mon directeur de thèse, Jean-Claude Lecron de m’avoir accueilli dans son laboratoire, sur un coup de tête réciproque, que je n’ai jamais regretté. Merci pour votre sympathie et la confiance que vous m’avez accordé tout au long de mon parcours. Merci également pour votre disponibilité malgré les nombreuses « casquettes » que vous portez et qui vous obligent parfois à choisir vos priorités, sans faillir à votre rôle de directeur de thèse. Merci pour votre enseignement et votre rôle essentiel et pléiotrope tout au long de ma formation, faisant de vous une sorte d’IL-1 au sein du laboratoire. Toutefois, malgré la confiance et le respect que je vous porte, vous ne me ferez JAMAIS monter dans votre avion…
J’adresse également mes remerciements au Pr. Elisabeth Solau-Gervais, pour sa sympathie et son implication dans l’étude des profils cytokiniques des pathologies inflammatoires. Votre arrivée au laboratoire nous a apporté, outre vos connaissances cliniques, un point de vue et un panel de pathologies dont l’étude préliminaire permet d’envisager une belle tournure à la suite de mon sujet.
Je remercie également toute l’équipe de Bioalternatives, ainsi que le Dr. Laurence Cuisset, le Pr. Gilles Grateau, le Dr. Nicolas Bourmeyster, le Dr. Isabelle Jéru, le Pr. Serge Amselem, et le Dr. Isabelle Couillin pour leur collaboration scientifique.
Un grand merci à Adriana pour sa gentillesse, ses conseils en ELISA et surtout son énorme coup de main pour les manips tout au long de ma thèse.
Merci à Laure, ma colocataire de bureau durant ses trois années, et ma geôlière sur la fin de l’écriture de ce manuscrit. Notre colocation a été idéale, se déroulant toujours dans la bonne humeur. Merci pour ton soutien et ta gentillesse, ainsi que pour tes nombreux conseils et ton implication dans nos manips de transfection. Malheureusement, et ce n’est pas faute de les avoir longuement priés, Sainte Ligation et Sainte Transfection ont eu raison de nous.
Merci à Benoit, mon prédécesseur, devenu un ami, ainsi qu’à sa compagne, Emmanuelle. Ton enseignement scientifique un peu particulier, souvent ponctué de métaphores dont toi seul as le secret, m’aura énormément apporté. Bien que le temps des gloutons barjots soit révolu, nos
Merci à Karline, ma petite sœur (plus grande que moi) de labo. Embarqués sur le même radeau, qui finalement s’est révélé être un paquebot de luxe, les échéances liées à notre parcours auront facilité, et c’est tant mieux, notre rapprochement. Merci pour ton amitié et tous les moments, bons ou mauvais, partagés au sein ou en dehors du labo. Merci pour ton rire dont la discrétion n’aura échappé à personne du labo (ni du PBS peut-être…). Tu resteras une de mes meilleures rencontres poitevines et j’espère que notre amitié durera au delà des murs du PBS, même si je n’en doute pas. Merci également à ton doudou pour les soirées passées chez vous, ou chez nous à essayer d’attraper mon chat.
Merci à Manu, ancien doctorant au laboratoire et ami depuis maintenant 10 ans (que le temps passe vite…) ainsi qu’à sa femme Vanessa. De nos parties de Kamoulox au baptême de Florent, tous les moments passés ensemble resteront de très bons souvenirs. Merci de votre fidélité amicale et de l’honneur de m’avoir fait parrain de votre fils. Cette attention m’a profondément touché et témoigne de la sincérité de nos relations.
Merci à l’ensemble du LITEC dans lequel règne sympathie et bonne humeur. A Franck (pour ses nombreux conseils et son humour acerbe), à Isabelle (pour tout ce qui touche aux animaux, au labo ou non…), à Thierry (pour son aide technique, surtout quand il s’agit de réparer le laveur de plaques. Je te soupçonne d’ailleurs de l’avoir volontairement mis en panne à plusieurs reprises…), à Martine (pour son expertise en Western Blot), à Pascale (pour sa gentillesse et son sourire), à Jean-Philippe (pour sa sympathie), à Sandrine (pour sa gentillesse), à Jean-François (bien que nouveau, tu as su t’intégrer rapidement au laboratoire, bon courage pour la suite). Merci également à toute l’équipe microbiologie du LITEC pour leur convivialité, Christophe, Martine, Marie-Odile, Marilyse, Hristo, Nicolas, Charles et Estelle.
Je remercie également toutes les personnes responsables des différents services communs du PBS, Daniel Guyonnet, Anne Cantereau, et l’ensemble du personnel des animaleries du PBS et du CHU.
Merci particulier aux personnes m’ayant permis de faire de l’enseignement, notamment Martine Garcia, Thierry Ferreira et Véronique Ladevèze qui m’ont directement encadré, mais aussi Jacques Frère et Thierry Bergès, qui dirigent les départements d’enseignement associés. Merci aux personnes m’ayant accompagné, certaines par étape, d’autre tout au long de mon parcours universitaire. Particulièrement Jessica qui fut mon binôme durant de nombreuses années et avec qui les cours de DEUG étaient régulièrement remplacés par des babyfoot – bières. Nous avons franchi de nombreuses étapes ensemble, et si dorénavant, on se voit moins souvent qu’il y a quelques années, l’amitié que je te porte est toujours aussi grande. Merci également à ton Nini ainsi qu’à vos amis, devenus les nôtres, Ludos, Richard, Hélène,
Jean-physique…(je rigole bien sûr).
Merci à Hamid pour les moments passés et pour tes nombreux cours photocopiés… Merci de m’avoir fait connaitre la fin du Ramadan, associée à la dégustation de pâtisseries très sucrées. Ne t’inquiète pas, depuis ton départ sur Castres, Najete, que je tiens également à remercier, a très bien pris le relais de cette tradition culinaire. Merci à Tof pour ses nombreuses bourdes, son franc-parler et surtout son amitié.
Merci à tous les thésards croisés durant ces trois ans, Dominique, Sophie, Thomas, Adrienne, Marie, Nicolas, Arnaud, Julie, et tous ceux que je pourrais oublier.
Je tenais également à remercier ma famille, en particulier mes parents qui malgré leurs doutes lors des premières années de mon cursus, m’ont toujours permis de choisir mon chemin, à l’encontre de leurs conseils. Vous avez toujours été présents, aussi bien par votre soutien qu’au niveau financier et je vous remercie des sacrifices qu’il vous a fallut faire. Merci également à mon frère Damien qui, malgré les nombreux coups échangés lors de notre jeunesse, m’a soutenu durant les phases difficiles.
Merci également à Jean-Paul et Marie, mes beaux parents, qui m’ont toujours considéré comme leur propre fils et qui m’ont soutenu durant les phases difficiles. Une pensée particulière pour nos discussions politiques, parfois houleuses, mais toujours accompagnées d’un bon whisky…
Enfin merci, et c’est peu dire, à Ludivine, ma petite femme de Bourguignol, qui a toujours été d’un immense soutien. Merci pour ton sourire de tous les jours et de m’avoir fait réagir dans les moments où il le fallait, car comme le dit la chanson, « j’ai toujours plus de sentiments pour ceux qui m’ont botté les fesses ». En espérant que nos chemins se croisent encore longtemps, voire toujours…
Abréviations
ABC1 : ATP binding cassette 1Ac : “anticorps”
ATP : “adénosine triphosphate”
ASC : apoptosis-related speck-like protein containing a CARD CAPS: cryopyrin-associated periodic syndrome
CARD: caspase activation and recruitment domain
CD : cluster of differentiation ou antigène de différentiation CIAS-1: cold-induced autoinflammatory syndrome-1
CINCA: chronic infantile neurological and cutaneous syndrome COX2 : cyclooxygénase-2
CRP C-reactive protein ou protéine C réactive
EAE : encéphalomyélite auto-immune expérimentale induite FADD : Fas-associated via death domain
FCAS : familial cold autoinflammatory syndrome FMF : fièvre méditerranéenne familiale
FOXP3 : forkhead box protein 3 FPP : farnésylpyrophosphate
GGPP : géranylgéranylpyrophosphate
HIDS : hyper-IgD and periodic fever syndrome ou syndrome d'hyper-IgD HMGB1: high mobility group box 1
HMGR: “3-hydroxy-3-méthylglutaryl coenzyme A reductase” HSP: Heat shock protein
ICE: IL-1 converting enzyme IFN : interféron
Ig : immunoglobuline IKB : Inhibitor kB IL : interleukine
IL-18BP : IL-18-binding protein IL-1R : IL-1 récepteur
IL-1RA : IL-1 récepteur antagoniste IL-1RAcP : IL-1R accessory protein
IRAK : IL-1R-associated kinase iTreg : Lymphocyte Treg inductible JAK: janus tyrosine kinase
JNK : c-jun N-terminal kinase KO : knock out
LBP : LPS-binding protein LPS : lipopolysaccharide LRR : leucine-rich repeat
MA : mevalonic aciduria ou acidurie mévalonique MAP : mitogen-activated protein
MDP : muramyl dipeptide MEFV : mediterranean fever
Méso-DAP : acide mésodiaminopimélique MK : mévalonate kinase
MKD : MK deficiency ou déficit en mévalonate kinase MMP : matrix metalloproteinase
MSU : monosodium urate MWS : Muckle-Wells syndrome MyD88 : myeloid differentiation 88
NALP : NACHT-LRR-PYD-containing protein NBD : nucleotide binding domain, NACHT
NF-HEV : nuclear factor from high endothelial venules NF-κB : nuclear factor-κ B
NIK : NF-κB inducing kinase NLR: Nod-like receptor
NLRP: Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain NLS: Nuclear localization sequence
NO: monoxide d’azote
NOD: Nucleotide-binding oligomerization domain NOMID: multi-system inflammatory disease P2X7: purinorécepteur 7
PAF: platelet activating factor
PBMC: Peripheral blood mononuclear cells PGE2: prostaglandine E2
PMA : phorbol 12-myristate 13-acétate PRR: pattern recognition receptor PYD: pyrin domain
RIP 1: Receptor interacting protein 1 RIP 2: RIP-like interacting CLARP kinase RNS: Reactive nitrogen species
ROS: Reactive oxygen species
SIGIRR: single Ig IL-1 related receptor ST2: suppression of tumorigenicity 2 ST2s: ST2 soluble
STAT : signal transducer and activator of transcription TACE : TNFα converting enzyme
TAK1: Transforming growth factor-activated kinase 1 TANK: TRAF-associated NF-κB activator
TGF : transforming growth factor Th : T helper ou T auxiliaire TIM : TRAF-interacting motif TIR : Toll / IL-1R
TIRAP : TIR domain-containing adaptor protein TLR : Toll-like receptor
TNF : tumor necrosis factor TNFR : TNF récepteur
TRADD: TNFR-associated via death domain TRAF: TNF receptor-associated factor TRAM: TRIF-related adaptor molecule
TRAPS: TNFRI associated periodic syndrome Treg : lymphocyte T régulateur
TRIAD3A : TLR-Ubiquitinating Enzyme
Sommaire
Introduction ... 1
I. La réponse inflammatoire : un lien étroit entre systèmes immunitaires inné et adaptatif. ...3
1 Activation de l’inflammation. ...4
1.1 Signaux de danger exogènes : les PAMPs. ...4
1.2 Signaux de danger endogènes : les alarmines. ...4
1.3 La notion de pathologies auto-inflammatoires. ...5
1.4 Les récepteurs de l’immunité innée. ...6
1.4.1 Les TLR. ...6
1.4.1.1. Structure et localisation des TLR. ...6
1.4.1.2. Transduction du signal par les TLR. ...8
1.4.2 Les NLR. ... 11
1.4.2.1. Structure et localisation. ... 11
1.4.2.2. Transduction du signal par les NLR. ... 13
1.4.3 Récepteurs de l’immunité innée, initiation de l’inflammation et pathologies auto-inflammatoires. ... 16
2 Les différentes phases de l’inflammation ... 18
2.1 La phase précoce ... 18
2.2 Recrutement de cellules immunitaires sur le site inflammatoire : le passage de relais entre réponses immunitaires innée et adaptative. ... 19
2.2.1 Contribution du système immunitaire inné... 19
2.2.2 Contribution du système immunitaire adaptatif. ... 20
2.3 Résolution de l’inflammation. ... 23
2.3.1 Rôle des médiateurs lipidiques. ... 24
2.3.2 Rôle du système nerveux. ... 24
2.3.3 Rôle des lymphocytes T régulateurs. ... 24
2.3.4 Résolution de l’inflammation médiée par l’immunité innée. ... 25
3 Réseau cytokinique et inflammation... 26
3.1 La triade inflammatoire : IL-1 – IL-6 – TNFα... 26
3.1.2.1. Vue d’ensemble ... 29
3.1.2.2. Cytokines de la famille de l’IL-1 et caspase-1 ... 30
3.1.2.3. Récepteurs des cytokines de la famille de l’IL-1 et transduction du signal 33 3.1.2.4. Cytokines anti-inflammatoires de la famille de l’IL-1... 34
3.1.2.5. Cytokines de la famille de l’IL-1 et alarmines ... 34
3.1.2.6. Cytokines de la famille de l’IL-1 et polarisation des lymphocytes Th ... 36
3.1.2.7. Effets biologiques des cytokines de la famille de l’IL-1 ... 37
3.1.3 L’IL-6 ... 37
3.1.4 Les cytokines sécrétées par les lymphocytes Th17 ... 38
3.1.4.1. L’IL-17 ... 38
3.1.4.2. L’IL-22 ... 39
3.1.4.3. L’IFN-γ ... 40
3.1.4.4. L’IL-21 ... 40
3.1.5 Propriétés anti-inflammatoires des cytokines ... 41
3.1.5.1. L’IL-10 ... 41
3.1.5.2. Le TGF-β ... 42
3.1.5.3. Propriétés anti-inflammatoires de l’IL-1β et de l’IL-6 ... 42
II. Fièvres périodiques héréditaires ... 43
1 Le « TNFR-associates periodic syndrome » (TRAPS) ... 43
2 La fièvre méditerranéenne familiale (FMF) ... 44
3 Les cryopyrinopathies (CAPS) ... 45
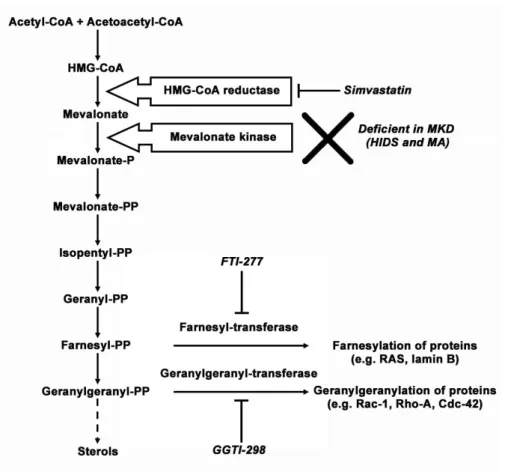
4 Les déficits en mévalonate kinase (MKD) ... 46
5 Des mutations des gènes codant NALP12 ou IL-1ra à l’appartition de nouvelles fièvres périodiques héréditaires liées à l’IL-1 ... 49
Objectifs ... 51
Résultats ... 55
Partie 1. Etude des mécanismes responsables de la sécrétion d’IL-1 lors de
l’inhibition de la voie du mévalonate... 57
I. La simvastatine augmente spécifiquement la sécrétion des cytokines de la famille de
l’IL-1 ... 58
II. La simvastatine induit la sécrétion d’IL-1β en augmentant l’activité de la caspase-1 59 III. La simvastatine influe sur l’activité et la localisation de Rac-1 ... 59
IV. L’activation de la caspase-1 induite par la simvastatine ne nécessite pas la présence de NALP3. ... 61
V. Les fibroblastes de patients MKD sécrètent plus d’IL-1β que les fibroblastes de donneurs sains ... 63
VI. Conclusion ... 63
Partie 2. Étude de la sécrétion d’IL-1β par des cellules de patients atteints
de fièvres périodiques héréditaires. ... 83
I. Etude de la sécrétion cytokinique spontanée et induite par le LPS par des PBMC issues d’un patient Muckle Wells. ... 83
II. Etude de la sécrétion cytokinique ex vivo des PBMC issues de patients présentant des mutations de NALP12. ... 84
III. Conclusion ... 87
Partie 3. Étude de la production d’IL-1α et d’IL-1β par des kératinocytes
de donneurs sains activés. ... 89
Conclusions et perspectives ... 93
Bibliographie ... 101
3
I. La réponse inflammatoire : un lien étroit entre systèmes
immunitaires inné et adaptatif.
La réponse inflammatoire est une réponse adaptative engendrée en réponse à des stimuli nocifs telle une infection ou une agression tissulaire. Elle nécessite une régulation fine, généralement bénéfique, elle conduit à l’élimination d’éventuels pathogènes et au retour à l’homéostasie du tissu lésé. Une régulation défectueuse peut engendrer des dommages irréversibles. Une réponse insuffisante conduit à une immunodéficience pouvant entrainer une infection secondaire ou un cancer. Exacerbée, au contraire, elle augmente la morbidité et la mortalité dans des pathologies comme l’arthrite rhumatoïde, la maladie de Crohn ou encore le diabète. Mal contrôlée, l’inflammation peut s’étendre au reste de l’organisme via la circulation sanguine. Elle peut alors conduire à des dommages tissulaires irréversibles locaux ou généralisés, parfois à un choc septique entrainant dans les cas les plus graves le décès (dans Nathan, 2002; et dans Tracey, 2002; Revue dans Barton, 2008). L’agression d’un tissu doit donc induire une réponse rapide et organisée afin de réparer les dommages, tout en assurant l’intégrité du reste de l’organisme.
La réponse inflammatoire est associée au système immunitaire, qui peut-être divisé en deux branches interconnectées. L’immunité innée est la plus ancienne. Elle est présente chez tout organisme pluricellulaire. Les cellules du système immunitaire inné possèdent des récepteurs (PRR pour Pattern Recognition Receptors) et des voies de signalisation hautement conservés pour détecter et réagir face à une infection ou à une blessure. La détection de ces signaux exogènes d’origine microbienne, les PAMPs (Pathogen-associated molecular pattern), ou endogènes, les alarmines (Revue dans Bianchi, 2007), va conduire à l’initiation de la cascade inflammatoire et à l’activation d’une réponse immunitaire acquise ou adaptative (Revue dans Barton, 2008; Revue dans Medzhitov, 2008). Classiquement, la réponse inflammatoire est décrite selon quatre étapes : la reconnaissance des signaux de danger, le recrutement de cellules sur le site d’infection, l’élimination du pathogène et la résolution de l’inflammation conduisant à un retour à l’homéostasie et à la cicatrisation du tissu lésé (Revue dans Barton, 2008). En absence de résolution, s’installe une inflammation chronique.
4
1 Activation de l’inflammation.
Les signaux de danger d’origine exogène ou endogène vont stimuler les récepteurs de l’immunité innée, les PRR, et activer la phase aigue de l’inflammation. A coté de ces signaux, le processus inflammatoire peut également être déclenché en absence de toute infection ou traumatisme, notamment lors de pathologies dites « auto-inflammatoires », parmi lesquelles les fièvres périodiques héréditaires. Cet aspect sera développé dans la deuxième partie de l’introduction.
1.1 Signaux de danger exogènes : les PAMPs.
Le principal mécanisme d’induction de l’inflammation est la reconnaissance des PAMPs par les PRR, notamment par les TLR (Toll like receptors) présents à la membrane des macrophages et des cellules dendritiques pré-infiltrés dans le tissu lésé et par les NLR (Nod- like receptors ou NACHT-domain and leucine rich repeat containing) qui sont cytoplasmiques. Les protéines du complément, des protéines liant le mannose ou le lipopolysaccharide (LPS), ou encore des « scavenger receptors » appartiennent également à cette famille (Revue dans Lee and Kim, 2007).
1.2 Signaux de danger endogènes : les alarmines.
Les PRR sont également capables de détecter des signaux de dangers endogènes. C’est en 2007 qu’apparait le terme « alarmine » pour définir ces signaux (Oppenheim et al., 2007). Les alarmines sont libérées rapidement après la mort cellulaire non programmée (nécrose) mais non par les cellules apoptotiques. De plus, les cellules immunitaires peuvent les sécréter sans mourir. Les alarmines permettent le recrutement de cellules immunitaires exprimant les PRR, notamment des cellules dendritiques, et favorisent ainsi indirectement la réponse immunitaire adaptative. Enfin, elles participent au retour à l’homéostasie en promouvant la reconstruction du tissu lésé (Revue dans Bianchi, 2007). Le chef de file des alarmines est HMGB1 (Hight mobility group box 1), dont l’étude a servi à définir les propriétés des alarmines. HMGB1 est un facteur de transcription qui lie les nucléosomes et permet le repliement de l’ADN (Revue dans Agresti and Bianchi, 2003; et dans Klune et al., 2008). Lors d’une blessure, HMGB1 est libéré dans le milieu extracellulaire (Revue dans Bianchi, 2007) après la nécrose de nombreuses cellules, telles les neurones, les entérocytes, les
5 cellules musculaires lisses ou les cellules endothéliales (Rauvala et al., 1988; Liu et al., 2006; Porto et al., 2006). Si les cellules apoptotiques sont incapables de libérer HMGB1 (Scaffidi et
al., 2002), leur phagocytose par les macrophages induit sa sécrétion (Qin et al., 2006). Fait
intéressant, HMGB1 peut interagir avec certains TLR, notamment TLR2 et TLR4 (Park et al., 2004; Park et al., 2006).
D’autres protéines ont rejoint la famille des alarmines, telles les protéines de la famille S100. Sécrétées par les phagocytes (Revue dans Foell et al., 2007) et les kératinocytes (Boniface et al., 2005a; Guilloteau et al., 2009). Elles attirent spécifiquement les leucocytes sur le site inflammatoire (Revue dans D'Ambrosio et al., 2003). Egalement, les protéines de choc thermique (HSP : Heat shock protein), libérées par les cellules nécrotiques, qui interagissent avec de nombreux récepteurs dont les TLR, induisent la sécrétion de cytokines pro-inflammatoires par les macrophages (Revue dans Bianchi, 2007; Revue dans Tsan and Gao, 2009).
L’interleukine-(IL)-1α et de l’IL-33 sont également considérées comme des alarmines (Werman et al., 2004; Moussion et al., 2008). Ce point sera développé dans la partie consacrée aux cytokines de la famille de l’IL-1.
Enfin l’acide urique, soluble dans le milieu intracellulaire, précipite lorsqu’il est libéré dans le milieu extracellulaire pour donner des cristaux d’acide urique. Ces cristaux sont capables d’activer l’inflammasome à cryopyrine NALP3 (pour NACHT-LRR-PYD containing protein ou NLRP3 pour Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain), un complexe multiprotéique dont l’oligomérisation entraine la maturation de cytokines pro-inflammatoires de la famille de l’IL-1. Dans la goutte, les cristaux d’acide urique déposés au niveau articulaire déclenchent une réaction inflammatoire (Revue dans Choi et al., 2005; Revue dans Petrilli and Martinon, 2007). Ce point sera développé dans la partie consacrée aux récepteurs de l’immunité innée.
1.3 La notion de pathologies auto-inflammatoires.
Comme décrit ci-dessus, la goutte fait partie d’une classe de pathologies dont l’origine du processus inflammatoire semble indépendante de toute infection ou traumatisme : les « maladies auto-inflammatoires ». Ce concept récent a été proposé, en écho avec celui de « maladie immune », pour décrire des pathologies, qui à l’inverse des pathologies auto-immunes, apparaissent en l’absence d’auto-Anticorps (Ac) et de lymphocytes T auto-réactifs
6
(Galon et al., 2000). Notons parmi celles-ci, les fièvres périodiques héréditaires dont l’origine génétique a été démontrée. La plupart de ces maladies auto-inflammatoires sont induites par des mutations de gènes codant des protéines impliquées dans la sécrétion et la maturation des cytokines de la familles de l’IL-1 (Revue dans Grateau, 2006; Revue dans Touitou and Kone-Paut, 2008; Revue dans Jeru et al., 2009). Cette approche génétique de l’inflammation sera développée dans le deuxième chapitre.
1.4 Les récepteurs de l’immunité innée.
Le système immunitaire inné constitue la première ligne de défense de l’hôte après une agression pathogène. Il joue un rôle crucial dans la reconnaissance des signaux de danger et dans l’orientation de la réponse inflammatoire. En 1989, Janeway est le premier a suggérer l’existence d’une classe de récepteurs de l’immunité innée capables de reconnaitre des structures microbiennes conservées, avant l’identification moléculaire de ceux-ci ( Janeway, 1989). L’activité pro-inflammatoire de l’ADN ou de l’ARN dérivés de pathogènes avait déjà été montrée. Ceux-ci induisent la production d’IFN par des fibroblastes, bien qu’aucun récepteur de ces acides nucléiques n’ait alors été décrit (Jensen et al., 1963; Rotem et al., 1963). Il faudra attendre 1997 pour que l’hypothèse émise par Janeway soit vérifiée, grâce à la découverte du premier TLR humain, en homologie avec le récepteur Toll de la drosophile responsable de la différenciation dorso-ventrale de l’insecte durant l'embryogénèse, et jouant un rôle prépondérant dans la réponse antifongique (Anderson et al., 1985; Lemaitre et al., 1996; Medzhitov et al., 1997). La caractérisation des NLR, dont la structure est semblable à celle des protéines codées par les gènes de résistance (R) des plantes viendra compléter la famille des PRR (Revue dans Inohara et al., 2005).
1.4.1 Les TLR.
1.4.1.1. Structure et localisation des TLR.
Parmi les PRR, la famille des TLR est la plus étudiée, et contient 12 membres. Ces récepteurs sont caractérisés par un domaine intracytoplasmique d’environ 200 acides aminés, le domaine TIR (Toll / IL-1 Receptor), qu’ils partagent avec les récepteurs de la famille de l’IL-1 (IL-1R) (Revue dans O'Neill and Bowie, 2007). Le domaine extracellulaire des TLR est constitué de répétitions riches en leucines (motifs LRR pour leucine rich repeats), à la
7 différence des IL-1R qui présentent trois domaines de type Ig (Revue dans Akira and Takeda, 2004).
Figure 1 : Structure simplifiée des récepteurs à domaine TIR (Modifié d'après Akira and Takeda, 2004)
Les TLR sont des protéines transmembranaires qui possèdent un domaine TIR intracellulaire qu’ils partagent avec les récepteurs de l’IL-1. Ils divergent par leur domaine extracellulaire, composé de séquences répétées riches en leucines pour les TLR et de trois domaines Ig pour les IL-1R.
Les TLR peuvent être divisés en deux sous-familles en fonction de la nature des PAMPs reconnus : la première inclut les TLR1, 2, 4 et 6 qui reconnaissent des motifs conservés des pathogènes, et la seconde comprend les TLR3, 7, 8 et 9 qui reconnaissent des acides nucléiques de micro-organismes (Revue dans Mogensen, 2009). Les TLR interagissent soit directement avec les PAMPs, soit indirectement grâce à des molécules adaptatrices. Par exemple, le TLR4 reconnait le LPS grâce à la protéine MD2 (Shimazu et al., 1999; Nagai et
al., 2002). Les TLR forment principalement des homodimères mais aussi des hétérodimères,
ce qui explique la diversité de reconnaissance des PAMPs. Par exemple, le TLR2 est capable de s’associer au TLR1 ou au TLR6 (Ozinsky et al., 2000). Les TLR peuvent également être classés selon leur localisation subcellulaire. Les TLR1, 2, 5, 6 et 10 sont exprimés à la membrane de la cellule, et reconnaissent des produits bactériens spécifiques, non produits par les cellules hôtes. Les TLR3, 7, 8 et 9 sont exprimés à la surface des organites intracellulaires, notamment des lysosomes et des endosomes. Ils sont spécialisés dans la reconnaissance d’acides nucléiques de micro-organismes, avec une discrimination du soi et du non soi apportée exclusivement par la localisation des ligands (Revue dans Mogensen, 2009).
8
Malgré leurs fortes homologies de séquence et de structure, les TLR sont capables de reconnaitre des ligands variés. Dans le cadre de ce travail, nous nous sommes particulièrement intéressés au TLR4, impliqué dans la reconnaissance du LPS, mais aussi d’alarmines comme la protéine de choc thermique HSP70 (Revue dans Akira et al., 2006) ou des peptides antimicrobiens, notamment S100A8 et S100A9 (Revue dans Ehrchen et al., 2009).
Table 1 : Ligands et localisation des TLR (Revue dans Kumar et al., 2009)
Les TLR peuvent être localisés soit à la membrane de la cellule, soit à la membrane des endosomes et des lysosomes. Bien qu’ayant de fortes homologies structurales, les TLR sont capables de reconnaitre un large panel de ligands d’origines endogène ou exogène.
1.4.1.2. Transduction du signal par les TLR.
Lors de l’engagement des TLR par des PAMPs, plusieurs voies de transduction sont induites. Cette diversité est rendue possible grâce à l’utilisation d’un panel de molécules adaptatrices qui déterminent en partie la spécificité de la réponse (Revue dans O'Neill and Bowie, 2007). Après activation des TLR, le recrutement d’une ou plusieurs molécules adaptatrices va engendrer une signalisation intracellulaire incluant des voies de phosphorylation, d’ubiquitinylation ou d’interactions protéine-protéine. Cette cascade conduit
9 à la translocation nucléaire de facteurs de transcription régulant l’expression de gènes impliqués dans la réponse inflammatoire, notamment des cytokines, ou dans la défense antimicrobienne, tels des peptides antimicrobiens (Revue dans Mogensen, 2009). La principale protéine adaptatrice utilisée par les TLR est MyD88 (pour Myeloid Differentiation 88). La réponse aux TLR est classée selon l’implication ou non de la voie de signalisation dépendante de MyD88, activée en aval de tous les TLR, excepté du TLR3. Le TLR4 a la possibilité d’induire simultanément les deux voies (Revue dans Akira et al., 2006), mais ne peut pas directement reconnaître le LPS. Il doit préalablement être opsonisé par la protéine circulante LBP (pour LPS-binding protein), et ce complexe LBP-LPS peut alors être reconnu par le CD14, une protéine membranaire que l’on retrouve essentiellement à la surface des monocytes. C’est l’association moléculaire LPS – LBP – CD14 qui va interagir avec le TLR4 par l’intermédiaire de la protéine adaptatrice MD2 (Schumann et al., 1990; Shimazu et al., 1999; Akashi et al., 2000).
Figure 2 : Protéines impliquées dans la reconnaissance du LPS par le TLR4 (Revue dans Aderem and Ulevitch, 2000)
Le LPS est opsonisé par la protéine LBP circulante, et ce complexe est reconnu par la protéine membranaire CD14 située à la surface des monocytes / macrophages. Le complexe ternaire LPS – PBP – CD14 est alors reconnu par le TLR4 par l’intermédiaire de la protéine MD2, induisant la transduction du signal via l’activation ou non de MyD88.
- La voie dépendante de MyD88.
En réponse au LPS, le TLR4 s’homodimèrise et, grâce à son domaine TIR, interagit avec la protéine sous-membranaire MyD88 qui recrute des kinases de la famille IRAK (pour IL-1R associated kinase). Cette signalisation initiée par MyD88 est analogue à celle engendrée par l’engagement des récepteurs de l’IL-1 (Wesche et al., 1997; Burns et al., 1998). Après interaction avec MyD88, IRAK4 est phosphorylée, puis phosphoryle IRAK1 ou
10
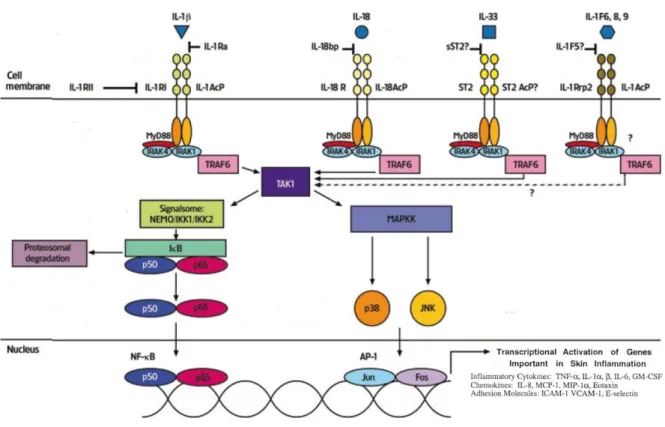
IRAK2. Ceux-ci pourront s’associer avec TRAF6 (TNF receptor associated factor 6), permettant l’activation de la kinase TAK1 (Transforming growth factor-activated kinase 1). TAK1 induit ensuite deux voies distinctes : l’une conduisant à l’activation de NF-κB via l’inhibition d’IκB (Inhibitor κB) qui le séquestrait jusqu’alors dans le cytoplasme, l’autre à l’activation des kinases JNK (c-jun N-terminal kinase) et p38 via l’activation des MAP (Mitogen-activated) kinases (Revue dans Mogensen, 2009). La translocation de ces facteurs nucléaires va conduire à l’expression de cytokines pro-inflammatoires, tels l’IL-6, IL-1β ou le TNF-α (tumor necrosis factor α).
- La voie indépendante de MyD88.
L’activation d’une voie de transduction indépendante de MyD88 a été montrée grâce à des souris déficientes pour MyD88, pour lesquelles la stimulation des TLR3 et 4 n’altère pas la production d’IFN-β (Kawai et al., 2001). Bien qu’incapable d’engendrer l’activation rapide de NF-κB ou des MAP kinases, la stimulation au LPS des macrophages de souris déficientes pour MyD88 induit la production d’IFN-β, ce qui a permis l’hypothèse d’une voie de signalisation parallèle. De plus, une activation tardive de NF-κB a tout de même été observée (Kawai et al., 1999). La protéine TRIF (TIR domain-containig adaptator inducing IFN-β) est l’adaptateur de cette voie MyD88-indépendante (Yamamoto et al., 2003a). Le TLR4 n’interagit pas directement avec TRIF via leurs TIR respectifs, mais par l’intermédiaire de TRAM (TRIF-related adaptator molecule), une molécule adaptatrice (Yamamoto et al., 2003b). TRIF pourra alors interagir avec TRAF6, induisant l’ubiquitinylation de TAK-1 et conduisant, mais plus tardivement que dans la voie dépendante de MyD88, à l’activation de NF-κB (Sato et al., 2003). TRIF et TRAF6 interagissent via la protéine RIP1 (receptor interacting protein 1) (Cusson-Hermance et al., 2005). TRIF peut également interagir avec TRAF3, permettant le recrutement de TANK (TRAF-associated NF-κB activator), et conduisant à la translocation du facteur de transcription IRF3 (IFN regulatory factor 3), responsable de l’expression du gène codant l’IFN-β (Fitzgerald et al., 2003; Sharma et al., 2003).
11 Figure 3 : Signalisation engendrée par l’activation du TLR4
(Revue dans Mogensen, 2009)
L’activation du TLR4 par le LPS engendre deux voies de signalisation distinctes : une voie dépendante de MyD88 conduisant à la translocation du facteur de transcription NF-κB et à l’activation des MAP kinases, l’autre indépendante de MyD88, conduisant à l’activation tardive de NF-κB et à la translocation d’IRF3.
1.4.2 Les NLR.
1.4.2.1. Structure et localisation.
Les NLR forment une famille de récepteurs de l’immunité innée présents dans le cytoplasme, et donc capables de reconnaitre des PAMPs intracellulaires. On compte aujourd’hui 22 membres de cette famille chez l’homme et au minimum 33 chez la souris (Revue dans Chen et al., 2009a). Bien qu’exprimés principalement dans les cellules
12
immunitaires, les NLR sont également exprimés dans des cellules non-immunitaires, notamment dans des cellules épithéliales et mésothéliales (Revue dans Chen et al., 2009a).
Figure 4 : Structure des NLR (Modifié d'après Martinon et al., 2009)
Les NLR sont caractérisés par 3 domaines: un domaine LRR, capable de lier le ligand, un domaine central NACHT nécessaire à leur oligomérisation, et un domaine de liaison protéine-protéine (PYD ou CARD). Ils peuvent être classés en sous-familles en fonction de ce domaine effecteur. La sous-famille IPAF/NAIP ne sera pas détaillée dans ce manuscrit. On note la forte homologie des NLR avec le récepteur R des plantes, annoté NLR sur ce schéma.
Les NLR sont constitués de trois parties (Creagh and O'Neill, 2006; Fritz et al., 2006; Chen et
al., 2009a):
- un domaine N-terminal d’interaction protéine-protéine qui peut être :
un domaine de recrutement de caspase (CARD pour caspase recruitment domain) pour les protéines NOD (pour nucleotide-binding oligomerization domain)
un domaine de liaison à la pyrine (PYD pour pyrin domain) pour les protéines NLRP (pour Nucleotide-binding oligomerization domain, Leucine rich Repeat (LRR) and Pyrin domain containing) également appelés NALP (pour NACHT-LRR-PYD-containing protein) ou PYPAF (pour pyrin-containing apoptotic protease-activating factor).
- un domaine central NOD.
- une partie C-terminale riche en leucine. Ces LRR permettent, comme pour les TLR, de reconnaitre les PAMPs ou les alarmines.
13 NOD1 et NOD2 furent les premiers membres de cette famille à être découverts et sont les mieux décrits (Inohara et al., 1999; Ogura et al., 2001b). NOD1 est exprimé de façon ubiquitaire alors que l’expression de NOD2 est restreinte à quelques types cellulaires, notamment, les macrophages (Ogura et al., 2001b), les cellules dendritiques (Tada et al., 2005), les kératinocytes (Voss et al., 2006) et les cellules épithéliales, principalement de l’intestin (Hisamatsu et al., 2003).
1.4.2.2. Transduction du signal par les NLR.
Bien qu’ayant une forte homologie de séquence, les NLR peuvent être divisés en deux sous-familles selon leurs différences de transduction du signal. Cette différence est liée à leur structure, particulièrement à leur partie N-terminale, contenant un domaine de liaison à la pyrine pour les NALP et un domaine de recrutement de caspase pour les NOD (Revue dans Chen et al., 2009a). Elles activent soit la voie NF-κB, soit la caspase-1.
- La sous-famille NOD.
De part leur forte homologie avec les TLR, surtout dans leur partie C-terminale, les NLR activent des voies de signalisation proches, notamment pour la sous-famille NOD. NOD1 et NOD2 sont capables de reconnaitre des molécules dérivées de la dégradation du peptidoglycan (Revue dans Mogensen, 2009). NOD1 est activé par le muramyl peptide, contenant des acides mésodiaminopiméliques (Méso-DAP), produit essentiellement par les bactéries à Gram négatif (Chamaillard et al., 2003; Girardin et al., 2003a) et NOD2 est capable de reconnaitre le muramyl dipeptide (MDP), produit par les bactéries à Gram positif et négatif (Girardin et al., 2003b). En absence d’infection, les NLR sont maintenus dans le cytoplasme sous forme inactive grâce à des interactions intraprotéiques entre leurs parties LRR et NOD. Lorsque la partie LRR est tronquée ou mutée, les NLR sont constitutivement actifs (Tanabe et al., 2004). Lors de la reconnaissance de PAMPs, les protéines NOD vont devenir actives, rompant ainsi leurs liaisons intra-protéiques. Leur domaine NOD devient alors accessible , rendant leur dimérisation possible (Revue dans Franchi et al., 2009). Ensuite, les protéines NOD s’associent via l’interaction directe de leurs domaines CARD, avec la kinase RIP2 (ou RICK pour RIP-like interacting CLARP kinase). RIP2 se lie ensuite directement avec NEMO (NF-κB essential modulator) qui promeut la translocation de NF-κB (Inohara et al., 1999; Revue dans Franchi et al., 2009). RIP2 peut également interagir avec
14
TAK1 conduisant à l’activation des MAP kinases, comme décrit précédemment pour les TLR (da Silva Correia et al., 2007).
- La sous famille NALP.
Les NALP diffèrent des NOD par leur domaine PYD N-terminal. De ce fait, leur signalisation ne conduit pas à la translocation de NF-κB, mais à l’activation de la caspase-1 (également appelée ICE pour IL-1β converting enzyme), l’enzyme de maturation de l’IL-1β. NALP3, également appelé NLRP3 ou cryopyrine, est le mieux décrit des NALP (Revue dans Mariathasan and Monack, 2007). Lors d’une infection, la reconnaissance des PAMPs par les TLR induit la synthèse de la pro-IL-1β via la translocation de NF-κB. Cette pro-IL-1β est cytoplasmique et inactive jusqu’à l’arrivée d’un second stimulus qui va entrainer sa maturation. Un complexe protéique responsable de cette maturation, l’inflammasome, fut identifié en 2002 (Martinon et al., 2002). Il est formé de la protéine ASC (Apoptosis associated Speck-like protein containing a CARD), de la pro-caspase-1 (forme inactive de la caspase-1), et d’un membre de la famille des NALP, notamment NALP3 (Revue dans Martinon et al., 2002; Sutterwala et al., 2006; Mariathasan and Monack, 2007). L’oligomérisation de ces protéines grâce à des interactions PYD-PYD entre NALP et ASC et CARD-CARD entre ASC et pro-caspase-1, induit l’activation de la caspase-1 qui pourra cliver la pro-IL-1β accumulée dans le cytoplasme, conduisant à la sécrétion d’IL-1β mature (Agostini et al., 2004). Le recrutement de deux molécules de pro-caspase-1 (p45) permet leur clivage respectif, donnant naissance à des sous-unités p10 et p20. Elles vont s’associer sous la forme d’un hétérotétramère, composé de deux hétérodimères p10-p20, formant la caspase-1 active (Walker et al., 1994; Wilson et al., 1994; Yamin et al., 1996). L’oligomérisation de l’inflammasome est induite suite à la reconnaissance de nombreux signaux par NALP3, parmi lesquels l’ATP (Mariathasan et al., 2006), les cristaux d’acide urique (Martinon et al., 2006), l’ARN viral (Kanneganti et al., 2006a), l’ADN (Muruve et al., 2008) et l’ARN (Kanneganti et
al., 2006b) bactérien. Le MDP peut également induire l’activation de la caspase-1 via NALP1
(Martinon et al., 2004; Faustin et al., 2007). Malgré ces nombreux ligands, les mécanismes moléculaires impliqués dans l’activation de NALP3 restent à préciser. L’hypothèse d’un efflux potassique induit par l’ATP a été émise (Mariathasan et al., 2006). En effet, suite à l’activation du récepteur P2X7 (purinorécepteur 7) des macrophages par l’ATP, un influx calcique est observé, compensé par un efflux potassique généré par le canal panexine-1 (Kahlenberg and Dubyak, 2004; Mariathasan et al., 2006; Pelegrin and Surprenant, 2006). Un
15 rôle important de HSP90 dans l’activation de NALP3 par des cristaux d’acide urique a été suggéré, via son interaction avec les domaines LRR et NACHT de NALP3. De plus, NALP3 est instable en absence de HSP90, ce qui engendre sa dégradation par le protéasome (Mayor
et al., 2007; Revue dans Shirasu, 2009).
La caspase-1 peut aussi être maturée grâce à des complexes protéiques NOD2 – NALP1 ou NOD2 – NALP3 suite à la stimulation de macrophages au MDP (Pan et al., 2007; Hsu et al., 2008). Le concept de pyroptosome, un complexe formé de dimères d’ASC, vient également perturber ce schéma (Fernandes-Alnemri et al., 2007; Fernandes-Alnemri and Alnemri, 2008).
Bien que principalement pro-inflammatoires grâce à leur capacité à activer la caspase-1 ou à induire la translocation de NF-κB, NALPcaspase-12, également appelé Monarch-caspase-1 ou PYPAF7, est un NLR dont les propriétés pro ou anti-inflammatoires restent à déterminer. Son rôle pro-inflammatoire serait lié à sa capacité à activer la translocation de NF-κB et, via son interaction avec ASC, à activer la caspase-1, résultant en une sécrétion accrue d’IL-1β (Wang et al., 2002). Par ailleurs, la surexpression de NALP12 dans des cellules HEK293T inhibe la translocation de NF-κB induite par des agonistes TLR2 ou TLR4, via une inhibition d’IRAK1 (Williams et al., 2005). Cette inhibition de la translocation de NF-κB pourrait également être due à la dégradation de NIK (NF-κB inducing kinase) engendrée par NALP12, via son interaction avec HSP90 (Arthur et al., 2007; Lich et al., 2007). Lorsque NALP12 est réprimé, la translocation de NF-κB est plus importante qu’après stimulation avec un agoniste TLR2 ou TLR4 seul (Williams et al., 2005). La mutation non-sens de NALP12 décrite dans une fièvre périodique héréditaire suggérant la levée du rôle anti-inflammatoire de NALP12 dans cette pathologie vient appuyer cette théorie (Jeru et al., 2008). A l’avenir, l’étude de ligands potentiels de NALP12 pourrait nous aider à comprendre sa réelle implication dans le processus inflammatoire lié à l’IL-1.
16
Figure 5 : Influence des NLR sur la synthèse et la maturation de l’IL-1β (Modifié daprès Delbridge and O'Riordan, 2007)
NOD2 induit la translocation de NF-κB via l’activation de RIP2. L’oligomérisation de l’inflammasome NALP3 induit l’activation de la caspase-,1 entrainant la maturation puis la sécrétion d’IL-1β. NALP12 inhibe la translocation de NF-κB en se fixant à IRAK1 empêchant son interaction avec TRAF6 ou en promouvant la dégradation de NIK. Le rôle activateur ou inhibiteur de NALP12 sur la maturation de la caspase-1 reste à préciser.
1.4.3 Récepteurs de l’immunité innée, initiation de l’inflammation et pathologies auto-inflammatoires.
Nous avons vu que l’engagement des PRR par des signaux endogènes ou exogènes, joue un rôle important dans la phase aigue de la réponse inflammatoire, principalement par l’activation et la translocation de NF-κB, l’induction des l’IFN et l’activation de la caspase-1. NF-κB induit l’augmentation d’expression de gènes codant des protéines pro-inflammatoires, notamment des cytokines, dont la pro-IL-1β, l’IL-6 et le TNFα, des chimiokines telles IL-8 et RANTES (Ghosh et al., 1998), mais aussi la caspase-1 (Creagh and O'Neill, 2006). La translocation de NF-κB augmente également l’expression de molécules d’adhérence, des récepteurs de cytokines et de chimiokines, des immunoglobulines, de TLR et de molécules du complexe majeur d’histocompatibilité (Iwasaki and Medzhitov, 2004). Les IFN, induits par les PRR, vont se fixer sur leur récepteurs, entrainant les voies de signalisation JAK / STAT (Janus kinases / Signal transducers and activators of transcription), puis l’expression de
17 nombreux gènes codant des protéines impliquées dans la réponse inflammatoire (Der et al., 1998; Revue dans Stark, 2007). La stimulation des récepteurs à l’IFN de type I se traduira principalement par une réponse antivirale incluant des mécanismes d’inhibition de la synthèse protéique, de clivage de l’ARN viral et d’interférence avec la réplication virale (Revue dans Stark, 2007).
Au-delà de la reconnaissance de PAMPs, les PRR sont engagés dans les pathologies dites « auto-inflammatoires ». Le concept de maladie auto-inflammatoire fut proposé en 2000 par Galon et al. pour définir des pathologies inflammatoires chroniques apparaissant en l’absence d’infection et de mise en évidence d’auto-Ac ou de lymphocytes T auto-réactifs, à la différence des pathologies auto-immunes (Galon et al., 2000). Aujourd’hui, la différence entre maladie auto-immune et auto-inflammatoire ne semble pas aussi simple et l’existence d’un continuum entre les deux est proposé (McGonagle and McDermott, 2006). Certaines, comme la goutte et la pseudogoutte sont d'origine métabolique, d’autres, comme la maladie de Crohn, sont multifactorielles. Les fièvres héréditaires périodiques qui nous intéressent particulièrement, sont d'origine monogénique. Ces pathologies sont associées à une activation exacerbée du système immunitaire inné, conduisant principalement à la sécrétion d’IL-1, ou encore à un défaut de production des composants des récepteurs de l’immunité innée. La goutte et la pseudogoutte sont induites par des dépôts de cristaux d’acide urique au niveau articulaire, qui vont activer l’oligomérisation de l’inflammasome NALP3 (Revue dans Choi et
al., 2005; Martinon et al., 2006; Revue dans Petrilli and Martinon, 2007). Si la maladie de
Crohn a de nombreuses causes, l’existence de polymorphismes de NOD2 semble particulièrement influer sur la susceptibilité à développer cette pathologie (Hugot et al., 2001; Ogura et al., 2001a). Chez la souris, NOD2 influe sur le développement de la colite expérimentale (Watanabe et al., 2008). Suite à la pré-stimulation de NOD2 par le MDP, les monocytes murins voient leur réponse aux TLR fortement diminuer, ainsi que la synthèse de cytokines telles l’IL-12 et l’IL-6 (Watanabe et al., 2008). L’hypothèse qui en résulte est que la mutation de NOD2 chez les patients atteints de la maladie de Crohn provoquerait une diminution de cette régulation négative des TLR par NOD2, conduisant au développement d’une colite. Plus récemment, la découverte de polymorphismes de NALP3 et CARD8 chez les patients atteints de la maladie de Crohn complète les éventuelles causes de la production non régulée d’IL-1 chez ces patients (Schoultz et al., 2009; Villani et al., 2009).
Les fièvres périodiques héréditaires sont aussi des maladies auto-inflammatoires. Les mutations qui en sont responsables sont extrêmement variées, mais conduisent pour la plupart
18
à la sécrétion non régulée d’IL-1β. De ce fait, les protéines impliquées jouent un rôle prépondérant dans la synthèse et/ou la maturation de l’IL-1. Ces pathologies ainsi que les mécanismes moléculaires associés sont développés dans la seconde partie de cette introduction.
2 Les différentes phases de l’inflammation.
2.1 La phase précoce.Quelle soit due à la reconnaissance de signaux endogènes, exogènes, ou à une mutation génétique, l’initiation de l’inflammation passe principalement par l’activation du facteur de transcription NF-κB, conduisant à l’augmentation de protéines pro-inflammatoires (cytokines, chimiokines, etc…). Les mastocytes jouent un rôle important dans la phase précoce du processus inflammatoire, en sécrétant du TNFα, de l’histamine, de la sérotonine, du PAF (platelet activating factor), des prostaglandines et des leucotriènes (Revue dans Nathan, 2002). Fortement pro-inflammatoires, ces signaux vont avoir des actions sur les nombreux types cellulaires infiltrant la lésion. Ainsi, l’histamine et les prostaglandines favorisent la vasodilatation et l’extravasion (Revue dans Nathan, 2002), et le TNF permet la dégranulation des neutrophiles et augmente leur capacité à sécréter des protéases, des peptides antimicrobiens et des espèces oxygénées réactives (ROS pour Reactive oxigen species) (Klebanoff et al., 1986; Nathan, 1987). Les ROS sont ensuite capables d’activer des métalloprotéases matricielles (MMP pour matrix metalloproteinase) induisant la destruction locale de la matrice extracellulaire et permettent la libération de TNF séquestré au niveau de la membrane plasmique des monocytes et macrophages infiltrés (Weiss et al., 1985; Gearing
et al., 1994; Revue dans Goetzl et al., 1996; Revue dans Okamoto et al., 2004). Les
macrophages et les cellules dendritiques résidentes phagocytent les bactéries, relayant la sécrétion de cytokines et chimiokines par ces cellules phagocytaires ainsi que par les mastocytes (Revue dans Barton, 2008).
19 Figure 6 : Schéma simplifié de la phase précoce du processus inflammatoire suite à une
infection (Le système immunitaire, Parham, 2003)
Les macrophages sont activés suite à la reconnaissance de signaux de danger exogènes, et libèrent des cytokines et des chimiokines, qui augmentent la perméabilité des vaisseaux sanguins. L’adhésion des cellules endothéliales des vaisseaux sanguins est également modifiée, permettant l’extravasion des neutrophiles, puis des monocytes. Ces cellules vont également sécréter des cytokines et des chimiokines, amplifiant la réponse inflammatoire et permettant le recrutement, puis l’activation des lymphocytes T.
2.2 Recrutement de cellules immunitaires sur le site inflammatoire : le passage de relais entre réponses immunitaires innée et adaptative.
Suivant le type d’infection, la réponse inflammatoire va conduire à l’élimination des pathogènes, grâce au système immunitaire inné, ou non. Alors, une réponse spécifique est nécessaire à la destruction des organismes pathogènes, faisant intervenir la réponse immunitaire adaptative.
2.2.1 Contribution du système immunitaire inné.
Les cytokines et autres facteurs pro-inflammatoires sécrétés suite à la reconnaissance des PAMPs vont favoriser la vasodilatation et l’extravasion (Revue dans Nathan, 2002). Les cytokines, telles le TNF et l’IL-1 ainsi que des médiateurs lipidiques vont conduire à la migration de leucocytes sur le site d’infection, en particulier des neutrophiles puis des
20
monocytes (Revue dans Nathan, 2006). Les neutrophiles arrivent sur le site inflammatoire armés d’une batterie de protéines facilitant la défense de l’organisme, notamment des protéases capables de dégrader les organismes microbiens mais également le tissu hôte (Revue dans Nathan, 2006). Elles produisent également des ROS et des espèces réactives de l’azote (RNS pour reactive nitrogen species) qui participent à la dénaturation des protéines et de l’ADN (Revue dans Fialkow et al., 2007). Les neutrophiles phagocytent les pathogènes et dirigent directement leur granules vers les phagosomes. Si les neutrophiles détectent le TNF-α mais n’interagissent pas directement avec les pathogènes, ils libèrent leur granules dans le milieu extracellulaire afin de créer un environnement inconfortable pour les pathogènes avoisinants (Revue dans Nathan, 2006; Revue dans Barton, 2008). Le contenu de ces granules est également nocif pour le tissu hôte, notamment l’élastase, la cathepsin G et la protéinase 3 qui sont des protéases à sérine aptes à dégrader la matrice extracellulaire ainsi que les cellules hôtes, provoquant la liquéfaction des tissus à l’origine de la formation de pus. Ce processus permet de contenir l’infection (Revue dans Pham, 2006; Revue dans Korkmaz et al., 2008). Les macrophages contribuent également à l’élimination des pathogènes par les mêmes mécanismes que les neutrophiles : phagocytose et sécrétion de protéases, de peptides antimicrobiens, de ROS et de RNS (Revue dans Bilitewski, 2008). Les peptides antimicrobiens, notamment les β-défensines, sont sécrétés par les cellules épithéliales et les kératinocytes, et forment la ligne de défense principale contre les pathogènes (Revue dans O'Neil, 2003). Cependant, bien qu’efficace, la réponse immunitaire innée n’est pas toujours capable d’éliminer les agents pathogènes et la persistance d’agents infectieux au niveau de la lésion va conduire au développement d’une réponse immunitaire plus spécifique médiée par le système immunitaire adaptatif grâce au recrutement et à l’activation des lymphocytes.
2.2.2 Contribution du système immunitaire adaptatif.
En complément de leur aptitude à sécréter des médiateurs pro-inflammatoires, la reconnaissance des PAMPs par les macrophages et les cellules dendritiques augmentent leur capacité à présenter l’antigène (Ag) et à migrer au niveau des organes lymphoïdes, entrainant l’activation des lymphocytes T. En réponse à une stimulation antigénique, les cellules T CD4 naïves prolifèrent et se différencient en cellules T effectrices qui se distinguent par leur production de cytokines et leurs fonctions. C’est en 1986 que Mosmann et al. proposèrent un modèle de différenciation des cellules T CD4 auxiliaires, ou Th (T helper), qui permettait de relier la fonction des effecteurs T CD4 et la nature de la réponse immunitaire induite, en
21 fonction du profil des cytokines produites, en lymphocytes Th1 et Th2. (Mosmann et al., 1986). En réponse à l’IL-12 produite par les cellules dendritiques, les cellules T CD4 naïves se différencient en lymphocytes Th1, capables de sécréter de l’interféron-(IFN)-γ et de l’IL-2, induisant une réponse à médiation cellulaire (Revue dans Gately et al., 1998). Les lymphocytes Th2 se différencient en présence d’IL-4 et sont amenés à sécréter de l’IL-4, IL-5, IL-10 et IL-13 (Mosmann et al., 1986; Cherwinski et al., 1987; Kurt-Jones et al., 1987; Fiorentino et al., 1989). Les cellules Th1 activent les fonctions bactéricides des macrophages et peuvent détruire les cellules infectées. Elles jouent un rôle dans le contrôle des pathogènes intracellulaires. Les cellules Th2 favorisent la commutation isotypique et la production d’immunoglobuline-(Ig)G1, IgE et IgA qui sont importantes dans le contrôle de certains pathogènes extracellulaires et des helminthes (Revue dans Curtis and Way, 2009). Cette dichotomie a également été associée à des pathologies du système immunitaire. Ainsi, chez l’homme et la souris, les cellules Th2 ont été associées à l’asthme allergique (Revue dans Lloyd and Hawrylowicz, 2009). De nombreuses études suggéraient l’implication des cellules Th1 dans le développement de pathologies immunes telles que l’encéphalomyélite auto-immune expérimentale induite (EAE pour experimental auto-auto-immune encephalomyelitis) ou l’arthrite rhumatoïde (Revue dans Curtis and Way, 2009). Ceci venait de l’observation d’une amélioration des symptômes de l’EAE lors du blocage de l’IL-12 (Revue dans Aranami and Yamamura, 2008). L’IL-12 est une cytokine hétérodimérique composée d’une sous-unité p35 et d’une unité p40 (Revue dans Hunter, 2005). L’étude de souris KO pour ces sous-unités souleva un paradoxe : les souris KO pour la sous-unité p35 ne développaient une EAE, comme les souris sauvages, alors que, les souris KO pour la sous-unité p40 ne développaient pas cette pathologie (Becher et al., 2002). Ce paradoxe fut rapidement résolu grâce à l’étude de souris KO pour la sous-unité p19 de l’IL-23, incapables de développer une EAE (Cua et
al., 2003). L’IL-23 est une cytokine hétérodimérique possédant une sous-unité commune avec
l’IL-12, la sous-unité p40, et une sous-unité p19 qui lui est propre (Revue dans Hunter, 2005). Des résultats similaires furent obtenus pour l’arthrite rhumatoïde induite (Cua et al., 2003), mettant en avant l’intérêt particulier de l’IL-23 sur la différenciation des lymphocytes auxiliaires. La découverte d’une population de lymphocytes Th capable de produire de l’IL-17 suite à une stimulation par l’IL-23 a permis de caractériser les lymphocytes auxiliaires Th17 (Harrington et al., 2005; Park et al., 2005). Les lymphocytes Th17 sont également capables de produire de l’IL-22, de l’IL-21 et de l’IFNγ (Harrington et al., 2005; Park et al., 2005; Liang
22
l’inflammation tissulaire et de l’autoimmunité. Outre leur implication dans l’EAE et l’arthrite rhumatoïde, leur implication fut également démontrée dans des pathologies inflammatoires chroniques de l’intestin telle la maladie de Crohn (Yen et al., 2006; van Beelen et al., 2007; Holtta et al., 2008) et des pathologies inflammatoires cutanées tel le psoriasis en induisant l’expression de gènes pro-inflammatoires dans les kératinocytes (Boniface et al., 2005a; Boniface et al., 2007; Wilson et al., 2007; Guilloteau et al., 2009). Les lymphocytes Th-17,
via la sécrétion d’IL-17, jouent également un rôle important dans la résolution de nombreuses
infections bactériennes et fongiques (Revue dans Curtis and Way, 2009).
A l’instar de l’IL-23, l’IL-1β peut également promouvoir la différentiation des lymphocytes T CD4 naïfs en lymphocytes Th17. Son action est tout de même plus précoce que l’IL-23, qui intervient plus dans le maintien des populations Th17, que dans leur différenciation (Wilson et al., 2007). Cette action de l’IL-1β est particulièrement intéressante au vu de la sécrétion exacerbée de cette cytokine dans nombre de pathologies auto-inflammatoires telles les fièvres périodiques héréditaires (Revue dans Masters et al., 2009). Intéressés par les mécanismes moléculaires induisant la sécrétion d’IL-1β dans ces pathologies, nous reviendrons plus en détails sur ce point dans le second chapitre de cette introduction. Depuis la mise en évidence de la population lymphocytaire Th17, son implication dans de nombreux phénomènes inflammatoires tissulaires a été démontrée. Ces cellules semblent être un point de relais entre systèmes immunitaires inné et adaptatif, ainsi qu’entre les cellules immunitaires et les cellules du tissu lésé permettant ainsi la spécificité de la réponse inflammatoire.
23 Figure 7 : Modèle de polarisation des lymphocytes auxiliaires (D'après Peck and
Mellins, 2009)
Les cellules CD4+ naïves peuvent se différencier en lymphocytes T auxiliaires, Th1, Th2 ou Th17 ou en lymphocytes T régulateurs en fonction de la nature des cytokines en présence. Elles sécrètent à leur tour des cytokines spécifiques à chaque lignée.
2.3 Résolution de l’inflammation.
Comme nous l’avons évoqué précédemment, une réponse inflammatoire non régulée peut conduire à sa pérennisation et à la destruction du tissu lésé, ou encore entrainer un choc
24
septique. Nombre de mécanismes sont mis en place dans le but d’arrêter la réponse inflammatoire et induire un retour à l’homéostasie.
2.3.1 Rôle des médiateurs lipidiques.
Des signaux anti-inflammatoires sont produits pour conduire à la résolution de l’inflammation (Revue dans Levy et al., 2001). Les lipides, telles les lipoxines, générées par les lipoxygénases à partir de l’acide arachidonique (Revue dans Serhan and Savill, 2005), les protectines et les résolvines, jouent un rôle majeur dans la promotion de la résolution du processus inflammatoire et de la réparation tissulaire (Revue dans Serhan, 2007). Les lipoxines, sécrétées par les macrophages, stoppent l’influx des neutrophiles, facilitent la phagocytose des neutrophiles apoptotiques par les macrophages et permettent le recrutement de monocytes afin d’éliminer les cellules apoptotiques et les débris cellulaires (Revue dans Serhan and Savill, 2005). En complément des lipoxines, les macrophages, les neutrophiles et les cellules épithéliales libèrent des inhibiteurs de protéases inactivant ainsi les protéases sécrétées par les neutrophiles lors de la phase aigue de la réponse inflammatoire (Ashcroft et
al., 2000).
2.3.2 Rôle du système nerveux.
Le système nerveux joue également un rôle important dans l’arrêt de l’inflammation. En effet, il a été montré qu’une stimulation du nerf vague atténue la réponse inflammatoire engendrée par le LPS (Borovikova et al., 2000b). Le système nerveux parasympathique induit l’activation des macrophages via la libération d’acétylcholine, son principal neurotransmetteur (Revue dans Tracey, 2002). De plus, la stimulation du nerf vague chez un modèle murin d’arthrite inhibe la réponse inflammatoire et supprime le gonflement des pattes, suggérant un rôle essentiel du système cholinergique dans l’inhibition de l’inflammation (Borovikova et al., 2000a). Cette action pourrait être liée à la diminution de la sécrétion de cytokines pro-inflammatoires (TNF, IL-1 et IL-18) et de l’induction de l’IL-10, une cytokine anti-inflammatoire (Borovikova et al., 2000b).
2.3.3 Rôle des lymphocytes T régulateurs.
A côté des Th, les cellules T CD4 naïves peuvent être différenciées en lymphocytes T régulateurs (Treg). Deux populations de cellules Treg sont décrites : les lymphocytes Treg
25 naturels CD25+, exprimant le facteur de transcription Forkhead box protein (Foxp3), et dérivant directement du thymus, et les lymphocytes Treg inductibles (iTreg) par le TGF-β (Tumor growth factor β) (Revue dans Josefowicz and Rudensky, 2009) mais également par l’IL-10 et l’IFN-γ (Levings et al., 2001). Deux populations iTreg majeures ont été décrites : les cellules Tr1 (Groux et al., 1997; Cottrez et al., 2000) et les cellules Th3 (Revue dans Weiner, 2001). Les lymphocytes Treg sont associés aux maladies auto-immunes dans lesquelles une partie de leurs fonctions semble altérée notamment dans la sclérose en plaque, le psoriasis et la polyarthrite rhumatoïde. Dans ces pathologies, la diminution de leur nombre induit une mauvaise régulation des lymphocytes CD8 « auto-réactifs », ce qui provoque l’auto-immunité (Revue dans Taams et al., 2006). Leur déplétion chez la souris, abolit leur tolérance aux Ag du soi, et conduit à l’apparition de pathologies inflammatoires chroniques (Sakaguchi et al., 1995).
Les lymphocytes Treg exercent leur activité anti-inflammatoire principalement à travers la sécrétion d’IL-10 et de TGF-β mais également par contact cellulaire direct avec les lymphocytes T effecteurs et les cellules présentatrices de l’Ag, concourant ainsi à la résolution du processus inflammatoire (Josefowicz and Rudensky, 2009).
2.3.4 Résolution de l’inflammation médiée par l’immunité innée.
Après avoir initier la réponse inflammatoire, les TLR contrôlent leur propre expression, conduisant à la résolution de la réponse inflammatoire. En effet, si l’activation des TLR conduit principalement au recrutement de la protéine adaptatrice MyD88, maillon de la translocation nucléaire de NF-κB, celui-ci va permettre l’expression de cytokines pro-inflammatoires, mais également de protéines anti-inflammatoire telle MyD88s qui empêche la signalisation induite par MyD88 (Revue dans Liew et al., 2005), ou l’IL-10 (de Waal Malefyt
et al., 1991). De plus des protéases telle TRIAD3A (pour TLR-Ubiquitinating Enzyme) sont
activées et interagissent avec le domaine cytoplasmique des TLR, provoquant leur ubiquitinylation puis leur dégradation par le protéasome (Revue dans Liew et al., 2005). En plus de ces actions anti-inflammatoires, les TLR induisent l’apoptose des macrophages.
Notons également que l’éradication des agents infectieux, donc de leurs PAMPs, diminue l’activation des TLR, et de cytokines et chimiokines pro-inflammatoires réduisant ainsi le recrutement leucocytaire (Revue dans Nathan, 2002). Les neutrophiles rentrent ensuite en apoptose et sont phagocytés par les macrophages induisant un vaste programme