Protein crystallogenesis of the arrowhead protease inhibitor-A
Texte intégral
Figure
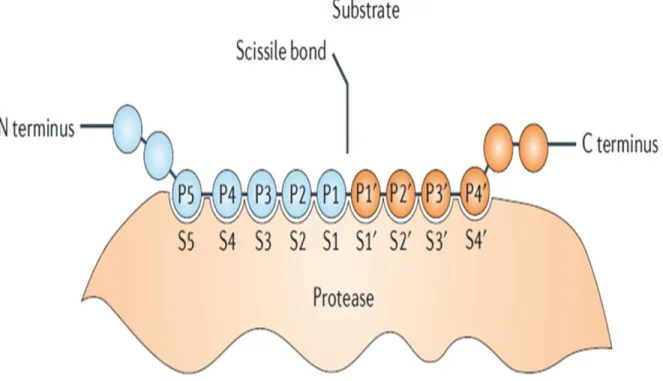
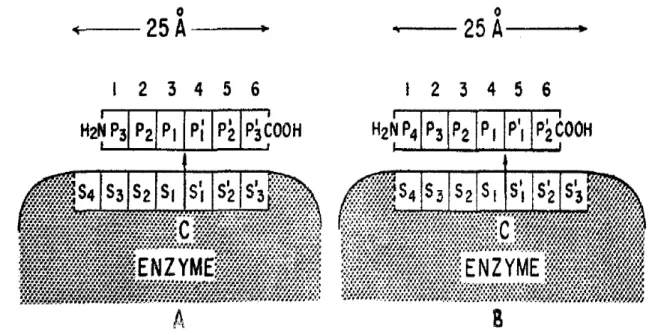
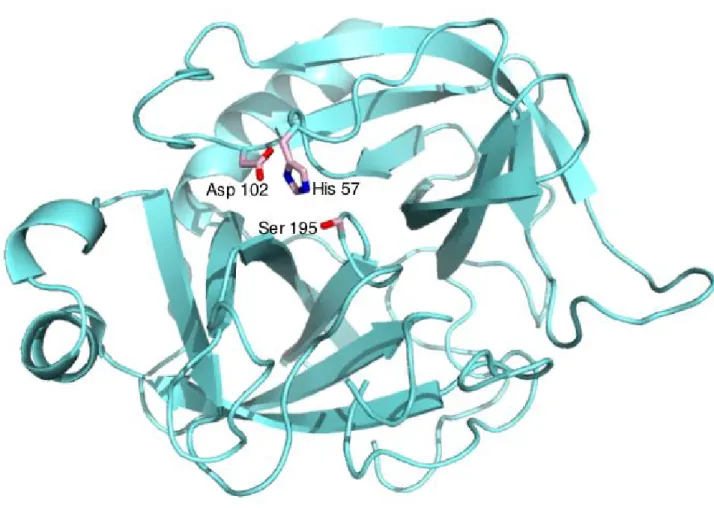
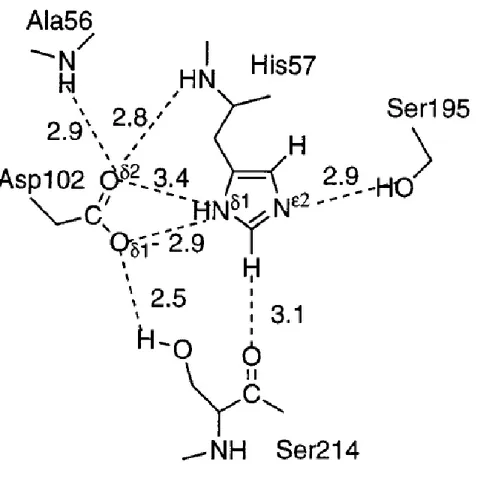
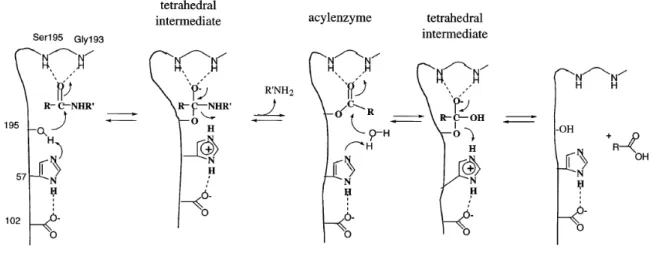
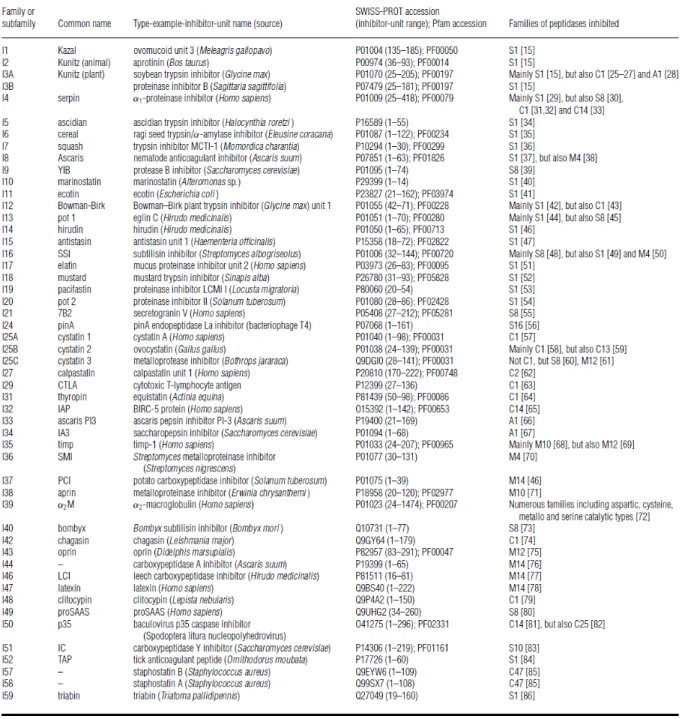

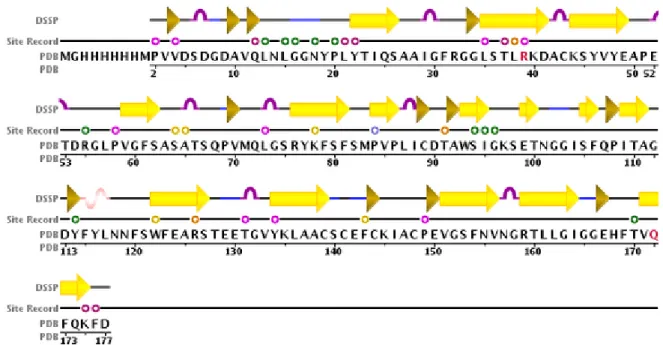

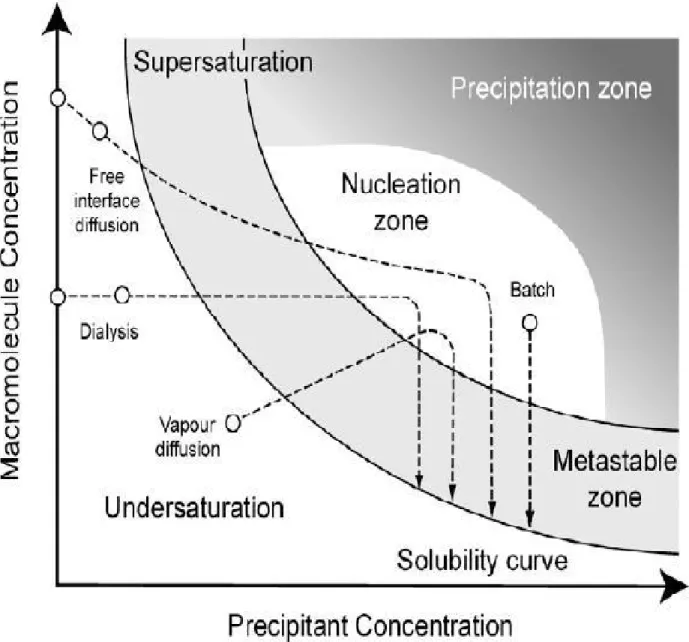
Documents relatifs
For all other treat- ments and the control, pollen of the corresponding non-transformed maize variety (Monumental) was fed to the bees. While pure sucrose solution was fed to bees
Prophenoloxidase system, lysozyme and protease inhibitor distribution in the common cuttlefish
The Immunomodulatory Effect of IrSPI, a Tick Salivary Gland Serine Protease Inhibitor Involved in Ixodes ricinus Tick
Here we show that the human intracellular serine protease inhibitor (serpin), protease inhibitor 9 (PI9), inhibits TNF-, TRAIL- and FasL- mediated apoptosis in certain
In this section, we present a method based on a GA to predict the graph whose vertices represent the SSE and edges represent spatial interactions between two amino acids involved in
[r]
Ces chocs organisationnels ont accru l'intensité du travail et se sont sou- vent accompagnés d'une perte d'autonomie : non seulement ils ont plus de tâches à accomplir, mais ils
The results of the current study indicated that in the case of ARVMs that underwent sI/R injury, the administration of exogenous rhSLPI, prior to or at the onset of sI significantly
