T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul SabatierDiscipline ou spécialité : Physiopathologie cellulaire, moléculaire et intégrée
JURY Dr Stéphane Germain Dr Daniel Henrion Dr Gilles Flouriot Dr Valérie Planat Dr Françoise Lenfant Pr Jean-François Arnal
Ecole doctorale : Biologie, Santé et Biotechnologie Unité de recherche : INSERM U858
Directeur(s) de Thèse : Dr Françoise Lenfant, Pr Jean-François Arnal Rapporteurs : Dr Stéphane Germain, Dr Daniel Henrion
Présentée et soutenue par Céline TOUTAIN Le 1er juillet 2009
Titre : EFFETS VASCULOPROTECTEURS DES ŒSTROGENES DANS L’ISCHEMIE CUTANEE ET APRES AGRESSION ARTERIELLE
Table des figures... 3
Principales abréviations ... 5
Résumé des travaux ... 7
Situation du sujet ... 9
Données bibliographiques... 11
I Œstrogènes et récepteurs des œstrogènes ... 13
I.1 Les ligands des récepteurs des œstrogenes ... 13
I.1.1 Œstrogènes et estradiol ... 13
I.1.1.1 Chimie, biosynthèse et métabolisme des œstrogènes ... 13
I.1.1.2 Physiologie des œstrogènes... 14
I.1.2 Les modulateurs sélectifs du récepteur des œstrogènes (SERM)... 18
Le récepteur des œstrogènes ... 19
I.2.1 Organisation génique ... 19
I.2.2 Structure protéique... 20
I.2.3 Modifications post-traductionnelles... 21
I.2.4 Mécanismes d’action ... 22
I.2.4.1 Le mécanisme classique : activité transcriptionnelle génomique ERE dépendante ... 22
I.2.4.2 Activité transcriptionnelle génomique non liée aux ERE... 22
I.2.4.3 Activité transcriptionnelle indépendante du ligand ... 22
I.2.4.4 Activation à partir d’un récepteur localisé à la membrane ... 23
Le récepteur des œstrogènes ... 27
I.4 Modèles d’invalidation génique des récepteurs des œstrogènes ... 28
I.4.1 Invalidation génique totale... 29
I.4.2 Génération de souris ERαAF-1° et ERαAF-2° ... 31
I.4.3 Invalidation génique cellule-spécifique ... 32
II Estradiol, ischémie et cicatrisation cutanée ... 35
II.1 Problématique clinique : nécrose des lambeaux cutanés ... 35
II.1.1 Définition, étiologie ... 35
II.1.2 Stratégies favorisant la survie des lambeaux ... 36
II.1.2.1 Approches pharmacologiques ... 36
II.1.2.2 Administration de facteurs de croissance ... 37
II.2 Estradiol et protection vis-à-vis de l’ischémie ... 37
II.2.1 Estradiol et ischémie cérébrale ... 38
II.2.2 Estradiol et ischémie myocardique ... 38
II.2.3 Estradiol et ischémie de membre postérieur ... 39
II.3 Estradiol et cicatrisation cutanée ... 39
II.3.1 Œstrogènes et physiologie cutanée ... 39
II.3.2 Œstrogènes et cicatrisation cutanée ... 39
II.3.3 Œstrogènes et inflammation ... 42
II.4 Les modèles de lambeaux cutanés ... 43
III Estradiol et cicatrisation vasculaire... 45
III.2 Stratégies pour favoriser la cicatrisation vasculaire... 47
III.3 Estradiol et endothelium ... 48
III.3.1 Production de monoxyde d’azote... 48
III.3.2 Prévention de l’apoptose ... 48
III.3.3 Régulation de l’expression des molécules d’adhérence leucocytaire... 48
III.3.4 Effet sur l'angiogenèse ... 49
III.3.5 Accélération de la réendothélialisation ... 50
III.3.6 Estradiol et progéniteurs endothéliaux ... 52
Revue... 55
Résultats expérimentaux ... 57
Article 1 ... 59
Editorial accompagnant l'article 1... 65
Article 1 : Résultats complémentaires ... 67
Article 2 ... 75
Article 2 : Résultats complémentaires ... 79
Discussion générale et Perspectives... 87
Annexe ... 97
TABLE DES FIGURES
Figure 1 : Voie principale de biosynthèse des œstrogènes... 16
Figure 2 : Principaux effets physiologiques et thérapeutiques des œstrogènes. ... 17
Figure 3 : Représentation schématique de l’organisation génique et protéique d’ERα. ... 20
Figure 4 : Mécanisme d’action des récepteurs aux œstrogènes. ... 25
Figure 5 : Activation de la voie d'EGFR par ERα membranaire... 26
Figure 6 : Structures protéiques d'ERα et ERβ... 27
Figure 7 : Modèles d'invalidation génique du récepteur aux œstrogènes α. ... 30
Figure 8 : Génération des souris ERαAF-1° ... 32
Figure 9 : Effets des œstrogènes sur la cicatrisation cutanée. ... 41
Figure 10 : Modèle de lambeau cutané. ... 44
Figure 11 : Stents actifs et retard de cicatrisation endothéliale... 47
Figure 12 : Accélération de la régénération endothéliale par l’E2... 52
Figure 13 : Effet de l'E2 chez des souris mâles... 72
Figure 14 : Effet de l'E2 chez des mâles ERα-/-... 73
Figure 15 : Effet de l'administration de testostérone et de flutamide sur l'apparition de la nécrose, chez des souris mâles, à jour 8. ... 74
Figure 16 : La fonction AF2 de ERα n’est pas nécessaire dans l’effet de l’E2 sur la réendothélialisation. ... 81
Figure 17 : Effet de l’E2 en présence ou non d’un inhibiteur d’EGFR. ... 83
PRINCIPALES ABREVIATIONS
AF-1/ AF-2 Activation function 1/2
AR Récepteur des androgènes
BrDU 5-bromo-2-deoxyuridine
CML Cellules Musculaires Lisses
CSX Castration
DBD DNA Binding Domain
E2 Estradiol
EGF Epidermal Growth Factor
eNOS NO synthase endothéliale
EPC Cellules Progénitrices Endothéliales
ER Récepteur des œstrogènes
ER α- Sous-type alpha ou bêta du récepteur des œstrogènes
ERE Estrogen Responsive Element
FGF-2 Fibroblast Growth Factor-2
FGF-2 hmw Fibroblast Growth Factor-2 high molecular weight
FGF-2 lmw Fibroblast Growth Factor-2 low molecular weight
GFP Green Fluorescent Protein
HSP Heat Shock Protein
IGF-1 Insulin-like Growth Factor-1
KO Knock Out
LBD Ligand Binding Domain
MIF Macrophage migration inhibitory factor
MMP Matrix Metalloproteinases
NO Monoxyde d’Azote
NS Non significatif
OVX Ovariectomie
PDGF Platelet Derived Growth Factor
RE Reendothelialized Area
REGEN Regenerative Endothelial Area
RetroP Retrograde Proliferating Zone
SERM Selective Estrogen Receptor Modulator
TGF Transforming Growth Factor
TNFα Tumor Necrosis Factor α
RESUME DES TRAVAUX
L’estradiol (E2) est une molécule multifonctionnelle exerçant majoritairement ses effets sur la physiologie sexuelle mais ayant également de nombreux effets sur des tissus non reproducteurs. Dans le cadre de mon travail de thèse, je me suis plus particulièrement intéressée aux effets de l’E2 sur l’ischémie cutanée et la cicatrisation vasculaire, dans deux modèles murins d’agression cutanée et vasculaire.
Mon travail a tout d’abord consisté à mettre au point un modèle d’ischémie cutanée, permettant de décrire et d’expliquer les principaux effets de l’E2 impliqués dans la prévention de la nécrose des lambeaux cutanés.La réalisation de lambeaux cutanés en chirurgie reconstructrice permet de disposer d’une surface tissulaire importante qui posséde sa propre vascularisation. Cependant, la nécrose des lambeaux est une complication majeure, car elle compromet leur survie. Son apparition est le plus souvent d’origine ischémique et il n’existe actuellement aucune thérapie réellement efficace. L’E2 est impliqué dans la physiologie cutanée et plus particulièrement dans les phénomènes de cicatrisation. De plus, l’E2 s’est révélé particulièrement efficace dans la prévention des lésions tissulaires d’origine ischémique. Dans un modèle de lambeaux cutanés, nous avons démontré un effet protecteur majeur de l’E2 vis-à-vis de la nécrose, associé à une meilleure survie tissulaire et à une préservation du réseau vasculaire cutané. Nous avons également montré que l’effet protecteur de l’E2 implique le récepteur aux œstrogènes (ER) α et que l’expression d'ERα dans les cellules médullaires n’est pas nécessaire pour conserver cet effet.
La préservation de ce réseau vasculaire permet une reperfusion plus rapide du lambeau cutané et limite par conséquence l’apparition de la nécrose.
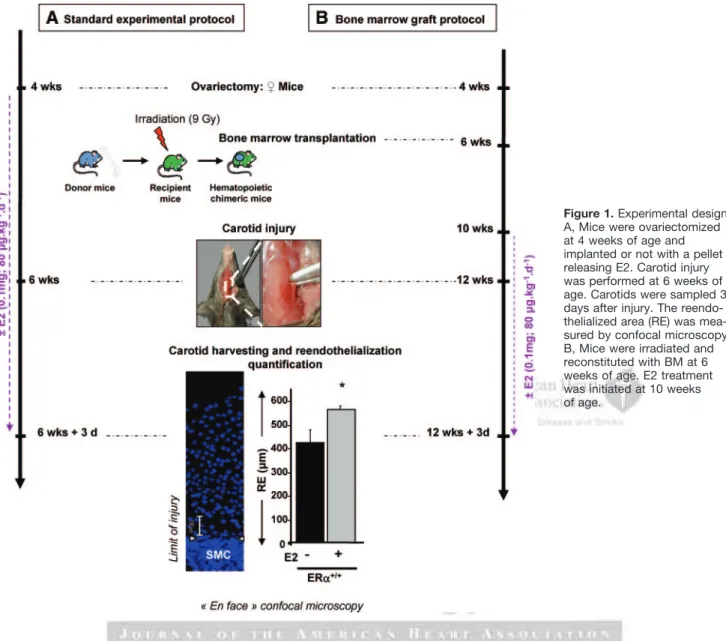
La deuxième partie de mon projet de thèse a consisté à déterminer les cibles cellulaires de l’E2 impliquées dans la réendothélialisation, en utilisant une technique originale de microscopie confocale "en face", permettant de visualiser la totalité de l’endothélium carotidien en régénération.Résumé
d’angioplasties. Ces stents inhibent la prolifération des cellules musculaires lisses et permettent de réduire les sténoses artérielles. Cependant, ils empêchent également la prolifération des cellules endothéliales. Le retard de cicatrisation artérielle ainsi induit augmente alors le risque de thrombose et de mort subite. L’optimisation de la cicatrisation vasculaire en clinique apparait donc nécessaire.
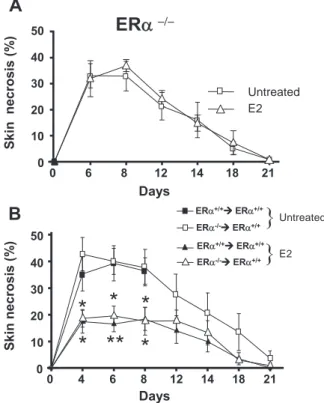
Dans un modèle d’agression électrique de la carotide, il a précédemment été montré au laboratoire que l’E2 accélère la régénération de l’endothélium et que la migration des cellules endothéliales précède leur prolifération, sous la dépendance d'ERα. Nous démontrons ici dans un modèle d’inactivation spécifique d’ERα et de souris chimères hématopoïétiques, que la coopération entre ERα dans les cellules endothéliales et ERα dans les cellules hématopoïétiques est indispensable à l’effet accélérateur de l’E2.
SITUATION DU SUJET
Les maladies cardiovasculaires représentent aujourd’hui la première cause de mortalité en France et dans le monde. Le nombre de décès imputables à une maladie cardiovasculaire a été estimé à 17.5 millions pour 2005, soit 30 % de la mortalité mondiale totale. Parmi ces décès, on estime que 7.6 millions sont dus à une cardiopathie coronarienne et 5.7 millions à un accident vasculaire cérébral (données de l’Organisation Mondiale de la Santé,
www.who.int).
Bien que de nombreuses données expérimentales et épidémiologiques démontrent des effets bénéfiques des œstrogènes sur le système cardiovasculaire, l’évaluation des bénéfices et des risques d’un traitement hormonal substitutif chez la femme ménopausée a soulevé de nombreuses questions et incertitudes. Alors que les études épidémiologiques suggèrent une diminution de l’incidence des maladies cardiovasculaires chez des femmes recevant un traitement hormonal substitutif, les résultats des essais randomisés en prévention primaire (WHI : Women’s Health Initiative Study) (Rossouw, Anderson et al. 2002) et secondaire (HERS : Heart and Estrogen/Progestin Replacement Study) (Hulley, Grady et al. 1998) ont montré une aggravation de la morbi-mortalité par maladie cardiovasculaire et une augmentation de l’incidence du cancer du sein.
Dans ce contexte, notre équipe travaille depuis de nombreuses années à la compréhension et à la caractérisation des mécanismes cellulaires et moléculaires responsables des effets vasculaires des œstrogènes. L’estradiol (E2) est la principale hormone stéroïde sexuelle œstrogènique chez les mammifères. L’E2 est une molécule multifonctionnelle exerçant majoritairement ses effets sur la physiologie sexuelle mais ayant également de nombreux effets sur des tissus non impliqués dans la physiologie de la reproduction et en particulier sur le système cardiovasculaire.
L'objectif de ma thèse est orienté vers une meilleure compréhension des effets, bénéfiques ou délétères, des œstrogènes. Au cours de ce travail, je me suis plus particulièrement intéressée à deux tissus cibles de l’E2 : la peau et les vaisseaux.
Situation du sujet
Dans la première partie bibliographique de ce manuscrit, nous exposerons la structure et les mécanismes d’action d’ERα puis nous ferons un bilan des effets vasculaires et cutanés des œstrogènes. La seconde partie sera consacrée à l’exposé des travaux expérimentaux, qui ont pour objet l’étude de l’effet protecteur de l’E2 sur l’ischémie cutanée et la caractérisation des cibles cellulaires impliquées dans l’effet accélérateur de l’E2 sur la régénération endothéliale.
I ŒSTROGENES ET RECEPTEURS DES OESTROGENES
L’objectif de mon travail de thèse a été d’évaluer les effets vasculaires de l’E2 dans deux modèles murins. Dans la première partie de cette étude bibliographique, nous résumerons certains éléments de physiologie des œstrogènes (Ganong 2001; Guyton and Hall 2003; Widmaier, Raff et al. 2004) et du mécanisme d’action de leurs récepteurs.
Les récepteurs des œstrogènes (ER) appartiennent à la superfamille des récepteurs nucléaires et plus précisément à la classe regroupant les récepteurs des hormones stéroïdes. Ils agissent en tant que facteurs de transcription. Il existe deux types d'ER, ERα (également appelé NR3A1) cloné en 1985 (Walter, Green et al. 1985) puis séquencé en 1986 (Green, Walter et al. 1986) et ER (NR3A2) cloné dix ans plus tard (Kuiper, Enmark et al. 1996). Une attention particulière sera portée aux modèles d’inactivation génique des récepteurs, dont je me suis servie au cours de mon étude.
I.1 LES LIGANDS DES RECEPTEURS DES OESTROGENES
I.1.1 Œstrogènes et estradiol
I.1.1.1 Chimie, biosynthèse et métabolisme des œstrogènes
Les œstrogènes sont des hormones sexuelles stéroïdes à 18 atomes de carbones (C18), dérivées du cholestérol. Les trois principaux œstrogènes naturels sont : (i) l’estradiol (E2, forme majoritaire secrétée par l’ovaire chez la femelle non gestante, obtenue par aromatisation de la testostérone), (ii) l’estriol (E3, produit principalement au cours de la gestation par le placenta) et (iii) l’estrone (E1, produit pendant la ménopause par aromatisation de l’androstènedione). Les œstrogènes, lipophiles, diffusent rapidement à travers les membranes cellulaires pour agir sur leurs récepteurs, appartenant à la superfamille des récepteurs nucléaires. Ils sont produits principalement par les gonades et les surrénales.
Chez la femme, les œstrogènes sont synthétisés dans les ovaires et en moindre quantité dans le cortex surrénalien. Au cours de la gestation, une quantité considérable est également produite par le placenta. La voie de biosynthèse des œstrogènes (Figure 1) est
Œstrogènes et Récepteur des œstrogènes
initiée à partir du cholestérol et passe par la formation de progestérone puis d’androgènes. Les androgènes, précurseurs des œstrogènes, sont synthétisés dans les cellules thécales puis ils diffusent ensuite dans les cellules de la granulosa où ils sont convertis en œstrogènes par une aromatase. Pendant la phase lutéale, le corps jaune sécrète également de grande quantité de progestérone et d'œstrogènes. La production des œstrogènes est stimulée par les hormones antéhypophysaires, la FSH (Follicle-stimulating hormone) et la LH (Luteinizing hormone). Après la ménopause, les œstrogènes proviennent de la transformation des androgènes surrénaliens, sous l'action de l'aromatase des tissus périphériques.
Chez l’homme, les œstrogènes sont formés dans le testicule et dans les glandes corticosurrénales. Dans le testicule, la voie de biosynthèse des œstrogènes est initiée dans les cellules de Leydig et les androgènes sont aromatisés en œstrogènes dans les cellules de Sertoli.
Chez la femme, les œstrogènes sont transportés dans le sang, liés à des protéines de transport : 2% seulement de l’E2 circulant est sous forme libre, le reste étant soit lié de façon non spécifique à l’albumine (60%) soit de façon spécifique à la globuline liant les stéroïdes sexuels (GBG : gonadal steroid-binding globulin 38%). Notons que chez la souris, il existe très peu de protéines porteuses : les concentrations plasmatiques totales en E2 sont certes plus faibles mais les concentrations libres sont presque identiques.
Les œstrogènes sont convertis en métabolites glucuro- ou sulfoconjugés dans le foie. Ces composés sont excrétés majoritairement dans l’urine. Un cinquième environ est secrété dans la bile pour être ensuite réabsorbé dans la circulation.
I.1.1.2 Physiologie des œstrogènes
Les œstrogènes sont présents aussi bien chez la femme que chez l’homme, bien qu’ils soient produits en plus grande quantité chez la femme en période d’activité génitale. Ils agissent sur le développement des caractères sexuels secondaires et le contrôle du cycle menstruel. Outre leur rôle paracrine dans les ovaires, leurs effets sur l’axe hypothalamo-hypophysaire et la stimulation de la croissance des organes sexuels, les œstrogènes agissent également sur de nombreux autres tissus (Figure 2).
La sécrétion des œstrogènes durant le cycle menstruel présente deux pics : juste avant l’ovulation puis vers le milieu de la phase lutéale. Les concentrations plasmatiques d’E2 chez la femme en période d’activité génitale sont les suivantes :
Phase folliculaire : 11-165 pg.mL-1 (0.04-0.6 nM)
Pic ovulatoire : 146-526 pg.mL-1 (0.5-1.93 nM)
Phase lutéale : 33-196 pg.mL-1 (0.12-0.72 nM)
Chez la femme ménopausée, les concentrations plasmatiques d’E2 sont inférieures à 37 pg.mL-1 (0.14 nM). Chez l’homme, les concentrations plasmatiques d’E2 sont d’environ 20-50 pg.mL-1 (0.07-0.18 nM). Chez la souris, les concentrations plasmatiques d’E2 sont comprises entre 1 et 5 pg.mL-1 (3.67 10-3-0.018 nM).
Œstrogènes et Récepteur des œstrogènes
Estriol (E3) Estradiol (E2) Estrone (E1) Cholestérol Prégnélonone 17-Hydroxyprénélonone Progestérone 17-Hydroxypogestérone Déhydroépiandrostérone Androstènediol Androstènedione Testostérone Placenta Granulosa (Sertoli) Th èqu e (L eydi g) Cor ps j au ne
Figure 1│Voie principale de biosynthèse des œstrogènes.
Les œstrogènes sont synthétisés à partir du cholestérol dans les gonades (ovaires ou testicules) où il est convertit en prégnénolone puis en progestérone. L’aromatase catalyse la conversion des androgènes en œstrogènes. (Modifié d’après(Hall and Phillips 2005)).
Croissance des organes génitaux à la puberté Développement des caractères sexuels secondaires Croissance des ovaires et des follicules ovariens Changement cyclique de l’endomètre, du col et du vagin
• Utérus: prolifération des cellules stromales et épithéliales (phase proliférative), développement glandulaire , stimulation de la croissance du muscle lisse, contraction myométriales, sensibilité accrue à l’ocytocine
• Trompes: stimulation de l’activité ciliaire • Vagin: épithélium devient kératinisé • Col: mucus cervical fluide et alcalin
• Glande mammaire: croissance des canaux, dépôt de graisse, inhibition de la sécrétion lactatée
Comportement d’œstrus, instinct maternel
Effets de rétrocontrôle sur l’axe hypothalamo-hypophysaire Apprentissage, mémoire…
Activité ostéoblastique
Soudure des cartilages de conjugaison Prévention de l’ostéoporose
Texture fine et souple Vascularisation
Sécrétion liquidienne des glandes sébacées
Réduction du cholestérol plasmatique
Vasodilatation, augmentation de la production de NO Protection contre l’athérosclérose
Favorisent les thromboses veineuses Comportement sexuel, fertilité Testicule: spermatogenèse
Canaux efférents: concentration du sperme, différentiation épithéliale
Figure 2│ Principaux effets physiologiques et thérapeutiques des œstrogènes. (Modifié d’après (Widmaier, Raff et al. 2004)).
Œstrogènes et Récepteur des œstrogènes
I.1.2 Les modulateurs sélectifs du récepteur des œstrogènes (SERM)
Les modulateurs sélectifs du récepteur des œstrogènes (Selective Estrogen Receptor Modulator, SERM) sont une famille de ligands synthétiques, analogues structuraux de l’E2 et fonctionnant comme des agonistes de l’E2 dans certains tissus ou bien comme antagonistes dans d’autres (Degrelle 2004; Luzuy 2004). La découverte du tamoxifène il y a plus de 40 ans a ouvert la voie à la recherche de substances possédant les effets recherchés des œstrogènes, sans leurs effets délétères. L'activation du récepteur des œstrogènes par un ligand œstrogènique entraîne un changement de conformation du récepteur. Le récepteur activé peut alors se lier sur les ERE (Estrogen Responsive Elements) et déclencher la transcription de gènes spécifiques. L'activité transcriptionnelle dépend de la présence de facteurs tissus- ou cellules-dépendants (co-activateurs ou co-inhibiteurs). La transcription génique dépend donc à la fois de la nature du SERM et de l’environnement protéique de la cellule cible (Levenson and Jordan 1999). Le complexe SERM-ER peut également activer la transcription génique grâce à une interaction protéine-protéine avec les proto-oncogènes fos et jun sur les sites AP-1 (Activator Protein-1) (Jordan, Gapstur et al. 2001).
Le tamoxifène possède des activités antagonistes sur la glande mammaire et agonistes sur l’os et le système cardiovasculaire, si bien qu’il s’est révélé particulièrement efficace dans le traitement adjuvant du cancer du sein hormono-dépendant (Love and Koroltchouk 1993) mais aussi dans la prévention de l’ostéoporose et des maladies cardiovasculaires (Love 1992; Love, Mazess et al. 1992). Le tamoxifène module également le métabolisme des lipides en diminuant le cholestérol plasmatique et en augmentant les triglycérides (Osborne, Zhao et al. 2000). Cependant, les effets agonistes du tamoxifène sur l’utérus, favorisant le cancer de l’endomètre, la survenue de bouffées de chaleur et le risque thromboembolique ont rendu impossible son utilisation dans la prévention de l'ostéoporose et des maladies cardiovasculaires (Love 1992).
Le raloxifène agit comme agoniste sur le tissu osseux en augmentant la densité osseuse et en diminuant la résorption osseuse, sans effet sur l’endomètre. Il a donc été développé dans le cadre de la prévention de l’ostéoporose (Osborne, Zhao et al. 2000). Par contre, il n'a pas d'efficacité dans la prévention des maladies cardiovasculaires (Clarkson, Anthony et al. 1998).
Le développement de nouveaux SERM implique donc une meilleure compréhension des effets moléculaires et cellulaires des œstrogènes, afin de proposer de nouvelles substances
pour la prévention des complications liées à la ménopause (maladies cardiovasculaires, ostéoporose, bouffées de chaleur) et dans le traitement du cancer du sein.
LE RECEPTEUR DES ŒSTROGENES
Les effets de l'E2 dépendent de l'activation de récepteurs nucléaires. Il existe deux types d'ER, codés par deux gènes distincts : ERα et ER. Les travaux de notre équipe ont permis de montrer que la majorité des effets vasculaires des œstrogènes sur la production de monoxyde d’azote (NO) (Darblade, Pendaries et al. 2002) ou la réendothélialisation (Brouchet, Krust et al. 2001) étaient médiés par ERα et non pas par ER, tandis que le rôle de ER reste à l’heure actuelle moins bien connu.
I.2.1 Organisation génique
Le gène d’ERα, nommé ESR1 (estrogen receptor locus 1), est localisé sur le locus chromosomique 6q25.1 chez l’homme (Gosden, Middleton et al. 1986; Menasce, White et al. 1993). Ce gène s’étend sur plus de 140 kilobases et comporte 8 exons séparés par 7 régions introniques. L’exon 1 code pour le domaine A/B, les exons 2 et 3 pour une partie du domaine C, l’exon 4 pour une partie du domaine C, le domaine D ainsi qu’une partie du domaine E. Le domaine E est codé par la séquence exonique comprenant une partie de l'exon 4 et 8 et les exons 5,6 et 7. La fin de l’exon 8 code pour le domaine F (Ponglikitmongkol, Green et al. 1988) (Figure 3).
A l’exception de l’extrémité 5’ non traduite, la séquence de ce gène est fortement conservée entre les espèces. Cette variabilité au niveau de l’extrémité 5’ s’explique par l’existence de promoteurs multiples permettant une régulation ayant une sélectivité tissulaire pour l’expression de variants d’ARNm codant pour ERα (Kos, Reid et al. 2001).
Œstrogènes et Récepteur des œstrogènes
5’-UTR Ex1 Ex2 Ex3 Ex4 Ex5 Ex6 Ex7 Ex8 //
// -66KDa Domaine transactivateur ligand-indépendant DBD Domaine transactivateur ligand-dépendant LBD Dimérisation AF - 2 AF-2 AF-1 N C
Figure 3│ Représentation schématique de l’organisation génique et protéique d’ERα. (Modifié d’après (Billon-Gales, Fontaine et al. 2009)).
I.2.2 Structure protéique
Comme les autres membres de la superfamille des récepteurs nucléaires, ERα présente une structure protéique composée de domaines fonctionnels désignés de A à F, depuis l’extrémité N-terminale vers l’extrémité C-terminale (Figure 3). Le domaine de liaison à l’ADN (DNA-binding domain : DBD, situé dans la région C) et le domaine de liaison de l’hormone (ligand-binding domain : LBD, situé dans la région E) sont hautement conservés entre les espèces. Le domaine A/B situé en région N-terminale et la région D, sont quant à eux moins conservés. La région C-terminale F contiguë avec le domaine E a un rôle moins bien connu (Germain, Staels et al. 2006).
La région A/B porte une fonction de transactivation appelée AF-1 qui est impliquée dans des interactions protéines-protéines et dans l’activation transcriptionnelle de gènes cibles. Cette fonction peut être activée aussi bien de manière dite "ligand indépendante", après phosphorylations par exemple, que de façon "ligand dépendante". Cette région peut également interagir avec divers cofacteurs (co-activateurs ou facteurs de transcription).
La région C de domaine de liaison à l’ADN contient une structure dite en "doigt de zinc" qui joue un rôle important dans la dimérisation du récepteur et dans la liaison aux séquences spécifiques d’ADN, appelées "Estrogen Responsive Elements" (ERE).
Le domaine D sert de "charnière" entre le DBD et le LBD, permettant la rotation du DBD et l’adoption de différentes conformations du DBD et du LBD. Ce domaine
possède également un "Nuclear Localization Signal", permettant une localisation nucléaire du récepteur.
Le domaine E/F de liaison du ligand est fonctionnellement plus complexe car il permet la fixation du ligand, la dimérisation du récepteur, sa translocation nucléaire et la transactivation de gènes cibles, via la fonction de transactivation AF-2. La fonction AF-2 est une région complexe dont la structure et la fonction sont gouvernées par la liaison du ligand. La surface d’interaction d’AF-2 est composée d’acides aminés provenant des hélices 3, 4, 5 et 12. La fixation de l’E2 sur le récepteur entraine un basculement de l’hélice 12 qui referme la poche de liaison du ligand. Ce réarrangement s’accompagne d’une compaction générale du LBD, entraînant la stabilisation du dimère de récepteurs sur les séquences régulatrices des promoteurs, une dissociation des corépresseurs et la création de nouvelles surfaces d’interaction pour des coactivateurs. Différents ligands peuvent induire des conformations différentes du récepteur et le positionnement de l’hélice 12 est à l’origine des effets agonistes ou antagonistes des œstrogènes (Kumar, Green et al. 1987; Nilsson, Makela et al. 2001; Gronemeyer, Gustafsson et al. 2004; Germain, Staels et al. 2006).
I.2.3 Modifications post-traductionnelles
ERα peut être le siège de plusieurs types de modifications post-traductionnelles, impliquées dans ses régulations fonctionnelles. Par exemple, l'acétylation des résidus lysine 266 et 268 augmente sa capacité de fixation à l'ADN (Kim, Woo et al. 2006). Inversement, la nitrosylation des résidus cystéine au niveau des motifs en doigt de zinc par le NO diminue la fixation à l'ADN (Marino, Ficca et al. 2001). La méthylation de l'arginine 260 localisée dans le domaine de liaison à l'ADN a lieu dans le cytoplasme et est à l'origine d'une réponse non génomique. De même, la palmitoylation de la cystéine 447 située dans la région E est indispensable à la localisation membranaire d'ERα en association avec la cavéoline-1 et à son activité non génomique (Marino, Ascenzi et al. 2006). ERα est également le siège de phosphorylations sur de multiples résidus aminés, tels que les sérines 118, 104 ou 106. Ces phosphorylations résultent de l'activité de différentes kinases et augmentent l'activité transcriptionnelle du récepteur (Lannigan 2003). Enfin, l'ubiquitination permet la régulation de la dégradation du récepteur dans le protéasome (Reid, Denger et al. 2002).
Œstrogènes et Récepteur des œstrogènes
I.2.4 Mécanismes d’action
Après leur diffusion à travers la membrane plasmique, les œstrogènes peuvent activer la transcription de leurs gènes cibles par diverses voies de signalisation, génomiques ou bien extra-génomiques (Nilsson, Makela et al. 2001) (Figure 4).
I.2.4.1 Le mécanisme classique : activité transcriptionnelle génomique ERE dépendante
En absence de ligand, le récepteur se présente sous forme monomérique, complexé avec des protéines chaperonnes HSP (Heat Shock Proteins), principalement HSP70 et HSP90 et localisé dans le noyau ou le cytoplasme. La liaison du ligand entraine un changement conformationnel du récepteur, qui se dissocie des HSP, se dimérise avec un autre récepteur puis est transféré vers le noyau (Reid, Denger et al. 2002). Le dimère peut alors se fixer à des ERE situés en amont des gènes cibles puis active leur transcription via ses fonctions de transactivation AF-1 et AF-2, qui recrutent des cofacteurs impliqués dans le remodelage de la chromatine et l’activation transcriptionnelle (McKenna and O'Malley 2002). En fonction du type cellulaire et des promoteurs mis en jeu, le récepteur peut alors exercer une activité stimulatrice ou inhibitrice vis-à-vis de l’expression des gènes cibles (Tora, White et al. 1989).
I.2.4.2 Activité transcriptionnelle génomique non liée aux ERE
Le récepteur des œstrogènes est capable d’induire la transcription de gènes dépourvus de séquence ERE, par liaison indirecte de l’ADN, via l’interaction avec un complexe formé de deux facteurs de transcription fos et jun également appelé AP-1 (Activator Protein 1) (Safe and Kim 2008). Cette voie de signalisation est impliquée dans la transcription des gènes tels qu’IGF-1 (Insulin-like Growth Factor-1). L’expression de certains gènes contenant des séquences promotrices riches en bases GC, est activée via l’interaction avec le complexe SP1 (Stimulating Protein 1) (Hall, Couse et al. 2001). Un dernier exemple est l’interaction entre ERα et la sous-unité c-rel du complexe NF-B, qui inhibe l’expression de l’interleukine-6 (Galien and Garcia 1997).
I.2.4.3 Activité transcriptionnelle indépendante du ligand
En parallèle d’une activation hormono-dépendante, le récepteur des œstrogènes peut être modulé par des signaux extracellulaires, en absence d’E2. Les facteurs de croissance EGF
(Epidermal Growth Factor) et IGF-1 sont capables d’activer le récepteur et d’induire l’expression de gènes cibles (Ignar-Trowbridge, Pimentel et al. 1995).
L’activation de MAP kinases par des facteurs de croissance (EGF, IGF-1, insuline) entraîne la phosphorylation de la serine 118 localisés au niveau de la fonction AF-1 et permettent au récepteur de potentialiser la transcription de gène cibles (Kato, Endoh et al. 1995; Kato, Kitamoto et al. 1998; Kato 2001).
I.2.4.4 Activation à partir d’un récepteur localisé à la membrane
A côté des effets génomiques, il existe des effets dits "rapides", usuellement appelés effets "non génomiques", et pouvant cependant être à l'origine d'effets transcriptionnels et survenant quelques secondes ou quelques minutes après l’ajout d’E2. Ces effets font notamment intervenir l’activation de kinases et de phosphatases.
Le récepteur membranaire est localisé au niveau de cavéoles, associé à des protéines comme la cavéoline (Evinger and Levin 2005). Il a été proposé que la localisation membranaire du récepteur soit permise grâce à des modifications post-traductionnelles, en particulier la S-palmitoylation de la cystéine 447 (Marino, Ascenzi et al. 2006). La liaison du ligand met en jeu diverses cascades de signalisation telles que l’activation des MAP kinases, des phophatidyl-inositol 3-kinases (PI3K), de la phospholipase C (PLC), de la protéine kinase C (PKC), de la NO synthase endothéliale (eNOS), ou encore de la voie de l'EGFR (Epidermal growth factor receptor) (Levin 2002).
L’E2 est en effet capable d’activer le récepteur de l’EGF à partir de la stimulation d’ERα membranaire, conduisant à la libération d’EGF (Figure 5). Le ERα adressé à la membrane cytoplasmique est couplé à une protéine G et active, via la protéine Src, les métalloprotéinases MMP-2 et MMP-9, qui libérèrent l’heparin binding epidermal growth factor (HB-EGF). Ce dernier va ensuite induire la phosphorylation de son récepteur (EGFR) et ainsi activer la voie de signalisation ERK/MAPK. L’activation d’EGFR induit l’activation en cascade de kinases (Razandi, Alton et al. 2003). Il s’agit de l’un des effets non génomiques d’ERα (Levin 2003).
En plus de ces mécanismes d'action, en absence d'E2, ERα "non liganté" est capable de moduler la croissance et la différentiation cellulaire. Par exemple, le ERα non liganté inhibe la croissance neuronale par une régulation négative de la voie des MAPK et PI3K/Akt. Cette
Œstrogènes et Récepteur des œstrogènes
action d'ERα non liganté n'implique pas les domaines de transactivation et de liaison à l'ADN, suggérant qu'elle serait indépendante de toute activité transcriptionnelle (Merot, Ferriere et al. 2009).
ER Hs p70 Hs p90 ER + ERE P P Fos Jun AP1 / SP1 Kinases P P IGF-1 EGF 1- Classique 2- ERE indépendante 3- Ligand indépendante Kinases Phosphatases P P activité transcriptionelle stabilité 4- Effets rapides Noyau Cytoplasme E2 ERE
Figure 4│Mécanisme d’action des récepteurs aux œstrogènes.
1- Mécanisme d’action classique (direct) : après activation par le ligand, les dimères de récepteurs se lient à l’ADN au niveau de séquences spécifiques (ERE).
2- Mécanisme d’action ERE indépendant (indirect) : les dimères de récepteurs se lient à l’ADN via des interactions protéiques.
3- Mécanisme d’action ligand-indépendant : des facteurs de croissance activent des kinases qui phosphorylent les récepteurs et se fixent à l’ADN au niveau de séquences spécifiques (ERE).
4- Mécanisme d’action non-génomique : les récepteurs localisés à la membrane activent des kinases, conduisant à des modifications rapides de protéines cytoplasmiques ou bien à des régulations transcriptionnelles.
Œstrogènes et Récepteur des œstrogènes
Figure 5│ Activation de la voie d'EGFR par ERα membranaire.
LE RECEPTEUR DES ŒSTROGENES
Le gène ER, nommé ESR2 (estrogen receptor locus 2), est localisé sur le chromosome 14 chez l’homme (Enmark, Pelto-Huikko et al. 1997). Comme les autres membres de la superfamille des récepteurs nucléaires, ER possède une structure protéique similaire à celle d’ERα. La région C est la plus conservée, avec 97% d’homologie avec ERα. Les autres régions sont moins conservées : A/B (20-30%), D (30%), E (59%) et F (18%) (Mosselman, Polman et al. 1996; Morani, Warner et al. 2008) (Figure 6). ER peut également former des hétérodimères avec ERα pour se fixer à l'ADN (Cowley, Hoare et al. 1997).
Figure 6│Structures protéiques d'ERα et ER.
Le pourcentage indique le degré d'homologie entre ERα et ER (Marino, Galluzzo et al. 2006).
ER possède une distribution tissulaire différente de celle d’ERα. Chez les rongeurs, ER est particulièrement abondant dans l’épithélium prostatique, les cellules de la granulosa ovarienne (Sar and Welsch 1999) et le parenchyme pulmonaire (Koehler, Helguero et al. 2005; Dahlman-Wright, Cavailles et al. 2006).
ER est abondamment exprimé dans le lobe ventral de la prostate de souris et de rat. L’étude de souris déficientes en ER montre initialement la présence de multiples foyers hyperplasiques, associés à une augmentation de la prolifération épithéliale et à une diminution de l’apoptose dans la prostate (Krege, Hodgin et al. 1998; Weihua, Makela et al. 2001; Imamov, Morani et al. 2004). Il a également été suggéré que la perte de l’expression d’ER était associée au développement du cancer de la prostate chez l’homme (Horvath, Henshall et al. 2001).
L’hypofertilité des animaux ER
Œstrogènes et Récepteur des œstrogènes
folliculaire) (Weihua, Saji et al. 2000; Cheng, Weihua et al. 2002) a été associée à un défaut de vascularisation de la thèque, diminuant la maturation folliculaire (Inzunza, Morani et al. 2007). De plus, ER serait impliqué dans la parturition (Wu, Geimonen et al. 2000) et la placentation (Tessier, Deb et al. 2000). ER favorise également la différentiation cellulaire dans la glande mammaire (Forster, Makela et al. 2002) et réduirait la prolifération de cellules de cancer du sein induite par l’estradiol (Palmieri, Cheng et al. 2002; Strom, Hartman et al. 2004).
Il a également été rapporté qu’ER jouerait un rôle dans le développement post-natal et l’homéostasie pulmonaire. Il est en particulier impliqué dans la formation et la régénération des alvéoles ainsi que dans la maintien de l’élasticité pulmonaire (Patrone, Cassel et al. 2003; Massaro and Massaro 2004; Morani, Barros et al. 2006).
ER serait impliqué dans l’hématopoïèse : des souris ER-/-
âgées de plus de 1 an développent une maladie myéloproliférative analogue à une leucémie myéloïde chronique humaine (Shim, Wang et al. 2003).
Cependant, les effets attribués à ER et les phénotypes observés sont différents selon les modèles de souris transgéniques étudiés et le "lieu" où les souris sont étudiées, et demeurent donc controversés. Des donnés utilisant les mêmes modèles murins indiquent que ER jouerait un rôle minime sur l’utérus, le système hypothalamo-hypophysaire, le squelette alors qu’il joue un rôle évident dans les ovaires, le cerveau (anxiété, plasticité synaptique (Krezel, Dupont et al. 2001), mémoire et apprentissage (Andreescu, Milojkovic et al. 2007)) ainsi que dans des modèles d’inflammation chronique (maladie inflammatoire chronique de l’intestin, arthrite rhumatoïde), de métrites ou de septicémie (Harris 2007).La description récente d’un nouveau mutant d’ER (ERSTL-/L-) montre que mis à part ses effets sur la
reproduction, ER ne serait pas impliqués dans le développement et l’homéostasie de la plupart des organes (Antal, Krust et al. 2008) et remet en cause bon nombre des données précédentes.
I.4 MODELES D’INVALIDATION GENIQUE DES RECEPTEURS DES OESTROGENES
Avec l’avènement de l’inactivation génique par recombinaison homologue, la souris est devenue un modèle de choix pour l’identification des mécanismes moléculaires et cellulaires impliqués dans les effets des œstrogènes. Afin d’étudier l’implication d’ERα dans
la prévention de la nécrose des lambeaux cutanés et les cibles cellulaires mises en jeu dans l’effet accélérateur de l’E2 sur la réendothélialisation, plusieurs lignées de souris transgéniques ont été utilisées.
I.4.1 Invalidation génique totale
La première lignée de souris invalidées pour ERα (appelées ERα-Neo KO) a été générée en 1993 en insérant d’une cassette de résistance à la néomycine dans le premier exon codant du gène ERα (Lubahn, Moyer et al. 1993) (Figure 7). Cependant, les travaux de notre équipe ont révélé la production de deux formes tronquées du récepteur chez ces souris : une isoforme de 55 kDa (protéine chimérique déletée pour 64 acides aminées dans le domaine B remplacées par 7 acides aminées codées par la casette néomycine) détectée dans l’utérus et l’aorte et une isoforme de 46 kDa (déletée pour le domaine A/B mais possédant le DBD et le LBD) dans l’utérus. Ces deux isoformes sont dépourvus de la fonction transactivatrice AF-1 (Pendaries, Darblade et al. 2002).
Pour la suite de nos protocoles, nous avons utilisé une deuxième lignée (appelée ERα-2KO puis ERαKO ou ERα
ensuite) dont l’invalidation a été obtenue par excision de l’exon 2 du gène ERα, codant pour le premier doigt de zinc du domaine de liaison à l’ADN. Cette nouvelle construction ne permet l’expression d’aucune protéine ERα fonctionnelle (Dupont, Krust et al. 2000) (Figure 7).
Plusieurs mutants déficients pour le gène d’ER ont également été générés, par délétion de l’exon 3 codant pour une partie du DBD (ERKOCH, ERKOKI, ERKOST) ou par
insertion de codons stop et troncation des exons 1 et 2 codant pour le domaine A/B et une partie du DBD (ERKOWY). Cependant plusieurs transcrits ont pu être détectés dans ces
différents variants (Antal, Krust et al. 2008). Plus récemment, une lignée de souris dépourvue d’activité transcriptionnelle pour ER (ERKOSTL-/L-), a été obtenue par l’excision de l’exon 3
Œstrogènes et Récepteur des œstrogènes
A
B
Figure 7│Modèles d'invalidation génique du récepteur aux œstrogènes α (Gourdy, Bayard et al. 2005). A : Organisation du gène du récepteur des œstrogènes α (ERα) en 8 exons. La première construction d’inactivation d'ER (ER Néo KO) a consisté à insérer une cassette néomycine dans le premier exon codant du gène ER (Lubahn, Moyer et al. 1993). La deuxième construction d’inactivation d'ER (ER 2 KO) a consisté à exciser le deuxième exon du gène ER codant pour le domaine de liaison du récepteur à l’ADN (Dupont, Krust et al. 2000).
B : Le récepteur des œstrogènes comporte 6 domaines (de A à F), avec des domaines de liaison à l’ADN, à l’hormone et des fonctions transactrivatrices AF-1 et AF-2. La construction de la lignée ER 2 KO ne permet l'expression d'aucune protéine fonctionnelle en l’absence de domaine de liaison à l’ADN. En revanche, nous avons mis en évidence chez les souris ER Néo KO, à la faveur de mécanismes d’épissage alternatif, l’existence de deux formes tronquées de récepteurs : une isoforme de 46 kDa et d'une molécule chimère de 55 kDa contenant 7 acides aminés de la cassette Néomycine (Pendaries, Darblade et al. 2002). Ces deux récepteurs sont certes dépourvus de la fonction transactrivatrice AF1, mais conservent la fonction transactrivatrice AF2.
I.4.2 Génération de souris ERαAF-1° et ERαAF-2°
Afin d’étudier le rôle de la fonction transactivatrice AF-1, un modèle murin a été construit grâce à une stratégie "knock in", dans lequel 441 nucléotides appartenant à l’exon 1 ont été excisés (Figure 8). La protéine tronquée obtenue par recombinaison homologue est dépourvue du domaine A et des trois motifs constituant AF-1 (AF-1 boxes 1–3) situés dans le domaine B, aboutissant à la synthèse d’une protéine de 451 acides aminés et de 49 kDa. Comme attendu, les isoformes ERα-66 et ERα-46 sont détectées dans les utérus de souris sauvages. En revanche, dans les utérus provenant de souris ERαAF-10, seule l’expression d’une isoforme de 49 kDa, correspondant à la protéine ERα tronquée de son domaine A/B, ainsi que l’isoforme physiologique de 46 kDa ont pu être observées. Comme pour l’isoforme naturelle de pleine taille ERα-66kD, l’expression de l’isoforme ERα-49 est initiée à partir premier codon ATG. L’expression de l’isoforme de 49 kDa chez les animaux ERαAF-10 est comparable à celle de l’isoforme de 66 kDa chez les animaux sauvages (Billon-Gales, Fontaine et al. 2009).
Chez des souris sauvages ERα+/+, le traitement par l’E2 induit une hypertrophie utérine, absente chez des souris ERα−/−. Chez les souris ERαAF-10, une très faible augmentation du poids utérin a été relevée en réponse à l’E2, ce qui démontre un rôle crucial d’AF-1 dans l’hyperplasie de l’utérus (Billon-Gales, Fontaine et al. 2009).
Les souris ERαAF-20 ont été obtenues en utilisant une stratégie similaire, par recombinaison homologue et délétion des acides aminés 543 à 549. Chez ces souris, l’utérus est également hypoplasique.
Œstrogènes et Récepteur des œstrogènes
TGA ATG1 ATG2
5’-UTR Ex1 Ex2 Ex3 Ex4 Ex5 Ex6 Ex7 Ex8 //
// -D B -D L B D A F -2 D B D L B D A F -2 D B D L B D A F -2 Met (ATG1) ATG1 ATG2
Ex1 Ex2 Ex3 Ex4 Ex5 Ex6 Ex7
TGA // -5’-UTR // -Ex8 1 AF -1 DBD LBD AF -2 AF -AF-1 DBD LBD Met (ATG1) AF-2 ERαAF-1° 49kDa ERαAF-2° AF - 2
AF-2
AF-1
N
C
66kDaERα TGA ATG 1 ATG 25’-UTR Ex1 Ex2 Ex3 Ex4 Ex5 Ex6 Ex7 Ex8 //
// -DBD LBD AF-2 DBD LBD AF-2 DBD LBD AF-2 ATG2 DBD LBD AF-2 DBD LBD AF-2 DBD LBD AF-2 ATG2 ERα 46kDa
Figure 8│Génération des souris ERαAF-1°(Billon-Gales, Fontaine et al. 2009)
Représentation schématique du gène codant pour ERα. La surface grisée dans l'exon 1 correspond à la séquence déletée pour générer des souris ERαAF-10. Les deux isoformes physiologiques d'ERα de 66 kDa (isoforme de pleine taille) et de 46 kDa (déficiente en AF-1) et la protéine de 49 kDa exprimée chez les souris ERαAF-10 sont représentées. Chez les souris ERαAF-10 les acides aminés 2 à 148 ont été supprimés. Chez les souris ERαAF-20 les acides aminés 543 à 549 ont été supprimés.
I.4.3 Invalidation génique cellule-spécifique
L’inactivation d’ERα spécifiquement dans l’endothélium a été obtenue par croisement de souris flanquées d'un site LoxP (séquence d’ADN de 34 paires de bases comprenant aux extrémités 13 nucléotides palindromiques) de part et d’autre du deuxième exon du gène ERα, avec des souris exprimant la Cre recombinase, sous la dépendance du promoteur endothélial Tie2. La Cre est une recombinase issue d’un bactériophage, qui catalyse la recombinaison entre deux sites de reconnaissance, les sites LoxP.
Plusieurs études ont montré que l’expression du récepteur à tyrosine kinase Tie2 concerne la lignée endothéliale (Isermann, Hendrickson et al. 2001; Kisanuki, Hammer et al. 2001; Koni, Joshi et al. 2001; Yao, Yokota et al. 2005) mais intéresse également les cellules hématopoïétiques (Shimoda, Mmanywa et al. 2006). Le croisement conduit alors à l’inactivation d’ERα dans les cellules endothéliales mais aussi dans la plupart des cellules de la moelle osseuse de la lignée myéloïde, erythroïde et lymphoïde (Takakura, Huang et al. 1998).
II ESTRADIOL, ISCHEMIE ET CICATRISATION CUTANEE
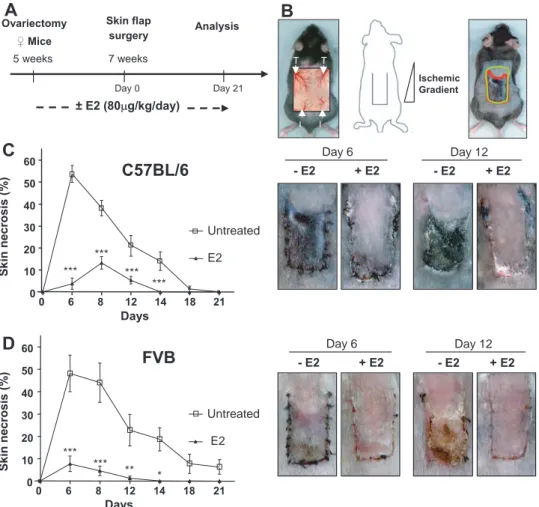
La première partie de mon travail expérimental a été l’étude des effets de l’E2 sur la prévention de la nécrose des lambeaux cutanés. Nous avons choisi de tester les effets de l’E2 dans un modèle de nécrose cutanée induite par ischémie, sur la base de deux arguments : (i) l’E2 exerce un rôle protecteur majeurs vis-à-vis des lésions d’origine ischémique et (ii) l’E2 favorise la cicatrisation cutanée. Ces deux points ainsi que le contexte clinique associé à la nécrose des lambeaux, sont détaillés dans le chapitre suivant.
II.1 PROBLEMATIQUE CLINIQUE : NECROSE DES LAMBEAUX CUTANES
II.1.1 Définition, étiologie
Parmi les techniques utilisées en chirurgie plastique et reconstructrice pour combler les pertes de substances (destructions cutanées étendues, recouvrements des plaies, reconstructions mammaires), la réalisation de lambeaux cutanés permet de disposer de surfaces tissulaires importantes et possédant une vascularisation autonome. Un lambeau cutané se définit comme étant un segment de peau et de tissu sous-cutané, partiellement découpé, séparé de son lieu d’origine, auquel il ne reste attaché que par un pédicule vascularisé. Cette structure tissulaire est d’emblée vivante puisqu’elle conserve sa propre vascularisation, contrairement aux greffes, dont la survie est liée à la revascularisation spontanée par la zone receveuse.
Une surface trop importante du lambeau, une compression ou une thrombose du pédicule vasculaire, une torsion du lambeau, une hypovolémie ou encore une infection, peuvent être des causes directes d’une insuffisance ou d’un arrêt de la circulation sanguine assurant la viabilité du lambeau. La nécrose cutanée qui résulte de cette ischémie constitue une source majeure de complication chirurgicale (Heller, Levin et al. 2001; Payette, Kohlenberg et al. 2005) et compromet la survie du lambeau, plus particulièrement dans sa partie distale qui est la plus faiblement vascularisée (Harder, Amon et al. 2004). Ainsi, 6 à 25% des lambeaux nécessitent une seconde intervention chirurgicale (Yuen and Feng 2000),
Estradiol, ischémie et cicatrisation cutanée
afin d’éliminer la zone nécrosée.
II.1.2 Stratégies favorisant la survie des lambeaux
Il n’existe actuellement que peu de thérapies efficaces de la nécrose des lambeaux cutanés. L’utilisation d’agents pharmacologiques telles que des agents sympatholytiques, des vasodilatateurs, des inhibiteurs calciques, anticoagulants ou anti-inflammatoires, a été proposée depuis maintenant une trentaine d’années (Waters, Pearl et al. 1989; Lepore, Knight et al. 1994; Zhang, Waller et al. 2004). Au cours des dix dernières années, le concept d’angiogenèse thérapeutique a également émergé, avec l’administration locale de facteurs de croissance (Zhang, Waller et al. 2004)
Notons cependant que la reperfusion après une ischémie prolongée peut être délétère. En effet, des radicaux libres oxygénés et des médiateurs de l'inflammation sont libérés au cours de la reperfusion. Ces radicaux libres sont toxiques et en présence d'oxygène, ils causent de multiples dommages aux membranes cellulaires, par peroxydation lipidique, entrainant finalement la mort des cellules (Karaaslan, Ulusoy et al. 2009). Le préconditionnement (cycles répétés d’ischémie-reperfusion avant une ischémie prolongée) (Salmi, Hong et al. 1999; Coskunfirat, Ozkan et al. 2006) et le postconditionnement (interruption intermittente de la reperfusion) (Moon, Lim et al. 2008) ont montré une certaine efficacité pour augmenter la survie des lambeaux soumis à une ischémie-reperfusion délétère, chez l'animal.
II.1.2.1 Approches pharmacologiques
De nombreuses substances ont été étudiées dans le cadre de la survie des lambeaux cutanés, afin de neutraliser les modifications physiopathologiques induites par les phénomènes d’ischémie-reperfusion, telles que l’inflammation ou les atteintes vasculaires (Knight 1994). En voici quelques exemples :
L’administration d’un anti-inflammatoire stéroïdien, la dexamethasone, améliore la survie cutanée (Willemart, Knight et al. 1998; Willemart, Knight et al. 1999) en permettant la diminution de l’œdème et des médiateurs pro-inflammatoires, alors que l’administation de celecoxib, un anti-inflammatoire non stéroïdien, s’est révélé inefficace (Wax, Reh et al. 2007).
L’administration de substances ayant des effets vasculaires : anticoagulants (héparine, urokinase) (Maeda, Fukui et al. 1991; Pantazi, Knight et al. 2000), vasodilatateurs
(L-arginine : précurseur du NO, bulfomedil, pentoxifylline, sildénafil) (Cordeiro, Santamaria et al. 1998; Hart, Baur et al. 2006; Mauad, Shimizu et al. 2006) ou des effets antioxydants (diméthyl sulfoxyde (DMSO) (Leite, Gomes et al. 2007), resveratrol (Hsieh, Huang et al. 2007) ont également été utilisé avec succès sur des modèles animaux.
II.1.2.2 Administration de facteurs de croissance
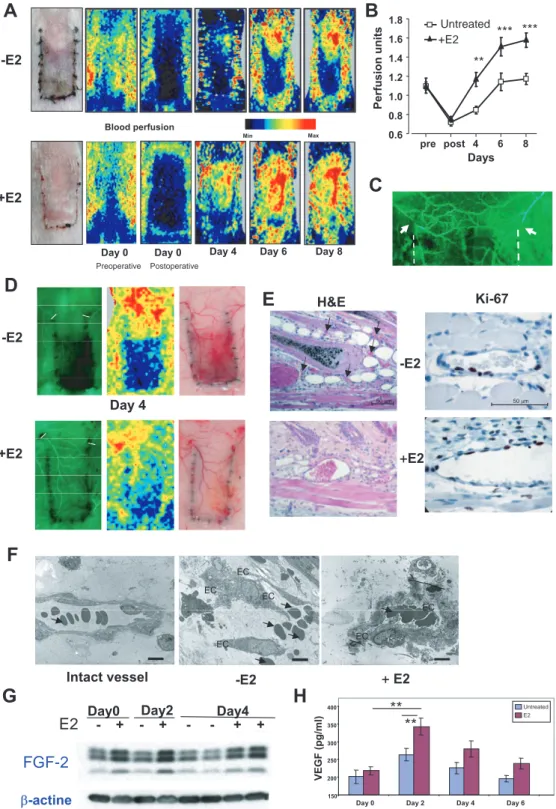
La survie des lambeaux cutanés dépend tout particulièrement de leur bonne vascularisation et donc d’une perfusion sanguine adéquate. Le concept d’angiogenèse thérapeutique, favorisant la formation de nouveaux vaisseaux par l’activation, la migration et la prolifération de cellules endothéliales à partir de vaisseaux préexistants, apparait comme une approche novatrice. L’administration locale de facteurs de croissance angiogéniques tels que le "Fibroblast Growth Factor" (FGF) (Fujihara, Koyama et al. 2005) ou le "Vascular Endothelial Growth Factor" (VEGF) (Zacchigna, Papa et al. 2005) a été proposée sur des modèles animaux de lambeaux cutanés et il a été démontré qu’elle permet d’améliorer la survie des lambeaux en diminuant significativement la zone de nécrose (Zhang, Waller et al. 2004). L’administration combinée de ces facteurs augmente encore la viabilité des lambeaux (Liu, Liu et al. 2005).
Cependant, l’innocuité des thérapies angiogéniques est encore mal connue et malgré l’enthousiasme suscité, il convient de rester prudent quant à leur utilisation chez l’homme en raison de leurs effets potentiellement néfastes, tels que le développement de vaisseaux anormaux ou de néo-angiogenèse tumorale (Epstein, Kornowski et al. 2001).
L’administration locale de "Transforming Growth Factor beta" (TGF), impliqué dans la synthèse de collagène, de protéoglycanes, d’élastine et de fibronectine, stimule la cicatrisation cutanée ainsi que la survie des lambeaux (Nall, Brownlee et al. 1996). Il a également été montré que l’administration d'IGF-1 permet d’augmenter la taille des lambeaux, ce qui est particulièrement intéressant lors de reconstructions étendues (Yuksel, Weinfeld et al. 2000).
II.2 ESTRADIOL ET PROTECTION VIS-A-VIS DE L’ISCHEMIE
Dans le cadre de la prévention des lésions tissulaires d’origine ischémique, de nombreux travaux ont révélé un rôle clé de l’E2. Il s’est révélé particulièrement efficace vis-à-vis de l’ischémie cérébrale, myocardique et dans le traitement de l’ischémie du membre
Estradiol, ischémie et cicatrisation cutanée
postérieur.
II.2.1 Estradiol et ischémie cérébrale
Comparativement aux hommes, les femmes sont protégées contre la survenue d'accidents vasculaires cérébraux jusqu’à l’âge de la ménopause. Les œstrogènes ont été largement étudiés et ont montré un effet neuropotecteur dans de nombreux modèles animaux d’ischémie cérébrale (McCullough and Hurn 2003; Brann, Dhandapani et al. 2007), grâce à leurs nombreux effets vasculaires et leurs effets sur la survie tissulaire.
L’E2 protège le cerveau de l’ischémie, notamment par ses propriétés vasoactives sur l’endothélium et sur les cellules musculaires lisses des vaisseaux sanguins cérébraux (Murphy, McCullough et al. 2004) qui favorisent la reperfusion. L’E2 agit également directement sur les cellules cérébrales, neurones et cellules gliales, en préservant leur intégrité (Hurn and Brass 2003), en inhibant les voies de l’apoptose (Jover, Tanaka et al. 2002; Jia, Guan et al. 2009) et en activant les voies de survie par l’expression de Bcl-2 (Alkayed, Goto et al. 2001; Won, Kim et al. 2006). De plus, l’E2 agirait en tant qu’antioxydant en prévenant la peroxydation des lipides membranaires (Green and Simpkins 2000; Dhandapani and Brann 2002; Ozacmak and Sayan 2008).
II.2.2 Estradiol et ischémie myocardique
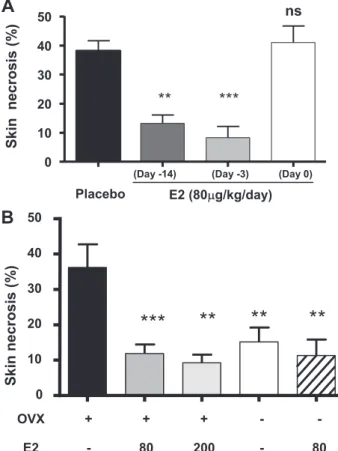
De nombreuses études ont également rapporté un effet cardioprotecteur de l’E2. L’administration prolongée d’E2 à des rats femelles ovariectomisées (pendant 14 jours avant l'ischémie) à des taux physiologiques, améliore la fonction contractile du cœur ex vivo, après un épisode d’ischémie d'une durée de 20 minutes, probablement par un effet direct sur les cardiomyocytes (Kolodgie, Farb et al. 1997; Beer, Reincke et al. 2002). La protection par l'E2 pourrait faire intervenir différents mécanismes, comme une action anti-oxydante, un effet inotrope négatif (par antagonisme calcique) ou encore des modifications du métabolisme glucidique (Beer, Reincke et al. 2002).
De même, l’administration aiguë d'E2, 30 minutes avant la reperfusion et pendant toute la durée de la reperfusion, est capable de réduire de façon significative la nécrose cardiaque et l'infiltration par les polynucléaires neutrophiles chez le chat (Delyani, Murohara et al. 1996). Dans un modèle d’infarctus du myocarde chez le rat (Wang, Tsai et al. 2006) et la souris (Patten, Pourati et al. 2004), les effets protecteurs de l’E2 ont été attribués à une action
anti-inflammatoire (diminution de la production de TNFα, d'IL-1 et d'IL-6) et anti-apoptique (diminution de l'activité de la caspase-3, activation de la kinase Akt) sur les cardiomyocytes.
D’autre part, l’E2 augmente également la mobilisation et l’incorporation des progéniteurs endothéliaux médullaires (EPC) vers les sites de néovascularisation après un infarctus du myocarde (Iwakura, Shastry et al. 2006).
II.2.3 Estradiol et ischémie de membre postérieur
Un petit nombre d’étude rapporte un rôle de l’E2 vis-à-vis l’ischémie du membre postérieur. L’administration d’E2 à des rats femelles ovariectomisées diminue les lésions musculaires et l’infiltrat neutrophilique dans un modèle d’ischémie-reperfusion (Stupka and Tiidus 2001). L’administration intramusculaire d’E2 favorise l’angiogenèse et la perfusion dans un modèle d’ischémie chronique chez le lapin (Kyriakides, Kremastinos et al. 2001).
II.3 ESTRADIOL ET CICATRISATION CUTANEE
II.3.1 Œstrogènes et physiologie cutanée
Les œstrogènes exercent une influence notable sur la physiologie cutanée. Chez l’animal, les œstrogènes stimulent la synthèse, la maturation et le renouvellement du collagène, augmentent la synthèse d’acide hyaluronique et inhibent la croissance des follicules pileux (Thornton 2002). Chez l’homme, ils augmentent également le contenu en collagène (Brincat, Versi et al. 1987), l’épaisseur cutanée et sa vascularisation. Enfin, les œstrogènes réduisent la taille et la sécrétion des glandes sébacées (Pochi and Strauss 1974).
La ménopause chez la femme est associée à de nombreux changements cutanés tels qu’à l’amincissement de l’épiderme, à la diminution de l’importance du collagène dermique, de l’hydratation de la peau et de son élasticité et surtout, à un défaut de cicatrisation (Brincat 2000; Hall and Phillips 2005).
II.3.2 Œstrogènes et cicatrisation cutanée
Les œstrogènes influencent la cicatrisation cutanée chez la femme comme chez l’homme ou encore dans les modèles animaux, par plusieurs mécanismes : modulation de la réponse inflammatoire, expression de cytokines, dépôt de matrice extracellulaire, reépithelialisation, angiogenèse, régulation de la protéolyse (Gilliver, Ashworth et al. 2007)
Estradiol, ischémie et cicatrisation cutanée
(Figure 9).
Une carence en œstrogènes chez des femelles ovariectomisées est à l’origine d’un retard de cicatrisation, associé à une augmentation de la réponse inflammatoire, à un défaut de reépithelialisation et à une diminution du dépôt de collagène (Ashcroft, Dodsworth et al. 1997), alors que la supplémentation en œstrogènes prévient ces effets délétères et accélère la cicatrisation.
Des retards de cicatrisation sont fréquemment observés chez les personnes âgées. Une étude menée chez des hommes et des femmes, âgés de plus de 70 ans, a montré une amélioration significative de la cicatrisation d’une plaie induite par une biopsie, chez des patients recevant un traitement topique d’E2 (patch libérant 25 μg d’E2 par 24h) initié 24 heures avant la biopsie. L’E2 réduit la taille de la plaie et augmente les niveaux de collagène et de fibronectine (Ashcroft, Greenwell-Wild et al. 1999).
a- Les œstrogènes modulent la réponse inflammatoire en diminuant le recrutement des neutrophiles et des macrophages ainsi que le niveau d’élastase d’origine neutrophilique (responsable de la dégradation de la fibronectine et de la jonction dermo-épidermique) (Mills, Ashworth et al. 2005). Ils diminuent également la production par les macrophages de cytokines pro-inflammatoires telles que le Macrophage Migration Inhibitory Factor (MIF) ou le Tumor Necrosis Factor-α (TNF-α) (Ashcroft, Mills, Lei et al. 2003). MIF est produit dans la peau par les macrophages, les neutrophiles, les cellules endothéliales et les kératinocytes. Cette cytokine pro-inflammatoire joue un rôle important dans la cicatrisation en régulant un grand nombre de gènes impliqués dans l’inflammation et les œstrogènes inhibent sa transcription via une inhibition de NF-B (Nuclear Factor B) (Hardman, Waite et al. 2005).
En plus de leurs effets sur la phase inflammatoire, les œstrogènes régulent la phase proliférative caractérisée par la restauration de la barrière cutanée (reépithélialisation) et de la vascularisation (angiogenèse), par la formation d’un tissu fibreux (fibroplasie) puis par la rétraction cicatricielle (Calvin 2000).
b- Les œstrogènes stimulent la migration et la prolifération des kératinocytes, permettant de favoriser la reépithélialisation (Ashcroft, Mills, Flanders et al. 2003; Merlo, Frasca et al. 2009) et diminuent leur apoptose in vitro (Kanda and Watanabe 2003).
c- Les œstrogènes stimulent largement l’angiogenèse en agissant directement sur les cellules endothéliales. La stimulation de la production de VEGF et de NO pourraient
intervenir dans les effets angiogéniques de l’E2 (Ashcroft, Dodsworth et al. 1997; Losordo and Isner 2001).
d- Les œstrogènes stimulent la production de PDGF (Platelet Derived Growth Factor) par les macrophages, qui stimule la prolifération des fibroblastes et la rétraction cicatricielle. Ils augmentent également la sécrétion par les fibroblastes de TGF-β qui est impliqué dans la synthèse de la matrice extracellulaire, la formation du tissu de granulation et le dépôt de collagène (Ashcroft, Dodsworth et al. 1997).
e- Enfin, les œstrogènes agissent sur la formation de la matrice extracellulaire et le remodelage cicatriciel, caractérisé par la synthèse, la dégradation et la réorganisation du collagène et la formation d’une cicatrice. Cette phase de remodelage est contrôlée entre autre par des enzymes protéolytiques, les MMP-2 et -9 (Matrix Metalloproteinases) (Pirila, Ramamurthy et al. 2001) et par leurs inhibiteurs, les TIMP (tissue inhibitor of metalloproteinase). Fibroblaste Kératinocyte Neutrophiles Macrophages Matrice Vaisseau sanguin Angiogenèse FGF-2 VEGF Prolifération, migration TGF-1, FGF-2 Collagène, TIMP MMP Matrice extracellulaire TNFα, MIF Cytokines proinflammatoires PDGF Recrutement Cytokines proinflammatoires Elastase Prolifération Apoptose Reépithélialisation Epiderme Derme Hypoderme a b c d e
Figure 9│Effets des œstrogènes sur la cicatrisation cutanée.
Les œstrogènes agissent sur les différentes phases de la cicatrisation : phase inflammatoire, proliférative et formation de la matrice extracellulaire.
Estradiol, ischémie et cicatrisation cutanée
II.3.3 Œstrogènes et inflammation
Comme nous venons de le voir précédemment, la cicatrisation cutanée met en jeu de nombreux acteurs cellulaires et moléculaires. En particulier, les cellules du système immuno-inflammatoire produisent de nombreux médiateurs, qui interviennent au cours de la cicatrisation. L'E2 est capable de moduler cette réponse inflammatoire.
Notre laboratoire a en effet démontré que l'E2 prévient le dépôt lipidique chez les souris hypercholestérolémiques immunocompétentes, alors qu’il n'a aucun effet chez les souris hypercholestérolémiques immuno-déficientes (RAG-2-/-, déficientes en lymphocytes T et B matures) (Elhage, Clamens et al. 2000; Elhage, Gourdy et al. 2005).
A la différence des effets anti-inflammatoires de l’E2 décrit à court terme et in vitro, l’administration d’E2 in vivo conduit à une augmentation de production des cytokines pro-inflammatoires dans diverses populations du système immuno-inflammatoire : les lymphocytes CD4+ T (Maret, Coudert et al. 2003), les lymphocytes Natural Killer T (Gourdy, Araujo et al. 2005) et les macrophages péritonéaux. La présence d’ER est indispensable à cet effet pro-inflammatoire et potentiellement délétère de l’E2 sur les macrophages (Calippe, Douin-Echinard et al. 2008), puisque cet effet est perdu dans le modèle de souris déficient pour ERR
-/-).
Les macrophages sont des acteurs clés dans les processus de régénération tissulaire, favorisant la phagocytose des débris nécrotiques et la cicatrisation tissulaire. Ils constituent une population cellulaire hétérogène, capables de changer leur état d'activation en fonction de leur environnement : les macrophages Ly-6C+ ont un phénotype plutôt pro-inflammatoire alors que les macrophages Ly-6C- ont un phénotype plutôt anti-inflammatoire (Gordon and Taylor 2005). Lors de régénération musculaire, des macrophages pro-inflammatoires sont exclusivement recrutés. Ils adoptent ensuite un phénotype anti-inflammatoire, dans le muscle, permettant sa régénération (Arnold, Henry et al. 2007). Lors d'une ischémie du membre postérieur chez des souris déficientes en thrombospondine-1 (protéine de la matrice extracellulaire, régulatrice des propriétés des macrophages, ayant un rôle dans le remodelage tissulaire), l'infiltration du tissu ischémique par des macrophages préférentiellement Ly-6C low, est associée à une prévention de l'apparition de nécrose (Brechot, Gomez et al. 2008). De plus, les macrophages sont impliqués dans le processus d'angiogenèse (Dirkx, Oude Egbrink et al. 2006), favorable lors d'une ischémie. Ainsi, les macrophages sont impliqués dans les différentes phases de la cicatrisation après ischémie : leurs propriétés pro-inflammatoires