Étude de la variabilité et de l'assise génétique de
l'architecture du système racinaire chez le soya (Glycine
max (L.) Merr.)
Mémoire
Waldiodio Seck
Maîtrise en biologie végétale - avec mémoire
Maître ès sciences (M. Sc.)
Étude de la variabilité et de l’assise génétique de
l’architecture du système racinaire chez le soya
(Glycine max (L.) Merr.)
Mémoire
Waldiodio Seck
Sous la direction de :
Résumé
L'architecture du système racinaire (ASR) est un aspect fondamental de la productivité des plantes, en particulier dans les environnements aux ressources limitées. Bien que l'importance de l’ASR soit connue, peu d'études ont exploré sa variabilité et son assise génétique chez les plantes, sans doute parce que les racines sont sous terre et sont difficiles à observer. Dans cette étude, nous avons étudié la variation naturelle de l’ASR au sein d’une collection de 137 lignées de soya hâtif représentative de ce qui est cultivé dans l’est du Canada. Nous avons utilisé des « rhizoboîtes », des enceintes constituées de plaques en acrylique, au sein desquelles nous avons documenté le développement du système racinaire en deux dimensions. Des photos ont été prises à l’aide d’une caméra et traitées à l’aide de logiciels d’analyse d’images pour mesurer différents caractères racinaires. Les analyses statistiques ont montré des différences phénotypiques significatives (P < 0,001) pour les caractères étudiés. Pour cette même collection de 137 lignées, nous avions des données génotypiques importantes issues de génotypage par séquençage et de reséquençage (2,18M de marqueurs SNP). Au moyen de ces données phénotypiques et génotypiques, nous avons effectué des analyses pangénomiques ou GWAS (« Genome-wide association study ») pour identifier des locus de caractère quantitatif (QTL) contrôlant les caractères racinaires à l’étude. Au total, 10 QTL sont détectés pour deux caractères importants : la longueur totale des racines et le diamètre de la racine principale. Au sein de ces régions génomiques, deux gènes candidats sont identifiés dont les fonctions sont connues pour avoir un impact majeur sur l’ASR chez les plantes et qui expliquent de 15 à 25 % de la variation phénotypique observée. Ces gènes pourront servir à développer de nouvelles variétés de soya dotées de meilleurs systèmes racinaires afin d’assurer de meilleurs rendements en conditions de stress.
Abstract
Root system architecture (RSA) is a fundamental aspect of plant productivity, particularly in resource-limited environments. Despite the importance of RSA, few studies have explored its variability and genetic basis in crops, because roots are underground and are so difficult to observe. In this work, we explored the phenotypic variation in RSA traits in a panel of 137 early soybean lines from Eastern Canada. We used rhizoboxes, transparent plastic enclosures, that allowed the study of root system development in two dimensions. Root systems were photographed using a camera and image analysis softwares were used to measure various components of RSA. Significant phenotypic differences for different RSA-related traits were found. The same panel of 137 lines had been characterized through a mixed genotyping approach (Genotyping by sequencing (GBS) and Whole genome sequencing (WGS)) to yield a catalog of 2.18M SNPs. The phenotypic and genotypic data were used for to perform a genome-wide association study (GWAS) to identify quantitative trait loci (QTL) controlling RSA-related traits. In total, 10 QTL regions were detected for two RSA-related traits, namely total root length and main root diameter. These genomic regions harbored two candidate genes whose predicted functions are known to play a role in RSA and which explained from 15 to 25% of the phenotypic variation. These genes can serve to develop new soybean varieties with better root systems to ensure productivity in stressful environments.
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des tableaux ... vi
Liste des figures ... vii
Liste des abréviations ... viii
Remerciements ... ix
Avant-propos ... x
Introduction générale ... 1
Chapitre 1 Revue de la littérature ... 3
1.1 Le soya ... 3
1.1.1 Les caractéristiques du génome du soya ... 3
1.1.2 L’importance du soya au Canada ... 3
1.1.3 Les utilisations du soya ... 4
1.1.4 Le changement climatique : risques accrus de stress chez le soya... 5
1.2 Le système racinaire ... 6
1.2.1 L’importance du système racinaire chez les plantes ... 6
1.2.2 L’architecture du système racinaire chez les plantes ... 7
1.2.3 L’ASR : quels moyens de mesure pour un compartiment difficile d’accès? ... 10
1.2.4 L’assise génétique de l’ASR: état actuel des connaissances ... 12
1.3 L’utilisation des marqueurs moléculaires ... 14
1.3.1 Les types de marqueurs moléculaires... 15
1.3.2 Les nouvelles technologies de génotypage à haut débit des SNP ... 15
1.3.2.1 Le Génotypage par séquençage (GBS) ... 16
1.3.2.2 Le reséquençage ou WGS (« Whole genome sequencing »)... 17
1.3.2.3 Approche hybride (GBS + WGS) ... 17
1.4 La cartographie QTL ... 18
1.4.1 L’analyse sur marqueurs individuels... 18
1.4.2 L’analyse par intervalles ... 18
1.4.3 Les analyses d’association pangénomiques : GWAS ... 19
1.4.3.1 Les facteurs qui influencent les analyses GWAS ... 20
1.4.3.2 Les modèles statistiques : avantages et inconvénients ... 22
En quoi notre travail pourrait-il être utile? ... 23
Hypothèse et objectifs de recherche ... 24
2.2 Abstract ... 28
2.3 Introduction ... 29
2.4 Material and Methods ... 31
2.4.1 Plant Material and RSA Phenotyping ... 31
2.4.2 Genotypic Data ... 32
2.4.3 Population Structure and Relatedness ... 33
2.4.4 GWAS Analysis on Traits Related to Root System Architecture ... 33
2.4.5 Identification of Candidate Genes ... 34
2.5 Results ... 34
2.5.1 Phenotypic Variation of RSA-Related Traits in Soybean ... 34
2.5.2 Genotypic Data and Population Structure ... 37
2.5.3 GWAS of RSA-Related Traits ... 37
2.5.4 RSA-Related Candidate Genes ... 39
2.6 Discussion ... 41
2.6.1 Significant Phenotypic Variation of RSA-Related Traits in Soybean ... 41
2.6.2 High and Significant Correlations among RSA-Related Traits ... 42
2.6.3 GWAS using Whole-Genome Data Revealed 10 QTLs Controlling RSA ... 42
2.6.4 Putative Candidate Genes for RSA-Associated QTL ... 43
2.7 References ... 45
2.8 Supplementary data ... 52
Conclusion générale ... 78
Liste des tableaux
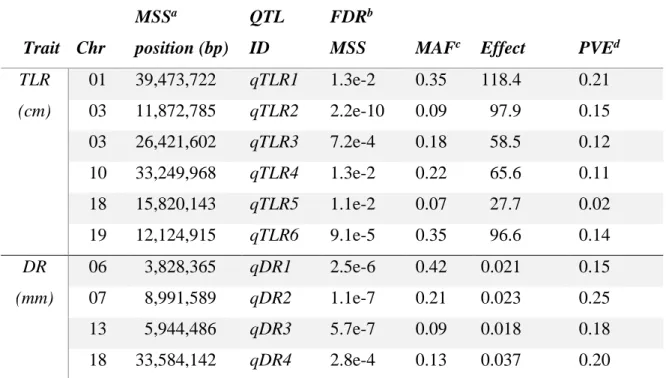
Table 2-1: Summary statistics of the twelve RSA-related traits. ... 35 Table 2-2 : List of quantitative trait loci (QTL). ... 39
Liste des figures
Figure 1-1 : Variation de l’architecture du système racinaire chez les plantes. ... 9
Figure 1-2 : Évolution des méthodes de mesures du système racinaire ... 12
Figure 1-3 : Procédure complète d’une approche GBS ... 17
Figure 1-4 : Diagramme Manhattan pour l’analyse d’association... 20
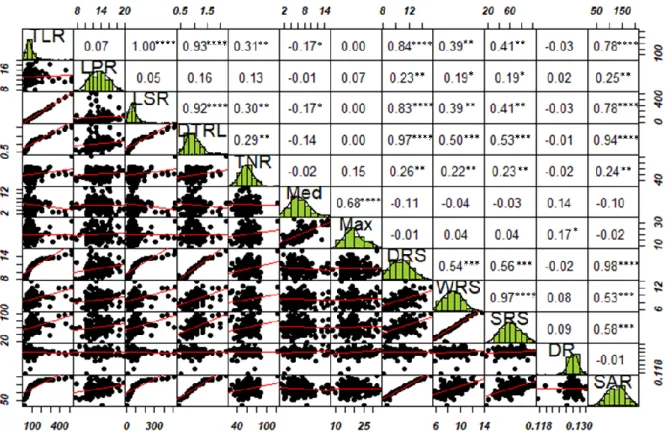
Figure 2-1: Correlations and frequency distributions of RSA-related traits... 36
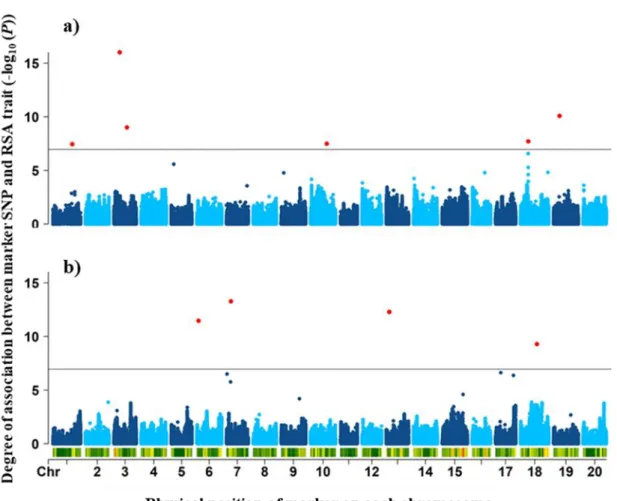
Figure 2-2: Manhattan plots showing genome-wide association results ... 38
Liste des abréviations
ADN : Acide désoxyribonucléique ANOVA : Analysis of varianceASR : Architecture du système racinaire
FarmCPU: Fixed and random model circulating probability unification FDR: False discovery rate
GBS: Genotyping by sequencing GLM: General linear model
GREML: Genome-based restricted maximum likelihood GWAS: Genome-wide association study
LD: Linkage disequilibrium LOD: Logarithm of the Odds MAF: Minor allele frequency MLM: Mixed linear model
NGS: Next-generation sequencing PCA: Principal Component Analysis PCR: Polymerase chain reaction QTL: Quantitative trait loci RSA: Root system architecture
SNP: Single nucleotide polymorphism WGS: Whole genome sequencing
Remerciements
Je commencerai par remercier mon directeur de recherche, François Belzile qui m’a offert la possibilité d’intégrer son équipe de recherche. Je te remercie beaucoup pour ton aide précieuse, ta disponibilité et pour l’excellence de ton encadrement tout au long de ce projet. J’ai sincèrement eu la chance de travailler avec toi. Je n’ai jamais été encadré ainsi (sincèrement). Merci beaucoup François!
Je tiens également à remercier Martine Jean, notre professionnelle de recherche, pour sa disponibilité à répondre à mes questions et pour ses suggestions constructives.
Je remercie très chaleureusement Davoud Torkamaneh. J’ai grandement apprécié ton aide, tes remarques, tes idées toujours à point. Tu as considéré ce projet comme le tien Davoud. Merci beaucoup!
Un grand merci à tous les membres du laboratoire François Belzile : Sidiki Malle, Marc André, Amina, Everton, Magdalena, Chiheb, Sébastien, Livia, Martin, Patricio. Merci beaucoup pour vos aides et conseils. Je garderai pour toujours mes souvenirs avec vous!
Merci à Serigne Mame Cheikh Anta Mbacké pour vos conseils et votre affection.
Merci à tous mes amis, du Sénégal au Canada.
Merci à Diodio pour ton amour sincère, ta confiance et ton aide.
J’arrive finalement au remerciement le plus important, ma chère famille qui sans eux je n’aurai jamais pu parcourir ce chemin, même s’ils étaient trop loin, leur soutien leur encouragement me donnaient chaque jour la force pour avancer. Merci ma très chère maman pour toute ton affection, toutes les peines endurées, tous tes sacrifices et toutes tes prières. Que Dieu vous accorde longue vie maman! Mon cher père, merci beaucoup pour ton amour. Je ne pourrai pas oublier mes chers oncles particulièrement Ibrahima, Saliou, Fallou et Baye Nar qui se sont toujours sacrifiés pour mon bien-être. Vous êtes pour moi ma plus grande source d’inspiration, symbole de courage, d'honnêteté, de discipline. Merci à mes sœurs Ndickou, Rokhaya et mon petit frère Yoro. Trouvez ici, le fruit de vos efforts!
Avant-propos
Ce mémoire est organisé en une introduction générale, deux chapitres et une conclusion générale. L’introduction générale décrit le cadre général, la problématique et les objectifs de ce projet de recherche. Le premier chapitre présente de manière générale l’état des connaissances du sujet abordé à savoir la plante (le soya), le phénotype (l’architecture du système racinaire), les méthodes de génotypage et la cartographie de QTL particulièrement les études d’association pangénomiques (GWAS). Le deuxième chapitre comporte un manuscrit intitulé « Genome-Wide Association Analysis Reveals the Genetic Basis of Root System Architecture in Canadian Soybean » pour fins de publication. Dans ce manuscrit, je présente le matériel et méthodes qui ont permis d’atteindre les objectifs de ce projet de recherche, les résultats des travaux et la discussion qui situe nos résultats dans un contexte plus large de la science avec des perspectives de recherche. Je suis le premier auteur du manuscrit et j’ai réalisé toutes les expériences qui y figurent. Davoud Torkamaneh et François Belzile ont aussi participé à l’élaboration de ce manuscrit. La conclusion générale qui termine ce mémoire résume l’ensemble des résultats obtenus et présentés avec des réflexions plus profondes dans pour leur utilisation dans la science.
Introduction générale
Le soya se situe actuellement au quatrième rang mondial des cultures en termes de superficie cultivée (FAOSTAT, 2018). Il constitue le principal oléo-protéagineux produit et échangé au monde. Sa production est l'une des cultures qui ont connu le plus grand essor avec une augmentation de 200 millions de tonnes de la consommation mondiale depuis les années 70 (Garrett et al., 2013). Il est loué d’abord pour les qualités nutritionnelles exceptionnelles de ses graines, lesquelles sont une source de protéines et d’huile tant pour l’alimentation humaine qu’animale. Le soya est aussi une culture très attrayante sur le plan environnemental, de par sa capacité à fixer l’azote de l’air avec l’aide de rhizobactéries. Cela conduit à une réduction de l’utilisation d’engrais azotés, une réduction de l’infestation par des adventices et une meilleure rentabilité pour les agriculteurs pour les rotations culturales, surtout avec les céréales.
Au Canada, le soya constitue la troisième plus grande culture en matière de revenus, ceux-ci étant supérieurs à 2,5 milliards de dollars par année. Le soya est la grande culture qui a affiché la plus forte augmentation au cours des cinq dernières années. Cette culture est établie depuis quelques décennies dans les provinces du Québec et de l’Ontario, tandis qu’elle est en pleine expansion dans l’ouest du Canada. En dépit de cette forte croissance, des défis restent à relever pour la sélection et le développement de cultivars de soya à haut rendement qui pourront bien s’adapter aux conditions climatiques canadiennes. Cependant, les nouveaux outils de la génomique constituent un excellent moyen pour relever ces défis.
C’est ainsi que notre équipe de recherche a pu bénéficier en 2015 d’un financement de Genome Canada (8,2 millions de dollars sur 4 ans) avec une contribution de l’industrie de près de 3,2 millions de dollars dans le cadre du projet SoyaGen. L’objectif central de ce projet était de développer des outils génomiques pour faciliter et accélérer le développement de cultivars améliorés combinant rendement élevé et maturité hâtive.
Des efforts considérables au sein de notre équipe et celles de nos collaborateurs ont été menés pour mieux caractériser la diversité génétique au sein du germoplasme canadien. Un des fruits de ces efforts de recherche a été la mise en place d’une collection de centaines de
lignées de soya chez laquelle la variation génétique a été caractérisée à des millions de marqueurs moléculaires SNP (« single nucleotide polymorphism »).
Une telle ressource a été mise à profit pour réaliser de nombreuses études visant à caractériser l’assise génétique de nombreux caractères d’intérêt au sein du soya canadien. Dans le cadre de ce projet, nous avons tiré profit de ce catalogue de marqueurs pour réaliser des analyses d’association pangénomiques (GWAS « Genome-wide association ») afin identifier pour la première fois des régions génomiques qui contrôlent l’architecture du système racinaire (ASR) chez le soya canadien.
Chapitre 1 Revue de la littérature
1.1 Le soya
Le soya ou soja (Glycine max (L.) Merr. ; 2n=40) est une espèce de plante annuelle de la famille des légumineuses (Fabaceae), originaire d’Asie de l’Est (Medic et al., 2014). Elle constitue la quatrième culture la plus importante en termes de superficie et de production au monde après le blé (Triticum aestivum L.), le maïs (Zea mays L.) et le riz (Oryza sativa L.) (FAOSTAT, 2018).
1.1.1 Les caractéristiques du génome du soya
Schmutz et al. (2010) ont séquencé pour la première fois le génome du soya. Ce dernier comprend 1,1 milliard de nucléotides avec plus de 50 000 gènes codant pour des protéines, soit 70 % de plus que la plante modèle Arabidopsis. Un nombre considérable de ces gènes (environ 75 %) sont présents en plusieurs copies. Selon (Schmutz et al., 2010), le génome du soya est paléopolyploïde et constitue le résultat de deux événements de duplication génomique complète qui seraient survenus il y a 59 et 13 millions d'années. Ces deux événements de duplication ont été suivis d'une diversification et d'une perte de gènes et de nombreux réarrangements chromosomiques. Environ 78 % des gènes prédits sont situés dans les extrémités des chromosomes, lesquelles représentent moins de la moitié du génome, mais correspondent à la fraction du génome où se déroule la quasi-totalité de la recombinaison génétique (Schmutz et al., 2010). Le génome de référence actuel du soya a été séquencé à partir du cultivar américain Williams 82. À l’aide de ce génome de référence, certaines stratégies de génotypage ont été facilitées.
1.1.2 L’importance du soya au Canada
Bien que le soya soit originaire d’Asie de l’Est, les plus grands pays producteurs de soya sont en Amérique. Actuellement, les États-Unis, le Brésil et l’Argentine sont responsables de plus de 80 % de la production mondiale de soya (Soystats, 2018). Le soya constitue une importante source de protéines et d’huiles : les protéines représentent près de 40 % du poids de la graine, et l’huile 20 % (Vollmann et Rajcan, 2010). Le soya est la plante
oléoprotéagineuse la plus importante au monde puisqu'elle représente plus de 60 % de la production totale de graines oléagineuses (Soystats, 2018).
Au Canada, la culture du soya n’a été introduite qu’au 19e siècle. Elle a progressé à pas de
géant au cours des dernières années pour devenir la troisième culture la plus importante, après le canola et le blé, avec une production de 6,05 millions de tonnes en 2019 (SOY Canada, 2019). La culture du soya est principalement pratiquée dans les provinces de l’Ontario, du Manitoba et du Québec, même si de nouveaux cultivars créés peuvent être cultivés ailleurs au Canada. Avant 1930, le soya était utilisé principalement pour la production fourragère. L'émergence d'une usine de broyage dans le sud-ouest de l'Ontario (la première en 1934, à Chatham) a amené la transition d'une culture de fourrage en une culture de grains. L'installation du moulin Victoria à Toronto en 1944 a encouragé l'établissement rapide de la culture du soya. L'évolution du soya à partir de 1949 reflète l'expertise des producteurs, l'amélioration de la technologie de production, l'amélioration des cultivars et l'apparition des cultivars plus hâtifs (Dorff, 2007). Le soya exporté a une valeur monétaire de plus de trois milliards de dollars (SOY Canada, 2019), et sa fève a plusieurs usages dans les secteurs alimentaire et industriel.
1.1.3 Les utilisations du soya
Près de 6 % des graines de soya sont consommées telles quelles (Dutch Soy Coalition, 2012), principalement dans les pays asiatiques comme la Chine, le Japon et l’Indonésie. On peut consommer les graines entières comme de l’édamame ou les broyer pour en faire des préparations comme le tofu, le lait de soya ou la sauce soya. La grande majorité des fèves de soya sont transformées pour leur huile. Les transformateurs prennent les fèves sèches et séparent l'huile du tourteau. L'huile de soya est utilisée dans la cuisson et la friture des aliments. La margarine, par exemple, est un produit qui peut être produit à base d'huile de soya. Les sauces à salade et les mayonnaises sont faites avec de l'huile de soya (WWF, 2014). En outre, certains aliments sont emballés avec de l'huile de soya (thon, sardines …). Les pains au four, les craquelins, les gâteaux, les biscuits et les tartes contiennent généralement de l'huile de soya.
La demande mondiale pour de la viande et des produits laitiers à moindre prix a augmenté et, avec elle, la demande en tourteau, aliment riche en protéines. Plus de la moitié des graines de soya transformées pour l'alimentation du bétail sont destinées à la volaille, un quart environ aux porcs et le reste aux bovins de boucherie, aux bovins laitiers et aux aliments pour animaux de compagnie.
L’huile de soya est parfois utilisée pour la production de biodiesel par le processus de transestérification, même si cela représente une part relativement petite (2 %) de la production mondiale. Les défenseurs de l’utilisation du soya comme agro-carburant avancent que comme la majeure partie du soya sert à la production d’aliments pour les animaux ou directement pour les humains, l’utilisation de l’huile restante pour produire de l’énergie est un meilleur compromis entre les utilisations alimentaires et énergétiques avec le soya qu’avec d’autres agro-carburants (United Soybean Board, 2009).
Malgré son importance et ses utilités, la culture de soya est confrontée à de nombreuses contraintes climatiques.
1.1.4 Le changement climatique : risques accrus de stress chez le soya
Le changement climatique pose déjà, et risque de poser encore davantage, de problèmes majeurs en agriculture. Il est prévu qu'il y aura une baisse du rendement des cultures y compris celle du soya en raison des événements climatiques extrêmes, que ce soient des précipitations excessives, des épisodes de sécheresse et de canicule ou de salinité élevée (Satari et al., 2020). Le soya est cultivé principalement aux latitudes 20-50° N et 10-40° S (Leff et al., 2004). La large distribution de sa culture suggère que le soya est très adaptable à différents environnements et climats. Ce degré d'adaptabilité peut ne pas être suffisant sous l'influence du changement climatique. En général, le soya peut survivre dans une large gamme de températures: de 10 à 40° C, selon le cultivar. Cependant, des températures extrêmes (inférieures à 12° C ou supérieures à 36° C) peuvent conduire à une germination réduite du soya (Tyagi et Tripathi, 1983). Elles peuvent aussi causer l'abolition de la germination et de l'allongement des tubes polliniques (Luo, 2012) ; ce qui entraîne une baisse de la fécondité et une perte de rendement. En même temps, les souches de Rhizobium couramment utilisées pour réaliser la fixation symbiotique de l’azote atmosphérique sont sensibles aux températures extrêmes, tant élevées que basses, et de telles températures
pourraient donc affecter la formation de nodules et la fixation de l'azote (Asadi Rahmani et al., 2009).
Au Canada, selon les outils de modélisation climatique, les régions où le soya est cultivé principalement (dans les provinces de l’Ontario, du Manitoba et du Québec) coïncident avec les zones qui devraient connaître les augmentations les plus sévères de l’incidence de journées très chaudes (plus de 30°C) entre 2021 et 2050 (https://climateatlas.ca). Une des clés majeures pour surmonter ces stress abiotiques réside dans l’exploration et l’exploitation des systèmes racinaires à travers des caractères architecturaux liés qui sont déjà connus pour jouer des rôles dans l’adaptation aux différents stress (Kano et al., 2011; Sánchez-Calderón et al., 2013; Wu et al., 2018).
1.2 Le système racinaire
Le système racinaire est une structure souterraine. Les racines sont d’habitude moins familières que les fleurs, les tiges et les feuilles qui sont les plus visibles, mais elles ne sont pas moins importantes pour la plante (Atkinson et al., 2014).
1.2.1 L’importance du système racinaire chez les plantes
Le système racinaire assure diverses fonctions dont l’absorption de l’eau et des éléments minéraux, l’ancrage dans le sol et l’établissement d’association bénéfique avec des microorganismes du sol. L'absorption se produit principalement par les poils racinaires, les fines structures en forme de doigts qui se développent entre les particules du sol pour extraire l’eau ainsi que les éléments minéraux du sol (Jungk, 2001). Ces derniers, une fois introduits dans la racine, sont transportés vers les tiges et les feuilles où les sucres et les glucides sont produits par le processus de photosynthèse (Eberhard et al., 2008). Une association symbiotique des racines est rencontrée spécifiquement entre les légumineuses comme le soya et des bactéries du genre Bradyrhizobium (Delamuta et al., 2013). Les bactéries pénètrent dans les cellules racinaires, se multiplient et forment ainsi des nodules où les bactéries ont accès aux glucides synthétisés par la plante. En retour, les bactéries fixent l’azote, convertissant l'azote gazeux de l'atmosphère en azote-ammoniacale pouvant être utilisés par les plantes. Les racines peuvent souvent former des associations symbiotiques avec les
le phosphore. Les plantes cultivées en l'absence de mycorhizes dans le sol se portent généralement moins bien que lorsque des mycorhizes sont présentes.
En résumé, le système racinaire fonctionne principalement pour acquérir l’eau et les nutriments, en plus d’assurer l’ancrage de la plante. Les symbioses assurent l’apport de nutriments. Il participe également à des fonctions secondaires comme le stockage de photoassimilats ou la synthèse de phytohormones. Au cours de l'évolution, l'organisation racinaire est progressivement passée de très simple, comme les rhizomes, à très hiérarchique, y compris les tissus spécialisés (Brundrett, 2002). Cette élaboration est probablement due au fait que le système racinaire est déterminant pour faire face à des contraintes majeures à la croissance et au succès de reproduction des plantes: la disponibilité de l’eau, des nutriments, l’ancrage dans le sol et la concurrence souterraine d'autres plantes pour ces ressources. Selon la composition du sol, des différences dans l'architecture du système racinaire (ASR) peuvent affecter la capacité de compétition pour les ressources du sol (Fitter, 1987). Seulement, les connaissances sur l’ASR sont limitées.
1.2.2 L’architecture du système racinaire chez les plantes
Il existe de nombreuses façons de définir l’ASR. Probablement, la définition la plus simple est que l’ASR est la configuration spatiale de l'ensemble du système racinaire dans le sol (Fitter, 1987; Lynch, 1995). Elle comprend ainsi la forme, la distribution et le mode de ramification des racines (Osmont et al., 2007), lesquelles propriétés jouent un rôle clé dans la détermination de la productivité des cultures dans des environnements aux ressources limitées (Bengough et al., 2011). L’ASR est généralement modulée par la promotion ou l'inhibition de la croissance des racines primaires, par la croissance des racines latérales et par la formation de racines adventives et de poils racinaires. La racine primaire se forme lors de l'embryogenèse, tandis que les racines latérales constituent des dérivés post-embryonnaires des racines existantes. Les racines latérales proviennent de la couche tissulaire du péricycle ou de l'endoderme, généralement adjacente aux cellules polaires du xylème chez les dicotylédones ou aux cellules polaires du phloème chez les monocotylédones (Casero et al., 1995). Chez de nombreuses espèces, le système racinaire s'élargit également par des racines adventives, qui peuvent être largement définies comme des racines provenant des structures des pousses, généralement des tiges. En outre, tout type de racine peut augmenter sa surface absorbante en augmentant les extensions des cellules épidermiques, les
poils racinaires (Schiefelbein, 2003). Il est bien admis que deux types récurrents de systèmes racinaires sont rencontrés chez les angiospermes : le type homorhizique, répandu chez les monocotylédones comme le maïs (Figure 1-1 (A)) et le type allorhizique chez les dicotylédones comme le soya (Figure 1-1 (B)). Malgré l’existence de ces grands types d’architecture, l’ASR varie d’une espèce à l’autre ou même entre cultivars d’une même espèce, car elle est déterminée par des facteurs génétiques inhérents (Osmont et al., 2007). Par conséquent, des cultivars peuvent avoir la même biomasse racinaire alors qu’ils sont dotés d’une ASR différente (Figure 1-1).
La ramification des racines constitue un élément clé pour augmenter la surface d’occupation du système racinaire, ce qui permet ainsi à la plante d'exploiter et d’aller puiser les réserves plus éloignées d'eau et de nutriments et d'améliorer l'ancrage dans le sol. D’ailleurs, Lynch (2007) a démontré que la longueur des ramifications latérales des racines influence directement les performances des plantes dans des conditions de faible teneur en azote par exemple. La formation des racines est un complément essentiel à la croissance des racines. Il contribue à élargir le volume de sol exploré, à augmenter la capacité d'extension du système racinaire. La formation de racines post-embryonnaires comprend la production de racines latérales et de racines adventives qui ont des propriétés physiques et physiologiques spécifiques (p. ex. comportements d'absorption, taux de survie). Par conséquent, les racines latérales et adventives contribuent de manière sensiblement différente aux multiples fonctions des systèmes racinaires et à l'efficacité du système racinaire. De même, les processus de formation des racines latérales et des racines adventives sont susceptibles d'être coordonnés afin d'atteindre une performance optimale à l'échelle de la plante entière. Il a été documenté que les systèmes racinaires de toutes les espèces végétales étudiées subissent des altérations de l’ASR en réponse à une carence en phosphore (P), probablement l'élément minéral le plus limitant pour la croissance racinaire et le moins mobile dans le sol (Kochian et al., 2005). En effet, pour extraire le P fixe du sol, la plante doit modifier son ASR pour placer plus de racines latérales dans le sol de surface afin de modifier la rhizosphère et ainsi solubiliser et absorber le P du sol. Des études récentes ont montré que l’ASR est parfaitement corrélée aux fonctions vitales d’absorption de l’eau et des éléments minéraux et à la
Bien que l'importance de l’ASR soit connue, peu d'études ont exploré sa variabilité chez les espèces cultivées (Prince et al., 2015; 2019). La cause principale de cette ignorance est que les racines ne sont pas directement accessibles et sont difficiles à observer parce qu’elles sont sous terre.
Figure 1-1 : Variation de l’architecture du système racinaire chez les plantes. (A)
Architecture racinaire de trois différents cultivars (M1, M2 et M3) de maïs dotés d’un système racinaire homorhizique typiquement trouvé chez la plupart des monocotylédones (Tötzke et al., 2017, image sous CC). (B) Architecture racinaire de trois différents cultivars (S1, S2 et S3) de soya, lequel est doté d’un système racinaire allorhizique typique trouvé chez la plupart des dicotylédones (image de Waldiodio Seck, Université Laval). Les poils racinaires ne sont pas représentés. RP : racine primaire; RL : racine latérale; RC : racine de couronne, RS : racine séminale.
1.2.3 L’ASR : quels moyens de mesure pour un compartiment difficile
d’accès?
Au cours des dernières années, un intérêt accru a été porté sur la mise en place et l’amélioration de plateformes ou d’approches de phénotypage et d'imagerie efficaces dans le sol ou d’autres milieux de culture pour évaluer et mesurer différents aspects de l’ASR (Kuijken et al., 2015; York et Lobet, 2017; Atkinson et al., 2019). Auparavant, les racines étaient généralement extraites et lavées pour leur comptage et leur mesure. Cette méthode est fastidieuse et délicate. Elle est de surcroît destructive et ne permet donc pas d’observer une dynamique de croissance sur les racines extraites. Par la suite, des systèmes d’acquisition d’images en deux dimensions (2D), à l’aide de caméras ou de numériseurs ont vu le jour (Kirchhof, 1992) pour des mesures automatisées. Les images peuvent être prises ex-situ après lavage et étalement des racines sur une surface plane, ou in situ dans des dispositifs adaptés. Ces dispositifs en 2D généralement connus sous le nom de rhizotrons permettent une observation non destructive (Desgroux, 2016). Il s’agit en général d’une construction d’une boîte remplie d’un substrat solide avec surface transparente généralement appelée « rhizoboîtes » et dont la partie transparence permet d'observer le développement du système racinaire. Conjointement à la baisse des coûts d’acquisition d’images, beaucoup d’efforts ont été déployés afin de développer des outils informatiques permettant de faciliter et automatiser la collecte de données à partir d’images en 2D. À ce jour, il existe près de 40 logiciels d’analyse d’image pour les systèmes racinaires (https://www.quantitative-plant.org/software). Certains sont accessibles gratuitement comme Automatic root image analysis (ARIA) (Pace et al., 2014) tandis que d’autres sont sous licence commerciale comme WinRHIZOTM (Arsenault et al., 1995).
Les systèmes d’imagerie en 2D sont peu dispendieux, efficaces mais sont souvent limitants, car ils ne permettent pas une observation parfaitement représentative d’une croissance en milieu naturel. De ce fait, de nouvelles plateformes de phénotypage du système racinaire en trois dimensions (3D) ont été mises au point et font appel à des numériseurs, des caméras, de la fluorescence ou des radiations (Piñeros et al., 2016). À noter que ces dernières technologies
car c’est un système 2D qui a été utilisé dans la présente étude pour phénotyper rapidement plus de 100 lignées de soya (voir l’article, chapitre 2). La Figure 1-2 ci-dessous illustre l’évolution des systèmes de phénotypage de l’ASR chez les plantes.
L’analyse des propriétés de l’ASR a progressé beaucoup moins rapidement que la caractérisation des parties visibles ou exposées de la plante, parce que les racines n’étaient pas directement observables. Toutefois, les avancées récentes en matière de plateformes de mesure des caractères liés à l’ASR constituent une étape très importante afin de mieux étudier la variabilité de l’ASR et de comprendre son assise génétique chez les plantes.
Figure 1-2 : Évolution des méthodes de mesures du système racinaire. (a) Tracé manuel du
système racinaire après prélèvement destructeur (Weaver, 1919). (b) Nettoyage des racines extraites au champ-plateforme de phénotypage CREAMD (« Core Root Excavation using Compressed-air ») (Zheng et al., 2020). (c) Images 2D représentatives de la tomodensitométrie par rayons X (CT) de plants de riz cultivés pendant 10 jours dans un substrat artificiel (Turface) et dans du sol (Piñeros et al., 2016). (d) Système de phénotypage par rhizoboîtes utilisé chez l’orge (Jia et al., 2019). (e) Système de tour à mailles verticales assemblé de plastique permettant une croissance racinaire illimitée, mais conservant l'architecture des racines en 3D (Piñeros et al., 2016). (f) Exemple d'une image 2D du système racinaire d’un plant de soya en culture hydroponique à l'aide du système de maillage en plastique (Piñeros et al., 2016).
1.2.4 L’assise génétique de l’ASR: état actuel des connaissances
L’ASR est très souvent contrôlée par des QTL à effets faibles (Burton et al., 2014; Dorlodot et al., 2007; Orman-Ligeza et al., 2014; Rogers et Benfey, 2015). Ces QTL détectés présentent souvent de fortes interactions avec l’environnement en lien avec la plasticité élevée du système racinaire (Desgroux, 2016).
La découverte de gènes impliqués dans la croissance des racines remonte à 1993 chez Arabidopsis lorsque Benfey et al. (1993) ont découvert les gènes SCARECROW
(SCR)/SHORT-ROOT, des facteurs de transcription de type GRAS qui sont des régulateurs
de la croissance et du développement des racines. Des mutants à racines courtes (shr/scr) ont établi un rôle pour ces gènes dans les altérations de l'organisation radiale de l'axe embryonnaire (Scheres et al., 1995). Meijón et al. (2014) ont aussi identifié et caractérisé un gène F-box, KURZ UND KLEIN (KUK), régulant le méristème et la longueur des cellules racinaires chez Arabidopsis. Ces auteurs ont montré que les polymorphismes dans la séquence codante sont les principales causes de la variation naturelle dépendante des allèles
KUK dans le développement des racines. Toujours chez Arabidopsis, un modulateur de la
voie des auxines (EXOCYST70A3) a été identifié comme provoquant ainsi une variation de l’ASR en agissant sur la distribution des protéines PIN4 (Ogura et al., 2019). La variation
gravitropiques des racines, entraînant des profondeurs d’enracinement différentes en conditions de sécheresse (Ogura et al., 2019).
Chez le riz, un QTL majeur (DRO1) contrôlant l’angle racinaire a été identifié (Uga et al., 2011). Le gène responsable de ce QTL a ensuite été cloné par cartographie à haute résolution (Uga et al., 2013). Le QTL DRO1 est régulé négativement par l'auxine et est impliqué dans l'élongation des cellules de l’apex de la racine, provoquant une croissance racinaire asymétrique et une flexion vers le bas de la racine en réponse à la gravité (Uga et al., 2013). L’introduction, par rétrocroisement, de l’allèle favorable de DRO1 chez un cultivar de riz à racines peu profondes a permis à la lignée résultante d'éviter la sécheresse en augmentant l'enracinement en profondeur avec une production de biomasse et des rendements en grains beaucoup plus importants en conditions de sécheresse modérée et sévère (Uga et al., 2013). Phung et al. (2016) ont aussi caractérisé la longueur des racines, la biomasse racinaire, l'épaisseur des racines et le nombre de racines sur un panel de 180 variétés de riz en utilisant des sacs en plastique remplis de sable. Parmi les gènes identifiés par cartographie d’association, OsIAA4 (signalisation de l’auxine) était retenu comme important pour la formation du méristème racinaire (Phung et al., 2016). Au total, 22 autres gènes candidats identifiés nécessitent une validation supplémentaire (Phung et al., 2016).
Chez le maïs, les études d’association sur une collection de 300 lignées haploïdes doublées ont permis de détecter des marqueurs significativement associés avec des caractères racinaires au stade plantule (Sanchez et al., 2018). Parmi ces marqueurs, un SNP était situé au sein du gène GRMZM2G021110, lequel est exprimé dans les racines (Sanchez et al., 2018).
Récemment chez colza, les travaux de He et al. (2019) sur un panel de cartographie d’association a permis d’identifier 295 gènes candidats associés à plusieurs caractères racinaires. Parmi ces gènes, huit sont des homologues de gènes connus pour avoir un impact majeur sur le développement du système racinaire chez Arabidopsis (He et al., 2019). Chez le blé, Manschadi et al. (2010) ont trouvé un génotype (SeriM82) très adapté à la sécheresse, lequel est doté d’un système racinaire plus compact avec plus de racines dans les couches de sol plus profondes et par conséquent associé à un angle d’enracinement plus important et à un plus grand nombre de racines séminales.
Chez le soya, peu d’études sur l’ASR ont été faites. Les études de cartographie traditionnelles du soya ont identifié de nombreux QTL (Kassem et al., 2006; Abdel-Haleem et al., 2011; Rong et al., 2011; Brensha et al., 2012; Manavalan et al., 2015), mais peu ont identifié des gènes prometteurs dans les QTL détectés (Brensha et al., 2012; Manavalan et al., 2015; Prince et al., 2015). Cependant, l'identification des gènes impliqués dans des processus biologiques spécifiques et la catégorisation subséquente des effets fonctionnels sur l’ASR reste toujours un défi chez le soya. Récemment, Prince et al. (2019) ont évalué une liste importante de caractères liés à l’ASR à l’aide de WinRhizo dans une collection de 397 accessions (élites et locales) de soya. Leurs analyses d’association basées sur des marqueurs SNP à haute densité ont permis d’identifier des gènes candidats. Les loci les plus significatifs identifiés indiquaient un gène inconnu sur le chromosome 16, associé au nombre de racines latérales. Une exploration supplémentaire des allèles a révélé deux sites polymorphes, identifiant les changements de séquence protéique qui nécessitent encore une validation supplémentaire (Prince et al., 2019).
Il reste plusieurs contraintes qui limitent les progrès entre la découverte de QTL jusqu’à la commercialisation de nouvelles variétés. Certaines de ces contraintes concernent la nature des gènes sous-jacents, la faible héritabilité causée par les petits effets des loci individuels, les interactions épistatiques (entre gènes) et les effets multiples d'un gène (pléiotropie). Des limitations supplémentaires sont également dues à la nature de l’ASR elle-même. Les caractères racinaires sont particulièrement sujets à la plasticité environnementale et sont difficiles à mesurer avec précision, notamment la chronologie des événements et les détails de la ramification racinaire. Enfin, ceux qui sont mesurés sont parfois d'une pertinence discutable pour les plantes sur le terrain (Dorlodot et al., 2007).
1.3 L’utilisation des marqueurs moléculaires
La détection de QTL repose sur la détection d’un lien entre la variation quantitative observée pour un caractère et la variabilité génétique au sein d’une population, laquelle est capturée
par des marqueurs moléculaires ou morphologiques balisant une carte génétique ou un génome (Desgroux, 2016).
1.3.1 Les types de marqueurs moléculaires
Une grande variété de marqueurs moléculaires a été utilisée durant les vingt dernières années pour les analyses génétiques des caractères complexes. Ces marqueurs présentent un polymorphisme au niveau de l’ADN (variabilité du code génétique dans une séquence d’ADN) basé sur la présence ou l’absence de sites de restriction (RFLP « Restriction Fragment Length Polymorphism »), la longueur de fragments amplifiés (AFLP « Amplified Fragment Length Polymorphism »), le nombre de séquences répétées en tandem (SSR « Single Sequence Repeat »), un nucléotide unique (SNP « Single Nucleotide Polymorphism ») (Kwok, 2003).
Les marqueurs SNP présentent en général deux allèles (bialléliques). Ils sont donc moins informatifs que des marqueurs multialléliques comme les SSR, mais en contrepartie, ils sont présents en très grand nombre dans le génome. De plus, avec l’évolution rapide des techniques de séquençage, les marqueurs SNP sont maintenant peu coûteux à développer et à analyser sur un grand nombre de lignées. Ils sont les plus couramment utilisés aujourd’hui.
1.3.2 Les nouvelles technologies de génotypage à haut débit des SNP
La génomique est actuellement au cœur d'un nombre extraordinaire de découvertes, d'innovations et d'applications grâce aux avancées technologiques de séquençage de nouvelle génération ou NGS (« Next-generation sequencing ») (Kumar et al., 2012). Les technologies de NGS ont réduit à la fois le coût et le temps requis pour générer des données de séquence facilitant ainsi le développement de méthodes de génotypage à haut débit pour générer un nombre considérable de marqueurs moléculaires, particulièrement les SNP (Torkamaneh et al., 2017). Certaines plateformes permettent de détecter des SNP dont les loci sont déjà connus. Les puces à ADN en sont un exemple. Chez le soya, SoySNP50K est une puce qui utilise la plate-forme Infinium d’Illumina et qui peut interroger jusqu’à 52 041 SNP simultanément (Song et al., 2013). D’autres plateformes, par contre, ne séquencent qu’une partie du génome en pratiquant ce qu’on appelle la « réduction de complexité ». Le plus souvent, des enzymes de restriction sont employées pour fragmenter le génome et on séquence l’ADN qui borde immédiatement ces sites de restriction. Ainsi, on peut identifier des SNP au sein de ces courts segments d’ADN dispersés çà et là dans le génome. Plusieurs protocoles assez semblables s’appuient sur ce principe, dont le séquençage des fragments
amplifiés provenant de locus spécifiques (SLAF-seq), le séquençage d’ADN lié à des sites de restriction (RAD-seq) et le génotypage par séquençage (GBS) (Elshire et al., 2011). Ce dernier est aujourd’hui l’approche la plus couramment utilisée chez les plantes à cause de sa simplicité, de sa rapidité et de son faible coût.
1.3.2.1 Le Génotypage par séquençage (GBS)
Le GBS est une approche hautement flexible. Il permet de génotyper des milliers de SNP répartis sur l’ensemble du génome et non pas seulement dans les régions codantes; ce qui augmente la probabilité d'identifier des SNP associés avec des caractères d'intérêt (Elshire et al., 2011; Poland et Rife, 2012; Torkamaneh et al., 2017). L’approche GBS emploie deux stratégies: une réduction de la complexité en ciblant un sous-ensemble du génome et un multiplexage des individus, produisant une librairie d’ADN de plusieurs individus (Elshire et al., 2011). La réduction de la complexité est assurée par des enzymes de restriction. Un système de codage des fragments de restriction est utilisé dans l’étape du multiplexage. Des adaptateurs sont ligaturés aux extrémités des fragments de restriction et ces adaptateurs incluent des séquences spécifiques à chaque individu (code-barres) et des séquences communes à tous les individus (Poland et al., 2012). Une quantité égale de produits de digestion/ligature de chaque individu est par la suite mélangée pour créer une seule librairie GBS. Les fragments de restriction du mélange sont par la suite amplifiés par PCR, les produits PCR purifiés et la taille des fragments de la librairie contrôlés avant d’effectuer le séquençage. Ce dernier permet de lire simultanément plusieurs séquences et de faciliter l’appel SNP grâce à des outils de bioinformatique (Sonah et al., 2013) pour trier les séquences selon leurs codes par une approche comparative entre les séquences des différents individus ou par l’alignement des séquences par rapport à un génome de référence (Schmutz et al., 2010).
Figure 1-3 : Procédure complète d’une approche GBS (image de Amina Abed, Université
Laval; figure adaptée des travaux d'Elshire et al., 2011 et de Poland et Rife, 2012).
1.3.2.2 Le reséquençage ou WGS (« Whole genome sequencing »)
Le séquençage du génome complet (reséquençage) a pour objectif majeur de répertorier les variations nucléotidiques et structurales et de comprendre leurs conséquences biologiques en identifiant la majorité des polymorphismes au sein d’un génome complet. En séquençant le génome entier, il est possible d’identifier la majorité des polymorphismes, soit des millions de polymorphismes (SNP), en comparaison de quelques dizaines de milliers de SNP obtenus par les approches de réduction de complexité. Cependant, le reséquençage demeure trop cher pour l’utilisation à grande échelle et est souvent utilisé pour les études approfondies d’un nombre limité de lignées.
1.3.2.3 Approche hybride (GBS + WGS)
Chez le soya, Torkamaneh et al. (2017) ont mis en place une approche alternative permettant de combiner les avantages du GBS et du WGS. En effet, dans l’approche hybride proposée, une collection de lignées représentatives d’une espèce (ou d’un sous-ensemble de l’espèce) est caractérisée de manière exhaustive par le biais du WGS, ce qui produit typiquement un catalogue contenant des millions de SNP. Ce catalogue sert ensuite de panel de référence pour des travaux d’imputation. En effet, à partir de ce moment, on peut génotyper un grand nombre de lignées par GBS et obtenir un catalogue de plusieurs dizaines de milliers de SNP
à faible coût. En faisant appel au panel de référence issu du WGS, on peut ensuite imputer les millions de SNP manquants avec une grande justesse. Cette imputation s’appuie sur les dizaines de milliers de SNP dérivés du GBS, lesquels permettent de guider avec justesse l’imputation des millions d’autres variants recensés dans le catalogue plus exhaustif issu du WGS. Au final, il devient donc possible d’obtenir un catalogue composé de millions de SNP, mais à un coût beaucoup moindre. Nous avons utilisé cette démarche dans la présente étude (voir l’article, chapitre 2).
1.4 La cartographie QTL
Depuis une trentaine d’années, la détection de QTL a été largement menée à l’aide de populations biparentales en ségrégation. Ces populations, issues du croisement entre deux parents contrastés pour un ou plusieurs caractères d’intérêt, comprennent des populations F2, d’haploïdes doublés, de lignées recombinantes fixées et de rétrocroisement avec un parent récurrent (backcross). Initialement, lorsqu’un petit nombre de marqueurs était disponible pour couvrir le génome, la détection de QTL était réalisée marqueur par marqueur.
1.4.1 L’analyse sur marqueurs individuels
Dans ce cas, il s’agissait de tester si les individus partageant un même génotype à un marqueur donné présentaient un contraste phénotypique statistiquement significatif avec les individus partageant un autre génotype (p. ex. AA vs CC, pour un locus SNP) à ce même locus (Jones et al., 2009). Une analyse de variance à un facteur (le marqueur) était alors réalisée. Or, si le marqueur n’était pas positionné très proche du QTL, la fréquence de recombinaison entre le marqueur et le vrai QTL n’était pas prise en compte, conduisant à des biais de détection (Desgroux, 2016). C’est pour remédier à ces lacunes qu’une nouvelle approche de cartographie a été mise au point : l’analyse par intervalles.
1.4.2 L’analyse par intervalles
Des méthodes basées sur l’étude d’intervalles génétiques (définis par un marqueur distinct à chaque extrémité) ont été développées. Dans une telle approche d’analyse par intervalles, on cherche à comparer le phénotype de lignées qui sont contrastées en matière de leur bagage
groupes d’individus contrastés présentent un phénotype significativement différent, on peut en conclure qu’il existe au sein de cet intervalle génétique un ou des QTL qui déterminent en partie le phénotype étudié (Mansur et al., 1993; Xue et al., 2017).
Ici, une approche statistique différente est employée pour estimer la position et l’effet du QTL : le maximum de vraisemblance (Lander et Botstein, 1989). La position du QTL est alors fixée et l’intervalle est exploré pas à pas de manière exhaustive. À chaque pas, le rapport entre la vraisemblance de la présence d’un QTL et son absence (hypothèse nulle) est calculé, puis représenté graphiquement par la fonction logarithmique de ce rapport, le « LOD score » (« Logarithm of the Odds »). La position la plus probable du QTL est celle qui maximise la vraisemblance du modèle avec le QTL, et donc celle où le score LOD est à son maximum. L’intervalle de confiance est souvent déterminé autour du maximum lorsque le LOD score est abaissé de 1 par rapport à sa valeur la plus élevée. Toutefois, en utilisant cette méthode, la variance interclasse comprend toutes les sources de variation non prises en charge par le QTL, y compris les QTL localisés en dehors de l’intervalle exploré (Desgroux, 2016). Chez le soya, des études d’analyse par intervalles pour de nombreux caractères d’intérêt agronomiques ont été rapportées (Zhao et al., 2015; Kandel et al., 2018). Malgré les avantages que présentent les analyses par intervalles par rapport à l’analyse sur marqueurs individuels, cette approche souffre néanmoins de certaines lacunes. En effet, elle demande beaucoup de temps pour développer une population de cartographie. De plus la diversité allélique est limitée à celle entre les deux parents de la population d’étude et les évènements de recombinaison sont limités lors de la construction de la population.
1.4.3 Les analyses d’association pangénomiques : GWAS
Les études d'association pangénomique (ou GWAS de l’anglais « Genome-Wide Association Study »), également connues sous le nom de cartographie de déséquilibre de liaison (ou LD pour « linkage disequilibrium », d’où le « LD mapping »), fournissent un autre moyen d'identifier les associations entre les caractères phénotypiques et les marqueurs moléculaires (Gupta et al., 2014).
Actuellement, l’approche GWAS s'est imposée comme la plus puissante pour identifier des gènes contrôlant des caractères complexes chez des espèces cultivées (Famoso et al., 2011; Huang et al., 2012; Courtois et al., 2013; Morris et al., 2013; Torkamaneh et al., 2020; Jia et al., 2019). En principe, le GWAS tire profit du grand nombre d'événements de recombinaison
survenus historiquement et relie ces événements au phénotype, permettant une cartographie à une échelle plus raffinée. Le GWAS présente des avantages par rapport à la cartographie traditionnelle : une résolution plus élevée et l'identification de plusieurs variants alléliques (Korte et Farlow, 2013). L’approche GWAS s’effectue au sein de collections de lignées non-issues d’un croisement entre deux parents. Elle permet de détecter un niveau ou degré d’association qu’a un ou des marqueurs SNP avec un caractère d’intérêt en se basant sur le LD (Korte et Farlow, 2013; Torkamaneh et al., 2020). Le diagramme Manhattan est une illustration d’un résultat GWAS.
Malgré tout, il est important de souligner l’existence de certains facteurs qui influencent très souvent le pouvoir avec lequel de réelles associations marqueur SNP-phénotype peuvent être identifiées avec les analyses GWAS.
Figure 1-4 : Diagramme Manhattan pour l’analyse d’association SNP-phénotype chez le
soya. Chaque point représente le degré d’association du marqueur SNP avec le phénotype. Sur l’axe des x, on indique la position physique de chaque marqueur le long des 20 chromosomes du génome et, sur l’axe des y, on représente la force de l'association SNP-phénotype sous forme de mesure de signification de l'association rencontrée (-log10(P)).Le
seuil de signification est indiqué par la ligne horizontale. Figure tirée et adaptée de Torkamaneh et al. (2020).
1.4.3.1 Les facteurs qui influencent les analyses GWAS
variation phénotypique significative est nécessaire pour une résolution élevée de la dissection allélique (Asimit et Zeggini, 2010; Gibson, 2012; Uchiyama et al., 2013). Un des facteurs critiques affectant les analyses GWAS est le LD à l’échelle du génome. Pour qu’un QTL soit identifié, au moins un marqueur devrait avoir un LD élevé avec ce QTL. L’étendu du LD permet de savoir à quand remonte le dernier « ancêtre commun », traduit le nombre de recombinaisons qui sont capturées au sein de la population de lignées et varie au long des chromosomes et même dans voisinage des gènes. Le LD s’étend sur une distance beaucoup plus courte au sein des régions euchromatiques, lesquelles sont riches en gènes et plus actives en matière de recombinaison. Ainsi, une densité élevée de marqueurs dans ces régions est nécessaire afin de capturer de fortes associations marqueurs-gènes.
Parfois, une variation d’un caractère peut être liée à la présence de sous-populations ou d’apparentements entre lignées. De telles associations « populationnelles » peuvent biaiser les analyses GWAS et amener à déclarer significative une association qui ne l’est pas (les « faux positifs »). Pour bien distinguer les vraies associations des fausses, les modèles d’analyse GWAS prennent en compte une matrice de structure de la population (Q ou P) et une matrice d’apparentement entre les individus (K) comme cofacteurs (Gupta et al., 2014). Il y a deux paramètres pour définir la structure de la population : le nombre de sous-populations et la probabilité d’appartenance des individus à une sous-population. Ces deux paramètres sont estimés par le logiciel STRUCTURE (Pritchard et al., 2000), lequel produit une matrice Q qui capture cette information. Une autre approche pour capturer la structure de population repose sur l’analyse en composantes principales (PCA de « Principal Component Analysis ») (Price et al., 2006). Cette seconde approche capture l’information sur la structure de la population sous la forme d’une matrice P. Ces deux méthodes d’analyse de la structure de la population sont réalisées à partir de données génotypiques des lignées pour des marqueurs pris au hasard dans la matrice de génotypage et répartis sur l’ensemble du génome (Desgroux, 2016).
Néanmoins, ces deux méthodes ne sont pas complètement efficaces pour capturer tous les types de relations génétiques qui existent au sein de populations complexes. L’apparentement (« kinship » en anglais) constitue un autre type de relation qui peut exister entre les paires de lignées et il mesure la probabilité que deux gènes homologues aient été hérités d’un ancêtre commun (Price et al., 2006). Il existe différentes méthodes qui permettent de calculer ce
degré de parenté génétique, et l’information qui en résulte est capturée sous la forme d’une matrice K. Dans des analyses GWAS, il est fréquent d’utiliser ces deux types de matrices (Q ou P et K) comme cofacteurs pour mesurer l’intensité de l’association entre un marqueur et le phénotype étudié (Zhao et al., 2007).
La définition du seuil de signification statistique approprié dans les études d'association à l'échelle du génome est également essentielle pour différencier les vrais positifs des faux positifs. Différentes méthodes statistiques prenant en compte plusieurs tests ont été proposées. La correction de Bonferroni et celle du « False discovery rate » (FDR) sont généralement utilisées dans les études d’association GWAS chez les plantes (Hommel, 1988; Hochberg, 1988; Benjamini et Hochberg,1995). Ces méthodes permettent de limiter le taux de faux positifs, mais elles augmentent parfois le taux de faux négatif (Perneger 1998). Récemment, une formule empirique basée sur l’héritabilité du caractère d’intérêt dans un environnement spécifique a été développée (Kaler et Purcell, 2019). Cette approche est moins conservatrice et capture plus de vrais positifs par rapport aux méthodes Bonferroni et FDR (Kaler et Purcell, 2019).
Plusieurs méthodes statistiques ont été développées dans les analyses GWAS pour l’identification des associations marqueur SNP-phénotype.
1.4.3.2 Les modèles statistiques : avantages et inconvénients
Pour un caractère quantitatif associé à un locus à effet fort, le modèle linéaire généralisé ou GLM (« General linear model ») peut être utilisé : Y = Q + S + e, où Y est le phénotype, Q la matrice de parenté (avec ou sans PCA), S est le marqueur génétique et e les résidus. En utilisant ce modèle, le caractère suit une distribution normale, sa variance reste la même dans chaque groupe d’échantillons et les sous-populations sont indépendantes (Bush et Moore, 2012). Par conséquent, ce modèle ne prend généralement pas en considération les liens de parenté complexes entre une population de lignées à l’étude même si des covariables sont utilisées pour contrôler la structure de la population. Donc le modèle GLM ne permet pas de détecter l’effet synergique des marqueurs sur le phénotype (Xiao et al., 2017).
Au contraire, le modèle linéaire mixte ou MLM (« Mixed linear model ») reste le modèle le plus efficace en corrigeant à la fois la structure de la population et les liens de parenté (Zhao
d’apparentement, u l’effet aléatoire de l’apparentement et ε la résiduelle. Ce modèle contrôle mieux les faux positifs que les modèles naïfs comme le t-test, qui n’inclut que les marqueurs testés. Le MLM contrôle les effets de biais d’un ensemble de loci ayant des effets faibles (Zhang et al., 2010). En utilisant les matrices Q et K dans le MLM, l’inflation des valeurs est contrôlée, mais les associations réelles marqueur-phénotype sont affaiblies.
La méthode FarmCPU « Fixed and random model circulating probability unification » réunit les avantages du MLM et surmonte son inconvénient en l’utilisant de manière itérative (Liu et al., 2016). FarmCPU effectue des tests de marqueurs avec des marqueurs associés en tant que covariables dans un modèle à effet fixe et une optimisation sur les marqueurs de covariants associés dans un modèle à effet aléatoire séparément. L’un des avantages de FarmCPU est le temps de calcul efficace qui est linéaire à la fois en nombre d'individus et de marqueurs (Liu et al., 2016).
En quoi notre travail pourrait-il être utile?
De nombreuses études ont estimé l’ASR comme un phénotype très important. En effet, l’ASR se veut un déterminant clé dans la performance et la productivité chez les espèces cultivées en conditions sous-optimales. Malgré son importance pour de nombreuses fonctions végétales, l’ASR n’a pas reçu toute l’attention qu’elle mérite. Chez le soya, peu d’études ont porté sur la caractérisation et le déterminisme génétique de l’ASR (Abdel-Haleem, et al., 2011; Rong et al., 2011; Brensha et al., 2012; Prince et al., 2015). Ces études sont principalement limitées par l’utilisation de plateformes de phénotypage imprécises (reflétant possiblement mal la réalité). De plus, elles s'appuyaient sur un nombre limité de marqueurs n'offrant pas une couverture exhaustive du génome et sont parfois limitées par une mauvaise résolution de la cartographie en raison d'une ségrégation et d'une recombinaison limitées (cartographie QTL traditionnelle). Par conséquent, aucun gène reconnu comme un candidat très prometteur contrôlant l’ASR n’a été identifié chez le soya jusqu’à présent, et peu chez les autres cultures. Maintenant, nous avons l’opportunité de faire une analyse plus approfondie de l’ASR chez le soya avec une meilleure couverture génomique afin de combler cette lacune.
Hypothèse et objectifs de recherche
L’hypothèse de ce projet de recherche est qu’il existe des déterminants génétiques qui contribuent à façonner le système racinaire et en à définir l’architecture chez le soya. Pour valider cette hypothèse, trois objectifs ont été fixés :
1) Caractériser l’ASR à l’aide d’un nouveau système de phénotypage racinaire en 2D en utilisant des rhizoboîtes, des enceintes au sein desquelles nous pouvons documenter le développement du système racinaire chez le soya.
2) Tirer profit des données génotypiques (marqueurs SNP) qui existent déjà pour les lignées canadiennes grâce aux travaux réalisés dans le cadre du projet SoyaGen aux fins d’analyses d’association pangénomiques (GWAS) pour identifier des marqueurs SNP associés à l’ASR.
Chapitre 2 Comprehensive Genome-Wide
Association Analysis Reveals the Genetic Basis
Comprehensive Genome-Wide Association Analysis
Reveals the Genetic Basis of Root System
Architecture in Soybean
Waldiodio Seck1,2, Davoud Torkamaneh1,2,3 and François Belzile1,2*
1 Département de phytologie, Faculté des sciences de l’agriculture et de l’alimentation
(FSAA), Université Laval, Québec (Québec), Canada
2Institut de biologie intégrative et des systèmes (IBIS), Université Laval, Québec (Québec),
Canada
2.1 Résumé
L’architecture du système racinaire (ASR) est un élément clé dans l'adaptation des plantes aux événements climatiques extrêmes. Ici, douze caractères associés à l'ASR ont été caractérisés au sein d'un panel de 137 lignées de soya canadien hâtif (collection de référence) à l'aide de rhizoboîtes et d'imagerie en 2D. Une variation phénotypique significative (P < 0,001) a été observée au sein de ces lignées pour différents caractères de l’ASR. Ce même panel a été génotypé au moyen d'une approche hybride combinant le génotypage par séquençage (GBS) et le reséquençage (WGS) pour donner un catalogue de 2,18M de SNP. Des analyses d’association pangénomique (GWAS) ont détecté au total 10 régions QTL pour la longueur totale de la racine et le diamètre de la racine principale. Ces régions QTL expliquaient entre 15 et 25 % de la variation phénotypique et deux gènes candidats putatifs sont rapportés sur la base de leur homologie.
2.2 Abstract
Breeding new crop cultivars with better and more efficient root system architecture (RSA) carries great potential to enhance resource-use efficiency and plant adaptation a changing and less stable climate. Roots being underground, their direct observation and detailed characterization are challenging. Here, were characterized twelve RSA-related traits in a panel of 137 early maturing soybean lines (Canadian soybean core collection) using rhizoboxes and 2D imaging. A significant phenotypic variation (P < 0.001) was observed among these lines for different RSA-related traits. This panel was genotyped with 2.18M genome-wide SNPs using a combination of genotyping-by-sequencing (GBS) and whole-genome sequencing (WGS). A total of 10 QTL regions were detected for root total length and root diameter through a comprehensive genome-wide association study (GWAS). These QTL regions explained from 15 to 25% of the phenotypic variation and contained two putative candidate genes with homology to genes previously reported to play a role in RSA in other species. This study was performed on a set of elite soybean lines providing fundamental insights into RSA and yielded a rich catalogue of QTLs and strong candidate RSA-related genes that will accelerate future efforts aimed to dissect genetic architecture of RSA and breed more resilient varieties.
2.3 Introduction
Plant root systems function as critical links between the growing shoot and the rhizosphere, by providing water and nutrients to the plant. Because roots grow underground and thus are not readily visible, they are often taken for granted as the “hidden half” of the plant, especially when it comes to crop improvement strategies (Kochian, 2017; Atkinson et al., 2019). In recent years, there has been a growing awareness of the importance of root structure and function in many important agronomic traits, especially those associated with crop adaptation to climate change and environmental stresses (Kochian, 2017). More recently, root system architecture (RSA), representing the two- and three-dimensional (2D and 3D) shape of the root system, has received greater attention. Root system architecture determines the ability of the plant to extract water and nutrients from the soil and thus has a direct impact on aboveground traits such as yield (Zhu et al., 2010; Chen et al., 2015; Chimungu et al., 2014; Postma et al., 2014; Saengwilai et al., 2014; Mutava et al., 2015; Liu et al., 2018; Robinson et al., 2018; Voss-Fels et al., 2018). Differences in RSA can lead to different root system archetypes, with some better adapted to acquiring nutrients that have relatively low mobility in the soil such as phosphorous, while other root architectures are better designed to acquire mobile soil nutrients (e.g. potassium nitrate) .
The root system originates from a primary root that develops during embryogenesis. This primary root produces secondary roots, which in turn produce tertiary roots. All secondary, tertiary, quaternary and further roots are referred to as lateral roots (Casero et al., 1995; Waidmann et al., 2020). RSA is generally characterized by measuring numerical variables that describe the size and abundance of components of the root system such as secondary lateral root number, root length and average diameter of root. In contrast, other measured variables focus on the topology or structure of the root system (such as the type and angle of connection between roots) (Hodge et al., 2009). The intrinsic determinants of RSA are those which are essential for lateral root initiation, developmental patterning of the primordium, and lateral root formation and growth (Malamy, 2005). In response to environmental stimuli, plants can optimize their RSA by initiating more or less lateral root primordia and influencing growth of primary or lateral roots (Waidmann et al., 2020). Despite the known importance of RSA-related traits, the quantification and analysis of RSA-related traits have evolved