Élaboration d’une méthodologie d’analyses bio-informatiques pour des données de séquençage Illumina dans un contexte de metabarcoding
95
0
0
Texte intégral
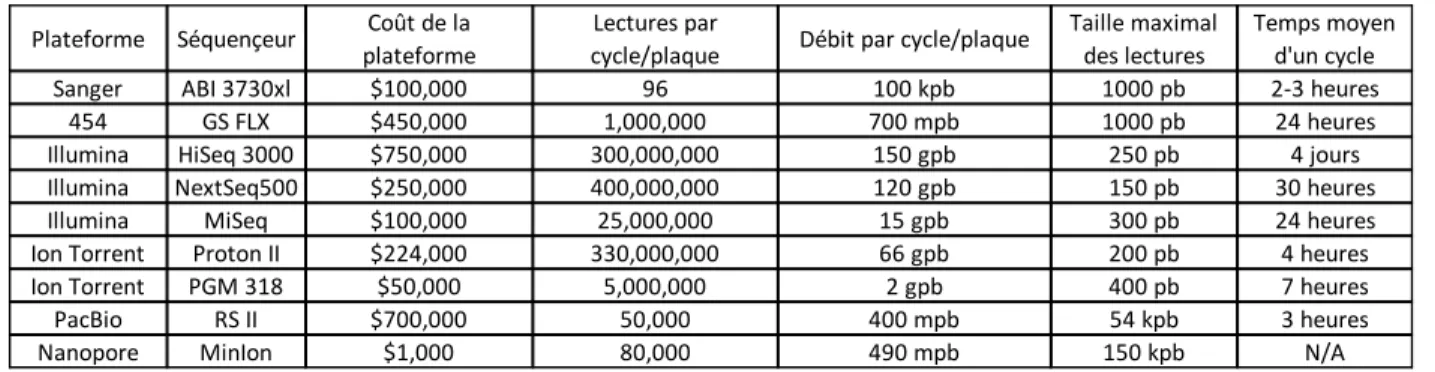
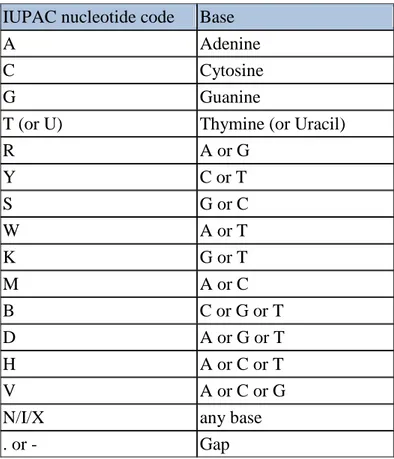
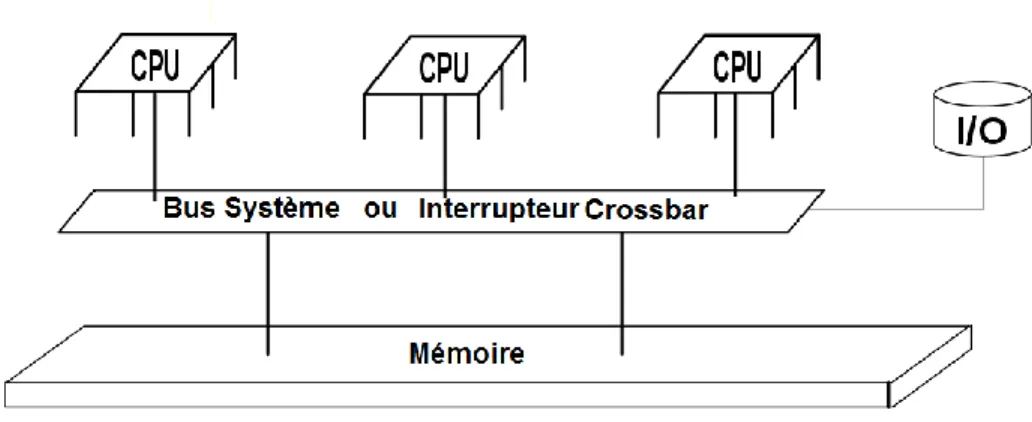
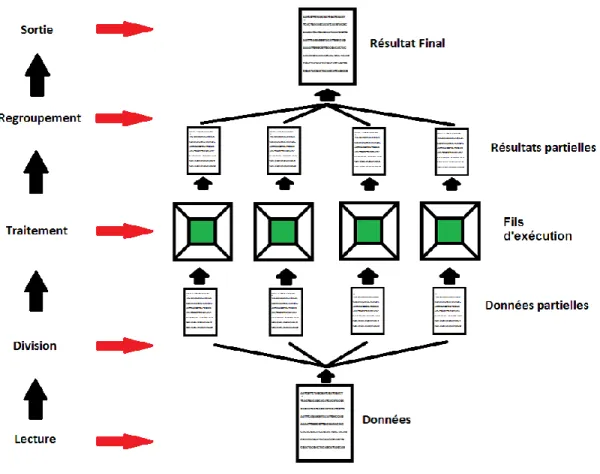
Figure


+7




Documents relatifs