UNIVERSITE TOULOUSE III – PAUL SABATIER
Ecole doctorale Biologie Santé et Biotechnologies
T H E S E
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITE TOULOUSE III
Physiopathologie cellulaire, moléculaire et intégrée présentée et soutenue par
Cédric FILIPE
Le 29 novembre 2007
ETUDE DE L’ EFFET DE L’OESTRADIOL SUR LA REENDOTHELIALISATION Directeur de thèse : Jean-François ARNAL
__________
JURY
Georges UZAN Rapporteur
Antonino NICOLETTI Rapporteur
Philippe COCHARD Examinateur
Alain-Pierre GADEAU Examinateur
TABLE DES MATIERES
TABLEDESMATIERES... 2
TABLE DES FIGURES ET DES TABLEAUX... 4
PRINCIPALESABREVIATIONSUTILISEES ... 6
SITUATION DU SUJET... 8
INTRODUCTION... 10
PRESENTATIONDELAPAROIVASCULAIRE ... 11
I. STRUCTURE DE LA PAROI ARTERIELLE ... 11
I.1. Intima ... 11
I.2. Média... 11
I.3. Adventice ... 12
II. FONCTION DE L’ENDOTHELIUM ... 12
ŒSTRADIOLETPHYSIOPATHOLOGIEVASCULAIRE... 16
I. ROLE PROTECTEUR DES ŒSTROGENES ... 16
Dans les modèles animaux... 17
II. MECANISMES CELLULAIRES ET MOLECULAIRES DE L’EFFET DES OESTROGENES SUR LE SYSTEME CARDIO-VASCULAIRE ... 18
II.1. Production du monoxyde d’azote ... 18
II.2. Régulation de l’expression des molécules d’adhérence leucocytaire ... 19
II.3. Effets anti-apoptotiques des œstrogènes... 20
III. IMPLICATION DES RECEPTEURS DES ŒSTROGENES ... 21
III.1. Modèles d’invalidation génique des récepteurs des œstrogènes... 21
III.1.a. Récepteurs des œstrogènes et effets endothéliaux ... 24
III.1.b. Récepteurs des oestrogènes et effet athéroprotecteur ... 24
RESTENOSEETREENDOTHELIALISATION ... 26
I. CONTEXTE ET IMPLICATIONS CLINIQUES ... 26
I.1. Mécanismes cellulaires de la resténose ... 26
I.2. Solutions pour limiter la resténose ... 28
I.2.a. Evolution de la technologie des stents ... 28
I.2.b. Blocage de l’inflammation... 29
I.2.c. Endothélium et resténose... 29
II. OESTROGENES ET REENDOTHELIALISATION ... 30
MODELISATIONDELADESENDOTHELIALISATION/REENDOTHELIALISATION... 32
I. BALLONISATION DE L’AORTE OU DE LA CAROTIDE DU RAT ... 32
II. ASPECTS MORPHOLOGIQUES DE LA DESENDOTHELIALISATION / REENDOTHELIALISATION ... 33
III. EPAISSISSEMENT INTIMAL... 35
IV. MODELES MURINS DE TRAUMATISME VASCULAIRE... 36
IV.1. Modèles endovasculaires... 37
IV.2. Modèles périvasculaires ... 38
CELLULESPROGENITRICES ... 40
I. DEFINITION ... 40
II. CARACTERISATION MOLECULAIRE... 41
III. ORIGINES ... 42
III.1. Cellules de la moelle osseuse ... 42
III.1.a. EPCs dérivant des cellules souches hématopoïétiques ... 42
III.1.b. Cellules myéloïdes... 42
III.1.c. Cellules souches mésenchymateuses ... 43
III.2.a. EPCs du tissu adipeux ... 43
III.2.b. Cellules souches tissus spécifiques... 44
IV. EFFETS BIOLOGIQUES ... 45
IV.1. Dans les modèles d’ischémie cardiaque ... 45
IV.2. Dans les modèles athéromateux ... 46
IV.3. Dans les modèles de stress vasculaire ... 47
V. MODE D’ACTION ... 48
VI. EPCs ET ŒSTRADIOL ... 49
FGF2(FIBROBLASTGROWTHFACTOR2)... 50
I. FAMILLE DES FACTEURS DE CROISSANCE FIBROBLASTIQUES ... 50
II. GENE ET SA TRANSCRIPTION ... 51
III. ARN MESSAGER ET SA TRADUCTION... 51
IV. ISOFORMES PROTEIQUES DU FGF2 ET LEUR LOCALISATION SUBCELLULAIRE ... 53
V. ACTIVITES BIOLOGIQUES DU FGF2... 54
V.1.a. FGF2 et développement embryonnaire... 54
V.1.b. FGF2 et angiogénèse ... 54
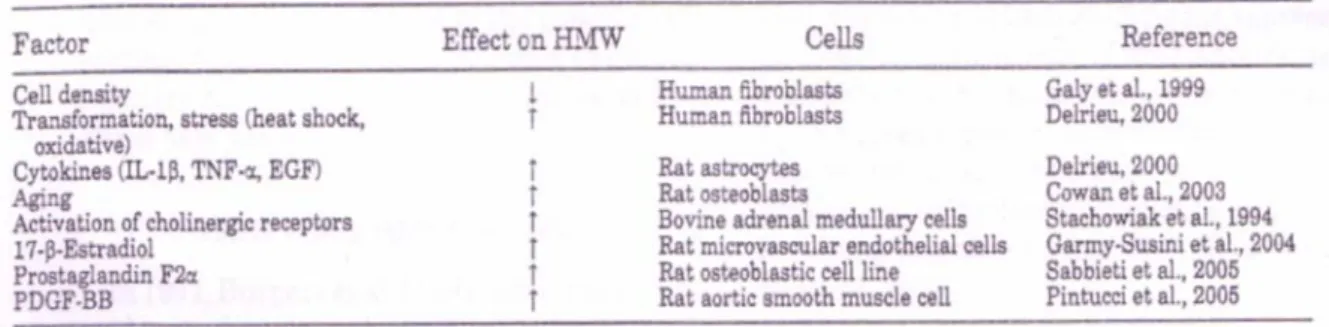
V.2. Rôle physiologique des isoformes de haut poids moléculaire (FGF2 HMW) ... 55
VI. INVALIDATION DU GENE FGF2... 56
VII. FGF2 ET ŒSTRADIOL... 57
VII.1. In vivo ... 57
VII.2. In vitro ... 58
MICROSCOPIECONFOCALE« ENFACE »... 60
I. INTRODUCTION A LA MICROSCOPIE CONFOCALE... 60
II. Developpement de la technique « en face »... 61
EXPOSE DES TRAVAUX EXPERIMENTAUX ... 66
ARTICLEN°1... 68 I. INTRODUCTION... 68 II. OBJECTIFS ... 69 III. RESULTATS ... 69 ARTICLE N°2... 73 I. INTRODUCTION... 74 II. OBJECTIFS ... 74 III. RESULTATS ... 75
III.1. Implication du FGF2 dans le processus de réendothélialisation (article 2) ... 75
III.2. Etude du rôle du FGF2 : HMW et LMW (Figure 13) ... 76
III.3. Quantification du nombre de cellules médullaires incorporées au niveau de la zone lésée (Figure 14)... 76
III.4. Caractérisation des cellules médullaires incorporées au niveau de la zone lésée ... 76
CONCLUSION... 81
ET... 81
DISCUSSION GENERALE ... 81
CONCLUSION ET DISCUSSION GÉNÉRALE... 82
I. Effet de l’oestradiol au niveau des cellules endothéliales... 82
I.1. Migration et prolifération des cellules endothéliales adjacentes à la lésion ... 82
I.2. Effet de l’E2 sur le profil d’expression des cellules endothéliales ... 83
I.2.a. eNOS ... 83
I.2.b. Facteur de von Willebrand (vWF) ... 84
I.2.c. CD31 ou PECAM (Platelet Endothelial Cell Adhesion Molecule) ... 84
I.3. Effet de l’E2 sur la prolifération... 86
I.4. Implication des CMLs dans l’effet de l’E2... 86
II.1.a. Quantification ... 88
II.1.b. Caractérisation ... 88
III. Effet de l’E2 sur les cellules médullaires incorporées au niveau de l’adventice... 89
IV. Implication du FGF2 ... 89
PERPECTIVES... 91
PERSPECTIVES... 92
I. Identification du type cellulaire de la moelle impliqué dans les effets de l’E2... 92
II. Rôle des cellules médullaires... 92
III. Implications cliniques de ce travail ... 93
III.1. Méthologie des stents ... 93
III.2. Effets des SERMs dans le modèle de réendothélialisation... 93
REFERENCES ... 95
BIBLIOGRAPHIQUES... 95
TABLE DES FIGURES ET DES TABLEAUX
Figure 1 : Représentation schématique de la paroi artérielle ... 14Figure 2 : Intima en microscopie électronique à transmission... 15
Figure 3 : Récepteur des œstrogènes α (ERα) ... 23
Figure 4 : Description des processus conduisant à la resténose ... 27
Figure 5 : Visualisation d’une zone de désendothélilisation... 33
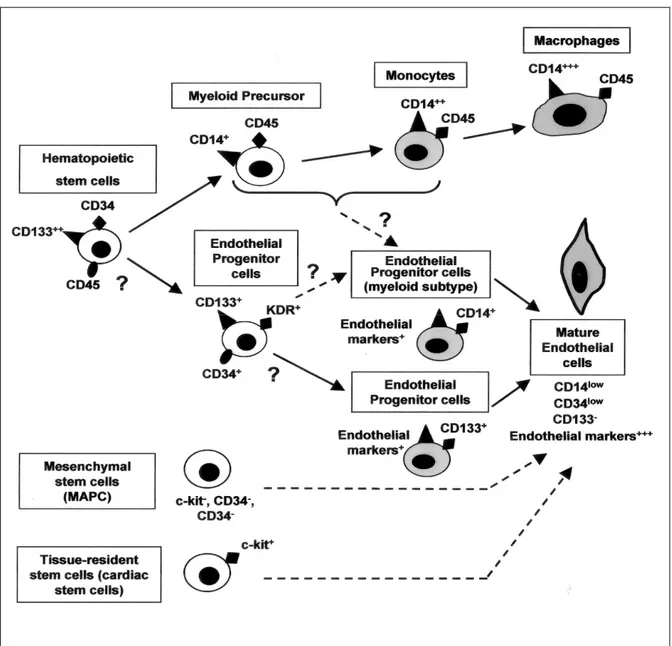
Figure 7 : Origine et différenciation des cellules endothéliales progénitrices... 45
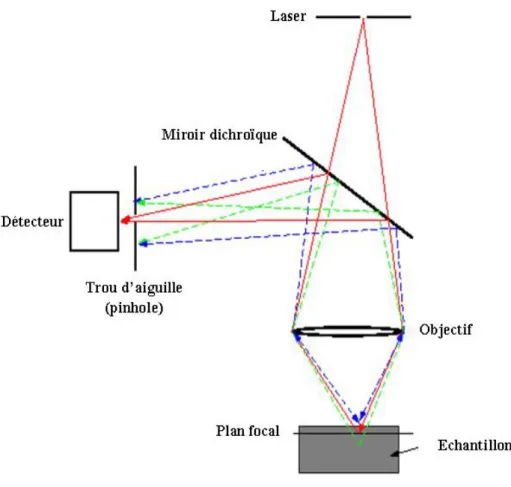
Figure 8 : Principe de la microscopie confocale ... 61
Figure 9 : Etudes de l'endothélium par coupes anatomopathologiques ... 63
Figure 10 : Etude de l'endothélium par imagerie confocale "en face"... 64
Figure 11 : Etude de l'endothélium par imagerie confocale "en face", exemple chez des souris chimères reconstituées par de la moelle GFP+... 65
Figure 12 : Quantification des cellules endothéliales dans la RE et la zone adjacente à la lésion... 72
Figure 13 : Etude de l'effet de l'E2 chez des souris lmw-/- ... 79
Figure 14 : Quantification de cellules d’origine médullaire ... 80
PRINCIPALES ABREVIATIONS UTILISEES
Ac LDL Acetylated Low Denstity Lipoprotein
AMPc Adénosine MonoPhosphate cyclique
AP-1 Activator Protein 1
ApoE Apolipoproteine E
BAECs Cellules Endothéliales Bovines
BrDU Bromo-Désoxy-Uridine
CD .. Cluster of Differentiation Molecule
CEs Cellules Endothéliales
CMLs Cellules Musculaires Lisses
EDHF Endothelium Derived Hyperpolarising Factor
E2 17β-œstradiol
EGF Epidermal Growth Factor
eNOS NO Synthase endothéliale (NOS III)
EPCs Cellules Progénitrices Endothéliales
ER α-β Sous-type alpha ou bêta du récepteur des œstrogènes
ERE Estrogen Response Element
ERK Extracellular signal-Regulated Kinases
FasL Fas ligand
FGF Fibroblast Growth Factor
FGFR Fibroblast Growth Factor Receptor
GFP Green Fluorescent Protein
HMW FGF2 High Molecular Weight en francais dans texte
HUVECs Human Umbilical Vein Endothelial Cells
HSP27 Heat Shock Protein 27 kDa
ICAM-1 Intracellular Adhesion Molecule-1
IL Interleukine
IFN-γ Interferon-γ
IRES Internal Ribosome Entry Site
ITAF IRES TransActing Factors
KO Knock-Out
LEI Limitante Elastique Interne
LMW FGF2 Low-Molecular Weight
L-NAME Nω-Nitro-L-Arginine Methyl Ester
MAC-1 Monocyte Chemoattractant Protein-1
MCP-1 Monocyte chemoattractive protein-1
NLS Nuclear Localisation Signals
NO Monoxyde d’azote ou Oxyde Nitrique
NOS NO Synthase
NF-κB Nuclear Factor-kappa B
PDGF Platelet Derived Growth Factor
PECAM Platelet Endothelial Cell Adhesion Molecule-1
PGI2 prostaglandine I2 (prostacycline)
PNN Polynucleaire Neutrophile
RE Reendothelialized Area
RetroP Retrograde Proliferating Zone
RT - PCR Reverse transcriptase – Polymerase Chain Reaction
SERM Steroid Estradiol Receptor Modulator
THS Traitement Hormonal Substitutif
TNF-α Tumor Necrosis Factor-α
uPA Urokinase-type plasminogen ativator
VCAM-1 Vascular Adhesion Molecule-1
VEGF Vascular endothelial growth factor
VEGF-A Vascular endothelial growth factor A
VEGFR-2 Vascular Endothelial Growth Factor Receptor 2
SITUATION DU SUJET
Depuis de nombreuses années, notre équipe s’intéresse à l’athérosclérose, première cause de mortalité dans les pays développés. Cette pathologie se caractérise par la formation d’un noyau lipidique au niveau sous endothélial dans la paroi vasculaire. Ce noyau, dans un premier temps réduit la taille de la lumière artérielle puis dans un second temps engendre une obturation des vaisseaux par rupture de la chape fibreuse ; libérant du matériel fortement thrombotique. Ce thrombus peut alors induire de fortes ischémies ou encore des accidents vasculaires cérébraux. Pour remédier au rétrécissement de la lumière des vaisseaux, une des solutions apportée est la dilatation de l’artère à l’aide d’un ballonnet gonflable (angioplastie), écrasant ainsi le noyau lipidique. Cette procédure est très souvent accompagnée de pose de stent. Cette grille métallique a pour but de maintenir le vaisseau dilaté. Mais l’angioplastie induit une mort massive des cellules endothéliales entraînant la proliférations des cellules musculaires lisses (CMLs). Ce processus, à la base des phénomènes de resténose, induit donc un nouveau rétrécissement de la lumière artérielle.
Notre laboratoire et d’autres équipes, ont mis en évidence les effets bénéfiques d’une hormone dans le développement athéromateux : l’œstradiol (E2). En effet, dans ce contexte physiopathologique, des études d’observation suggèrent un effet athéroprotecteur des œstrogènes. Cette hypothèse est confortée par l’effet protecteur de l’œstradiol dans tous les modèles animaux. De nombreuses études démontrent que l’œstradiol n’agit pas uniquement au niveau des tissus impliqués dans la reproduction (utérus, glandes mammaires….) mais également au niveau des différents types cellulaires constituant la paroi vasculaire.
Au vu des nombreux effets de l’œstradiol sur la paroi vasculaire, ma thèse a consisté à comprendre l’effet du 17ß-œstradiol (E2) non pas dans un contexte athéromateux, mais dans un contexte de lésion de l’endothélium généré par la pose de stent en clinique. La lésion et la perte de l’endothélium favorisent le phénomène de resténose. Accélérer la régénération de ce dernier (c’est à dire favoriser la réendothélialisation) limiterait donc les complications post-opératoires.
Notre laboratoire a ainsi développé une nouvelle technique permettant de modéliser cette agression vasculaire. Nous avons perfectionné un modèle dit d’agression périvasculaire. Cette lésion n’est pas, dans ce cas, créée par un stress mécanique mais par un stress électrique périvasculaire. A partir de ce modèle, notre équipe a démontré précédemment, grâce à des souris Knock-Out (ERα KO), que l’œstradiol accélère le processus de réendothélialisation via ce récepteur (Brouchet et al., 2001). Par ailleurs, des études ont démontré que ce processus de cicatrisation n’impliquait pas uniquement les cellules adjacentes à la zone stressée mais également des cellules de la moelle osseuse.
Au cours de ma thèse j’ai mis au point une technique d’imagerie confocale dite « en face », permettant d’explorer les mécanismes cellulaires et moléculaires impliqués dans ce processus de réendothélialisation. Nous avons ainsi pu comprendre le processus de cicatrisation dans ce modèle et certains mécanismes d’action de l’œstradiol. Nous démontrons par l’utilisation de souris chimères (irradiées puis reconstituées), que les cellules d’origine hématopoïétique jouent un rôle indispensable à l’effet accélérateur de l’œstradiol sur la réendothélialisation. Cette technique nous a permis de réaliser également une dissection des mécanismes moléculaires. Ainsi, nous avons mis en évidence : (i) une forte variation de protéines endothéliales (telles que la NO Synthase endothéliale (eNOS) et le facteur de von Willebrand (vWF)) et (ii) une forte activité proliférative au niveau de l’endothélium adjacent à la lésion. Enfin, nous démontrons que le Fibroblast Growth Factor-2 (FGF2) d’origine médullaire est un intermédiaire indispensable aux effets de l’E2 sur la réendothélialisation.
PRESENTATION DE LA PAROI VASCULAIRE
I. STRUCTURE DE LA PAROI ARTERIELLE
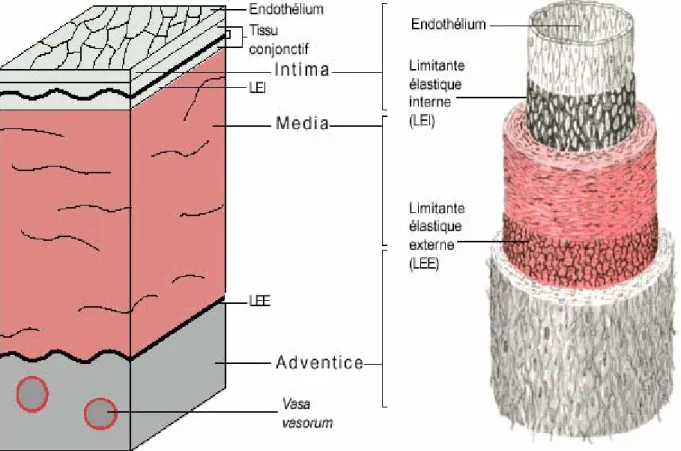
La paroi artérielle est constituée de trois tuniques concentriques qui sont, de l’intérieur vers l’extérieur : l’intima, la média, l’adventice. Etroitement réunies, elles assurent ensemble les différentes fonctions vasculaires (Figure 1).
I.1. Intima
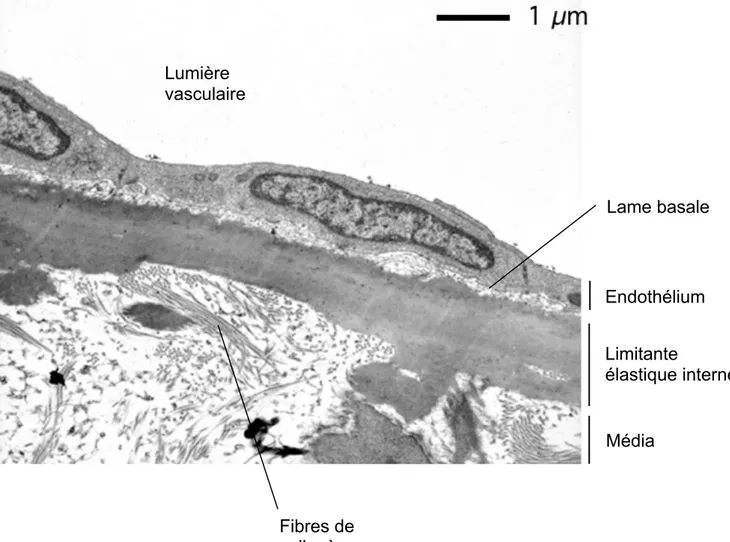
L’intima, composée de l’endothélium, repose sur une lame basale et la limitante élastique interne, couche fenêtrée de tissu élastique qui autorise des contacts entre les cellules endothéliales et la média sous-jacente. La lame basale est composée de microfibrilles de collagène et de glycoprotéines. Elle est synthétisée par les cellules endothéliales (CEs) et elle assure un rôle de support et de guide lors de la régénération endothéliale (Figure 2).
La monocouche de cellules endothéliales est une mosaïque de cellules pavimenteuses losangiformes de moins de 1 µm d’épaisseur et d’une dizaine de µm de longueur orientées dans le sens du flux sanguin. Elle constitue le revêtement interne des vaisseaux à l’interface du compartiment sanguin et la paroi. En étroite relation entre les éléments de la circulation et les composants de la média sous-jacente, l’endothélium n’est pas une surface inerte et joue un rôle majeur dans la régulation de la physiologie vasculaire.
I.2. Média
La média, tunique intermédiaire, est organisée en unités lamellaires concentriques. Chaque unité est constituée de deux lames élastiques fenêtrées composées de microfibrilles d’élastine entre lesquelles se trouve une couche de cellules musculaires lisses qui sécrètent les éléments de la matrice extracellulaire tels que des fibres de collagène, des microfibrilles d’élastine et des glycoaminoglycanes de la substance fondamentale qui les enveloppent. Le nombre d’unité lamellaire dépend du diamètre de l’artère et de l’espèce étudiée. Il est de 60 unités dans l’aorte humaine adulte, 20 unités dans l’aorte de lapin, 8 unités dans l’aorte de rat
La média assure : (i) la compliance des artères de conductance, c’est-à-dire leur capacité à se déformer sous l’effet de l’onde systolique et de reprendre leur forme initiale pendant la diastole ; (ii) la résistivité des artérioles. Les CMLs régissent le calibre du vaisseau sous contrôle d’une activité myotatique autonome, de signaux d’origine endothéliale (ou sanguine en cas de lésion de l’endothélium) et de signaux d’origine nerveuse. Elles ont aussi un rôle important dans le remodelage vasculaire par leur capacité à passer du phénotype quiescent contractile à un phénotype synthétique et prolifératif sous l’effet des facteurs physiologiques ou pathologiques.
I.3. Adventice
Couche la plus externe du vaisseau, l’adventice est une tunique d’interface entre la paroi artérielle et les tissus environnants. Elle est formée d’un tissu conjonctif lâche composé de fibres de collagène, de fibres élastiques et contient quelques fibroblastes. Ce tissu conjonctif est traversé par des petits vaisseaux, les vasa vasorum qui assurent la nutrition des couches les plus externes de la paroi dans les artères de gros calibre, par un réseau lymphatique et par des terminaisons nerveuses vasomotrices ortho- et parasympathiques. Les neurotransmetteurs libérés au niveau de ces terminaisons nerveuses participent au contrôle du tonus des cellules musculaires lisses et donc aux variations du calibre vasculaire et de ce fait à la régulation du débit sanguin.
II. FONCTION DE L’ENDOTHELIUM
L’endothélium est non seulement une surface hémocompatible qui empêche l’exposition du sous-endothélium thrombogène aux facteurs circulants de la coagulation et qui régule la fibrinolyse, mais il est aussi considéré comme une glande endocrine qui joue le rôle d’intégrateur et de relais des différents signaux neurohormonaux et mécaniques (flux sanguin) qu’il reçoit. Il module la réponse physiologique et pharmacologique à ces différents stimuli en synthétisant des agents qui vont réguler aussi bien le tonus vasculaire que la perméabilité transendothéliale vis-à-vis des cellules leucocytaires circulantes lors des processus inflammatoires. De façon plus générale, il constitue une barrière sélective du passage d’éléments circulants (figurés : leucocytes ; macromolécules : lipoprotéines ; molécules de la communication : hormones, cytokines, chimiokines, facteurs de croissance ; et enfin petites molécules : ions, eau, éléments nutritifs…) vers l’espace sous-endothélial.
L’endothélium présente de nombreuses vésicules non recouvertes de clathrine, les cavéoles, « petites cavités de la membranes plasmique ». Elles apparaissent plus abondantes dans la microcirculation qu’au niveau des gros vaisseaux. Les cavéoles assurent l’endocytose, la transcytose et la podocytose de macromolécules solubles, c’est à dire leur transport trans-cellulaire ou leur internalisation. Plus récemment, un rôle clé de la transduction du signal leur a été attribué. Elles participent ainsi aux fonctions de l’endothélium qui intègre et qui réagit aux différents stimuli qu’il reçoit.
En réponse à de nombreux stimuli, l’endothélium génère en particulier un puissant vasodilatateur : le monoxyde d’azote (NO). Il produit également d’autres molécules vasodilatatrices ou vasoconstrictrices selon les conditions physiologiques ou physiopathologiques. Ainsi, les dérivés de l’acide arachidonique, au premier rang desquels la prostacycline (PGI2), provoque localement, comme le NO, la relaxation des cellules
musculaires lisses et l’inhibition de l’agrégation plaquettaire. L’importance respective du NO et de la prostacycline varie selon les espèces, les territoires vasculaires et les stimuli physiologiques ou pharmacologiques utilisés. L’endothélium peut aussi générer une substance non encore caractérisée sur le plan biochimique , l’EDHF (Endothelium Derived Hyperpolarising Factor), qui est également un vasodilatateur à action locale (Feletou and Vanhoutte, 1988). Enfin, l’endothélium peut synthétiser et sécréter diverses substances vasoconstrictrices dont l’endothéline et l’angiotensine II, puisqu’il exprime l’enzyme de conversion de l’angiotensine I.
Figure 2 : Intima en microscopie électronique à transmission
Coupe longitudinale obtenue à partir d’une section d’aorte thoracique de lapin.
Endothélium Limitante élastique interne Lame basale Fibres de collagène Lumière vasculaire Média
ŒSTRADIOL ET PHYSIOPATHOLOGIE VASCULAIRE
L’observation de certaines populations ainsi que des études expérimentales animales ont montré que les œstrogènes exerçaient un effet « protecteur » sur le système cardio-vasculaire. Dans ce chapitre, nous résumerons les effets bénéfiques recensés de l’E2 au niveau du système cardio-vasculaire, et principalement dans un contexte athéromateux, domaine riche en données. Également, nous nous intéresserons tout particulièrement aux effets de cette hormone sur un type cellulaire clé de l’athérome et de la resténose : les cellules endothéliales.
I. ROLE PROTECTEUR DES ŒSTROGENES
Au cours des dernières décennies, plusieurs enquêtes épidémiologiques prospectives, dont l’étude de Framingham, ont affirmé que le risque d’évènements cardio-vasculaires observé chez les hommes était 2 à 4 fois supérieur à celui des femmes (Lerner and Kannel, 1986). Cet excès de risque chez l’homme, vérifié dans diverses populations ethniques, présente des taux d’incidence bas ou au contraire élevés. Après 50 ans, le taux d’incidence des maladies cardio-vasculaires observé chez les femmes rejoint progressivement celui des hommes. Cette dernière observation est en partie liée à une diminution de l’incidence des accidents vasculaires chez les hommes les plus âgés, mais a fortement suggéré un rôle protecteur des hormones sexuelles féminines, principalement des œstrogènes, vis à vis du risque cardio-vasculaire (Barrett-Connor and Stuenkel, 1999).
Afin de conforter cette hypothèse, plusieurs travaux se sont attachés à étudier les conséquences d’une ménopause précoce en terme d’athérosclérose coronarienne et de survenue d’accidents cardio-vasculaires. Les autopsies réalisées, chez des femmes ayant subi une ovariectomie bilatérale, ont donné des résultats discordants (Barrett-Connor and Bush, 1991). Par contre, dans la Nurses’ Health Study, l’existence d’une ovariectomie bilatérale, mais pas d’une ménopause « naturelle », était associée à une augmentation du risque de coronaropathie (Colditz et al., 1987).
Depuis les années 1970, de nombreuses études d’observation (prospectives ou cas/témoin) ont rapporté que les femmes ménopausées utilisatrices de différents modes de THS (Traitement Hormonal Substitutif) présentaient une réduction significative du risque cardio-vasculaire (Barrett-Connor, 1996; Stampfer et al., 1991). Les données d’observation publiées jusqu’en 1997 ont fait l’objet d’une méta-analyse montrant une diminution du risque d’évènements coronariens chez les utilisatrices de traitements œstrogéniques (RR : 0,70) et œstro-progestatifs (RR : 0,66) (Barrett-Connor and Grady, 1998). D’autres essais, non contrôlés, ont conforté l’hypothèse d’un effet protecteur du THS sur l’incidence des pathologies cardio-vasculaires (Varas-Lorenzo et al., 2000) et la progression des lésions d’athérosclérose, évaluée par coronarographie (Husak et al., 2004). Cependant, ces études d’observation sont soumises à de nombreux biais méthodologiques, principalement liés aux critères de recrutement des sujets et aux caractéristiques des femmes bénéficiant du THS (Barrett-Connor, 1991). Par conséquent, l’ensemble de leurs conclusions a été remis en cause au cours des dernières années.
Dans les modèles animaux
L’utilisation de différents modèles animaux d’athérosclérose a permis de mettre en évidence un effet protecteur des œstrogènes vis-à-vis du développement des lésions d’athérosclérose. Dans l’ensemble des travaux initialement rapportés, le développement des lésions était provoqué par des régimes hypercholestérolémiants (Smith and Breslow, 1997). Ainsi, les modèles de lapins, porcs et singes soumis à ce type de régime athérogène ont été particulièrement étudiés au cours des dernières décennies. Tous ces travaux concordent pour démontrer que les œstrogènes endogènes et exogènes exercent un effet protecteur significatif, caractérisé par une réduction de la constitution des lésions athéromateuses non seulement chez des femelles ovariectomisées, mais également chez des mâles castrés. En fonction des sites vasculaires étudiés (aortes ou coronaires) et du type de traitement administré (formulation orale ou parentérale des œstrogènes), cet effet protecteur varie de 35 % à 80 % (Hodgin and Maeda, 2002). Selon les études, l’effet préventif des œstrogènes est soit non modifié, soit atténué par l’association d’un progestatif.
De manière intéressante, chez des animaux présentant des lésions athéromateuses pré-existantes, l’effet protecteur du traitement hormonal est soit atténué (Bjarnason et al., 2001; Clarkson et al., 2001), soit inexistant (Hanke et al., 1999; Williams et al., 1995), suggérant que les œstrogènes préviennent électivement les stades précoces du processus athéromateux. Par ailleurs, dans un modèle d’agression de l’aorte thoracique de lapin, il a été montré que l’effet de l’œstradiol (E2) sur le développement de stries lipidiques est variable en fonction du statut de l’endothélium. En effet, l’E2 exerce un effet protecteur dans les zones non traumatisées, mais s’avère inefficace en présence d’un endothélium régénéré, et même aggravant dans les zones vasculaires demeurant désendothélialisées (Holm et al., 1999).
II. MECANISMES CELLULAIRES ET MOLECULAIRES DE L’EFFET DES OESTROGENES SUR LE SYSTEME CARDIO-VASCULAIRE
Compte tenu de la complexité physiopathologique de l’athérosclérose et de la resténose, il est probable que les mécanismes cellulaires et moléculaires médiant les effets des œstrogènes soient multiples. Nous proposons de décrire ici, dans l’état actuel de nos connaissances, les effets des œstrogènes sur un des acteurs cellulaires clé de l’athérosclérose et de la resténose : la cellule endothéliale.
II.1. Production du monoxyde d’azote
L’endothélium produit de nombreuses substances, dont le monoxyde d’azote (NO), un messager radicalaire qui joue un rôle vasculo-protecteur important du fait de ses propriétés vasodilatatrices et antiagrégantes. La potentialisation de la production endothéliale de NO par les œstrogènes a été mise en évidence pour la première fois au niveau de l’artère fémorale de lapin, puisque l’E2 stimule la relaxation endothélium dépendante en réponse à l’acétylcholine dans cette espèce (Gisclard et al., 1988). Depuis, il a été rapporté dans plusieurs espèces animales, que l’administration d’E2 chez des femelles ovariectomisées majore la production basale et/ou stimulée de NO, avec cependant une certaine variabilité en fonction des territoires vasculaires étudiés (Barbacanne et al., 1999; Cheng et al., 1994; Keaney et al., 1994; Williams et al., 1994). Dans ces modèles, plusieurs mécanismes peuvent participer à la majoration de l’activité du NO par l’E2 : l’augmentation de l’expression et/ou de l’activité de la NO synthase endothéliale (Hayashi et al., 1992; Weiner et al., 1994), l’augmentation de la biodisponibilité du NO (Arnal et al., 1996). Plus récemment, notre groupe a démontré chez la
souris, que l’E2 augmente également la production basale de NO par l’endothélium. Cet effet est médié par le récepteur des œstrogènes α (ERα) et non par le récepteur β (ERβ) (Darblade et al., 2002).
Il a été initialement proposé que l’augmentation de la production basale de NO par l’endothélium exerce un effet protecteur puissant vis-à-vis du processus athéromateux, et par conséquent pouvait contribuer à l’effet protecteur des œstrogènes. Cependant, le blocage pharmacologique de la production de NO par le L-NAME (Nω-Nitro-L-Arginine Methyl Ester) n’influence pas le développement de la strie lipidique chez la souris apoE-/-(apolipoproteine E )((Elhage et al., 1997b), à la différence de ce qui avait été observé chez le lapin, et n’altère en rien l’effet athéro-protecteur de l’E2 (Elhage et al., 1997a). De façon similaire, la surface des lésions est réduite de 75 % par l’administration d’E2 chez des souris apoE-/- déficientes en NO synthase endothéliale (Hodgin et al., 2002).
Ces données indiquent que l’augmentation de la production de NO par les œstrogènes n’est pas impliquée dans la prévention de la strie lipidique. Néanmoins, cet effet peut s’avérer bénéfique à des stades plus avancés de l’athérosclérose, grâce aux effets antispastiques et antiagrégants plaquettaires du NO.
II.2. Régulation de l’expression des molécules d’adhérence leucocytaire
Comme nous l’avons évoqué, le recrutement des cellules inflammatoires par l’endothélium est une étape clé du processus athéromateux. Également, nous le verrons par la suite, le système inflammatoire joue également un rôle important dans le processus de resténose. L’hypothèse, d’une régulation par les œstrogènes de l’expression endothéliale de plusieurs molécules d’adhérence leucocytaire, a été proposée sur la base d’observations épidémiologiques et de travaux expérimentaux.
En effet, les taux circulants de certaines molécules d’adhérence, en particulier la forme soluble d’ICAM-1 (Intracellular Adhesion Molecule-1), sont majorés chez les femmes ménopausées. Plusieurs études ont montré que l’administration d’un THS permet de réduire ces taux plasmatiques (Oger et al., 2001; Scarabin et al., 1999; Stork et al., 2002), bien que des résultats opposés aient aussi été rapportés (Jilma et al., 1994).
Les approches expérimentales in vitro ont également donné lieu à des résultats divergents. Ainsi, Cid et al. ont montré que des concentrations pharmacologiques d’E2 favorisaient l’adhérence des leucocytes sur des HUVECs (Human Umbilical Vein Endothelial Cells) stimulées par du TNF-α (Tumor Necrosis Factor-α) et de l’IFN-γ (Interferon-γ). Cet effet était partiellement inhibé en présence d’anticorps E-sélectine, ICAM-1 ou anti-VCAM-1 (Vascular Adhesion Molecule-1) (Cid et al., 1994), et l’expression de ces molécules significativement majorée dans les cellules traitées par l’hormone. A l’opposé de ces résultats, deux études plus récentes, utilisant des cellules endothéliales humaines stimulées par l’IL-1 (Interleukine-1) ont démontré que l’E2 inhibait l’induction de VCAM-1 et l’expression membranaire de E-sélectine et ICAM-1 (Caulin-Glaser et al., 1996; Simoncini et al., 2000). La diminution de l’expression du gène VCAM-1 a été imputée à une inhibition des activités de NF-κB (Nuclear Factor-kappa B), AP-1 (Activator Protein-1) et GATA (Simoncini et al., 2000). En accord avec ces observations, des expériences ex vivo chez le lapin hypercholestérolémique montrent que l’E2 diminue l’adhérence endothéliale des monocytes en inhibant l’expression de VCAM-1 (Nathan et al., 1999).
Enfin, il a été proposé que l’E2 favorise l’expression endothéliale de la molécule pro-apoptotique FasL (Fas ligand) et pourrait ainsi limiter le trafic transendothélial des leucocytes, favorisant en particulier l’apoptose des lymphocytes activés. En effet, chez le lapin, l’administration d’E2 prévient la diminution de l’expression constitutive de FasL par l’endothélium, induite par un régime hypercholestérolémiant (Amant et al., 2001).
II.3. Effets anti-apoptotiques des œstrogènes
Bien que les cellules endothéliales soient considérées comme résistantes à l’apoptose (Kockx and Herman, 2000), il est acquis que ce processus classique de mort cellulaire concerne l’endothélium (Tricot et al., 2000). A ce jour, l’effet des œstrogènes sur ce phénomène n’a pu être testé que dans des modèles de cellules endothéliales en culture, soumises à divers stimuli pro-apoptotiques. Spyridopoulos et al. ont montré que le traitement d’HUVECs par l’E2 entraîne une inhibition dose dépendante de l’apoptose induite par le TNF-α en inhibant la caspase 1 (Spyridopoulos et al., 1997). Deux études utilisant des BAECs (Cellules Endothéliales Bovines) ont confirmé l’effet anti-apoptotique de l’E2 dans des conditions d’hypoxie (Razandi et al., 2000) ou de modification des milieux de culture
(Alvarez et al., 1997).
Des travaux récemment réalisés dans notre groupe n’ont cependant pas permis de retrouver cet effet anti-apoptotique in vitro en utilisant des cellules endothéliales murines, mais également des HUVECs soumises à plusieurs types de stimuli apoptotiques (A. Billon, résultats non publiés).
III. IMPLICATION DES RECEPTEURS DES ŒSTROGENES
La majorité des effets de l’E2 résulte classiquement de la transcription génique de gènes cibles ; cependant des effets à court terme, indépendants de la transcription (effets extragénomiques), ont été rapportés plus récemment (Mendelsohn, 2000). Le récepteur des œstrogènes α (ERα), récepteur « classique » des œstrogènes, médie la majorité des effets de ces hormones au cours de la reproduction (Couse and Korach, 1999). Plus récemment, un deuxième récepteur appelé ERβ a été identifié, mais son rôle dans les tissus extra-reproducteurs demeure à l’heure actuelle mal connu (Couse and Korach, 1999). Les deux types de récepteurs sont exprimés (identification par immuno-cytochimie et/ou par RT-PCR (Reverse transcriptase – Polymerase Chain Reaction)) par les cellules endothéliales.
III.1. Modèles d’invalidation génique des récepteurs des œstrogènes
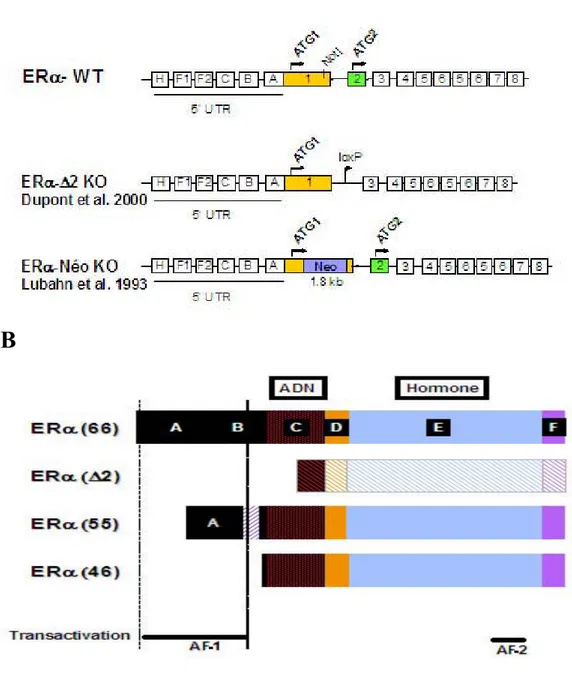
Afin d’étudier l’implication d’ERα, dans les effets vasculaires des œstrogènes, deux lignées de souris transgéniques invalidées pour ce récepteur nucléaire ont été utilisées au cours des dernières années (Figure 3). Pour générer la première lignée (ERα-Néo KO), une cassette contenant l’ADNc conférant la résistance à la néomycine a été insérée dans le premier exon du gène ERα (Lubahn et al., 1993). Dans la deuxième lignée (ERα-D2 KO), l’invalidation a consisté en l’excision du deuxième exon du gène ERα, codant pour une partie du domaine de liaison du récepteur à l’ADN (premier doigt de zinc) (Dupont et al., 2000). Comme attendu, la construction de la lignée ERα-D2 KO ne permet l’expression d’aucune protéine ERα. En revanche, chez les souris ERα-Néo KO, deux formes tronquées de récepteurs (une isoforme de 46 kDa et une molécule chimère de 55 kDa contenant 7 acides aminés de la cassette néomycine) ont été retrouvées, à la faveur de mécanismes d’épissages
fonction transactivatrice AF-1, conservent la fonction transactivatrice AF-2 (Flouriot et al., 2000).
Concernant ERβ, deux invalidations ont aussi été réalisées, mais les souris mutantes apparaissent phénotypiquement identiques (Krust et col., résultats non publiés).
Ces observations imposent une grande prudence dans l’interprétation des résultats obtenus chez les souris ERα-Néo KO. Par exemple, Iafrati et al. ont suggéré que l’effet préventif de l’E2 vis-à-vis de l’hyperplasie néo-intimale, dans les modèles d’agression vasculaire, puisse être indépendant de ERα et ERβ ; mais le modèle d’invalidation génique du récepteur α utilisé dans cette étude ne permet pas de conclure définitivement (Iafrati et al., 1997).
A
B
Figure 3 : Récepteur des œstrogènes α (ERα)
A : Organisation du gène du récepteur des œstrogènes α (ERa) en 8 exons. La première
construction d’inactivation de ERα (ERα Néo KO) a consisté à insérer une cassette néomycine dans le premier exon du gène ERα (Lubahn DB, PNAS 95). La deuxième construction d’inactivation de ERα (ERα ∆2 KO) a consisté à exciser le deuxième exon du gène ERα codant pour le domaine de liaison du récepteur à l’ADN (Dupont S, Development 2000).
B : Le récepteur des œstrogènes α comporte 6 domaines (de A à F), avec des domaines de liaison à l’ADN, à l’hormone et des fonctions transactrivatrices AF-1 et AF-2. La construction de la lignée ERα ∆2 KO ne permet l'expression d'aucune protéine fonctionnelle en l’absence de domaine de liaison à l’ADN. En revanche, nous avons mis en évidence chez les souris ERα Néo KO, à la faveur de mécanismes d’épissage alternatif, l’existence de deux formes tronquées de récepteurs : une isoforme de 46 kD et d'une molécule chimère de 55 kD contenant 7 acides aminés de la cassette Néomycine (Pendaries C, PNAS 2002). Ces deux récepteurs sont certes dépourvus de la fonction transactrivatrice AF-1, mais conservent la fonction transactrivatrice AF-2.
III.1.a. Récepteurs des œstrogènes et effets endothéliaux
L’effet de l’E2, sur la production endothéliale de NO, a été étudié dans les deux lignées de souris déficientes en ERα. Tandis que l’effet de l’E2 sur cette production est totalement aboli chez les souris ERα-∆2 KO, l’effet de l’E2 persiste chez les souris ERα-Néo KO et est identique à celui observé chez les souris sauvages (Pendaries et al., 2002). Ces résultats suggèrent que la fonction AF-1 du ERα n’est probablement pas nécessaire pour médier les effets de l’E2 dans l’endothélium et imposent l’utilisation des souris ERα-D2 KO pour caractériser les mécanismes endothéliaux dépendants de l’E2 (Pendaries et al., 2002). Dans ce dernier modèle, notre groupe a pu démontrer que l’effet de l’E2, non seulement sur la production endothéliale de NO, mais aussi sur la réendothélialisation, dépend exclusivement du ERα. En effet, ces effets persistent chez les souris déficientes en ERβ (Brouchet et al., 2001; Darblade et al., 2001), bien qu’une augmentation de l’expression de ERβ ait été rapportée dans la carotide de rat après agression endovasculaire (Makela et al., 1999).
III.1.b. Récepteurs des oestrogènes et effet athéroprotecteur
Afin de déterminer la contribution relative de chacun de ces deux récepteurs dans l’effet protecteur des œstrogènes, J.B. Hodgin et al. ont croisé les souris déficientes en ERα (ERα-Néo KO) ou ERβ avec des souris apoE-/-. L’E2 réduit les lésions d’athérosclérose des animaux apoE-/- également de 80%, mais cet effet inhibiteur de l’E2 est en grande partie aboli chez les souris doubles-mutantes ERα-/- apoE-/- (Hodgin et al., 2001). En revanche, l’E2 inhibe la progression des lésions d’athérosclérose chez les souris ERβ-/- apoE-/- de la même façon que chez les souris apoE-/-, démontrant que son effet protecteur est totalement conservé en l’absence d’ERβ (Hodgin and Maeda, 2002). L’effet athéroprotecteur du soja alimentaire, source de phyto-œstrogènes, s’exerce également par l’intermédiaire d’ERα puisque son effet est aboli chez les souris ERα-/- apoE-/-. De façon intéressante, l’E2 diminue la formation de la chape fibromusculaire chez les souris apoE-/- mais aussi chez les souris ERα-/- apoE-/- (Hodgin et al., 2001). Cette observation suggère la possibilité que ERβ puisse jouer un rôle protecteur dans la maturation des plaques tandis qu’il n’est clairement pas impliqué dans des stades plus précoces comme le développement de la strie lipidique.
l’expression persiste chez les souris ERα-/- dont l’invalidation est imparfaite (Pendaries et al., 2002). Cet effet de prévention de la formation de la chape fibromusculaire par l’E2 ne peut pas être considéré comme un effet bénéfique, mais pourrait au contraire être délétère, en empêchant le processus de cicatrisation de la strie lipidique et donc favoriser l’instabilité des plaques.
Les effets vasculaires des œstrogènes ne sont donc pas univoques, ils peuvent être à la fois bénéfiques et délétères. Il est donc impératif de progresser dans la compréhension de leur mécanisme d’action. Pour cela, la mise au point de modèles expérimentaux murins, qui offrent la possibilité d’une approche moléculaire in vivo, est donc un impératif de tout premier ordre, lorsqu’on connaît les limites de l’expérimentation in vitro.
RESTENOSE ET REENDOTHELIALISATION
I. CONTEXTE ET IMPLICATIONS CLINIQUES
L’agression de l’endothélium de la paroi vasculaire est une situation clinique quotidienne : dilatation de sténose artérielle, pose de stent, gestes de chirurgie vasculaire. La cicatrisation secondaire de l’endothélium peut prendre un caractère pathologique sous la forme d’une resténose et ou d’une thrombose du vaisseau. La diminution de ces complications post-opératoires représente un véritable enjeu médical. En effet, chaque année, sur 500 000 angioplasties coronariennes percutanées réalisées aux Etats-Unis, 150 000 patients présentent des complications de type resténose.
I.1. Mécanismes cellulaires de la resténose
Le processus conduisant à la resténose, lié à l’angioplastie et/ou à la pose de stent, est relativement bien documenté dans des modèles animaux de dénudation de l’endothélium par ballonisation. Ces divers travaux mettent en évidence le rôle de trois éléments clés : les CMLs, les CEs et les cellules du système inflammatoire (leucocytes, macrophages et plaquettes). Le déploiement du stent, dans un premier temps écrase la plaque athéromateuse rétablissant ainsi une circulation sanguine efficace. Mais cette action n’est pas sans conséquence sur la paroi artérielle. En effet le stent induit, dès les premières minutes, la mort des cellules endothéliales sur une large surface. Une couche de fibrine et de plaquettes recouvre alors la zone lésée. Cette dernière, par l’expression de molécules d’adhésion, permet l’encrage de leucocytes (via leur récepteur Mac-1 : Monocyte Chemoattractant Protein-1) et leur diapédèse. La migration des leucocytes, au travers de la couche plaquette/fibrine et du tissu, est alors favorisée par la synthèse de chimiokines par les CMLs et les macrophages résidents. La synthèse de facteurs de croissance par les plaquettes, les leucocytes et les CMLs elles-mêmes, stimule la synthèse de matrice extracellulaire facilitant ainsi la migration et la prolifération de ces dernières (Figure 4).
Figure 4 : Description des processus conduisant à la resténose
A. Plaque d’athérosclérose mature avant intervention. B. Réponse précoce induite après la pose de stent,
avec recrutement des plaquettes. C/D. Recrutement et infiltration des leucocytes accompagnés d’une prolifération des CMLs et un recrutement des macrophages. E. Progression de l’hyperplasie néo-intimale quelques semaines après la pose du stent, avec un maintien de la prolifération des CMLs et un recrutement des monocytes. F. Des semaines après l’opération, une forte hyperplasie néo-intimale s’accompagnant d’un rétrécissement de la lumière artériel s’est formée. (Welt and Rogers, 2002)
I.2. Solutions pour limiter la resténose
Au vu de la complexité des mécanismes mis en jeu, tant au niveau prolifératif qu’au niveau inflammatoire, de nombreux axes de recherche ont été développés afin de limiter ces complications post-opératoires.
I.2.a. Evolution de la technologie des stents
Compte tenu du pourcentage élevé de complications dues au phénomène de resténose, de nouvelles technologies ont été développées. Ces dernières reposent sur le recouvrement des stents par des molécules anti-mitotiques. Les deux plus utilisées sont le Sirolimus® et le Paclitaxel®. Le premier induit l’inhibition de la synthèse d’ADN par sa fixation à une molécule indispensable au passage de la phase G1 à la phase S. Également, le Sirolimus® possède des activités anti-inflammatoires. En effet, il a été démontré chez le porc (Oberhoff et al., 2002), que ce type de stent permettait une diminution de l’expression de MCP-1 (Monocyte Chemoattractant Protein-1) et IL-6 dans la paroi vasculaire. Le Paclitaxel® quant à lui, inhibe principalement la migration et la prolifération des cellules. Il se lie spécifiquement à la sous unité β de la tubuline des microtubules, empêchant ainsi tout réassemblage du cytosquelette. Les cellules sont alors bloquées entre la phase G2 et M. La pose de ces nouveaux stents est devenue très fréquente (80% des stents posés aux Etats-Unis concernent des stents « coatés »). Mais le bénéfice attendu est loin d’être atteint. L’ensemble des données récoltées depuis près de 4 ans ne montre aucun bénéfice à la pose de ce type de stent ; voire même dans certains cas une augmentation du nombre d’infarctus du myocarde et de thromboses, dans les 6 premiers mois. Enfin, la pose de stent implique un traitement antiplaquettaire double (aspirine + plavix) pendant au moins un an (Curfman et al., 2007; Kastrati et al., 2007; Lagerqvist et al., 2007; Maisel, 2007; Mauri et al., 2007; Spaulding et al., 2007).
En conclusion, le développement d’une approche aussi générale, qu’est de bloquer la prolifération de l’ensemble de la paroi artérielle, est discutable. En effet, le blocage de la prolifération des CMLs mais aussi de la réendothélialisation ne permet pas de régénérer un tissu vasculaire proche du natif, l’exposant ainsi à des risques multiples tels que la thrombose.
I.2.b. Blocage de l’inflammation
De nombreuses données expérimentales et cliniques démontrent le rôle essentiel des leucocytes lors de la lésion mécanique de l’artère. Recrutés très précocement, ils initient le rétrécissement vasculaire. Également, l’utilisation de modèles animaux a montré une très forte accumulation de neutrophiles et de monocytes/macrophages. Ces derniers, au cours du temps, s’accumulent dans la zone de resténose ; augmentent le niveau de cytokines pro-inflammatoires (IL-6, MCP-1 et IL-8) favorisant ainsi le phénomène de resténose (Welt and Rogers, 2002). Le blocage de cette inflammation représente un autre axe de recherche. Mais l’utilisation de molécules telles que les corticostéroïdes, le tranilaste et l’acide anthranilique, n’a pas permis de mettre en évidence un effet bénéfique sur le processus de resténose.
I.2.c. Endothélium et resténose
L’intégrité de l’endothélium est essentielle au maintien de l’homéostasie vasculaire. Des études démontrent que la dysfonction endothéliale contribue au développement de phénomènes de resténose (Thanyasiri et al., 2007). Les cellules endothéliales contrôlent également le passage inflammatoire, inhibent l’activité plaquettaire et la prolifération des CMLs (Badimon et al., 1992). Il a été démontré, que les stents « coatés » inhibent certes la prolifération des CMLs mais aussi celle des cellules endothéliales, favorisant ainsi une très forte inflammation et augmentant les phénomènes de thrombose. Dans ce contexte, il semble évident que stimuler la réendothélialisation est le meilleur moyen de prévenir la resténose.
La eNOS participe également au contrôle de la cicatrisation en inhibant l’inflammation, la migration et la prolifération des CMLs (Gurjar et al., 1999; Sato et al., 2000; von der Leyen et al., 1995). Par transfert de gène in vivo, la transfection du gène de la eNOS dans un modèle de dénudation de carotide de rat, restaure non seulement la contractibilité du vaisseau mais inhibe l’hyperplasie néo-intimale de 70%. Cette expérience montre ainsi l’importance des produits de synthèse issus des cellules endothéliales, et le rôle protecteur que ces cellules exercent.
Une autre molécule, le VEGF (Vascular Endothelial Growth Factor), a été étudiée pour ces effets sur la régénération endothéliale et sur l’angiogénèse. Des études ont montré que l’administration de protéines VEGF après ballonisation (Asahara et al., 1995), induit la régénération
supplémentaires, montrent une forte corrélation entre la vitesse de réendothélialisation et la diminution du processus de resténose. Mais les effets du VEGF sont très contradictoires. En effet, des travaux rapportent une augmentation des deux récepteurs du VEGF, VEGF-R1 (Flt-1) et VEGF-R2 (Flk-1), au niveau des lésions athéromateuses et de resténose. De plus, ce facteur de croissance est connu comme stimulant la migration et l’activité des monocytes.
II. OESTROGENES ET REENDOTHELIALISATION
Des données ont mis en évidence que l’E2 contribuait à la cicatrisation vasculaire, à la réduction de la prolifération et migration des CMLs et induisait une augmentation de l’angiogénèse dans des modèles animaux (New et al., 2002; White et al., 1997). Également, l’E2 permet une accélération de la réendothélialisation dans un modèle de dénudation chez le rat (Krasinski et al., 1997) et chez la souris (Brouchet et al., 2001; Iwakura et al., 2003; Strehlow et al., 2003). Ces effets sont dépendants du récepteur α des œstrogènes et de la eNOS (Brouchet et al., 2001; Iwakura et al., 2003).
De nombreuses études rapportent qu’une injection prolongée d’E2 en sous-cutané ou en intra musculaire permet une diminution de l’hyperplasie après lésion artérielle par ballonisation (Chen et al., 1996; Iafrati et al., 1997; Karas et al., 1999; Sullivan et al., 1995). En 2005, une étude de pose de stent chez le porc vient confirmer ces résultats. En effet, cette équipe montre chez des cochons traités ou non à l’E2, une forte diminution de la prolifération des CMLs et une forte accélération de la réendothélialisation chez les animaux traités. Mais de façon très intéressante, ces études démontrent une très forte corrélation entre l’hyperplasie néo-intimale et l’inflammation. La délivrance locale d’E2 au niveau du stent, entraîne une forte diminution de l’inflammation et une diminution de la resténose (Chandrasekar et al., 2005). A signaler que la pluplart de ces études se sont cantonés à l’étude de l’hyperplasie sans étude au niveau de l’endothélium.
Récemment, une explication à cet effet bivalent des œstrogènes été proposée. En effet, comment expliquer, qu’une même hormone soit capable à la fois de stimuler la prolifération des cellules endothéliales et d’inhiber celle-ci au niveau des CMLs ? L’étude chez le lapin hypercholestérolémique, de la pose de stent recouvert d’E2 a permis de mettre en évidence, par immunohistologie, une forte augmentation de la phosphorylation de ERK (Extracellular signal-Regulated Kinases) au niveau des CMLs alors que celle-ci est inhibée au niveau des cellules
endothéliales (Han et al., 2007).
Enfin, une étude préliminaire de pose de stent recouvert d’E2 (30 patients) a été menée chez l’homme. Cette étude montre que même si l’E2 n’abolit pas complètement l’hyperplasie, elle est fortement réduite chez les patients avec stent recouvert d’E2. Les auteurs proposent comme explication à cet effet partiel, un problème de diffusion de la molécule active. Également, le temps de demi-vie in vivo étant très court, l’E2 ne peut agir que très peu de temps (Abizaid et al., 2004).
En conclusion, les approches qui ont été développées au cours de ces dernières années, n’ont en rien apporté une solution définitive au problème de resténose. Il semble, au vu de ces arguments, qu’accélérer la réendothélialisation représenterait un effet protecteur majeur. Comme nous l’avons vu, l’E2 a de nombreux effets bénéfiques sur les cellules endothéliales. Des données obtenues au laboratoire ont mis également en évidence que l’E2 accélère la vitesse de réendothélialisation. Il est donc impératif de progresser dans la compréhension des mécanismes d’action de l’E2.
MODELISATION DE LA DESENDOTHELIALISATION /
REENDOTHELIALISATION
Une cicatrisation rapide d’un endothélium fonctionnel est donc indispensable au maintien de l’intégrité de la paroi. De nombreux modèles expérimentaux sur l’animal ont été développés afin d’étudier cette réendothélialisation : le singe, le porc, le lapin ou le rat. Parmis ceux-ci, le modèle de ballonisation de l’aorte ou de la carotide de rat a été particulièrement bien décrit.
I. BALLONISATION DE L’AORTE OU DE LA CAROTIDE DU RAT
Ce modèle a été décrit par Schwartz et col. (Schwartz et al., 1975). Sur un animal anesthésié, une sonde à ballonnet type cathéter de Fogarty® 2 French, est introduite dans l’aorte par la carotide primitive gauche. Le ballonnet est inflatté avec une pression contrôlée dans la partie distale de l’aorte thoracique, puis est ramené par une traction douce jusqu’à l’orifice de la carotide primitive. L’aorte thoracique est donc dilatée et désendothélialisée. Cette manœuvre détruit l’ensemble de l’endothélium aortique, du diaphragme jusqu’au pied de la carotide primitive gauche. Cette disparition du revêtement endothélial est mise en évidence par l’injection intraveineuse de bleu Evans 10 à 15 minutes avant le sacrifice. Ce colorant se fixe électivement à la matrice extracellulaire sur les zones désendothélialisées (figure 5). Une modification de ce modèle a été apportée en 1983 (Clowes et al., 1983). La manœuvre est identique, mais seule la carotide primitive est traumatisée. Le cathéter est introduit par la carotide externe puis le ballonnet est gonflé à l’orifice aortique de la carotide primitive et comme précédemment ramené jusqu’à la bifurcation carotidienne.
En fin de procédure, le cathéter est enlevé ; le vaisseau où siège l’orifice d’introduction est lié à la carotide primitive ou à la carotide externe selon le modèle choisi.
A
B A
B
Figure 5 : Visualisation d’une zone de désendothélilisation
A. Exemple de marquage au bleu Evans dans une carotide de souris. ; B. vue en microscopie électronique à
balayage de la zone frontière . La zone désendothélialisée est recouverte de plaquettes.
II. ASPECTS MORPHOLOGIQUES DE LA DESENDOTHELIALISATION / REENDOTHELIALISATION
Dans les premières heures suivant le traumatisme, la zone désendothélialisée est recouverte par une couche de plaquettes (figure 5) (Schwartz et al., 1978). Dès la huitième heure, les cellules endothéliales au contact de la zone désendothélialisée, en amont et en aval de la lésion, commencent à migrer vers la zone traumatisée.
Cette migration initiale dans l’axe du vaisseau est extrêmement rapide. Dans un modèle équivalent, mais où l’agression ne concerne qu’une bande circulaire de 2 à 3 rangées de cellules endothéliales, la zone désendothélialisée est entièrement recouverte en huit heures par des cellules endothéliales. Ces dernières ont un aspect plus large et ont perdu les interdigitations habituelles. L’administration de thymidine tritiée confirme l’absence de prolifération endothéliale dans les
Entre la 20ème et la 24ème heure après l’agression il apparaît une « zone de régénération endothéliale », cette zone est observable en aval et en amont de la zone traumatisée. Elle correspond à une bande de 60 à 100 cellules au contact de la zone désendothélialisée. Les cellules endothéliales présentent un noyau surélevé sur un cytoplasme allongé dans l’axe du vaisseau. L’observation en microscopie électronique montre également que le nombre de « gap jonctions » est diminué et les « jonctions serrées » ont disparu (Spagnoli et al., 1982). Cette observation a été confirmée en 2000, dans ce même modèle, par l’analyse de l’immuno marquage en microscopie confocale de 3 connexines (37,40 et 43). Leur expression est nulle immédiatement après le traumatisme, et en 28 jours elle revient progressivement à la normale, avec une surexpression pour les connexines 40 et 43 (Yeh et al., 2000). Au sein de la zone régénérée la densité cellulaire est multipliée par 3, excepté sur la « ligne de front » ou de « migration », où la densité est plus faible. Cette observation peut s’expliquer par le fait que la migration compense la prolifération (Schwartz et al., 1978). La vitesse de migration est estimée à 9 rangées de cellules par 24heures (environ 300 µm/24h dans ce modèle), elle est identique en amont et en aval de la lésion. Ces cellules sont en division comme en atteste l’incorporation de thymidine tritiée qui est intense sur une bande de 2 à 3 mm et absente en amont et en aval. Cette bande correspond à la zone de régénération. La prolifération endothéliale apparaît à 20-24 heures après le traumatisme au niveau des berges de la lésion, elle est maximale vers 48 heures et décroît après la 96ème heure. En arrière de cette zone de régénération les cellules endothéliales ont un aspect normal et ne prolifèrent pas.
Cette « migration-prolifération » est six fois plus rapide dans l’axe du vaisseau que dans la circonférence. Au niveau des artères intercostales, l’endothélium intact participe donc à la réendothélialisation ; la zone réendothélialisée prend un aspect ovale dont le grand axe est parallèle à celui du vaisseau (Schwartz et al., 1978). Dans un autre modèle, cité plus haut (Reidy and Schwartz, 1981), où cette fois-ci le microtraumatisme n’est plus circonférentiel mais longitudinal et concerne 3 à 5 rangées de cellules endothéliales, la zone désendothélialisée est recouverte en 48 heures (contre 8 heures en cas de traumatisme circonférentiel). Et dans ce cas, la réendothélialisation fait appel à la réplication des cellules endothéliales adjacentes. La zone traumatisée devient même une zone « hyperplasique » ou la densité cellulaire est plus importante. Cet aspect est toujours présent à 4 semaines, comme une cicatrice endothéliale.
Cette réendothélialisation peut être incomplète. Il semble exister une limite temporelle et spatiale au-delà laquelle l’endothélium ne progresse plus et laisse une zone désendothélialisée. Dans
ce modèle, au niveau de la carotide, la distance maximale parcourue par l’endothélium est de 10 mm en 6 semaines. Cette limite est variable suivant les espèces, chez le lapin la zone réendothélialisée ne s’étend que sur 3 mm et ne progresse plus au-delà de 15 jours (Reidy et al., 1983). Au niveau de l’aorte de rat, à 6 semaines, une zone désendothélialisée sur la face opposée aux artères intercostales persiste (Haudenschild and Schwartz, 1979). Les mécanismes de cet arrêt de la réendothélialisation ne sont pas encore élucidés. Il ne s’agit pas d’une limite du nombre de division, la réalisation d’un nouveau traumatisme montre que ces cellules peuvent encore se diviser (Reidy et al., 1983). Cette zone désendothélialisée persistante est le siège d’un épaississement intimal.
III. EPAISSISSEMENT INTIMAL
L’épaississement intimal correspond à une prolifération de CMLs issues de la média, associée à une matrice extra-cellulaire faite de fibres de collagène et d’élastine (Haudenschild and Schwartz, 1979). Il apparaît dans ce modèle au niveau des zones non recouvertes par l’endothélium en une semaine à 10 jours, c’est-à-dire à distance des artères intercostales ou au centre de la zone traumatisée dans le modèle carotidien. Dans les modèles expérimentaux, où la réendothélialisation est complète et réalisée en moins de 7 jours, l’épaississement intimal n’est pas observé (Hirsch and Robertson, 1977). Par contre, si cette cicatrisation dépasse ce délai, une hyperplasie peut être observée aux extrémités des zones lésées.
L’apparition des CMLs dans la partie luminale du vaisseau est précoce (Clowes et al., 1983). Au moment du traumatisme il y a une diminution du nombre de CMLs dans la média. Un quart de ces cellules disparait, cela concerne surtout les couches les plus internes de la média. En 2 à 4 jours elles sont remplacées et les premières CMLs apparaissent dans l’intima. Ces cellules franchissent la LEI (Limitante Elastique Interne) qui présente des zones de rupture, ces lésions sont observables 24 heures après le traumatisme (Haudenschild and Schwartz, 1979). L’épaississement intimal est à son maximum dès la 8ème semaine, et atteint sa plus forte épaisseur dans les zones désendothélialisées. Les fibroblastes de l’adventice artériel semblent également participer à cette néo-intima. Après une apoptose initiale suivant le traumatisme, ils prolifèrent et migrent de l’adventice vers la lumière du vaisseau, s’associant aux CMLs de la média qui elles aussi ont migré en se dédifférenciant (Sartore et al., 2001).
La réplication des CMLs est précoce, elle atteint son maximum en 48 heures dans la média et en 4 à 7 jours dans l’intima. Au niveau de cette néo-intima, la réplication persiste bien au-delà de 12 semaines, en dépit d’un nombre stable de cellules, et d’un rapport de surface entre l’intima et la média (calculé sur des coupes transversales) qui reste stable à un an. Il existe donc un « turn over » important, près de 6%, au niveau des couches les plus superficielles (Clowes et al., 1986). A la surface de cette zone désendothélialisée les CMLs ont une forme étalée proche des cellules endothéliales, elles ne sont pas jointives, elles n’expriment pas le facteur de Willebrand, et elles présentent, au niveau apical, des microvillosités prises dans une matrice extracellulaire qui a des propriétés anti-thrombotiques. En effet, les plaquettes présentes à la surface de la zone désendothélialisée disparaissent progressivement dès la 2ème semaine, pour ne représenter que quelques micro-amas épars entre les CMLs de surface vers la 6ème semaine (Reidy et al., 1983). Dans la zone désendothélialisée, dès les stades précoces, on observe des monocytes épars dont le rôle reste à préciser (Haudenschild and Schwartz, 1979).
Cet épaississement intimal n’est pas l’explication de l’arrêt de la progression endothéliale. En effet, une partie de cet épaississement est recouverte par un endothélium régénéré. D’autre part, des expériences de greffe vasculaire avec un segment de vaisseau décellularisé par la glutaraldéhyde et un autre présentant un épaississement intimal préalable, montrent que l’endothélium progresse de façon identique dans ces deux segments (Reidy, 1988).
IV. MODELES MURINS DE TRAUMATISME VASCULAIRE
Le développement de la transgènèse, chez la souris, a permis d’appréhender la biologie à l’échelon moléculaire in vivo, en générant des animaux chez lesquels un gène a été invalidé. En outre, ces modèles permettent de reproduire des pathologies humaines classiques comme le diabète et l’hypercholestérolémie. La principale difficulté de la modélisation du traumatisme vasculaire, chez la souris est liée à la taille des vaisseaux. Par exemple une carotide de souris a un diamètre de 500 microns. Plusieurs modèles ont été décrits, pouvant être classés en deux catégories : les modèles endovasculaires et les modèles périvasculaires. Les premiers sont de réalisation délicate et difficiles à mettre en œuvre, par contre ils sont voisins des modèles décrits chez le rat. Les modèles périvasculaires, bien qu’ils soient moins proches des situations physiopathologiques habituelles, sont de réalisation plus simple, rapide et reproductible.
IV.1. Modèles endovasculaires
En 1993, Linder et col. ont décrit un modèle d’agression de la carotide commune de souris. Par la branche externe de la carotide, ils passent à plusieurs reprises un fil semi-rigide de 0,35 mm de diamètre (Lindner et al., 1993). A la fin de l’expérience la carotide externe est liée (Figure 6).
Figure 6 : Modèle de traumatisme endovasculaire
Le guide semi-rigide est passé à trois reprises dans la carotide primitive (Lindner et al., 1993).
La réparation vasculaire, dans ce modèle, est tout à fait comparable à celle observée chez le rat. Le traumatisme dénude la totalité de l’endothélium, quelques CMLs sont également traumatisées. La zone désendothélialisée est rapidement recouverte par une monocouche de plaquettes ; la réendothélialisation initiée à partir des berges est totale en 3 semaines. La zone centrale de la carotide, siège d’une hyperplasie néo-intimale (15% dans les travaux de Mendelshon), est stoppée par la cicatrisation endothéliale. L’auteur décrit également la présence de quelques leucocytes, CD45+, adhérant au niveau de la zone traumatisée. Ce modèle est utilisé par plusieurs équipes notamment celle de Mendelsohn et coll.
Une variante de ce modèle a été publiée en 2003 (Goukassian et al., 2003). Le traumatisme est réalisé en passant un guide de 0,36 mm par la carotide externe à trois reprises. Un segment distal de 4mm de la carotide primitive, isolé de la circulation générale, est traumatisé puis remis en circulation. La réendothélialisation est achevée en une à deux semaines, les auteurs n’observent pas
Ce modèle est applicable à l’artère iliaque (Roque et al., 2000). Un guide de 0,25 mm est introduit par l’artère hypogastrique jusqu’à l’iliaque, trois passages sont réalisés, le guide est alors retiré et l’hypogastrique liée. Durant la procédure un clamp est positionné sur l’artère fémorale. La réendothélialisation est complète en 4 semaines ; 80% de la zone traumatisée constituent le siège d’une hyperplasie néo-intimale. Les auteurs décrivent la présence de nombreux leucocytes et polynucléaires au niveau de la zone désendothélialisée dans les premières heures qui suivent le traumatisme. Les polynucléaires neutrophiles (PNN) disparaissent en 24 heures, les macrophages persistent au-delà d’une semaine. Le principal écueil de ce modèle, outre sa difficulté de mise en œuvre, est le risque de thrombose vasculaire (21% des vaisseaux traumatisés sont thrombosés dès la deuxième semaine).
Le traumatisme vasculaire peut également être réalisé au niveau de l’artère fémorale. L’équipe de Sata et col. décrit un modèle de dilatation endovasculaire (Sata et al., 2000). Le segment d’artère fémorale est isolé de la circulation par des lacs, les collatérales sont liées et un guide de 0,38mm est passé par la branche musculaire de la fémorale. Ce guide, qui a un calibre deux fois supérieur à l’artère, est laissé en place une minute. Les auteurs décrivent une apoptose initiale des CMLs, suivie d’une hyperplasie néo-intimale qui atteint son maximum à 4 semaines, la réendothélialisation est achevée à 4 semaines. Ici aussi l’auteur observe des macrophages dans l’intima et l’adventice. La dilatation artérielle persiste à la huitième semaine et comme dans le modèle précédent la thrombose vasculaire est fréquente
La modélisation du traumatisme endovasculaire est donc possible, mais les difficultés techniques observées et les écueils de ces expériences (longueur des manipulations, thromboses, hémorragies) font que d’autres équipes ont développé des modèles plus simples.
IV.2. Modèles périvasculaires
En 1997, l’équipe qui avait décrit le premier modèle d’agression endovasculaire, propose un autre modèle de réalisation beaucoup plus rapide (Kumar and Lindner, 1997). Ils décrivent la ligature distale de la carotide, au niveau de la bifurcation. Cette ligature provoque une réduction irréversible du calibre de l’artère de 28%. Il n’existe pas de lésion endothéliale mais l’intima et la média sont le siège de remaniements. Après une réduction initiale du nombre de CMLs dans la média, ces dernières se multiplient au niveau de l’intima et de la média. Cette prolifération décroît
dès le 5ème jour mais persiste à 4 semaines et même au-delà, ce qui suggère un renouvellement permanent des cellules de cette lésion. Les auteurs de cette étude notent également la présence, aux temps précoces, de cellules CD45+ au niveau de l’adventice, de la lumière et de l’intima.
Une autre expérience consiste à mettre en place un manchon de polyéthylène (« cuff »), de 2mm, autour de l’artère fémorale (Moroi et al., 1998). Une hyperplasie intimale apparaît de façon reproductible en deux semaines ; les auteurs observent un ratio intima/média deux fois plus important chez les mâles ; et l’absence d’épaississement intimal chez les femelles en gestation. Dans ce modèle, l’endothélium n’est pas lésé, les cellules présentes dans l’intima et la média sont marquées par un anticorps anti-alpha smooth actine. L’adventice est le siège d’un infiltrat inflammatoire important.
Ces deux modèles de réalisation simple ne lèsent pas l’endothélium ; cependant les mécanismes impliqués dans cette hyperplasie ne sont pas bien compris. Dans le modèle du « cuff », les auteurs avancent l’hypothèse d’une inflammation ou la suppression des vasa-vasorum, mais ces derniers n’existent pas au niveau des artères qui n’ont que 2 ou 3 couches de CMLs dans leur média.
En 1997 l’équipe de Carmeliet et col. a décrit un nouveau modèle d’agression périvasculaire de la fémorale de souris (Carmeliet et al., 1997). Ce modèle consiste en l’application, grâce à une pince bipolaire, d’un courant électrique produit par un générateur chirurgical. Un courant de 160 µA est appliqué pendant deux secondes tous les mm. Ce traumatisme détruit l’ensemble des cellules de la paroi artérielle sans induire de thrombose, et provoque des ruptures partielles au niveau de la limitante élastique interne. A partir du 7ème jour, des CMLs « repeuplent » la média et s’accumulent dans l’intima, ces cellules colonisent la zone traumatisée à partir des berges intactes de la média. Au 14ème jour, il existe une hyperplasie néo-intimale constituée de cellules exprimant l’alpha-smooth actine. L’endothélium détruit est remplacé initialement par un thrombus mural (plaquettes), puis le segment d’artère fémorale est complètement réendothélialisé en 14 jours. Des cellules CD45+ et des PNN présents au niveau de l’intima et de l’adventice, dès la 2ème heure représentent 17% de la population cellulaire. Les PNN disparaissent au deuxième jour, et au 14ème jour les cellules CD45+ représentent moins de 5% des cellules au niveau de l’intima et de la média.