UNIVERSITE DE GENÈVE
Département de biochimie FACULTÉ DES SCIENCES
Professeur S. Edelstein
Département de psychiatrie FACULTÉ DE MÉDECINE
Professeur J. Guimón
Professeur J.A. García-Sevilla
____________________________________________________________________________________________________
modulation et mécanismes régulateurs du récepteur
µ-opioïde, des
voies de signalisation associées et des neurofilaments
dans l’addiction aux opiacés
THÈSE
présentée à la Faculté des sciences de l’Université de Genève pour obtenir le grade de Docteur ès sciences, mention biochimique
par
Marcelino FERRER ALCÓN de
Valencia (Espagne)
Thèse N° 3322
GENÈVE 2002
Caminante, son tus
huellas
el camino, y nada
más;
caminante, no hay
camino,
se hace camino al
andar.
Al andar se hace
camino,
y al volver la
vista atrás
se ve la senda que
nunca
se ha de volver a
pisar.
Caminante, no
hay camino,
sino estelas en
la mar.
Antonio Machado
(1875-1939)
“Proverbios y
cantares”
Remerciements
Cette Thèse s’est déroulée au sein du Département de Psychiatrie de la Faculté de Médecine, dans l’Unité de Recherche Clinique.
Je tiens à exprimer ma très vive reconnaissance au Professeur José Guimón, qui m’a permis de réaliser ce travail et qui m’a témoigné une grande confiance durant ces trois années.
Je souhaiterais exprimer toute ma gratitude au Professeur Jesús A. García-Sevilla qui dès le début, s’est occupé de ma formation et a dirigé mes projets. Par ses compétences scientifiques et sa disponibilité il a toujours répondu à mes demandes. La quantité d’idées, de suggestions et de conseils, qu’il m’a prodiguée pendant toutes ces années continuent encore de m’impressionner. De plus, son optimisme et son enthousiasme resteront toujours un exemple à suivre pour moi.
Mes remerciements s’adressent également au Professeur Stuart Edelstein qui a accepté de co-diriger et de présenter ce travail à la Faculté des Sciences, en Biochimie.
Je voudrais aussi exprimer toute ma reconnaissance au Docteur Claude Walzer pour son amitié, sa patience et ses conseils, ainsi que pour son accueil chaleureux dans le laboratoire.
Je remercie les Professeurs Anne Kato et Karl-Heinz Krause d’avoir accepté de faire partie de mon comité de parrainage.
Je remercie aussi le Docteur Philippe Jaquet pour son aide dans la réalisation de l’étude de la modulation des NFs et surtout, pour son amitié.
Un grand merci au Docteur Muriel Grange-Midroit pour la lecture de mon travail de Thèse, pour sa patience et pour les discussions intéressantes que nous avons eues ensemble.
Mes remerciements vont également aux autres membres de ce groupe : Mesdames Pascale Marin, Béatrice Pastori, Christiane Aubry et Mariuccia Cathieni, pour leur précieuse assistance technique et morale.
A l’extérieur de l’Unité, je remercie les Docteurs Javier Meana, Pablo Escribá et Yuji Odagaki pour les discussions scientifiques que nous avons eues ensemble, mais aussi pour leur amitié et leur bonne humeur.
Finalement, j’aimerais remercier du fond du cœur les personnes qui en dehors du milieu professionnel m’ont soutenu et aidé durant toutes ces années. Merci à mes amis et à ma mère, qui malgré la distance, a toujours été à mes côtés.
Enfin rien n’aurait été possible sans la compréhension et l’amour de ma femme Encarni, qui m’a soutenu et encouragé tout au long de mon travail. Une dernière pensée pour ma fille Gizane. Sa tendresse et son innocence m’ont permis de travailler avec plus de courage et persévérance. Un grand bisou.
En souvenir de mon père.
Je remercie le Fond National de la Recherche Suisse (FNRS) qui a financé ce travail de Thèse par le subside n° : 31-52242.97.
TABLE DES MATIERES
1. LISTE DES ABREVIATIONS………...10
2. INTRODUCTION ET OBJECTIFS………..12
3. RAPPEL BIBLIOGRAPHIQUE………14
3.1. MORPHINE ET SES DERIVES.
3.2. RECEPTEURS AUX OPIACES.
3.3. OPIACES ENDOGENES.
3.4. ADDICTION AUX OPIACES.
3.5. MECANISMES MOLECULAIRES DE LA DEPENDANCE ET DE LA
TOLERANCE.
3.5.1. Dépendance.
3.5.2. Tolérance.
3.5.3. GRKs et arrestines.
3.5.3.1. Protéines régulatrices et opiacés
3.5.4. Régulation homologue des récepteurs aux opiacés.
3.5.5. Régulation hétérologue des récepteurs aux opiacés.
3.5.6. MAPKs (mitogen activated protein kinases).
3.6. PLASTICITE NEURONALE.
3.7. NEUROFILAMENTS (NFs).
3.7.1. Structure des NFs.
3.7.2. Croissance et transport axonaux.
3.7.2.1.Croissance axonale et NFs. 3.7.2.2.Transport axonal. 3.7.3. Assemblage des NFs. 3.7.3.1.Dimère. 3.7.3.2.Oligomère. 3.7.4. Phosphorylation des NFs.
3.7.4.1.Cdk5 (cyclin dependent kinase 5) et NFs.
3.7.4.2.MAPKs et NFs.
3.7.5. Phosphatases régulant la phosphorylation des NFs.
3.7.6. Pathologies.
3.7.6.1.Maladies des motoneurones.
3.7.6.2.Maladies neurodégénératives.
3.7.6.3.Neuropathies produites par des neurotoxines.
3.7.6.4.Modulation de NFs dans l’addiction aux opiacés.
4. MATERIELS ET METHODES ………50
4.1. APPAREILS.
4.2. REACTIFS.
4.4. SELECTION DE CERVEAUX HUMAINS (CONTROLES ET ADDICTS
AUX OPIACES).
4.5. ANIMAUX.
4.5.1. Traitement des rats avec de la morphine et de l’héroïne.
4.5.2. Traitement des souris avec de la morphine.
4.6. PREPARATION D’HOMOGENAT TOTAL DE CORTEX PREFRONTAL
HUMAIN ET DE CORTEX CEREBRAL DE SOURIS.
4.6.1. Homogénéité des préparations de cortex frontal humain.
4.7. PREPARATION DES PROTEINES MEMBRANAIRES DE CORTEX
CEREBRAL DE RATS.
4.8. ETUDE DU DELAI POST-MORTEM (PMD) ET DE L’AGE SUR LE
CERVEAU HUMAIN.
4.9. PREPARATION DE NFs PURIFIES.
4.10. DEPHOSPHORYLATION DES NEUROFILAMENTS (NFs)
PHOSPHORYLES AVEC LA REACTION A LA PHOSPHATASE
ALCALINE.
4.11. CULTURE CELLULAIRE.
4.11.1. Traitement des cellules SH-SY5Y avec de la morphine.
4.11.2. Inhibition de l’activité des kinases ERK1/2 par l’inhibiteur des kinases
MEK1/2, PD98059.
4.12. OBTENTION D’UN CULOT CELLULAIRE POUR ELECTROPHORESE.
4.13. QUANTIFICATION DES PROTEINES.
4.14. ELECTROPHORESE (SDS-PAGE).
4.16. TRANSFERT DES PROTEINES SUR MEMBRANES DE
NITROCELLULOSE.
4.17. IMMUNODETECTION DES PROTEINES.
4.18. QUANTIFICATION DES PROTEINES PAR DENSITOMETRIE.
4.19. LINEARITE DE LA CONCENTRATION DE PROTEINES POUR LE
WESTERN BLOTTING.
4.20. STATISTIQUE.
5. RESULTATS………72
5.1. ETUDES DANS LES CERVEAUX HUMAIN POST-MORTEM.
5.1.1. Statut du récepteur µ-opioïde dans les cerveaux d’addicts aux opiacés.
5.1.1.1.Caractérisation de l’antisérum Mu/2EL.
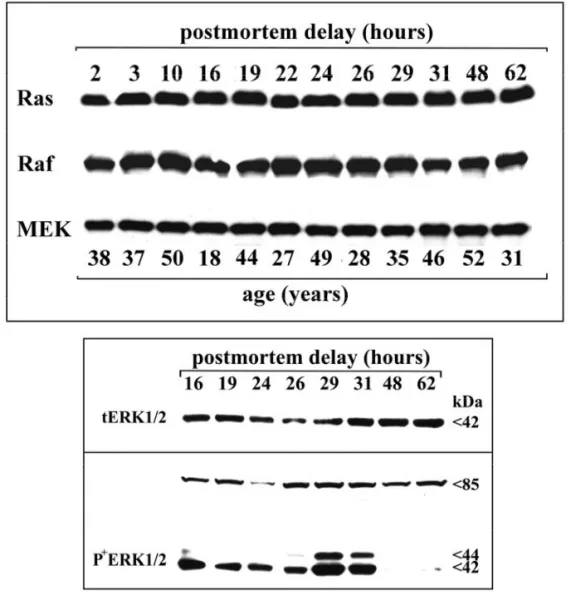
5.1.1.2. Effet du PMD et de l’âge sur l’immunoréactivité du récepteur µ.
5.1.1.3.Densité du récepteur µ dans les cerveaux d’addicts aux opiacés.
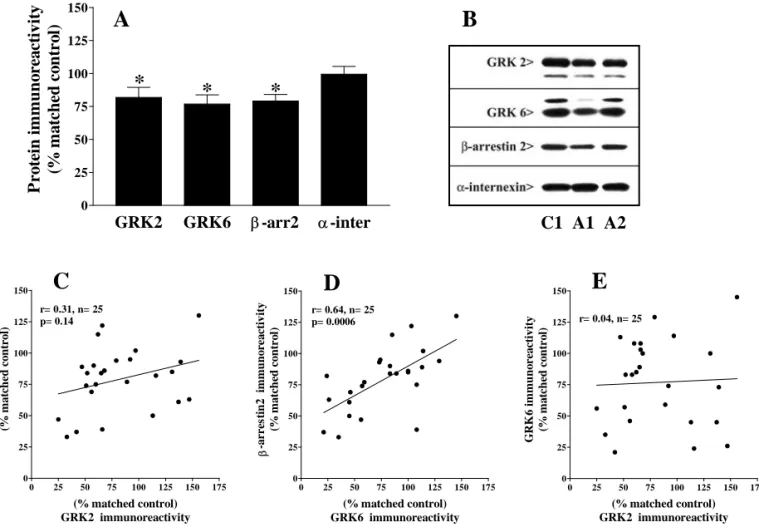
5.1.2. Régulation du récepteur µ-opioïde par les protéines GRKs et de la protéine β-arrestin2 dans le cerveau humain.
5.1.2.1.Caractéristiques des anticorps pour l’immunodétection des kinases
GRKs et de la protéine β-arrestin2 dans le cerveau humain.
5.1.2.2.Effet du PMD et de l’âge sur l’immunoréactivité des kinases GRKs et
de la protéine β-arrestin2.
5.1.2.3.Densité des protéines régulatrices du récepteur µ dans les cerveaux
5.1.2.4.Relation entre le récepteur µ-opioïde et les protéines impliques dans la
régulation homologue du récepteur.
5.1.3. Statut de la voie de l’AMPc dans le cerveaux d’addicts aux opiacés.
5.1.3.1.Caractérisation des anticorps utilisés pour l’immunodétection des
protéines appartenant à la voie de l’AMPc.
5.1.3.2.Effet du PMD et de l’âge sur les protéines cibles de la voie de l’AMPc.
5.1.3.3.Densité des protéines PKA, tCREB et pCREB dans les cerveaux
d’addicts aux opiacés.
5.1.3.4.Relation entre PKA et CREB.
5.1.4. Etat de la voie des MAPKs dans les cerveaux d’addicts aux opiacés.
5.1.4.1.Caractérisation des anticorps pour l’identification dans le cerveau
humain des protéines appartenant à la voie des MAPKs.
5.1.4.2.Effet du PMD et de l’âge sur les protéines appartenant à la voie des
MAPKs.
5.1.4.3.Densité des protéines de la voie des MAPKs dans le cerveau d’addicts
aux opiacés.
5.1.5. Etude de la modulation des neurofilaments dans le cerveau d’addicts
aux opiacés
5.1.5.1.Caractérisation des anticorps contre les différentes sous-unités de NFs
dans le cerveau d’addicts aux opiacés.
5.1.5.2.Effet du PMD et de l’âge sur les NFs dans le cerveau humain.
5.1.5.3.Densité des formes non-phosphorylées de NFs dans les cerveaux
d’addicts aux opiacés.
5.1.5.4.Densité de NFs phosphorylés et de la phosphatase PP2A dans les
5.1.5.5.Densité de la GFAP dans les cerveaux d’addicts aux opiacés.
5.1.6. Régulation du complexe p35/cdk5 dans le cerveaux d’addicts aux
opiacés : Relation avec les NFs.
5.1.6.1.Caractérisation des anticorps pour la détermination des densités des
protéines p35 et de cdk5 dans le cerveau humain.
5.1.6.2.Effet du PMD et de l’âge sur p35 et cdk5 dans le cerveau humain.
5.1.6.3.Densité de p35, de cdk5 et de NFs dans les cerveaux d’addicts aux
opiacés.
5.2. ETUDES DANS LES CERVEAUX DE RATS ET DE SOURIS.
5.2.1. Effet de la morphine sur la phosphorylation de NF-H dans les cerveaux
des rats.
5.2.1.1.Caractérisation des anticorps pour l’immunodétection des NFs dans les
cerveaux des rats
5.2.1.2.Effet des traitement chroniques et aigus de morphine et de naloxone sur
les protéines NF-L et NF-H phosphorylée dans les cerveaux des rats.
5.2.2. Spécificité de l’effet de la morphine sur les NFs. Souris Knockout pour
le récepteur µ-opioïde
5.2.2.1.Caractérisation des anticorps utilisés pour la détermination de la
spécificité de l’effet de la morphine sur les souris KO pour le récepteur µ
5.2.2.2.Détermination des taux de NF-H phosphorylée et de NF-L dans les
cerveau des souris WT et de souris KO pour le récepteur µ. Effet de la
morphine.
5.2.3. Effet de la morphine sur la voie de MAPKs dans le cerveau des rats.
5.2.4. Effet de la morphine sur le complexe p35/cdk5 dans les cerveaux des
5.3. MODULATION DE LA PHOSPHORYLATION DE NF-H ET DE LA VOIE
DES MAPKs PAR LES OPIACES DANS LA LIGNEE CELLULAIRE
SH-SY5Y.
5.3.1. Caractéristiques des anticorps utilisés pour la détermination des
kinases ERK1/2 et des NFs.
5.3.2. Estimation de la réponse d’ERK1/2 et de NF-H à la morphine.
5.3.3. Spécificité de l’effet de la morphine sur la voie des MAPKs dans les
cellules SH-SY5Y.
5.3.4. Relation de l’activation des ERK1/2 et la phosphorylation de NF-H.
6. DISCUSSION……….132
7. CONCLUSIONS GENERALES………...153
8. BIBLIOGRAPHIE……….155
1. LISTE DES ABREVIATIONS
AC : Adénylyl cyclase
AMP cyclique : 3'-5' Adénosine monophosphate -cyclique APS : Persulfate d’ammonium
BCA : Acide bicinchoninic BSA : Albumine de sérum bovine Cdk5 : Cyclin dependent kinase 5 CO2 : Dioxyde de carbon
CREB : cAMP response element-binding protein DMSO : Diméthylsulfoxide
DPBS : Tampon phosphate salin disodique avec EDTA ECL : Enhanced luminol chemiluminescent
EDTA : Ethylenediamine-tetraacetic acid
E64 : Trans-époxysuccinyl-L-leucylamido-(4-guanidino)-butane FCS : Sérum de veau foetal
GFAP : Protéine acide des fibrilles gliales
GRKs : Kinases spécifiques des récepteurs couples aux protéines G HCl : Acide chlorhydrique
IFs : Filaments intermédiaires
IOD : Densité optique intégrée
KCl : Chlorure de potassium
KH2PO4 : Dihydrogéno phosphate de potassium
KO : Knockout
KSP : Lysine-Sérine-Proline
MAPKs : Protéines kinases activées par les mitogènes
MgCl2 : Chlorure de magnésium
MoHCl : Chlorhydrate de morphine
Na2HPO4xH2O : Hydrogéno phosphate disodique dihydraté
NaCl : Chlorure de sodium NFs : Neurofilaments PA : Phosphatase alcaline PBS : Tampon phosphate salin PKA : Protéine kinase A
PKC : Protéine kinase C PMD : Délai post-mortem
PMSF : Phényl méthyle sulfoxy fluoride SDS : Dodécyl sulfate de sodium SNC : Système nerveux central SP : Pyrophosphate de sodium
TEMED : N,N,N',N'-Tétraméthylethylène-diamine TRIS : Tris (hydroximéthyl)-aminométhane
2-INTRODUCTION ET OBJECTIFS
L’addiction aux drogues d’abus comme les opiacés, est un phénomène complexe avec d’importantes causes et conséquences tant au niveau psychologique que social. L’exposition répétée aux opiacés produit des changements des fonctions cérébrales qui peuvent durer un certain temps après l’interruption de l’administration chronique de la drogue. En conséquence, les troubles de comportement, qui caractérisent l’addiction aux opiacés, sont intimement liés aux troubles fonctionnels des neurones du système nerveux central (SNC), donnant lieu à l’altération des circuits neuronaux dont ils font partie. A niveau biochimique, l’action des opiacés dans le cerveau se réalise par l’intermédiaire des récepteurs aux opiacés, principalement le récepteur µ. Ces récepteurs appartiennent à la famille des récepteurs aux sept domaines transmembranaires couplés aux protéines G ; ces protéines sont responsables de la liaison entre les récepteurs et les différentes voies effectrices qui vont réguler la réponse cellulaire aux opiacés.
Dans ce travail de thèse, les objectifs suivants sont proposés dans le but de mieux comprendre les mécanismes moléculaires de l’addiction aux opiacés ainsi que les altérations possibles induites par les opiacés tant au niveau structurel que fonctionnel :
1-Etudier, dans le cortex préfrontal des cerveaux des addicts aux opiacés décédés par overdoses, l’état du récepteur µ ainsi qu’un des mécanismes de régulation homologue, représenté par les kinases GRKs et la protéine β-arrestine.
2-Etudier, dans ces mêmes sujets addicts aux opiacés, la voie classique de transduction des systèmes opiacés correspondant à la voie de l’AMPc, où l’état de la kinase PKA et celui du facteur de transcription CREB sont deux paramètres importants.
3-Etudier, dans les cerveaux d’addicts aux opiacés, dans les cerveaux des rats traités à la morphine et dans la lignée cellulaire SH-SY5Y, une deuxième voie effectrice mise récemment en relation avec le récepteur µ. Cette voie correspond à la voie des kinases ERKs appartenant à la famille des MAPKs.
4-Finalement, dans le but d’étudier les effets possibles ainsi que la spécificité des opiacés sur le cytosquelette neuronal, une des principales protéines structurelles du
cytosquelette, le triplet de neurofilaments sera étudié dans le cerveau des addicts aux opiacés, dans les cerveaux des rats et des souris knockout pour le récepteur µ traités à la morphine et dans la lignée cellulaire SH-SY5Y. Dans ces modèles l’implication éventuelle de la kinase dépendante de cyclines cdk5 et de son activateur neuronal p35 sera aussi étudié en relation avec les processus d’addiction aux opiacés.
Pour atteindre ces objectifs nous avons utilisé trois modèles expérimentaux différents :
1- Le modèle expérimental le plus important dans cette étude est le cerveau humain post-mortem. Nous avons utilisé des biopsies de l’aire 9 de Brodmann du cortex préfrontal de sujets addicts aux opiacés (état tolerant) décédés d’une overdose d’héroïne ou de méthadone, appariés par le sexe, l’âge et le délai post-mortem à des sujets contrôles (sans aucune histoire d’addiction à une drogue quelconque) décédés accidentellement. Dans cette première partie du travail, nous avons effectué des comparaisons entre les addicts aux opiacés et les contrôles portant sur : (a) les taux des différentes protéines impliquées dans la régulation du récepteur µ, (b) les taux des protéines participant à la signalisation cellulaire contrôlée par ce récepteur ainsi que (c) les taux et l’état des certaines protéines appartenant au cytosquelette neuronal.
2- Le deuxième modèle exploré dans cette étude est le modèle animal, dans lequel des rats ainsi que des souris transgèniques n’exprimant pas le récepteur µ ont été utilisés. Les rats ont été utilisés pour étudier (a) si les altérations observées dans le cerveau humain sont des altérations générales du cerveau des mammifères et (b) si elles sont dues à l’effet chronique des opiacés ou si au contraire, elles sont dues à l’effet aigu de la dernière dose mortelle, prise par les addicts aux opiacés.
Les animaux transgèniques ont été utilisés pour confirmer si les modifications des protéines du cytosquelette neuronal (neurofilaments) observées dans les processus d’addiction aux opiacés, sont médiées par le récepteur µ.
3- Finalement, pour étudier un des mécanismes potentiellement responsables des modifications de l’état de neurofilaments en présence de la morphine, la lignée cellulaire SH-SY5Y, employée habituellement comme un modèle neuronale in vitro, a été utilisée.
3. RAPPEL BIBLIOGRAPHIQUE
L’opium est l’exsudat laiteux et desséché de la capsule des graines du Papaver
somniferum et il est certainement l’un des agents pharmacologiques les plus
anciennement connus. Sertürner parvient en 1806 à isoler à partir de l’opium une substance cristalline qu’il appela morphine, du grec Morpheus, dieu du sommeil. La morphine fut donc le premier alcaloïde isolé à l’état pur. On connaît maintenant plus de 20 alcaloïdes extraits de l’opium. Ces alcaloïdes représentent approximativement 25% du poids de l’opium et sont classés en deux catégories chimiques distinctes : (1) les dérivés du phénanthrène et (2) les dérivés benzylisoquinoléines.
3.1. MORPHINE ET SES DERIVES
La morphine est le principal dérivé du phénanthrène (10% de l’opium) et a été surtout utilisée pour ses propriétés antinociceptives, mais s’est révélée être une drogue dont l’usage chronique peut engendrer tolérance et dépendance. L’effort des chimistes et des pharmacologues fut donc dirigé vers le développement de substances dérivées de la morphine, possédant des propriétés analgésiques et antidiarrhéiques mais dépourvues de la capacité d’induire tolérance et dépendance. Cet effort aboutit aux dérivés morphiniques semi-synthétiques (molécules préparées en modifiant la structure de la morphine) et synthétiques (synthèse complète de molécules originales). On les regroupe sous le terme de substances opiacées (ou opioïdes).
Deux composés d'intérêt pharmacologique et thérapeutique sont à retenir : l’héroïne et la méthadone. L’héroïne ou diacétylmorphine est le premier exemple d'un opiacé semi-synthétique. L’héroïne est un dérivé de la morphine plus soluble que la morphine et donc permet le passage de la barrière hémato-encéphalique plus rapidement. Elle agit par conséquence plus rapidement, mais dans un temps plus court. La méthadone (opiacé synthétique) fut synthétisée en Allemagne durant la deuxième guerre mondiale; son intérêt thérapeutique réside dans la possibilité de l’administrer par voie orale et dans sa longue demi-vie. Ces propriétés la rendent particulièrement intéressante dans le traitement de la pharmacodépendance aux opiacés.
La plupart des opiacés actuellement disponibles possèdent, à différents degrés, les propriétés pharmacologiques de la morphine, qui produit ses effets pharmacologiques principaux au niveau de trois systèmes de l’organisme : le système nerveux central (SNC), le système gastro-intestinal et le système cardio-vasculaire (Jaffe et Martin, 1990).
Chez l’homme, l’administration de morphine a plusieurs effets sur le SNC : (1) des altérations de l’humeur, (2) une analgésie, (3) des nausées et vomissements, (4) une dépression de la respiration (elle est la cause principale des décès par intoxications aux opiacés) et (5) des myosis. D’une manière générale, on peut dire que la morphine augmente le tonus de la musculature lisse de tous les segments du tractus gastro-intestinal, tout en diminuant considérablement les contractions propulsives. Cette double action résulte en un ralentissement prononcé du temps de passage du bolus alimentaire et une réabsorption d'eau. L’effet global de la morphine est donc constipant, d’où son utilisation thérapeutique comme antidiarrhéique. A doses thérapeutiques, la morphine produit une dilatation artériolaire et veineuse. Cette action, conjointement avec l’effet inhibiteur sur les barorécepteurs, conduit à l’hypotension orthostatique que l’on peut parfois observer chez les patients sous morphine.
3.2. RECEPTEURS AUX OPIACES
Pour les opiacés, l’existence de récepteurs fut postulée au début de ce siècle déjà et confirmée dans les décennies suivantes. En effet, de nombreuses études sur les relations structure-activité révélèrent l’existence d’une stéréospécificité stricte pour les actions des opiacées (Pasternak, 1993). On développa ensuite des antagonistes compétitifs de la morphine qui démontrèrent son action dose-dépendante, aboutissant à un effet maximal (saturabilité), sur différentes préparations in vitro. Enfin, l’induction d’un syndrome de sevrage par l’administration d’un antagoniste compétitif, après traitement chronique aux drogues opiacées, ainsi que l’apparition d’une tolérance croisée entre opiacés possédant une structure moléculaire différente, constituèrent des arguments ultérieurs plaidant en faveur de l’existence de récepteurs spécifiques. La preuve définitive ne fut toutefois apportée qu’au début des années 70. Goldstein et al. (1971) découvrirent avec l’utilisation de lévorphanol marqué radioactivement (études de marquages radioactifs)
des sites de fixation aux opiacés dans une fraction subcellulaire du cerveau de souris. En 1973 différents groupes purent directement identifier et caractériser par la méthode de fixation spécifique utilisée par Goldstein et al., les récepteurs aux opiacés, grâce au développement de ligands radioactifs à très haute affinité (de l’ordre de la nanomole) et porteurs d’une activité spécifique élevée (Pert et Snyder, 1973 ; Terenius, 1973 ; Simon et al. 1973). La première indication de l’existence des multiples types de récepteurs opioïdes fut postulée lors de l’examen des actions d’un dérivé N-allyle de la morphine, la nalorphine (revu dans Pasternak, 1993). En effet, cet opiacé se comportait, en présence de morphine, comme un antagoniste, mais possédait les propriétés analgésiques d’un agoniste lorsqu’il était administré seul. Ces travaux permettaient à Martin (1967) de proposer le concept de « dualisme du récepteur ». L’auteur postula l’existence de deux types de récepteurs, l’un pour la morphine (appelé M), et l’autre pour la nalorphine (appelé N). Avec la découverte de nouveaux opiacés de synthèse, le modèle à deux récepteurs se révéla insuffisant pour expliquer la diversité et la sélectivité des actions de nombreuses molécules. Martin et al. (1976) distinguèrent entre le récepteur du type µ (pour morphine), le récepteur du type κ (pour ketocyclazocine) et le récepteur du type σ (pour SKF10,047 ou N-allylnormetazocine). Plus tard Lord et al. (1977) ont décrit le récepteur du type δ (pour vas deferens). Des recherches postérieures montrèrent que le récepteur du type σ n’était pas un récepteur aux opiacés, et la classification des récepteurs opioïdes s’est maintenue aux trois sous-types, µ, δ, et κ (Dhawan et al., 1996). L’utilisation d’antagonistes irréversibles et d’agonistes spécifiques a suggéré l’existence de différentes sous-types des récepteurs aux opiacés. Ces sous-types pourraient correspondre formellement à différents états d’un même récepteur, dépendant de son couplage avec les protéines G de transduction. D’après Pasternak (1993), il y aurait deux sous-types de récepteurs µ, appelés µ1 et µ2. Le profil
pharmacologique du récepteur µ cloné semble correspondre à celui du récepteur µ1. Le
sous-type µ1 a une haute affinité par la morphine et semble médier ses effets
analgésiques. Par contre le sous-type µ2 a une plus faible affinité pour la morphine et
semble être le responsable de la dépression respiratoire induite par la morphine. Certaines données suggèrent que l’héroïne, le fentanyl et l’un des métabolites de la morphine, le 6 β-glucuronide (M6G) agissent via un sous-type de récepteur µ distinct de ceux par l’intermédiaire desquels la morphine exerce ses effets. Une seule protéine présentant le profil pharmacologique d’un récepteur δ a été clonée. Toutefois de
nombreuses données suggèrent l’existence de récepteurs δ1 et δ2. Un certain nombre
d’agonistes et d’antagonistes des récepteurs δ sont d’ailleurs aujourd’hui considérés comme des ligands spécifiques, ou sélectifs, de l’un ou de l’autre sous-type. L’existence de sous-types des récepteurs κ est moins bien établie, mais ils sont actuellement séparés en trois sous-types : κ1, κ2et κ3 en fonction de leurs affinités respectives pour différents
opiacés (Dhawan et al., 1996). Des données récentes semblent indiquer que les récepteurs aux opiacés, de la même manière que d’autres récepteurs de la famille de récepteurs couplés aux protéines G, peuvent former des hétérodimères, lesquels auront des propriétés pharmacologiques différentes des homodimères, et de ce fait pourraient contribuer à leur pharmacologie complexe, même s’il n’existe pas qu’un seul gène pour chaque récepteur (Williams et al., 2001). La classification des récepteurs aux opiacés sur une base pharmacologique est encore en évolution, et certaines observations suggèrent l’existence d’autres types de récepteurs des opioïdes. Ainsi le récepteur ε, le récepteur ξ et un site de haute affinité λ, pourraient en faire partie (Van Ree et al., 1999). On peut trouver aussi le récepteur N/OFQ (pour récepteur orphelin) qui a un haut degré d’homologie structurelle avec les récepteurs aux opiacés mais qui a une réponse pharmacologique différente aux agonistes classiques des récepteurs aux opiacés. Ce récepteur peut être considéré comme un récepteur homologue plutôt que un récepteur aux opiacés (Alexander et Peters, 2000). Le comité sur les récepteurs aux opiacés de l’IUPHAR (International Union of Pharmacology) a proposé une autre terminologie pour différencier les récepteurs aux opiacés : récepteurs MOP, DOP et KOP (par Mu
Opioid Peptide, Delta Opioid Peptide et Kappa Opioid Peptide). D’autres
nomenclatures sont OP3 et MOR pour les récepteurs µ, OP1 et DOR pour les récepteurs δ, et OP2 et KOR pour les récepteurs κ. (Tableau I) (Dhawan et al., 1996 ; Alexander et Peters, 2000).
Tous ces récepteurs ont été clonés par des techniques de biologie moléculaire (Reisine et Bell, 1993) et possèdent des séquences d’environ 370 acides aminés. Ils font partie de la famille des récepteurs aux sept domaines transmembranaires couplés aux protéines G, avec une extrémité N-terminale extracellulaire et une extrémité C-terminale intracytoplasmique. La comparaison de séquence des structures primaires des récepteurs µ, δ, et κ montre une homologie de 65-70% entre eux, avec une très haute homologie dans les domaines transmembranaires, les boucles intracellulaires et dans une petite portion de l’extrémité C-terminale proche du septième domaine transmembranaire. Par
contre, la deuxième et la troisième boucle extracellulaire sont très hautement divergentes (Jordan et Devi, 1998). Ces récepteurs possèdent également des sites de phosphorylation, permettant de moduler leur activité, par exemple leur degré de désensibilisation, comme cela sera discuté ultérieurement.
Tableau I-. Nomenclature des récepteurs aux opiacés (Van Ree et al., 1999 ; Alexander et Peters, 2000)
Récepteurs aux opiacés
Recommandation de l’IUPHAR Nomenclature classique Autres nomenclatures
Ligands opiacés endogènes préférentiels
DOP δ OP1, DOR Enképhalines, β-endorphines
KOP κ OP2, KOR Dynorphines
MOP µ OP3, MOR Endomorphine-1
3.3. OPIACES ENDOGENES
La découverte de récepteurs spécifiques pour la morphine et ses dérivés dans le système nerveux posa la question suivante : comment se faisait-il que le système nerveux de l’homme possédât des récepteurs spécifiques pour un alcaloïde extrait d’un pavot originaire d’Asie Mineure et d’Extrême Orient ? La réponse était simple : le système nerveux produit des opiacés endogènes. La première indication de l’existence d’opioïdes endogènes venait des travaux qui montraient que des extraits de cerveau contenaient une activité semblable à celle produite par les opiacés (revu dans Van Ree et al., 2000). D’autres investigations conduisaient à l’isolement et la caractérisation des enképhalines, les premiers opiacés endogènes découverts (Hughes et al., 1975), qui apparaissaient formés par deux pentapeptides, la leucine–enképhaline et la méthionine-enképhaline, dont les actions, semblables à celles de la morphine, sont neutralisées par
la naloxone, un antagoniste spécifique des récepteurs aux opiacés. Au cours de la dernière décennie, on a identifié et caractérisé dans le système nerveux de nombreux autres opiacés endogènes. Très récemment a été montré que ces peptides peuvent être regroupés en trois familles, chacune dérivée d’un précurseur à poids moléculaire élevé (Van Ree et al. 1999). Les différents ligands opioïdes endogènes montrent des préférences pour les différents récepteurs aux opiacés : endomorphines pour les récepteurs µ, enképhalines et β-endorphines pour les récepteurs δ et dynorphines pour les récepteurs κ (Tableau I). Ces peptides, et les précurseurs dont ils sont dérivés sont contenus dans des groupes de neurones définis ; certains sont également présents à l’extérieur du SNC, notamment au niveau du tractus gastro-intestinal (Jaffe et Martin, 1990). Il est intéressant de relever que certains peptides sont présents dans des régions du SNC dont on connaît le rôle dans l’expression des actions de la morphine.
3.4. ADDICTION AUX OPIACES
L’addiction aux drogues d’abus comme les opiacés, est un phénomène complexe avec d’importantes causes et conséquences tant au niveau psychologique que social. Néanmoins, à la base de ce processus, il y a une implication biologique : l’effet de l’exposition répétée à un agent biologique (drogue) sur un substrat biologique (cerveau) (Nestler et Aghajanian, 1997). Le terme d’addiction, peut être défini comme la perte de contrôle sur l’utilisation d’une drogue ou la recherche et la prise de drogues d’une manière compulsive, malgré les conséquences négatives qui sont générées. L’addiction se produit par les actions d’une drogue d’abus sur un cerveau vulnérable et a besoin normalement d’une prise répétée de la drogue. Ces processus sont influencés par les caractéristiques génétiques de l’individu, son état psychologique ainsi que par le contexte social qui entoure la prise de la drogue. En dépit de quelques aspects de l’addiction qui se traduisent par une réponse rapide à une administration aiguë de drogues, la plupart des changements des fonctions cérébrales, qui sont associés à l’addiction, ont une réponse graduelle après une longue exposition aux drogues. Ces changements peuvent persister un certain temps après l’interruption de l’administration chronique de la drogue, c’est à dire, l’addiction produit des changements très stables dans le cerveau et de ce fait peut être vu comme un inducteur de la plasticité neuronale
et servir comme un modèle pour étudier les mécanismes neurobiologiques qui sont impliqués (Nestler, 2001).
Bien que les drogues d’abus soient des molécules chimiquement différentes et avec des activités initiales différentes, l’addiction qui en résulte montre des traits très semblables. Cela peut-être expliqué parce que chaque type de drogue, malgré les différents actions qu’elle entraîne sur le cerveau, converge dans la production d’effets similaires, par exemple l’activation du système mésolimbique de la dopamine, qui implique une augmentation de l’activité des neurones dopaminergiques dans l’aire tegmentale ventrale ou VTA (par aire tegmental ventral) du cerveau moyen et par voie de conséquence une augmentation de la libération de la dopamine vers le noyau accumbens et d’autres régions du cerveau antérieur limbique, comme le cortex frontal (Figure I).
Figure I-. Circuits neuronaux fondamentaux de l'addiction. Les lignes en pointillés
montrent les afférentes limbiques au noyau accumbens (NAc). Les lignes en bleu représentent les efférentes du NAc que l’on pense impliqués dans les phénomènes de récompense dans les processus d'addiction. Les lignes de couleur rouge indiquent les projections du système mésolimbique dopaminérgique qui pourraient être des substrats importants pour le phénomène de récompense aux drogues. Parmis les neurones dopaminérgiques dont l’origine est dans la VTA et qui sont dirigés vers le NAc et vers d’autres structures limbiques, ont trouve ceux du bulbe olfactif (OT), du domaines ventrales du caudate-putamen (C-P), de l’amygdale (AMG) et du cortex préfrontal (PFC). En vert sont montrés les neurones qui contiennent les peptides opioïdes, lesquels sont impliqués dans les processus de récompense aux opiacés, à l’éthanol et à la nicotine. Dans ces
systèmes des peptides opioïdes sont compris les circuits locaux des enképhalines (segments courts) ainsi que le circuit de β-endorphine du cerveau moyen hypothalamique (segment long). Les régions colorées en jaune indiquent la distribution approximative des complexes des récepteurs GABAA qui pourraient être impliqués dans les processus de récompense à
l’éthanol. Les petites structures de couleur jaune représentent les récepteurs nicotiniques dans des neurones que contiennent des peptides opioïdes et de la dopamine. (ARC, noyau arque; Cer, cervelet; DMT, thalamus dorsomédian; IC, collicule inférieur; LC, locus cœruleus; LH, hypothalamus latéral; PAG, substance gris periaqueduquale; SC, collicule supérieur; SNr, substance noire; VP, pallidum ventral) (modifications de la figure de Nestler, 2001).
Le système mésolimbique de la dopamine et ses cibles dans le cerveau antérieur font partie du système motivationnel qui régule la réponse à renforçateurs naturels, comme la nourriture, les boissons, le sexe et les interactions sociales. Les drogues d’abus altèrent cette voie d’une manière plus importante que les renforçateurs naturels. En conséquence un mécanisme possible de l’addiction pourrait être que dans ces neurones, la stimulation forte et répétée par des drogues d’abus induise des changements qui amèneront à des altérations dans les mécanismes de renforcement et dans l’état motivationnel de l’individu (Nestler, 2001). Plusieurs types d’altérations ou mécanismes adaptatifs déclenchés par la prise de drogues et qui caractérisent l’état d’addiction ont été décrits : (a) la tolérance est la nécessité d’augmenter les doses de la drogue pour obtenir un effet semblable à la dose précédente ; (b) la sensibilisation est la situation opposée à la tolérance, où des doses constantes produisent une augmentation des effets ; (c) la dépendance est un état psychologique perturbé induit par une exposition chronique à des drogues et qui nécessite la prise répétée de ces drogues afin de ne pas subir les effets pervers physiques et psychiques de son absence, qui caractérisent le syndrome d’abstinence (Nestler et al., 1993). En conséquence, ces troubles de comportement qui caractérisent l’addiction, sont intimement liés aux troubles fonctionnels des neurones du SNC, donnant lieu à l’altération des circuits neuronaux dont ils font partie. Cependant les mécanismes moléculaires fondamentaux de la dépendance et de la tolérance restent partiellement inconnus (Nestler et Aghajanian, 1997). Par contre, le rôle crucial du récepteur µ dans le développement et l’expression de l’addiction à la morphine est maintenant clair. L’utilisation des souris transgéniques qui n’expriment pas le gène du récepteur µ a été déterminante (Matthes et al., 1996). Ces souris avaient une perte du phénomène d’analgésie induit par la morphine. De plus ces souris avec un traitement chronique de morphine n’avaient pas de syndrome de sevrage
en présence de naloxone (antagoniste du récepteur µ capable d’induire le sevrage chez les rats traités chroniquement avec de la morphine (Ventayol et al., 1997)). Dans ce contexte, l’implication de la désensibilisation du récepteur µ-opioïde et/ou la
down-régulation du récepteur a été longtemps postulée dans la tolérance et la dépendance à la
morphine. Gabilondo et al. (1994) ont réalisé une étude de fixation de radioligands aux récepteurs µ dans le cerveau d’animaux dépendants aux opiacés et une mesure de la densité des récepteurs µ dans les cerveaux d’héroïnomanes décédés par overdoses. Ils ont montré qu’il n’y avait pas de modulation du récepteur µ dans aucun de ces deux modèles expérimentaux. De la même manière, les niveaux d’ARNm des récepteurs µ et δ restent invariables dans les cerveaux des rats traités chroniquement avec de la morphine (Brodsky et al., 1995 ; Buzas et al., 1996). Les niveaux d’immunoréactivités du récepteur µ dans les cerveaux d’héroïnomanes décédés par overdoses n’avaient pas de changements significatifs (García-Sevilla et al., 1997a). Finalement Meana et al. (2000) ont montré avec une étude de fixation de [35S]GTPγS que dans le cerveau d’addicts aux opiacés décédés par overdoses le récepteur µ semble avoir un comportement normal avec le couplage des protéines Gi/G0. Toutes ces données
suggèrent que le récepteur µ n’est pas down-régulé pendant le processus d’addiction aux opiacés et que le système des messagers intracellulaires, c’est à dire le système de postrécepteurs, devient le mécanisme principal pour comprendre les processus d’adaptation neuronale induits par l’action des drogues opiacées. Il a été démontré que l’exposition chronique aux opiacés provoque l’adaptation de quelques systèmes intracellulaires qui sont induits par l’exposition à des drogues. D’ailleurs un tel processus d’adaptation a été impliqué dans les phénomènes de la tolérance et de la dépendance, et a pu être démontré au niveau de neurones individuels.
3.5. MECANISMES MOLECULAIRES DE LA DEPENDANCE ET DE LA TOLERANCE
3.5.1. Dépendance.
Le mécanisme de la dépendance le mieux connu dans les cellules individuelles, correspond à l’up-régulation de la voie de l’AMP cyclique (adénosine monophosphate
de neuroblastome x gliome (Sharma et al., 1975), et plus tard dans des neurones du locus cœruleus (Nestler et al., 1993) ainsi que dans les plaquettes d’héroïnomanes et le cerveau d’héroïnomanes décédés par overdose (García-Sevilla, et al., 1987 ; Thilo et Burnet, 1992 ; Escribá et al., 1994). L’exposition aiguë aux opiacés inhibe les neurones du locus cœruleus directement à travers une augmentation de la conductance de canaux de K+, via la protéine liée au GTP ou protéine G sensible à la toxine de Pertussi (Giα
et/ou Goα) et indirectement par la diminution du courant intérieur dépendent de Na+
aussi via Giα ou Goα . Ces phénomènes entraînent l’inhibition de l’adénylyl cyclase (AC)
et la diminution de la production basale de l’AMPc. La réduction des taux d’AMPc diminue l’activité de la protéine kinase dépendante de l’AMP cyclique (PKA) et la phosphorylation des canaux ioniques et des phospho-protéines associés. Entre autre, l’inhibition de la voie de l’AMPc diminue la synthèse de catécholamines par une réduction de la phosphorylation de la tyrosine hydroxylase, ainsi que par des altérations de l’expression des gènes via l’altération de facteurs de transcription. Par contre, l’exposition chronique aux opiacés conduit à une up-régulation de la voie de l’AMPc des différentes régions du système nerveux central et périphérique. Cette up-régulation implique l’augmentation des taux de quelques sous-types de l’AC et de la quantité des protéines G associées (Giα et/ou Goα), une augmentation du niveau basal de l’AMPc ainsi qu’une augmentation de l’activité de la PKA et de quelques-uns de ses substrats comme la cyclic-AMP response-element-binding protein (CREB) (Figure II). CREB est un facteur de transcription qui régule la transcription de gènes qui contiennent, à l’intérieur de régions régulatrices, un site de reconnaissance appelé CRE (pour cAMP
response element ; séquence consensus TGACGTCA). Cette séquence a été décrite dans
la plupart des gènes qui s’expriment dans le système nerveux. CREB est capable de s’unir à CRE sous forme d’un dimère et active la transcription seulement quand les deux sous-unités sont phosphorylées sur la sérine 133 par différentes protéines kinases parmi lesquelles on trouve la PKA. Le rôle de CREB dans les processus d’addiction a été démontré avec des souris qui avaient de mutations dans le gène codant pour CREB. Ces animaux montraient une diminution du développement de la dépendance après un traitement chronique à la morphine, et une atténuation du syndrome de sevrage après l’administration d’antagonistes du récepteurs aux opiacés (Maldonado et al., 1996). Les différences entre les effets produits par le traitement aigu et par le traitement chronique, représentent une forme de tolérance physiologique ; quand la prise de drogue
est arrêtée, le système up-regulé devient complètement fonctionnel et contribue ainsi aux caractéristiques essentielles de la dépendance et du syndrome d’abstinence (Nestler et Aghajanian, 1997 ; Nestler, 2001).
3.5.2. Tolérance.
Les mécanismes sous-jacents de la tolérance restent inconnus, mais probablement chevauchent avec les mécanismes fondamentaux de la dépendance. L’up-régulation du système de l’AMPc peut probablement contribuer à la tolérance en limitant l’intense inhibition du flux intérieur de Na+ induite par les opiacés, via l’inhibition du système de l’AMPc. Il est aussi possible que l’up-régulation de la voie de l’AMPc puisse induire une augmentation de la quantité des récepteurs aux opiacés qui ont été désensibilisés, par phosphorylation. Pour promouvoir la désensibilisation des récepteurs dans l’état tolérant, le système up-regulé de l’AMPc pourrait mener à une réduction de la capacité des opiacés à activer intensément les protéines G, les canaux de K+ et le courant de Na+ (Nestler et al., 1993). Au niveau cellulaire, la tolérance peut-être vue comme une forme persistante de désensibilisation du récepteur, associée avec l’administration répétée de drogues.
Deux formes de régulation rapide ont été caractérisées : la régulation homologue et la régulation hétérologue ou non-spécifique de l’agoniste. La régulation homologue indique que lorsqu’un récepteur est activé par l’agoniste, ce mécanisme désensibilise seulement la réponse ultérieure de ce récepteur et son interaction spécifique avec les voies de transduction de signalisation, sans aucun effet sur d’autres types de récepteurs présents dans la même cellule. Par contre, la régulation hétérologue indique que la stimulation d’un agoniste diminue la réponse d’agonistes multiples et différents qui agissent avec différents types de récepteurs (Chuang et al., 1996).
Comme cela a été déjà décrit, le récepteur µ appartient à la famille des récepteurs aux sept domaines transmembranaires couplés aux protéines G, et partage avec eux des mécanismes de régulation similaires. Lefkowitz (1993) a montré un de mécanismes possibles de la régulation homologue avec le récepteur β-adrénergique. Ses résultats peuvent être extrapolés aux autres récepteurs de la même famille, parmi lesquels se trouvent les récepteurs aux opiacés. Deux types de protéines semblent jouer un rôle très important dans ce mécanisme : les G protein-coupled receptor kinase (GRKs) et les arrestines.
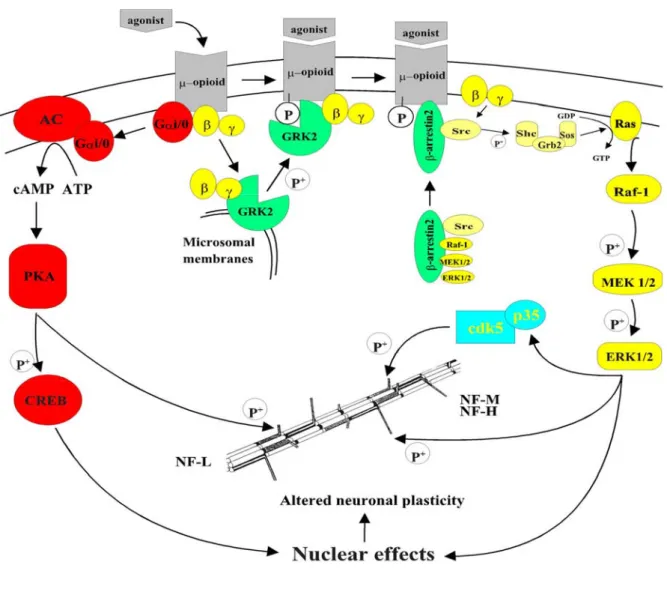
Figure II.- Complexité des voies de transduction de signalisation dans les processus
d'addiction aux opiacés. L'activation du récepteur µ par la morphine induit un changement
de conformation du récepteur qui conduit à une activation des protéines G et de ce fait à leur dissociation du récepteur. Les différentes sous-unités des protéines G peuvent activer plusieurs voies de transduction du signal. En couleur rouge est montrée la voie de l'AMPc. La sous-unité Gαi/o active l'adénylate cyclase (AC) qui est responsable de la production
d'AMPc, qui activera à son tour à la protéine kinase dépendant de l'AMPc (PKA). Cette kinase peut phosphoryler différents substrats, comme le facteur de transcription nucléaire CREB ou le triplet de neurofilaments (NFs). Les sous-unités Gβγ sont impliquées dans la
désensibilisation du récepteur via les kinases GRKs (vert) ou peuvent activer directement la voie des MAPKs (jaune). Les sous-unités Gβγ provoquent la translocation à la membrane de
la kinase GRK2, qui peut phosphoryler le récepteur et le rendre inactif. Le récepteur phosphorylé peut s'unir avec une très haute affinité à la β-arrestine2. Le complexe ainsi formé peut être internalisé pour être resensibilisé ou dégradé. La β-arrestine2 peut jouer aussi un rôle de protéine d'ancrage, et peut aussi se lier aux différents composants de la voie des MAPKs, et induire leur activation. Cette voie peut aussi être activée par les sous-unités Gβγ lesquelles peuvent activer la protéine Ras qui déclenchera l'activation des MAPKs. Ces
kinases pourraient phosphoryler, entre autres substrats, les NFs. Finalement, le complexe p35/cdk5 (en bleu) peut aussi phosphoryler les NFs.
3.5.3 GRKs et arrestines.
Sept gènes différents de GRKs ont été décrits, nommés GRK1 jusqu’à GRK7 (Tableau II), et classés en trois sous-familles différentes sur la base de la structure du gène, la similarité de séquence, leur fonction et leur régulation. Cette classification est la suivante : les kinases du type GRK1 (GRKs 1 et 7, aussi nommées opsin ou rhodpsine kinase), les kinases du type GRK2 (GRKs 2 et 3, autrefois nommées β-adrenergique
receptor kinase, βARK1/2) et les kinases du type GRK4 (GRKs 4, 5 et 6) (Pitcher et al.,
1998 ; Krupnick et Benovic, 1998 ; Premont et al., 1999).
Tableau II-. Propriétés moléculaires des GRKs (Krupnick et Benovic, 1998 ; Ferguson, 2001)
Kinase Taille (kDa) Nom classique Association à la membrane Distribution tissulaire Caractéristiques et régulation
GRK 1 63 RK* Farnesylation Rétine Autophosphorylation GRK 2 79 βARK1 Gβγ, PL acides Ubiquitaire Domaine PH, PKC,
calmoduline
GRK 3 80 βARK2 Gβγ, PL acides Ubiquitaire Domaine PH GRK 4 66 IT-11 Palmitoylation Testicules Quatre variants d’épissage GRK 5 68 Fixation aux PL Ubiquitaire Autophosphorylation, PKC,
calmoduline GRK 6 66 Palmitoylation Ubiquitaire Calmoduline
GRK 7 62 N.D. Rétine (cône)
*RK, rhopsine kinase ; PL, phospholipides ; PH, domaine d’homologie à la pleckstrin ; N.D., non-déterminé ; PKC, protéine kinase C.
Les kinases GRK2/3 ont un domaine d’homologie à pleckstrine (important pour l’union à la PKC) et sont régulées par les sous-unités Gβγ de protéines G, mais par contre les kinases GRK4/5/6 sont insensibles à la régulation par les sous-unités Gβγ. Les sous-unités Gβγ et les phospholipides ont un rôle important dans la médiation de la translocation des kinases GRK2/3 du cytosol à la membrane ; néanmoins la fixation de
phospholipides aux kinases GRK4/5/6 induit la liaison aux récepteurs activés sans le concours de sous-unités Gβγ (Pitcher et al., 1998). Les cinq kinases GRKs extra-rétinales
(GRKs 2-6) sont largement exprimées (aussi bien au niveau de l’ARNm que de la protéine) dans le cerveau et dans d’autres tissus. L’identification de différents récepteurs qui peuvent être des substrats pour les kinases GRK (c’est à dire si une seule GRK peut phosphoryler un seul type particulier de récepteur) est importante pour comprendre la régulation du récepteur et le rôle physiopathologique possible de ces kinases.
Les arrestines sont des protéines solubles qui fonctionnent conjointement avec les kinases GRK pour arrêter le signal intracellulaire. En présence continue d’un stimulus, les arrestines assurent que chaque récepteur activé couplé aux protéines G, sera mis
“hors de service” pour éviter le renouvellement du signal de transduction. Ces protéines
sont divisées en deux sous-types en fonction de leur localisation tissulaire : visuelle ou non-visuelle (Tableau III).
Tableau III-. Propriétés moléculaires des arrestines
(Krupnick et Benovic, 1998 ; Ferguson, 2001)
Arrestine Taille
(a.a.)* Spécificité de récepteur
Distribution tissulaire
Caractéristiques et régulation
Arrestine visuelle 404 rho<<β2AR~m2mAChR Bâtonnets Trois variants d’épissage
Arrestine des cônes (ou X-arrestine)
410 Inconnue Cônes Inconnue
β-arrestine1 388 β2AR~m2mAChr>rho ubiquitaire Deux variants d’épissage,
liaison à la clathrine β-arrestine2
(ou arrestine3)
418 β2AR~m2mAChr>rho ubiquitaire Deux variants d’épissage,
liaison à la clathrine
*a.a., acides aminés; rho, rhodopsine ; β2AR, β2-adrenocepteur
Au premier groupe appartient l’arrestine et la X-arrestine ou arrestine des cônes qui se localisent respectivement dans les bâtonnets et les cônes de la rétine et sont impliquées dans la régulation de photorécepteurs comme la rhodopsine. Les arrestines non-visuelles ont été décrites initialement dans le système de régulation du récepteur β-adrénergique
et correspondent à la β-arrestine1 et la β-arrestine2 ou arrestine3. Les arrestines non-visuelles ont 45% d’homologie avec la séquence globale des arrestines non-visuelles et elles ont aussi 75% de similitude dans la région des résidus d’acides aminés 16 et 349. A la différence des arrestines visuelles, les arrestines non-visuelles sont distribuées dans la plupart de tissus mais avec une importante présence dans les tissus neuronaux. Cette expression ubiquitaire peut suggérer que les arrestines non-visuelles aient moins de spécificité dans la sélectivité des récepteurs (Krupnick et Benovic, 1998).
3.5.3.1 Protéines régulatrices et opiacés.
Des études récentes ont montré une augmentation de la quantité de la kinase GRK2 et de β-arrestine2 dans le locus cœrelus de rats traités chroniquement à la morphine (Terwilliger et al, 1994). Les taux de la kinase GRK2 sont augmenté dans le cortex frontal des rats traités chroniquement à la morphine et/ou à la méthadone et dans le cortex préfrontal d’addicts aux opiacés décédés par overdose (Ozaita et al., 1998). Dans cette même ligne de résultats Hurlé (2001) a aussi montré une augmentation des quantités des kinases GRK2 et GRK6 ainsi que de β-arrestine2 dans le cortex des rats traités chroniquement avec de sufentanil (agoniste sélectif du récepteur µ). Des études très récentes avec des souris transgéniques qui n’expriment pas le gène de β-arrestine2 ont montré que ces animaux présentaient une augmentation de l’analgésie induite par la morphine ainsi qu’une perte de l’effet de la tolérance, tout en continuant à être dépendants à la morphine (Bohn et al., 1999, 2000). Ces souris avaient une détérioration de la désensibilisation des récepteurs, fait qui prouve dans un modèle animal une implication directe de la protéine β-arrestine dans la désensibilisation de récepteurs µ et dans le développement de la tolérance qui apparaît dans les processus d’addiction chronique à la morphine. Cependant, dans des lignées cellulaires transfectées avec le récepteur µ (Keith et al., 1996 ; Whistler et Von Zastrow, 1998 ; Zhang et al., 1998) la morphine n’induit pas la phosphorylation du récepteur µ par les kinases GRK ainsi que la formation du complexe récepteur-arrestine et de ce fait, est incapable d’induire l’internalisation du récepteur µ. Cette différence pourrait être due à l’utilisation dans ces études in vitro de récepteurs µ de rat ou de souris (MOR1), qui ne sont pas très riches en sites potentiels de phosphorylation. Plusieurs variants d’épissage du récepteur µ sont présents dans le cerveau de souris avec plusieurs sites potentiels de phosphorylation. L’implication de ces récepteurs pourrait éclaircir les différences entre les deux modèles
(Bohn et al. 1999). Les résultats obtenus dans les souris knockout pour la protéine β -arrestine, semblent aussi indiquer que les phénomènes de la tolérance et de la dépendance aux opiacés peuvent être dissociés, et que les mécanismes biochimiques fondamentaux qui les dirigent sont différents (Bohn et al., 2000).
L’ensemble de ces résultats in vivo et in vitro suggèrent que les kinases GRK2 et GRK6 ainsi que la β-arrestine2 peuvent contribuer à la désensibilisation du récepteur µ et ainsi jouer un rôle important dans les processus de tolérance et de dépendance aux opiacés.
3.5.4. Régulation homologue des récepteurs aux opiacés.
Les étapes essentielles de la régulation homologue des récepteurs aux opiacés peuvent être résumées selon le mécanisme décrit par Lefkowitz (1993) pour le récepteur β-adrénergique (Figure II) : l’agoniste se lie aux récepteurs et induit un changement de la conformation du récepteur qui fini par une activation des protéines G et de ce fait par sa dissociation du récepteur. Ensuite, les sous-unités Gβγ aident la kinase GRK à se transloquer du cytosol à la membrane, où elle est colocalisée avec le récepteur activé, dont elle phosphoryle des résidus sérine et thréonine dans la région C-terminale du récepteur. Finalement le récepteur phosphorylé, se lie avec une très haute affinité à la protéine arrestine, liaison qui empêche l’association avec d’autres protéines G inactives. Le complexe désensibilisé récepteur-arrestine fini par une internalisation et séquestration à travers des vésicules de clathrine. Ces récepteurs inactifs, suite à l’endocytose peuvent être déphosphorylés et recyclés vers la membrane, donc resensibilisés ou dirigés vers les mécanismes cellulaires de dégradation, lesquels finiront, en partie, en la down-régulation des récepteurs. Cette down-régulation implique une diminution dans le nombre de récepteurs avec ou sans atténuation de la réponse (Law et Loh, 1999 ; Williams et al., 2001 ). Bien que d’autres composants cellulaires, comme la dynamine, puissent être impliqués dans les processus d’internalisation du récepteur, l’étape critique pour la désensibilisation et l’internalisation du récepteur est l’union du récepteur phosphorylé à l’arrestine (Law et Loh, 1999). La plupart des détails décrits dans ce mécanisme ont été confirmés pour le récepteur µ, mais quelques points restent à clarifier.
3.5.5. Régulation hétérologue des récepteurs aux opiacés.
La régulation hétérologue implique principalement les protéines kinases dépendant de seconds messagers. Ces kinases ne phosphorylent pas seulement les récepteurs activés par agonistes, elles phosphorylent aussi au hasard d’autres récepteurs non-activés. Les kinases représentatives de ce processus sont la PKA et la PKC. Bien que la désensibilisation induite par la PKA ait été démontrée pour quelques récepteurs couplés aux protéines G, son rôle dans la désensibilisation du récepteur µ n’a été pas démontré (Williams et al., 2001). Par contre les récepteurs aux opiacés sont également couplés, par certaines protéines G, aux phospholipases C/D et conduisent à l’activation des PKC dans diverses cellules et tissus neuronaux (Chen et Huang, 1991 ; Mangoura et Dawson, 1993 ; Chen et Yu, 1994 ; Smart et al., 1995). La phosphorylation du récepteur µ par la PKC induit une perte de la sensibilité aux opiacés (Ueda et al., 1995), mais le processus est indépendant de l’occupation des récepteurs, donc il est hétérologue.
Busquets et al. (1994, 1995) ont montré une diminution de l’immunoréactivité des isoformes α et β de la PKC dans les cerveaux d’héroïnomanes décédés par overdose et dans les cerveaux de rats traités à la morphine. Ces données semblent confirmer l’implication de la voie de la PKC dans les processus d’addiction aux opiacés. La diminution de l’immunoréactivité des isoformes α et β de la PKC ainsi que l’augmentation de la quantité de la protéine Gαi1/2 (Escribá et al., 1994) suggèrent une
interconnexion entre la voie de la PKC et la voie de l’AMPc dans les mécanismes d’addiction.
3.5.6. MAPKS (mitogen activated protein kinases).
La régulation d’événements cellulaires à travers des protéines activées chroniquement par les récepteurs aux opiacés, est une question critique pour comprendre les phénomènes de la tolérance et de la dépendance aux opiacés. Les voies de signalisation qui conduisent à l’expression altérée des gènes sont des systèmes effecteurs. Au contraire des systèmes effecteurs délimités à la membrane cellulaire tel que les canaux K+ ou Ca2+, la voie des MAP kinase (par mitogen activated protein kinases) qui sert d’intermédiaire à la signalisation nucléaire et d’activateur à d’autres voies à travers de la cellule, implique de nombreuses interactions protéine-protéine, des translocations et la phosphorylation de protéines (Williams et al., 2001). Les MAPK sont des kinases qui phosphorylent des résidus sérine et thréonine. Ces kinases sont restés très conservées
dans l’évolution. Les MAPK sont activées par les MAPKKs (MAPK kinase), lesquelles sont activées à leur tour par les MAPKKK (MAPK kinase kinase). Ces trois kinases constituent un module de signalisation universel. La particularité des MAPKs, c’est que contrairement aux autres kinases du module-qui ne sont phosphorylées qu’en sérine/thréonine- pour être actives, les MAPKs doivent être doublement phosphorylées au niveau des résidus sérine/thréonine et tyrosine du motif d’activation TXY.
Il y a au moins trois familles présentes dans les cellules de mammifères : les
extracellular signal-regulated kinase (ERKs) ; les cJun N-terminal kinases (JNKs) ou
les stress-activated protein kinases (SAPKs) ; et les p38-MAPKs. La cascade d’ERK est en relation avec la croissance du cycle cellulaire et avec la différenciation cellulaire. Par contre les familles de JNKs/SAPKs et p38-MAPKs se trouvent impliquées dans la réponse cellulaire au stress environnemental (Derkinderen et al., 1999).
La cascade d’ERK (de la même manière que les autres familles de MAPKs) consiste en une séquence de trois protéines kinases (Figure II): (1) la sous-famille Raf de MAPKKKs qui chez les mammifères est formée par trois protéines, cRaf-1, A-Raf et B-Raf ; la famille MEK de MAPKK formée par MEK1 et MEK2 ; et la sous-famille ERK de MAPKs, formée par cinq protéines, ERK1-5. Le signal d’activation peut venir en amont de Raf, par exemple à travers de l’interaction avec Ras (ou p21ras) qui est une protéine G de petit poids moléculaire (Ras.GDP dans l’état inactif et Ras.GTP dans l’actif) ou converger au même niveau, par exemple avec la phosphorylation de Raf-1 par la PKC. L’activation de Ras peut être induite par la tyrosin kinase c-Src. c-Src va phosphoryler des protéines adaptatrices comme Shc, qui recrutera d’autres protéines adaptatrices comme Grb2 (par
growth-factor-receptor-bound protein 2). Grb2 à son tour amènera à la membrane des facteurs d’échange de
GDP/GTP tels que Sos (par son of sevenless) que induira l’échange GDP/GTP de la protéine Ras (van Biesen et al., 1996). Une fois Ras est activée se produit une succession de phosphorylation des protéines que finira par l’activation d’ERK. Par la suite il y a une divergence dans les voies de signalisation. La kinase ERK peut phosphoryler un grand nombre de substrats (Sugden et Clerk, 1997) parmi lesquels se trouvent des factors de transcription comme CREB (ERK1/2 doivent être transloqués au noyau pour les phosphoryler), des protéines de la même cascade d’ERK (comme Raf et MEK) que pourrait être un mécanisme de feedback négatif, et de protéines structurales du cytosquelette (les neurofilaments et les microtubule-associated protein ou MAP) qui
pourraient être impliquées dans la régulation de la structure et de la morphologie cellulaire.
3.5.6.1. ERKs (extracellular signal-regulated kinases) et opiacés.
Les récepteurs aux opiacés ont été impliqués dans l’activation de la cascade ERK à travers la morphine (Ortiz et al., 1995). L’activation de cette cascade par les récepteurs aux opiacés, qui sont des récepteurs couplés aux protéines Gi/o, implique principalement
les sous-unités Gβγ plutôt que les sous-unités Gαi/o (van Biesen et al., 1996). La
surexpression des sous-unités Gβγ active Ras, Raf-1 et ERK1/2, fait qui semble indiquer
le rôle important de ces sous-unités dans l’activation de la cascade ERK. Belcheva et al. (1998) ont récemment impliqué les sous unités Gβγ des protéines Gi/o ainsi que la
protéine Ras dans la modulation d’ERK par les opiacés dans des cellules COS-7 transfectées avec des récepteurs aux opiacés. Ces données avec celles obtenues par Escribá et al. (1994) qui ont montré une up-régulation de la densité de la sous-unité Gβ
dans le cerveau d’addicts aux opiacés décédés par overdose ainsi que dans le cerveau de rats traités chroniquement à la morphine, semblent donner un argument moléculaire in
vivo pour comprendre l’activation de la cascade ERK dans les processus d’addiction aux
opiacés.
Un de modèles pour expliquer l’activation de la cascade ERK par les récepteurs aux opiacés est basé sur les résultats obtenus avec le récepteur β2-adrénergique (Lutrell et al., 1999, 2001). Le mécanisme proposé est que l’union de l’agoniste au récepteur induit la libération des sous-unités Gβγ qui activeront la phosphorylation du récepteur par les
GRK et de ce fait induiront la liaison du récepteur phosphorylé aux protéines β-arrestines. Le complexe ainsi formé va se lier à la tyrosine kinase c-Src dans la membrane et peut activer la cascade ERK. De plus la β-arrestine peut interagir directement avec les composants de la cascade ERK. En conséquence la β-arrestine fait la liaison entre le récepteur, le signal d’initiation de la cascade ERK et la cascade elle-même. Donc la β-arrestine et le récepteur peuvent jouer aussi un rôle de scaffold
proteins ou protéines d’ancrage (Fergusson, 2001). DeFea et al. (2000) ont montré que
le complexe de signalisation formé par le récepteur, la protéine β-arrestine et les composant de la cascade ERK, Raf-1 et ERK1/2, peuvent assurer une localisation correcte (cytosolique ou nucléaire) pour l’activation d’ERK par le récepteur. Ces données montrent que les β-arrestines peuvent réguler la formation des complexes de
transduction du signal et peuvent aussi réguler les différentes localisations de ces complexes dans la cellule (Ferguson, 2001).
La cascade ERK peut aussi interagir avec la voie de l’AMPc. Dans plusieurs types cellulaires l’effet de l’AMPc s’oppose à l’activation d’ERK1/2, donc il a été proposé que la diminution du taux d’AMPc intracellulaire (par exemple dans le traitement aigu à la morphine) diminuera l’inhibition de Raf par l’intermédiaire de l’AMPc, et permettra l’augmentation de l’activité d’ERK1/2 (van Biesen et al., 1996). Inversement, l’augmentation des taux d’AMPc (par exemple dans les traitements chroniques à la morphine) pourrait résulter en une diminution de l’activité d’ERK1/2. Récemment il a été démontré que l’activation d’ERK par le Ca2+ induit l’activation de la PKA qui déclenche l’activation de CREB (Impey et al., 1998). Ce résultats suggèrent une interconnexion entre la cascade ERK et la voie de l’AMPc avec un rôle important dans les processus d’addiction aux opiacés. D’autre part la cascade ERK peut être activée aussi à travers de la phosphorylation de Raf-1 par la PKC et montre ainsi un mécanisme d’activation de cette cascade indépendant de la protéine Ras, qui pourrait avoir de même un rôle important dans l’addiction (van Biesen et al., 1996).
3.6. PLASTICITE NEURONALE
L’exposition répétée aux drogues d’abus a été impliquée dans des changements structurels de certains types de neurones. Par exemple l’exposition répétée aux opiacés induit une diminution de la taille et du calibre des dendrites et du corps cellulaire des neurones dopaminérgiques de la VTA (Sklair-Tavron, et al., 1996). Pezawas et al. (1998) ont montré avec des techniques de neuro-imagerie, une diminution du volume cérébrale dans les addicts aux opiacés. D’ailleurs le traitement chronique aux opiacés à été impliqué dans des altérations de protéines du cytosquelette neuronal. Beitner-Johnson et al. (1992) ont montré une diminution des trois sous-unités des neurofilaments (NFs) dans la VTA de rats traités chroniquement à la morphine, ainsi qu’une augmentation de l’état de phosphorylation de deux sous-unités, le NF-H et le NF-M. La sous-unité NF-L était diminuée dans le cortex frontal de cerveau d’héroïnomanes décédés par overdoses (García-Sevilla et al., 1997b). Les NFs ont été impliqués comme déterminants intrinsèques du calibre axonal (Hoffman et al., 1984) et
une réduction du taux des NFs est associée avec une diminution du calibre axonal et de la vitesse de conduction de l’influx nerveux (Sakaguchi et al., 1993). Des preuves directes de perturbation du transport axonale dans les traitements chroniques à la morphine ont été démontrées (Beitner-Johnson et Nestler, 1993). Ces changements structurels et fonctionnels pourraient refléter une détérioration neuronale induite par l’exposition chronique aux opiacés chez les rats (Nestler et al., 1996) et chez les humains (García-Sevilla et al., 1997b). En effet, le traitement chronique à la morphine a été aussi associé avec une augmentation des taux de la glial fibrillary acidic protein (GFAP) dans la VTA (Beitner-Johnson et al., 1993), altérations qui ont été associées avec des détériorations neuronales (O’Callaghan, 1994). Dans ce contexte l’exposition aux opiacés réduit la création des neurones dans l’hippocampe des rats adultes (Eisch, et al., 2000). Cette altération pourrait être en relation avec une détérioration des certaines formes d’apprentissage et de mémoire (Nestler, 2001).
L’ensemble de ces résultats semble indiquer probablement que la prise chronique d’opiacés pourrait induire des adaptations et des altérations neuronales permanentes tant au niveau structural qu’au niveau fonctionnel, changements qui pourraient être impliqués directement dans le développement d’une plasticité neuronale dans le cerveau. Une des protéines qui pourraient avoir un rôle important dans ces modifications est le triplet de neurofilaments (NFs).
3.7. NEUROFILAMENTS (NFs)
Au cours du XIX siècle, plusieurs anatomistes furent les premiers à décrire un réseau de fibres à l’intérieur des neurones, dénommés neurofibrilles. Avec le développement des techniques de coloration à l’argent les neurofibrilles ont été plus clairement visualisées (revu dans Lee et Cleveland, 1996). Schmitt (1968a, b), en microscopie électronique, découvrit que les neurofibrilles étaient composées de filaments de 10 nm de diamètre qui furent dénommés neurofilaments.
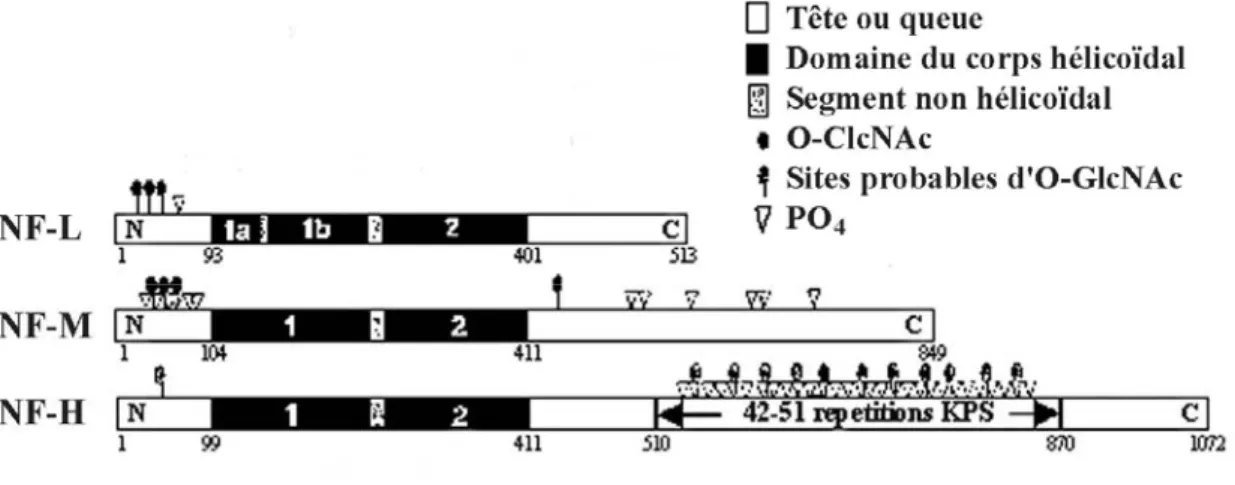
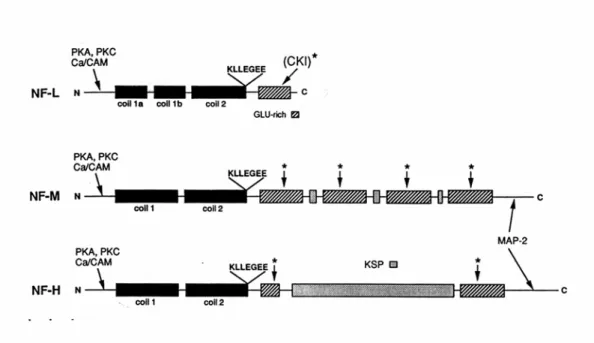
Les NFs appartiennent à la famille des Filaments Intermédiaires (IFs) de type IV, et avec les microtubules et les microfilaments d’actine forment le cytosquelette des neurones. Chez les mammifères et les oiseaux, les NFs sont formés par trois polypeptides ; leur masse moléculaire apparente sur gel de polyacrylamide en