© Marina Lorente Picon, 2020
Neuroprotective effect of stomatin-like protein 2
overexpression in A53T-a-synuclein parkinson disease
mice model
Mémoire
Marina Lorente Picon
Maîtrise en neurosciences - avec mémoire
Maître ès sciences (M. Sc.)
NEUROPROTECTIVE EFFECT OF STOMATIN-LIKE PROTEIN 2
OVEREXPRESSION IN
A53T- α-SYNUCLEIN PARKINSON DISEASE MICE MODEL
Mémoire
Marina Lorente Picón
Sous la direction de :
Dr. Martin Lévesque
ii
RÉSUMÉ
Une des principales caractéristiques histologiques de la maladie de Parkinson (MP) est l’apparition d’agrégats de protéines anormaux, principalement constitués d’α-synuclein (α-syn), appelés Lewy Bodies. Ces structures sont impliquées dans les processus toxiques conduisant à un dysfonctionnement mitochondrial et augmentent la vulnérabilité des neurones dopaminergiques à la dégénérescence. Stomatin-like protein 2 (SLP-2) est une protéine située dans la membrane mitochondriale interne et agit comme un échafaudage membranaire régulant les fonctions mitochondriales et la bioénergétique. Le but de notre étude est de tester le potentiel neuroprotecteur de SLP-2 dans un modèle de souris de la MP. Notre hypothèse est que la surexpression de SLP-2 protégera les neurones dopaminergiques contre la toxicité de l’α-syn et les troubles de la locomotion associés. La quantification par immunofluorescence a été utilisée pour mesurer les niveaux de SLP-2 et de phosphoSer129-α-syn présents dans les neurones dopaminergiques de cerveaux contrôles atteints de la MP. Chez la souris, nous avons utilisé une administration de vecteurs viraux de type adénoassocié codant pour A53T-α-syn humain mutée et nous avons simultanément surexprimé SLP-2 dans la SNc par injections stéréotaxiques. L'activité motrice a été évaluée à l'aide du « open field test » et du « cylinder test ». La survie des cellules dopaminergiques a été quantifiée par comptage stéréologique. La respiration mitochondriale de l'oxygène a également été étudiée par respirométrie à haute résolution (OROBOROS). L'expression de la SLP-2 dans les neurones dopaminergiques de la SNc s'est révélée être diminuée dans les échantillons de PD par rapport aux cerveaux contrôles humains. La surexpression de α-syn dans la SNc a entraîné une diminution significative de la respiration mitochondriale de l’oxygène pouvant être renversée par une expression forcée de la SLP-2. L'analyse histologique a également révélé que SLP-2 pourrait protéger les neurones dopaminergiques contre la toxicité de l'α-syn. Nos résultats préliminaires indiquent que sur expression de SLP-2 a permis de sauver les anomalies mitochondriales et la neurodégénérescence induite par la surexpression de α-syn. Globalement, nos résultats suggèrent que SLP-2 pourrait représenter une nouvelle cible thérapeutique contre la MP.
iii
ABSTRACT
One of the main histological hallmarks in Parkinson’s disease (PD) is the apparition of abnormal protein aggregates, mainly constituted by α-synuclein (α-syn) known as Lewy bodies. These structures are involved in toxic processes leading to mitochondrial dysfunction and increase the vulnerability of dopaminergic neurons to degeneration. Stomatin-like protein 2 (SLP-2) is a protein located in the inner mitochondrial membrane and acts as a membrane scaffold regulating mitochondrial functions, integrity and bioenergetics. The aim of our study is to test the neuroprotective potential of SLP-2 in PD mice models. Our hypothesis is that SLP-2 overexpression will protect dopaminergic neurons against α-syn toxicity and associated locomotion impairment. Immunofluorescence quantification was used to measure SLP-2 and phosphoSer129-α-syn levels present in dopaminergic neurons of control and PD human brains. In mice, we used viral-vector based delivery of adeno-associated viruses encoding human mutated A53T-α-syn, and we simultaneously overexpressed SLP-2 in the SNc by stereotaxic injections. Motor activity was assessed using the open field test and cylinder test. Dopaminergic cells survival was quantified by stereological counting. Mitochondrial oxygen respiration was also studied by high-resolution respirometry (OROBOROS). SLP-2 expression in dopamine neurons of the SNc was found decreased in PD samples when compared to human control brains. Overexpression of α-syn in SNc leaded to a significant decrease mitochondrial oxygen respiration that could be rescued by SLP-2 forced expression. Histological analysis also revealed that SLP-2 could protect dopamine neurons against α-syn toxicity. Our preliminary results indicate that enhancement of SLP-2 expression rescued mitochondrial defects and neurodegeneration induced by α-syn overexpression. Altogether, our results suggest that SLP-2 could represent a novel therapeutic target against PD.
iv
TABLE OF CONTENTS
RÉSUMÉ ii
ABSTRACT iii
TABLE OF CONTENTS iv
LIST OF FIGURES, TABLES, ILLUSTRATIONS vi
LIST OF ABBREVIATIONS AND ACRONYMS vii
REMERCIEMENTS ix INTRODUCTION 1 1. Dopaminergic system 1 1.1 Dopamine neurotransmitter 2 1.2 Dopamine receptors 3 1.3 Basal ganglia 5
1.3.1 Circuit model: Direct and indirect pathways 5
1.3.2 Dopamine’s role in the Basal Ganglia Circuit 7
2. Parkinson’s Disease 8
2.1 History, epidemiology and clinical features 8
2.2 Etiology 8
2.3 Pathology 9
2.3.1 Compensatory mechanisms 9
2.4 Treatments and therapies for Parkinson’s Disease 11
2.5 Alpha-synucleinopathy 11
2.5.1 Physiological function 12
2.5.2 Pathological function 13
3. Mitochondria and PD 14
3.1 Structure and functions 15
3.2 Mitochondrial dynamics 16
3.3 Mitochondrial dysfunction in PD 18
4. The SPFH protein family: structure, localization, and function. 19
4.1 Stomatin-like protein-2 (SLP-2) 19
PROBLEMATIC, HYPOTHESIS AND OBJECTIVES 21
Chapter 1: METHODOLOGY 22
Animals 22
v
Stereotaxic injections 23
Stereological counting 24
Open field behavior 24
Cylinder test 24
Oxygen consumption 25
Statistical analysis 25
Chapter 2: RESULTS 27
4.1 SLP-2 levels are reduced in Parkinsonian human brains. 27
4.2 Characterization of the A53T-α-syn mouse model. 29
4.3 Characterization of the effects of our viral vector encoding SLP-2 31 4.4 SLP-2 overexpression protects DA cells from A53T-α-syn-toxicity 33 4.5 SLP-2 overexpression compensates mitochondrial respiration dysfunction caused by A53T-α-syn
overexpression 35
4.6 AAV-mediated SLP-2 overexpression does not significantly rescue axonal degeneration induced
by A53T-α-syn overexpression 37
4.7 Locomotor dysfunction caused by A53T-α-syn is not rescued by AAV-mediated SLP-2
overexpression 38
Chapter 3: DISCUSSION 40
1. AAV-A53T- α-syn model validation 40
2. SLP-2 as a new neuroprotective target to slow down disease progression 41
CONCLUSION 44
vi
LIST OF FIGURES, TABLES, ILLUSTRATIONS
Figure 1. Localization of dopamine cell in a rodent brain 1
Figure 2. Dopamine synthesis 2
Figure 3. Schematic representation of key steps in GPCRs signaling, homologous desensitization and
internalization 4
Figure 4. Schematic summary of the classical circuit model of direct and indirect pathways in
physiological condition 6
Figure 5. Schematic summary of the improved complex circuit model of basal ganglia in physiological
condition 7
Figure 6. Schematic of the main nigro-striatal compensatory mechanisms in PD. 10
Figure 7. Schematic of α-synuclein physiological function 13
Figure 8. Schematic of physiological and pathological structure of α-synuclein 14
Figure 9. The mitochondrial ETC 16
Figure 10. Mitochondrial dynamics 17
Figure 11. SLP-2 immunofluorescent signal is reduced and pS129 signal is increased in PD human
brains 28
Figure 12. AAV-A53T-α-syn injection in the SNc causes widespread α-syn expression,
neurodegeneration of the nigrostriatal pathway and motor impairment 31 Figure 13. AAV-SLP-2 injection in the SNc causes widespread SLP-2 expression in a dose dependant
manner. 32
Figure 14. SLP-2 OE neuroprotects DA cells in the SNc from A53T-α-syn toxicity 34 Figure 15. SLP-2 OE increases mitochondrial oxygen consumption 36 Figure 16. Striatal DA axons degeneration cannot be rescued by SLP-2 OE 37 Figure 17. SLP-2 OE is not enough to rescue impaired behavior caused by A53T- α-syn OE 39
vii
LIST OF ABBREVIATIONS AND ACRONYMS
6-OHDA: 6-hydroxydopamine
AAV: Adeno-associated virus
AC: Adenylate cyclase
Acb: Accumbens nucleus
ADP: Adenosine diphosphate
ATP: Adenosine triphosphate
βArr: β-arrestins
BSA: Bovine serum albumin
Ca2+: Calcium
cAMP: Cyclic adenosine monophosphate
CCCP: Carbonyl cyanide m-chlorophenylhydrazone
CN: Caudate nucleus
CNS: Central nervous system
CTCF: Corrected total cell fluorescence
DA: Dopaminergic
DAT: Dopamine transporter
DBS: Deep brain stimulation
DNA: Deoxyribonucleic acid
DRP1: Dynamin-related protein 1
ER: Endoplasmatic reticulum
ETC: Electron transport chain
FADH2: Flavin adenine dinucleotide hydroquinone
GABA: Gamma-Aminobutyric acid
GC: Genome copy
GDP: Guanosine diphosphate
GPCRs: G protein-coupled receptors
GP: Globus palidus
GPe: External segment of the Globus palidus GPi : Internal segment of the Globus palidus
GRK: GPCRs kinases
GTP: Guanosine triphosphate
IMM: Inner mitochondrial membrane
IMS: Intermembrane space
KO: Knock-out
L-AADC: L-Aromatic amino acid decarboxylase L-DOPA: L-dihydroxyphenylalanine
LBs: Lewy bodies
LRRK2: Leucine-rich repeat Kinase 2
MFN1: Mitofusin 1
MFN2: Mitofusin 2
MPTP: 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine
mtDNA: Mitochondrial DNA
MTS: Mitochondrial targeting sequence
NAC: Non-amyloid-beta component
NADH: Nicotinamide adenine dinucleotide NMDA: N-methyl-D-aspartate receptor
viii
O2: Oxygen
OE: Overexpression
OMM: Outer mitochondrial membrane
O/N: Over night
OPA: Optic atrophy1
OXPHOS: Oxidative phosphorylation
PARK2: Parkin RBR E3 ubiquitin protein ligase PARK7: Parkinson protein 7-DJ-7
PARL: Presenilins-associated rhomboid-like protein
PBS: Phosphate-buffered saline
PD: Parkinson’s disease
PFA: Paraformaldehyde
PINK1: Phosphatase and tensing homolog-induced Kinase 1
PKA: Protein kinase A
PHB-1: Prohibitin-1
PHB-2: Prohibitin-2
Put: Putamen
ROS: Reactive oxygen species
RNA: Ribonucleic acid
rRNAs: Ribosomal ribonucleic acids SLP-1: Stomatin-like Protein 1 SLP-2: Stomatin-like Protein 2 SLP-3: Stomatin-like Protein 3
SN: Substantia nigra
SNARE: Soluble N‐ethylmaleimide-sensitive factor attachment protein receptor SNc: Substantia nigra pars compacta
SNCA/PARK1: α-synuclein gene
SNr: Substantia nigra pars reticulata SPN: Spiny projection neurons
STN: Subthalamic nucleus
TH: Tyrosine hydroxylase
TIM: Translocase of the inner membrane
TOM: Translocase of the outer membrane tRNAs: Transfer ribonucleic acids
VDAC: Voltage-dependent anion channel VMAT-2: Vesicular monoamine transporter
VPS35: Vacuolar protein sorting-associated protein 35 VTA: Ventral tegmental area
ix
REMERCIEMENTS
This « mémoire » is a compilation of a rollercoaster of emotions.
Behind all the work done during these 2 years, there is a big boat called LEVLAB who helped me to stay afloat. This amazing boat is constructed by Tess, Caro, Axelle, Vero, Anne-Marie, Julia, Charleen, François, Charles, Francis, Marcos, Tiago, Victoria and of course, as all the big boats, there is a great captain Martin, who is always guiding us for not to sink into the immense ocean of ignorance, despair and surrender. Thanks Martin to give me the opportunity of this incredible experience.
Maddy, Gamze, Javi and Romi you’re the best family I could chose. Even though, my family is the best too and has been supportive in all my decisions. Papa, Mami, Sergio os quiero a todos infinito! Gràcies xurrukas i Luce per no deixar-me marxar mai dels seus cors i per sempre demostrar-me tant d’amor encara que estigui tan lluny,
Mavrick, je t’aime amore, merci cariño pour me réjouir en ce dernier sprint.
1
INTRODUCTION
1. Dopaminergic system
Dopaminergic (DA) neurons are the major source of dopamine neurotransmitter in the mammalian central nervous system (CNS). DA neurons are a heterogeneous group of cells, anatomically and functionally, that are localized in nine distinctive cell groups, distributed from the mesencephalon to the olfactory bulb. The neuronal cell groups containing dopamine can be divided from A8 to A16 (Figure 1). Even though, a recent cell group, A17 has been added and is located in the retina. (Chinta and Andersen 2005; Björklund and Dunnett 2007)
Figure 1. Localization of dopamine cell in a rodent brain
Schematically illustration of a sagittal rodent brain where nine distinctive DA cell groups can be identified. They are distributed from the mesencephalon to the olfactory bulb in the mammalian brain. (Chinta and Andersen 2005)
This DA neurons present in those regions, constitute the DA system which is one of the several neurotransmitter systems that controls different brain processes and it has been traditionally divided in four main different pathways: the mesolimbic, the mesocortical, the nigrostriatal and the tuberoinfundibular pathways (Cox and Lee 2016; Craenenbroeck et al. 2005; Björklund and Dunnett 2007). The mesolimbic pathway is constituted of DA neurons that originate in the ventral tegmental area (VTA) in the midbrain. The mesolimbic pathway links the VTA to different components of the limbic system including the amygdala, the hippocampus and the accumbens nucleus. It is reported to be implicated in motivation and reward processes (Ikemoto 2010; Cox and Lee 2016; Alcaro, Huber, and Panksepp 2007). Very much alike the mesolimbic, the mesocortical pathway is also constituted by DA neurons originate in the VTA. However, these DA neurons project to the frontal cortex and they are implicated in cognition, motivation, mood, memory and attention (Craenenbroeck et al. 2005). The nigrostriatal pathway is composed by DA neurons that originate in the substantia nigra pars compacta (SNc) and they project to the basal ganglia including the striatum or putamen (Put) - caudate nucleus (CN). The nigrostriatal pathway is involved in the control of voluntary motor movement,
2
consequently loss of DA neurons in this pathway is one of the main neuropathological characteristics of Parkinson’s disease (PD) (Craenenbroeck et al. 2005; VERNIER et al. 2004; Parent et al. 2000). Finally, the tuberoinfundibular dopamine pathway is constituted by DA neurons located in the dorsomedial arcuate nucleus. Those neurons are involved in the hypothalamo-pituitary system regulating prolactin production (Stagkourakis et al. 2019; Grattan 2015).
1.1 Dopamine neurotransmitter
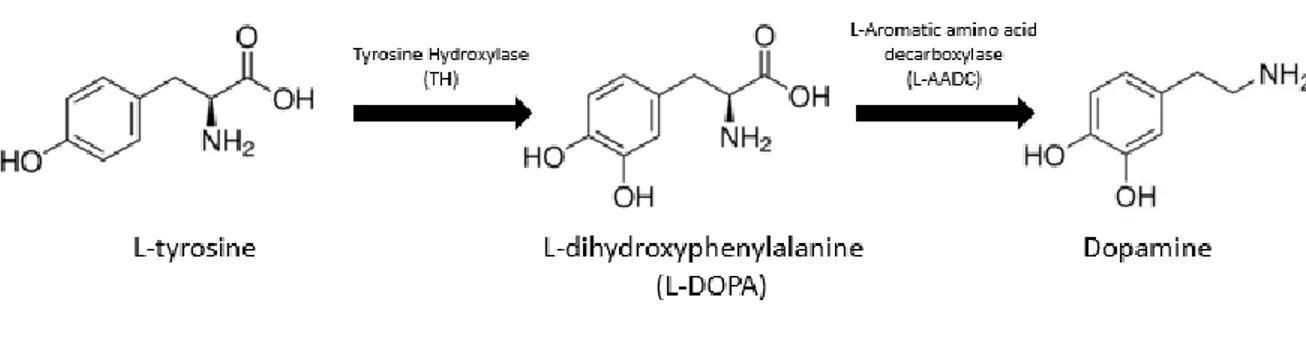
Dopamine is the main neurotransmitter synthetized by DA neurons and is one of the most behaviorally powerful neuromodulators. Dopamine is implicated in the regulation of electrical and biochemical aspects of neuronal function such as synaptic transmission, excitability, plasticity and protein trafficking (Tritsch and Sabatini 2012). Dopamine is synthetized in two steps starting by the hydroxylation of L-tyrosine by the enzyme tyrosine hydroxylase (TH) producing L-dihydroxyphenylalanine (L-DOPA). L-DOPA is then decarboxylated by the L-Aromatic amino acid decarboxylase (L-AADC) transforming it into dopamine (Figure 2) (Meiser, Weindl, and Hiller 2013).
Figure 2. Dopamine synthesis
The main pathway for dopamine biosynthesis starts at tyrosine which is hydroxylated by TH. tyrosine is hydroxylated to form L-DOPA, which is decarboxylated by L-AADC leading to dopamine (adapted from Meiser et al., 2013).
New synthesized dopamine is transported by the vesicular monoamine transporter (VMAT-2) from the cytoplasmic space into synaptic vesicles in the presynaptic terminals. Once dopamine is released in the synaptic cleft, different actions can occur. Dopamine can bind and activate both pre-synaptic and post-synaptic DA receptors, but it can also be repackaged into post-synaptic vesicles and be reuptake into the presynaptic DA terminals via the dopamine transporter (DAT) or it can be degraded (Vaughan and Foster 2013; German et al. 2015). DAT is a plasma membrane protein that is responsible of translocate released dopamine from the extracellular space into the presynaptic terminals in order to clear extracellular dopamine and control spatial and temporal dynamics of dopamine neurotransmission (Vaughan and Foster 2013).
3
1.2 Dopamine receptors
Dopamine interacts and activates G protein-coupled receptors (GPCRs) that are classified into D1-like and D2- like receptors classes based on their structural, pharmacological and signaling properties (Hisahara and Shimohama 2011). The D1-like receptor subfamily includes D1 and D5. Whereas D2, D3 and D4 receptors are grouped into the D2-like receptor class. The role of GPCRs is critical to normal brain function as their activation by the presence of dopamine, modulate the biochemical and electrical state of the cells via different signaling elements including kinases, phosphatases, transcription factors, ion channels and membrane receptors (Tritsch and Sabatini 2012). Downregulation or overactivity of the GPCR system may contribute to pathological conditions. GPCRs are constituted by seven trans-membrane-spanning domain architecture by which the amino terminal is extracellular located, and the carboxyl terminal is intracellular. There is a segment which interacts and activates the G-proteins made up of three protein subunits, α-syn, β and γ. The α-syn-subunit contains a guanine nucleotide binding site, whereas β and γ form a tightly associated βγ-complex. When there is no activation, the α-syn-subunit is bound to a guanosine diphosphate (GDP) and to βγ-complex forming a trimeric protein complex. When dopamine binds to the receptor and activates it, promotes the replacement of the GDP by guanosine triphosphate (GTP) which will bind to the α-syn-subunit and will activate it (Figure 3.A). The activation of the α-syn-subunit promotes the dissociation from the βγ complex. Both can then transduce the signal and activate intracellular effectors (Figure 3.B) (Hisahara and Shimohama 2011; Gainetdinov et al. 2004; Premont and Gainetdinov 2007).
Each class of dopamine receptor have their specific signaling cascades depending on which subunit are they coupled. Those subunits are the ones in charge of transduce the signal and activate many effector systems (Hisahara and Shimohama 2011). In the D1 subfamily, the activation of some of the G protein subunits by dopamine stimulates adenylate cyclase (AC) which stimulates the production of the second messenger cyclicadenosine monophosphate (cAMP) activating the protein kinase A (PKA) (Figure 3.B). On the contrary, in the D2 subfamily, the activation of some of the G protein subunits, inhibits the AC downregulating the production of cAMP, resulting in a decrease in PKA activity (Tritsch and Sabatini 2012; Beaulieu, Espinoza, and Gainetdinov 2015). These receptors present a high complexity of signal cascade. Apart from the well-known cAMP-mediated cascade signalling, there are other signalling mechanisms using non-G protein coupling mechanisms such as ion channels, or proteins like β-arrestins (βArr) and GPCRs kinases (GRK) and phosphatases. βArr act as a scaffold for different members of the GRK family and both are involved in the desensitization and internalization mechanisms which allow to control GPCR responsiveness (Gainetdinov et al. 2004; Del’guidice, Lemasson, and Beaulieu 2011).
The homologous desensitization mechanism in GPCR is a crucial mechanism to protect cells against over-stimulation of the receptors (Del’guidice, Lemasson, and Beaulieu 2011). This process starts when activated
4
GPCR acts as a substrate for protein phosphorylation by GRK (Figure 3.C). However, GRK-phosphorylation of receptors is not enough for desensitization, but it promotes the binding of βArr which prevents the receptor from exchanging GTP for GDP on the G protein α-syn-subunit, thus interdicting further G protein activation and ensuring desensitization (Claing et al. 2002). Finally, the GRK- βArr system promotes the internalization of inactivated receptors and subsequent recycling of resensitized receptors back to the cell surface. The recruited βArr bind the clathrin adaptor protein AP2 and to Clathrin itself in facilitating the entry of desensitized receptors intro clathrin-coated pits for subsequent internalization (Figure 3.D) (Gainetdinov et al. 2004).
Figure 3. Schematic representation of key steps in GPCRs signaling, homologous desensitization and internalization
A. Dopamine binds to the receptor promoting the replacement of the GDP by GTP and subsequent activation of the α-syn-subunit. B.
Activated α-syn-subunit dissociates from the βγ complex. Both can then transduce the signal and activate intracellular effectors. C. The activated state of GPCRs serves as well as a substrate for protein phosphorylation by GRKs. Thus, activated receptor regulation by GRKs results in homologous desensitization. D. GRK promotes βArr binding to the receptors preventing further G protein activation and ensuring desensitization. Moreover, the GRK- βArr system promotes the internalization of inactivated receptors and subsequent recycling of re-sensitized receptors back to the cell surface (Adapted from Gainetdinov et al., 2004).
5
1.3 Basal ganglia
The basal ganglia are a group of subcortical nuclei controlling voluntary actions such as motor control, as well as other roles such as motor learning, executive functions and behaviors, and emotions (Lanciego, Luquin, and Obeso 2012). Basal ganglia refer to nuclei embedded deep in the brain hemispheres such as the CN and Put (collectively they are called the striatum) and the globus pallidus (GP) (Báez-Mendoza and Schultz 2013; Parent et al. 2000). Whereas related nuclei are the structures located in the diencephalon like the subthalamic nucleus (STN), the mesencephalon like the substantia nigra (SN) that can be divided into SNc and SN pars reticulata (SNr) (Lanciego, Luquin, and Obeso 2012). The basal ganglia and related nuclei can be divided as: input nuclei, output nuclei and intrinsic nuclei. Input nuclei includes the CN, the Put, and the acumbens nucleus, they receive incoming information mainly from cortical, thalamic, and nigral areas. The output nuclei are the internal segment of the GP (GPi) and the SNr. Those structures send basal ganglia information to the thalamus. Finally, intrinsic nuclei are the structures located between the input and output nuclei in the relay of information and includes the external segment of the Gp (GPe), the STN and the SNc (Lanciego, Luquin, and Obeso 2012; Haber 2014; Parent et al. 2000). For the basal ganglia system to work properly, it requires the release of dopamine at the input nuclei. Dopamine dysfunction is associated with several basal ganglia movement disorders such as PD.
1.3.1
Circuit model: Direct and indirect pathways
There are two well described circuit models for transmission of signals through the striatum and basal ganglia outputs, which explain the interaction between glutamatergic and DA neurotransmission. Those models include the direct and indirect pathways (Calabresi et al. 2014).
The direct pathway starts with cortical activation and release of glutamate activating the principal striatal neurons, the spiny projection neurons (SPNs) which are inhibitory neurons that use gamma-Aminobutyric acid (GABA) as neurotransmitter and they contain D1-like receptors. SPNs project directly to the GPi and the SNr, thus exerting an inhibitory action on the present neurons which are also GABAergic. When the SNr is inhibited it leads to a disinhibition of the thalamic glutamatergic neurons in the thalamus that project to the cortex. Thus, resulting in the regulation of the tonic level of excitation in the premotor cortex that is involved in planning and initiation of movement. On the other hand, the indirect pathway consists on the activation of striatal SPNs expressing the D2-like receptors that innervate the GPe. They inhibit the GABAergic neurons present in the GPe triggering the disinhibition of the glutamatergic neurons of the STN. The activation of the STN excitatory neurons consecutively activates the GPi and SNr GABAergic neurons projecting to the thalamus. Eventually, this effect results in the reduction of locomotor activity and movement (Lanciego, Luquin, and Obeso 2012; Calabresi et al. 2014; Obeso et al. 2008; Gerfen and Surmeier 2011; Sonne and Beato 2019) (Figure 4).
6
Figure 4. Schematic summary of the classical circuit model of direct and indirect pathways in physiological condition
The circuit is composed of a cortico-striatal projection, two major striatal projection systems starting the direct and indirect pathways, and the efferent pallido–thalamo–cortical projections that close the motor loop (Lanciego, Luquin, and Obeso 2012).
Although the original functional organization of the basal ganglia was considered as a loop that facilitates or inhibits motor activity by the direct and indirect pathway projections from striatum, having approximately opposite behavioral effects, recent studies proposed a new way to see this functional model. The basal ganglia network is composed by different and parallel loops that interact dynamically between them forming a complex network of signal transmission (Figure 5). Moreover, single neuron tracing study showed that the large majority of striatal neurons project to the GPi, GPe and SNr challenging as well this dual system (Lévesque and Parent 2005). In addition, this network is not just involved in movement control, is it also related to associative learning, planning, working memory, and emotions (Lanciego, Luquin, and Obeso 2012; Obeso et al. 2008; Calabresi et al. 2014; Freeze et al. 2013; Cui et al. 2013).
7
Figure 5. Schematic summary of the improved complex circuit model of basal ganglia in physiological condition
Schematic of the main circuits linking the basal ganglia nuclei in the complex circuit model. Various and parallel loops interact and regulate signal transmission creating a complex network (Lanciego, Luquin, and Obeso 2012).
1.3.2
Dopamine’s role in the Basal Ganglia Circuit
The SN is a midbrain DA nucleus which has a critical role in modulating motor movement and reward functions as part of the basal ganglia circuitry (DeLong and Wichmann 2007). It can be morphologically and functionally divided into the SNr and the SNc. The SNr is populated largely by a majority of GABAergic neurons and some DA neurons (Parent et al. 2000). This nucleus works in conjunction with the GPi as the final output of the basal ganglia’s direct and indirect pathways (Sonne and Beato 2019).
The major cell type in SNc are the DA neurons whose axons innervate mainly the striatum, forming the nigrostriatal DA pathway (Parent et al. 2000). Dopamine released by SNc DA neurons have a complex and different modulatory effect on the normal functioning of the striatum, they regulate the excitability and synaptic connectivity of striatal output neurons (Gerfen and Surmeier 2011; Sonne and Beato 2019). SNc DA neurons are autonomous pacemakers, maintaining the DA release in the striatum (Gerfen and Surmeier 2011). They innervate the SPNs present in the striatum that are expressing D1-like and D2-like family receptors. When D1-like receptors are activated in the direct pathway, increase the SPNs excitability and promote long-term potentiation of excitatory synapses. On the other hand, when D2-like receptors are activated in the indirect pathway, decrease the excitability of SPNs and promote long-term depression of excitatory synapses (Sonne and Beato 2019; Frank 2005; Kerr and Wickens 2001). This dual modulation of SPNs activity by dopamine, can be translated into changes in cortico-striatal networks facilitating action selection (Gerfen and Surmeier 2011).
8
2. Parkinson’s Disease
2.1 History, epidemiology and clinical features
In 1817, James Parkinson first clinically described Parkinson’s disease (PD) in “An Essay on the Shaking Palsy”. He defined it as “Involuntary tremulous motion, with lessened muscular power, in parts not in action and even when supported; with a propensity to bend the trunk forwards, and to pass from a walking to a running pace: the senses and intellects being uninjured” (Parkinson 2002). Following this publication, the name Parkinson was attributed to this disease in his honor.
PD is the second most common chronic, progressive neurodegenerative disease that affects nearly seven to ten million people worldwide. PD is an age-related disease predominantly affecting people over age 60, although some people have been diagnosed as young as 18 (Pringsheim et al. 2014). The prevalence of the disease is 1.5 times higher in men than women and causes of the disease change with age. They may be sex-related risk and protective factors playing different roles (Moisan et al. 2016).
The symptoms of PD are characterized by the deterioration of some of the motor and non-motor capacities. The classical motor features are bradykinesia, muscular rigidity, rest tremor, and postural and gait impairment. Moreover, there are also non-motor features including cognitive impairment, psychiatric symptoms, sleep disorders, autonomic dysfunction, depression, pain and fatigue (DeMaagd and Philip 2015).
2.2 Etiology
There are two forms of PD including familial and idiopathic. The etiology of PD is still largely unknown, most of the cases are sporadic believed to develop from gene-environment interactions. Only about 10% of patients had a genetic mutation in the family history (Ball et al. 2019; Klein and Westenberger 2012). In recent years, seven different genes have been identified as causing familial PD. Specifically, mutations in α-synuclein gene (SNCA/PARK1), glucocerebrosidase, leucine-rich repeat Kinase 2 (LRRK2), vacuolar protein sorting-associated protein 35 (VPS35), parkin RBR E3 ubiquitin protein ligase (PARK2), phosphatase and tensing homolog-induced Kinase 1 (PINK1) which gene is PARK6, and Parkinson protein 7-DJ-7 (PARK7) (Ball et al. 2019). Idiopathic PD cases, as said before, are understood to be caused by gene-environment interactions. The combined effect of genetic and environmental factors may influence the onset of the disease. However, each individual present a unique genotype and environmental factors can affect different resulting in distinct phenotypes (Schlossmacher et al. 2017). Risk factors contribute to the development of PD by being involved in PD-associated mechanisms such as mitochondrial dysfunction, oxidative stress and protein degradation impairment. Some cases may be influenced by age, gender, and ethnicity while other cases may be attributed
9
to environmental, occupational, or residential exposure to neurotoxins including heavy metals, pesticides, herbicides and illicit drugs (Ball et al. 2019; Fleming 2017; Cannon and Greenamyre 2013).
2.3 Pathology
The neuropathological characteristics of PD is the progressive and irreversible degeneration and loss of DA neurons present in the SNc and their axon projections causing the loss of the nigrostriatal neuronal pathways triggering dopamine depletion in the basal ganglia (VERNIER et al. 2004; Dennis W Dickson 2012; Forno 1988). DA neurons contain a dark-brown cytoplasmic pigment called neuromelanin, the loss of this cells produces the depigmentation of the SNc, a classic neuropathological characteristic of PD (Carballo-Carbajal et al. 2019). In contrast, the very similar DA neurons in the VTA demonstrate a higher degree of resistance to degeneration (Brichta and Greengard 2014). It is estimated that between 70% and 80% of dopamine axons innervating the striatum are lost at the onset of the disease and there is about 60% neuronal loss in the SNc (Agid 1991). The loss of dopamine in the striatum, causes the onset of the disease's motor symptoms. However, motor symptoms are gradual, suggesting the existence of different compensatory mechanisms that delay the appearance of the first symptoms despite the massive loss of DA innervation (Blesa et al. 2017).
2.3.1 Compensatory mechanisms
PD, as many clinical disorders, tend not to appear until later life. It is usually not diagnosed until at least the sixth decade and it can not be studied as a discontinuous pathology because the patients don’t pass suddenly from a normal to a full parkinsonian state. Despite the DA neuronal death in the SNc, their efficiency is maintained during the preceding period before the appearance of clinical signs. The common justification to explain this event, is the presence of compensatory mechanisms which serve to delay the onset and aggravation of motor behavioural abnormalities (Zigmond 1997; Blesa et al. 2017; Bezard, E., & Gross 1998; Obeso et al. 2008). It is understood that the basal ganglia network adapts to the functional effects of dopamine depletion in several ways beyond the nigrostriatal system. Once the dopamine deficit surpasses a given threshold, the GPe‐STN‐GPi network becomes hyperactive losing its normal modulatory function leading to abnormalities in the desired movement (Obeso et al. 2008). There are different factors involved in these compensatory processes such as intrinsic properties that DA nigrostriatal neurones must possess in order to preserve dopamine availability in the striatum, DA neuronal plasticity, morphological modifications in the SPNs spines, variances in the transmission of dopamine in the striatum and the different inputs to SNc (Bezard, E., & Gross 1998; Villalba and Smith 2018; Huot and Parent 2007).
The main nigro-striatal compensatory mechanisms are activated by the surviving DA neurons which go through different functional changes (Blesa et al. 2017). Those changes can be alterations in TH activity following an increase in dopamine synthesis, release and turnover (Nandhagopal et al. 2011; De La
Fuente-10
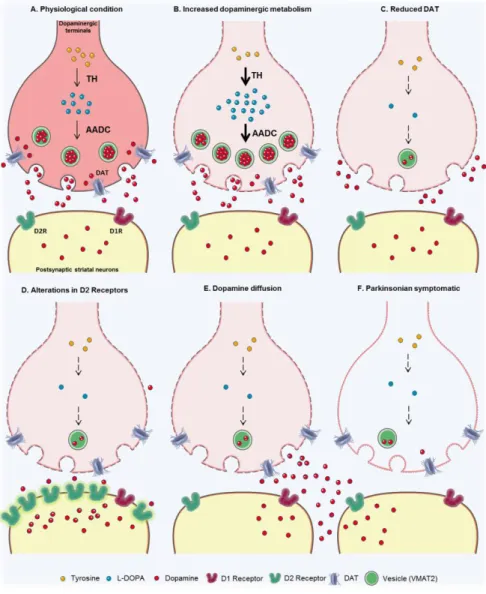
Fernández et al. 2001; Kozina et al. 2014). DAT levels may be reduced in order to decrease dopamine uptake into presynaptic terminals (Vaughan and Foster 2013; Vezoli et al. 2014). Moreover, there can also be changes in DA receptors in the postsynaptic terminals (Bezard et al. 2001) or dopamine diffusion between terminals (Bezard, E., & Gross 1998) (Figure 6).
Figure 6. Schematic of the main nigro-striatal compensatory mechanisms in PD.
A. Physiological condition of DA synaptic terminals. B. Increased dopamine metabolism, release and turnover. C. Dopamine re-uptake
is reduced due to lack of DAT. D. D2-like receptors presence is altered. E. Dopamine diffusion and passive balance. F. Synaptic terminals in the parkinsonian state (Blesa et al. 2017).
11
2.4
Treatments and therapies for Parkinson’s Disease
Nowadays, the purpose of the current treatments or therapies for PD is to reduce and mitigate the effect of the symptoms related to parkinsonian phenotypes in order to improve the patient’s daily life. Present treatments neither cure PD nor stop it from neurodegeneration progression. For that reason, it is extremely important that treatment is found to block, stop or prevent DA neurons from degeneration. In addition, the search for biomarkers related with PD is same or even more important in order to be able to detect and thus stop the progression of the disease.
The common and more used treatments are dopamine agonists for dopamine replacement therapy resulting in an improvement of motor symptoms in the early stage of the disease. However, they don’t influence the rate of clinical disease progression and eventually they cause serious side effects such as motor complications and dyskinesias. One example of the most common treatment is Levodopa, which is the common name for the drug. Levodopa penetrates DA neurons and replace the depleted endogenous neurotransmitter by providing the dopamine precursor L-Dopa (Brooks 2000; Brichta, Greengard, and Flajolet 2013; Salat and Tolosa 2013). There are other treatments that don’t use dopamine agonists such as deep brain stimulation (DBS) which consists on the placement of high frequency stimulating electrodes in the region of the ventral intermediate nucleus of the thalamus and stimulation of the STN or GPi to reduce tremor and also decrease bradykinesia, rigidity and gait impairment (Groiss et al. 2009; Perlmutter and Mink 2006).
2.5 Alpha-synucleinopathy
From a pathophysiological point of view, the death of DA neurons is preceded by the formation of abnormal intracellular protein aggregates known as Lewy bodies (LBs), in the DA neurons in the SNc. Those aggregates appeared to be mainly constituted by the protein α-synuclein (α-syn) (D. W. Dickson 2018; Schulz-Schaeffer 2010; Spillantini et al. 1997; Wakabayashi et al. 2007). Moreover, approximately 90% of the α-syn deposited in those LBs is extensively phosphorylated at Ser129. In contrast, only 4% or less of total α-syn is phosphorylated at this residue in healthy brains (Arawaka et al. 2017). This post-translational modification may play an important role in the regulation of α-syn aggregation, LBs formation and neuronal degeneration (Oueslati 2016).
As mentioned before, α-syn is encoded by the SNCA/PARK1 gene that consists 140 amino acids. The protein is divided into three main domains: N-terminal binding domain, central hydrophobic non-amyloid-beta component (NAC) domain which is relevant to α-syn aggregation, and a hydrophilic C-terminal domain with chaperone-like activity (Siddiqui, Pervaiz, and Abbasi 2016). At least 30 SNCA gene mutations have found to cause familial PD. Examples of those mutations are: A30P (Krüger et al. 1998), E46K (Zarranz et al. 2004),
12
H50Q (Appel-Cresswell et al. 2013), G51D (Kiely et al. 2013) and A53T and, gene multiplications such as duplication and triplication (Polymeropoulos et al. 1997; Nishioka et al. 2009).
2.5.1 Physiological function
In physiological conditions, α-syn is found forming monomers or tetramers even though it’s is not well elucidated which conformation state it has in the different compartments of the cell (Bartels et al. 2011; W. Wang et al. 2011; J Burré et al. 2013). Those structures are involved in different processes; however, it remains unknown the exact roles that α-syn has in the CNS. They are still ambiguous and still need to be clarified for a better understanding of the physiological and pathological functions of α-syn.
One of the main roles seems to be its involvement in the trafficking of synaptic vesicles and in the regulation of vesicle exocytosis and synaptic vesicle-mediated release of neurotransmitter. α-syn is mainly located at presynaptic terminals and it is associated with the distal reserve pool of synaptic vesicles (H. J. Lee et al. 2011; Bellani et al. 2010; Lautenschläger et al. 2018; Lashuel et al. 2013; Scott and Roy 2012). More specifically, monomeric α-syn plays an important role in the docking, priming and fusion steps of vesicle fusion and exocytosis probably by interacting and serving as a non-classical chaperone that facilitate SNARE (soluble N‐ethylmaleimide-sensitive factor attachment protein receptor) complexes assembly (Jacqueline Burré et al. 2010; Jacqueline Burré 2015; Lashuel et al. 2013; Huang et al. 2019). Endocytosis is directly related to exocytosis, and both are critical for the maintenance of presynaptic structural integrity. In the process of endocytosis, α-syn mediates membrane curvature and is also involved in the process of receptor internalization in order to regulate DAT homeostasis and N-methyl-D-aspartate receptor (NMDA) receptor through clathrin-mediated endocytosis (Kisos, Ben-Gedalya, and Sharon 2014; Cheng et al. 2011; Huang et al. 2019). In addition, α-syn also enhances microtubule formation and axonal transport through interactions with tubulin (Koprich, Kalia, and Brotchie 2017; Cartelli et al. 2016; Alim et al. 2004). Those possible different functions of α-syn may be due to its conformational flexibility allowing the protein to adopt different forms when it interacts with biological membranes, other proteins or protein complexes (Lashuel et al. 2013).
Different studies shown that α-syn knock-out (KO) mice are viable and they don’t have impairment in basic brain functions. Moreover, there is no significant changes in the structure of the synapses, in the synaptic plasticity or in the size of the recycling synaptic vesicles. These results suggest that α-syn is not essential for neurotransmitter release but may contribute to the regulation and maintenance of presynaptic function (Chandra et al. 2004; Abeliovich et al. 2000).
13
Figure 7. Schematic of α-synuclein physiological function
Different roles of α-syn at the presynaptic terminal in the regulation of vesicle trafficking and vesicle refilling (α-syn blue). α-syn interacts with membrane-associated t-SNARE and the vesicle-associated v-SNARE proteins and neurotransmitter release. When α-syn accumulates (α-α-syn red) induces an impairment in the neurotransmitter release, vesicle recycling and trafficking between α-synaptic buttons (Lashuel et al. 2013).
2.5.2 Pathological function
The problem appears when α-syn shifts from its monomer form to higher-order structures such as protofibrils, fibrils and oligomers (Koprich, Kalia, and Brotchie 2017; Winner et al. 2011). Those higher-order structures seem to be the ones involved in the toxic processes increasing the vulnerability of the cell. Cytosol and membrane-bound ring-like oligomers can form transmembrane pores compromising the cell membrane integrity, intracellular calcium homeostasis and cell-cell communication (Huang et al. 2019). They also cause impairment in protein clearance and degradation by the ubiquitin-proteasome and autophagy-lysosomal systems (Hinault et al. 2010; Zhang, Tang, and Liu 2008; Cuervo et al. 2004; Mak et al. 2010); cause mitochondrial dysfunction leading to oxidative stress and also, they bind mitochondrial membranes contributing to mitochondrial fragmentation and degradation (Nakamura et al. 2011; Mullin and Schapira 2013; Rocha, De Miranda, and Sanders 2018; B. J. Ryan et al. 2015; Park, Davis, and Sue 2018; Maio et al. 2016); disrupt endoplasmic reticulum (ER) and Golgi network (Colla et al. 2012; Arduíno et al. 2009; Calì, Ottolini, and Brini 2011); impair axonal transport (Lamberts, Hildebrandt, and Brundin 2015; Volpicelli-Daley 2017) and increase neuroinflammation (Q. Wang, Liu, and Zhou 2015; Vivekanantham et al. 2015). Moreover, they can result in loss of function effects such as impaired neurotransmitter release (Shen 2010; Plowey and Chu
14
2011) and disrupted microtubule formation (Carnwath, Mohammed, and Tsiang 2018; Pellegrini et al. 2017; Wersinger and Sidhu 2005) (Figure 8).
Overall, an imbalance between those mechanisms, caused by dysfunction of one or more of these pathways, can result in abnormal levels of α-syn that might favour the formation and/or accumulation of oligomeric and fibrillar species, which may be toxic (Lashuel et al. 2013). Nevertheless, the molecular mechanisms by which α-syn aggregation contributes to DA neurodegeneration, the nature of the toxic forms of α-syn and the cellular pathways that are affected by α-syn remain largely unknown.
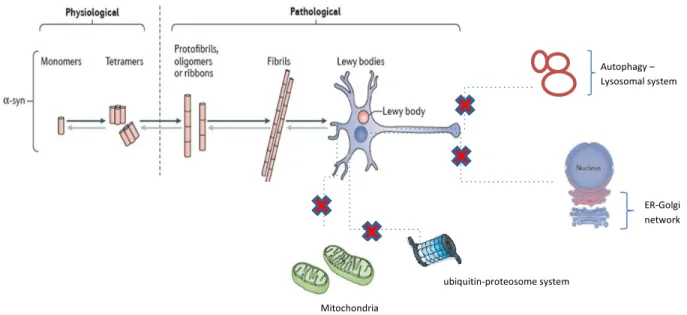
Figure 8. Schematic of physiological and pathological structure of α-synuclein
α-syn exists in different forms due to its conformational flexibility, that may be the reason of its different functions. Probably, monomeric and tetrameric are the native forms of α-syn in the physiological state. On the contrary, protofibrils, oligomers, ribbons and fibrils are the hypothesized α-syn forms in the pathological state in PD. Those pathological forms are involved in the toxic processes increasing the vulnerability of the DA neurons as they cause impairment (red cross) in mitochondria, the ubiquitin-proteasome and autophagy-lysosomal systems, disrupt the ER-Golgi network among other toxic processes not represented in the schematic. Moreover, LBs, which are mainly constituted by misfolded phosphorylated α-syn, start to accumulate inside the cytoplasm of the DA neurons (adapted from Koprich, Kalia, and Brotchie 2017).
3. Mitochondria and PD
Mitochondria are one of the major ancient endomembrane systems in eukaryotic cells. They are fundamental for the cell survival due to their role of supporting aerobic respiration and providing energy substrates such as adenosine triphosphate (ATP) for metabolic pathways (Friedman and Nunnari 2014). Moreover, they are also crucial for various other cellular processes such as regulation of intracellular Ca2+ homeostasis (Rizzuto et al. 2012), cell signaling (Tait and Green 2012), apoptosis (C. Wang and Youle 2009) and aging (Jang et al. 2018).
Mitochondria ubiquitin-proteosome system ER-Golgi network Autophagy – Lysosomal system
15
In addition, mitochondria are unique among the cytoplasmic organelles as they contain their own deoxyribonucleic acid (DNA), which encodes for transfer ribonucleic acids (tRNAs), ribosomal ribonucleic acids (rRNAs), and some mitochondrial proteins (Cooper 2000). Mitochondria should not be studied as isolated organelles as they can build a big interconnected network inside the organisms, thus they are highly dynamic organelles and this complexity can be reflected in their complex structures.
3.1 Structure and functions
Mitochondria are ubiquitous, semi-autonomous cellular organelles constituted by complex structures characterized by two lipid membranes delimitating four compartments: the outer mitochondrial membrane (OMM), the inner mitochondrial membrane (IMM), the intermembrane space (IMS), and the matrix (Cooper 2000).
The OMM is the compartment in charge of transporting the ATP synthesized in the IMM to be released out of the mitochondria and be available for cellular functions. The membrane protein responsible for ATP transport out of mitochondria is the voltage-dependent anion channel (VDAC) (Yeagle 2016; Zeth 2010). Moreover, the OMM must allow the passage of adenine nucleotides and metabolites from the cytoplasm without allowing leakage of proteins from the IMM (Nicholls and Ferguson 2013). The OMM also contains a multi-subunit machinery which is a translocase of the outer membrane (TOM) complex responsible for the specific recognition and translocation of precursors protein from the cytosol through the IMS. TOM consists of three receptor proteins, TOM20, TOM22 and TOM70, the channel protein TOM40, and several small TOM proteins (Model et al. 2002). In order to import nuclear encoded precursor proteins into the IMM, TOM interacts with the translocase of the IMM (TIM) complexes TIM23 and TIM22. The first complex mediates import of preproteins with a positively charged matrix-targeting signal whereas the second one, mediates the import of a class of integral IMM proteins which do not carry a matrix-targeting signal (Bauer et al. 2000).
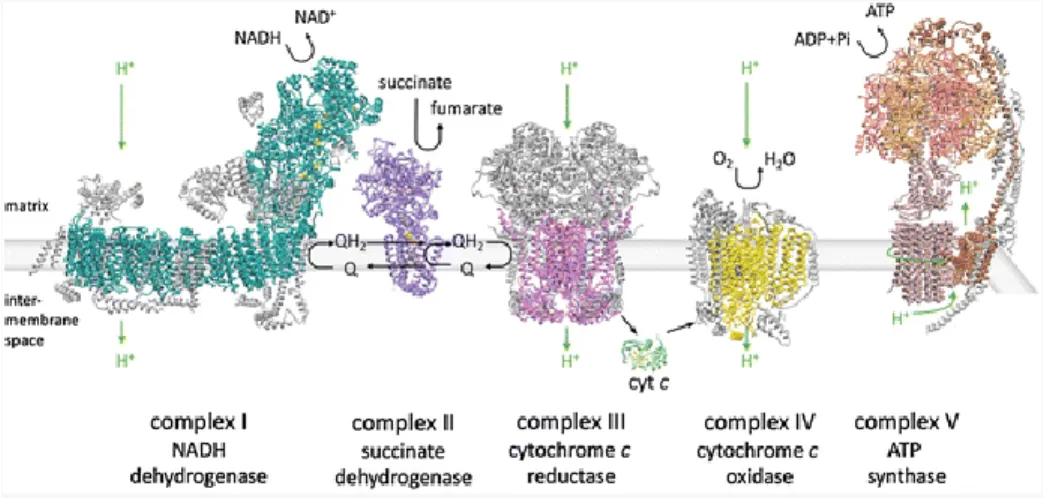
In the IMM is found the mitochondrial electron transport chain (ETC) that consists of five enzyme super complexes (Complex I, II, III, IV and V). It is characterized by invaginations called cristae, which give the representative structure to mitochondria and increase the surface area of the IMM thus increasing the efficiency of the ETC. The super complexes transfer electrons from donors like nicotinamide adenine dinucleotide (NADH) to oxygen, the ultimate electron acceptor, via several redox cofactors (Birsoy et al. 2015; Cooper 2000). First, Complex I oxidize NADH and transfers 2 electrons to ubquinone and translocates 4 protons across the membrane to Complex III. Complex II, which is part of both the citric acid cycle and the ETC, contributes additional electrons to ubiquinone that originate from succinate. Complex III transfers the electrons from ubiquinone to the peripheral electron carrier cytochrome C while translocating 4 protons across the membrane. Complex IV transfers electrons from cytochrome C to molecular oxygen and translocates 2
16
protons. In total 10 protons per NADH molecule are translocated across the IMM generating a gradient which flow back into the matrix via the ATP synthase or Complex V, actioning ATP production (Figure 9) (Kühlbrandt 2015; Sousa, D’Imprima, and Vonck 2018; Osellame, Blacker, and Duchen 2012). In neurons, since ATP cannot diffuse from the soma to distant synapses, it must be transported along the axons and dendrites by the mitochondria in order to fulfill the local demand. Throughout microtubules and actin, mitochondria are distributed to the distal parts from the cell body to provide the necessary energy and to regulate Ca2+ homeostasis during intense synaptic activity (Figure 10) (Chen and Chan 2009).
An aspect that should be considered is the production of reactive oxygen species (ROS) as consequence of the reaction between the electrons generated in the respiratory chain and oxygen. These species can interact with different biological molecules, causing protein, DNA, and RNA oxidation and lipids peroxidation. In order to balance the toxic effect of the free radicals, cells have a variety of antioxidant molecules and enzymes. Imbalance due to excess ROS or oxidants over the capability of the cell to provide an antioxidant response, the result is an accumulation of ROS within the cells, also known as oxidative stress (Ray, Huang, and Tsuji 2012; Schieber and Chandel 2014).
Figure 9. The mitochondrial ETC
The mitochondrial ETC in the IMM uses the energy of NADH and succinate to produce ATP through the different super complexes: Complex I, II, III, IV and V. Electrons enter the chain from NADH via complex I and from succinate via complex II. Complex I, III and IV pump protons across the IMM, which flow back into the matrix via the ATP synthase or Complex V (Sousa, D’Imprima, and Vonck 2018).
3.2 Mitochondrial dynamics
Balance between healthy and damaged mitochondria is crucial for maintaining proper cellular functions as they are essential for cellular energy homeostasis and cell death regulation. As mentioned before, mitochondria are highly dynamic structures and they go through continuous cycles of fusion and fission which are crucial for their proper function, maintenance and modulation of their morphology according to cell needs
17
(Figure 10) (Detmer and Chan 2007; Meyer, Leuthner, and Luz 2017). For the fusion process, which is the combination between two mitochondria to form a single organelle, there are three key GTPases needed: mitofusin 1 (MFN1) and mitofusin 2 (MFN2), which are OMM proteins, and the IMM protein, the optic atrophy 1 (OPA1) (Chen and Chan 2009). In addition, for the action of fission, which is the division of mitochondria into two or more parts, is required the dynamin-related protein 1 (DRP1) (Westermann 2010).
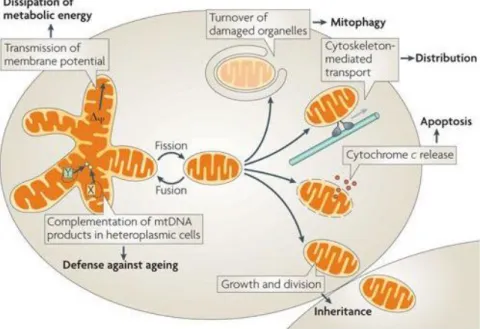
Figure 10. Mitochondrial dynamics
The mitochondrial life cycle starts with biogenesis and ends with degradation of impaired or extra organelles by mitophagy. This life cycle must be equilibrated in order to preserve cell viability. Through cycles of fusion and fission, the mitochondrial network is created depending on cell demand. On that way, they can dissipate metabolic energy through transmission of membrane potential and complement each other with mitochondrial DNA (mtDNA) gene products to prevent ageing dysfunctions. Moreover, mitochondria are intracellular distributed by cytoskeleton-mediated transport. Division is also a key mechanism required forinheritance and segregation of organelles during cell division. For quality control maintenance, mitochondria combine fusion, fission and mitophagy to promote the sequestration, sorting, and elimination of functionally impaired mitochondria. If some of the mechanisms fail, pro-apoptotic proteins are released into the cytosol such as cytochrome-c activating the apoptosis of the cell (Westermann 2010).
Another key mechanism exists for the removal of dysfunctional mitochondria through autophagy, called mitophagy (Figure 10) (Ding and Yin 2012). There are different pathways involved in mitophagy which comprise many proteins interacting between them. One of the pathways is PINK1-Parkin pathway which is a crucial amplifying mechanism that regulates mitochondrial quality control and promotes selective mitophagy (Pickrell and Youle 2015). Parkin is a cytosolic E3 ubiquitin ligase which plays a critical role in ubiquitination and it interacts with PINK1, which is a protein kinase with a mitochondrial targeting domain (Seirafi, Kozlov, and Gehring 2015; Truban et al. 2017). Dysfunctional mitochondria fail to import and degrade PINK1 by the TOM and TIM complexes and it accumulates on the OMM leading the recruitment of Parkin. Once recruited
18
to dysfunctional mitochondria, Parkin-dependent ubiquitination and proteasomal degradation of mitochondrial outer membrane proteins ultimately leads to mitophagy, involving the rupture of the outer mitochondrial membrane (Pickrell and Youle 2015; Narendra et al. 2010; Rakovic et al. 2010).
3.3 Mitochondrial dysfunction in PD
As a multifactorial disease, PD is caused by combination of genetic and environmental factors. However, among these factors, several observations indicate that mitochondrial dysfunction plays an important role in the pathophysiology of PD and the degeneration of DA neurons. Therefore, a correct mitochondrial function and a good mitochondrial quality control system are crucial for the maintenance and viability of the cells (Park, Davis, and Sue 2018). However, whether mitochondrial defects cause PD or occur because of the disease per se remains to be elucidated.
Defective mitochondria are related to some of the mutations linked to PD affecting genes that encode proteins with specific functions in mitochondrial dynamics such as fusion-fission, transport and mitophagy. Loss of function or mutations in PINK1, Parkin, DJ-1, LRRK2 and vacuolar protein sorting-associated protein 35 (VPS35) underlie the importance of mitochondrial dysfunction as a primary cause of DA cell death in PD (Pickrell and Youle 2015; B. J. Ryan et al. 2015; Shen 2010; Truban et al. 2017; J.-Y. Lee et al. 2010; Valente et al. 2004; Vilariño-Güell et al. 2011). The involvement of mitochondrial defects in PD pathology is also supported by evidence of reduced activity of ETC in the DA cells in the SNc by impairment of the Complex I caused by Complex I inhibitors such as rotenone, 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) and other pesticides (Schapira et al. 1990; Franco et al. 2010). The fact is that ETC activity is important because it impacts a variety of processes beyond energy balance such as ROS production (Sousa, D’Imprima, and Vonck 2018). Mitochondria are the main source of ROS and high orders of ROS production can be dangerous for cell viability and oxidative stress increases the chance of spontaneous mutations. In addition, DA cells in the midbrain are more susceptible to oxidative stress than other neurons present in other regions of the basal ganglia (Puspita, Chung, and Shim 2017). In addition, different studies have linked α-syn with mitochondrial dynamics and related it as one of the causes of mitochondrial impairment. α-syn has a mitochondrial targeting sequence (MTS) at its N-terminus giving the ability of being transported into the mitochondria. The MTS is recognized by TOM receptors, which are located on the OMM, which translocate the proteins through the TOM complex to TIM and finally into the matrix. Some toxic α-syn species like oligomers or phosphorylated forms can bind to the TOM20 receptor and prevent its interaction with its co-receptor TOM22, and inhibit mitochondrial protein import (Maio et al. 2016; Rocha, De Miranda, and Sanders 2018). Moreover, this toxic α-syn species impaired proper mitochondrial fusion-fission dynamics leading to mitochondrial fragmentation (Kamp et al. 2010). Clearance of dysfunctional mitochondria by mitophagy may also be affected by α-syn. Some studies shown that mutated α-syn interacting and accumulating in mitochondria is associated
19
with Complex I inhibition and decreased substrate-specific respiration along with increased lysosome mediated mitophagy (Chinta et al. 2010). It has to be well elucidated to determine the physiological role of α-syn in mitochondria as well as the pathological one.
4. The SPFH protein family: structure,
localization, and function.
The SPFH superfamily of proteins is a very diverse family of prokaryotic and eukaryotic membrane proteins that carry an evolutionarily conserved domain called the SPFH domain named after the proteins stomatin, prohibitin, flotillin, HflC/K (Hinderhofer et al. 2009). This family are scaffolds which assemble into ring-like structures and are anchored to different cellular membranes, including the plasma membrane, the membrane of early endosomes, the Golgi apparatus, mitochondria and ER (Zanon, Hicks, et al. 2017). However, even though all proteins containing a SPFH domain are connected to membranes through their N-terminal hydrophobic regions, they show some differences in their membrane topologies. Thus, their distribution reflects the key role of the SPFH domain in cellular membrane processes (Browman, Hoegg, and Robbins 2007; Kuwahara et al. 2009; Lapatsina et al. 2012). Their main functions are the mediation of lipid raft association, they are also involved in oligomerization and subcellular targeting, and in the organization of lipid raft microdomain formation at the ER or mitochondria (Zanon, Hicks, et al. 2017). These proteins show also other important functions such as ion channel regulation, membrane protein chaperoning, vesicle and protein trafficking, membrane-cytoskeletal association, formation of microdomains and also, they constitute specialized membrane structures (Langhorst, Reuter, and Stuermer 2005; Browman, Hoegg, and Robbins 2007).
4.1 Stomatin-like protein-2 (SLP-2)
The human stomatin protein family consists of five members: Stomatin, stomatin-like protein-1 (SLP-1), stomatin-like protein-2 (SLP-2), stomatin-like protein-3 (SLP-3) and podocin (Lapatsina et al. 2012). SLP-2 is a 357 amino acid evolutionarily conserved mitochondrial protein encoded by the STOML2 gene (Y. Wang and Morrow 2000; Green and Young 2008). It belongs to the SPFH family as they share a central domain that may mediate interactions with cell membranes. Moreover, SLP-2 has multiple phosphorylation sites indicating a possible role in cell signaling cascades (Kirchhof et al. 2008; Owczarek et al. 2001). It also presents a mitochondrial targeting sequence at the N-terminus indicating mitochondrial localization where is found predominantly in the IMM (Da Cruz et al. 2008; Hájek, Chomyn, and Attardi 2007). Moreover, SLP-2 interacts and forms a complex with MFN2 which is an OMM GTPase critical for mitochondrial fusion, mitochondrial dynamics and quality control. It also serves as a Parkin ubiquitination substrate (Filadi, Pendin, and Pizzo
20
2018; Hájek, Chomyn, and Attardi 2007). SLP-2 also interacts with the negatively charged phospholipid cardiolipin present in the IMM. The interaction of SLP-2 with cardiolipin may also help to recruit prohibitins-1 and 2 (PHB-1 and PHB-2) to form cardiolipin and PHB microdomains which are required for mitochondrial membrane formation and facilitate the assembly of the respiratory chain complexes (Da Cruz et al. 2008; D. A. Christie et al. 2011; Hájek, Chomyn, and Attardi 2007). Upon mitochondrial depolarization, rupture of the outer membrane or stress, cardiolipin is translocated to the OMM in order to provide an anchor for LC3 and initiate mitophagy (T. Ryan et al. 2018; D. A. Christie et al. 2011; Wei et al. 2017). Moreover, cardiolipin can also interact with mutant α-syn and facilitate the folding of its oligomers (T. Ryan et al. 2018). Overexpression (OE) of SLP-2 is associated with increased levels of cardiolipin which results into increased mitochondrial membrane formation and biogenesis (Zanon, Hicks, et al. 2017). In addition, SLP-2 forms part of a large protease complex composed of the presenilins-associated rhomboid-like protein (PARL) and the i‐ AAA protease YME1L (required for cardiolipin synthesis), termed the SPY complex (SLP2–PARL–YME1L) (Wai et al. 2016). The SPY complex facilitates PINK1 cleavage by PARL which regulates the activity of Complex I and mitophagy (Morais et al. 2014; Narendra et al. 2010; Jin et al. 2010). Moreover, SLP2 restricts OMA1 which is a stress‐activated peptidase ensuring protein quality control, mitochondrial bioenergetic function and controls cellular apoptotic resistance. The limitation of OMA1‐mediated processing of the dynamin‐like GTPase OPA1 allows mitochondrial hyperfusion under stress conditions (Wai et al. 2016). Overall, SLP-2 seems to have an important role as a membrane scaffold for the spatial organization of IMM proteases regulating mitochondrial dynamics, quality control and cell survival.
In vitro knockdown of SLP-2 leads to a decrease in mitochondrial membrane potential and a reduced Complex
I and IV activity including ATP production. Mitochondrial network appears to be fragmented and levels of PHB-1 and PHB-2 are also decreased (Zanon, Kalvakuri, et al. 20PHB-17; Da Cruz et al. 2008).
21
PROBLEMATIC, HYPOTHESIS AND OBJECTIVES
After over 200 years of research, we are still without any treatment to cure PD. Current treatments neither cure PD nor stop it from advancing, their purpose is to mitigate the symptoms and try to ameliorate patient’s daily life. PD the second most common age-related neurodegenerative disorder (Abbas, Xu, and Tan 2018) we need to find a treatment that slows down, blocks and/or prevents neurodegeneration of DA neurons. Discovery of SLP-2, a protein located in the inner mitochondrial membrane and acting as a membrane scaffold regulating mitochondrial functions, integrity and bioenergetics, (Zanon, Kalvakuri, et al. 2017; D. A. Christie et al. 2011) provides the rationale for testing the neuroprotective potential of SLP-2 for mitochondrial dysfunction induced by α-syn, as the exact link between α-syn and neuronal degeneration due to mitochondrial disfunction is unknown yet. Thus, we hypothesize that SLP-2 and its binding partners could be part of an interacting network for α-syn affected in α-synucleinopathies and SLP-2 OE will protect DA neurons against A53T-α-synuclein toxicity and locomotion impairment, characteristic of parkinsonian phenotypes. In order to verify our hypothesis, which is to find a neuroprotective target gene against neurodegeneration in PD by assessing in a preclinical model of PD the role and neuroprotective potential factor of SLP-2.
22
Chapter 1: METHODOLOGY
Animals
The mice in this study, were used in accordance with the Canadian Guide for the Care and Use of Laboratory Animals and were approved by the Université Laval Animal Protection Committee. They were maintained in the animal facility with a controlled temperature (22 ± 2°C) on a 12 h light–dark cycle with access to food and water ad libidum. Between 60-100 days old C57BL/6 male and female mice from Charles River (Laval, Quebec, Canada) were used for this study.
Tissue preparation and immunohistofluorescence
Animals were deeply anesthetized with ketamine-xylazine injection (10 mg/mL) and transcardially perfused with phosphate-buffered saline (PBS 1X, pH 7.4) followed by 4% paraformaldehyde (PFA) in PBS 1X, pH 7.4. Brains were removed, immersion-fixed in the same fixative overnight (O/N) at 4°C and subsequently cryoprotected in 30% sucrose-PBS 1X for 24 hours. Tissue samples were embedded in OCT compound (Tissue-Tek) and cryosectioned into 60 μm-thick coronal sections. Immunohistofluorescence was performed on free-floating brain sections. (Bachman 2013) The slices were blocked for nonspecific antibody binding with PBS 1X-NDS1%-Triton 0.2% for 30 min to 1 hour at room temperature, and subsequently incubated O/N at 4°C with primary antibodies at optimized concentrations. The slices were then washed in PBS 1X and placed in a solution containing the secondary antibodies for 40 min to 1 hour at room temperature.
The following primary antibodies were used: rabbit anti-TH (1/1000; Pel-Freez Biologicals, P40101), sheep anti-TH (1/1000; Pel-Freez Biologicals, P60101), mouse anti–phospho-Ser129 (1/2000; Wako Chemicals, 015-25191), mouse anti-NeuN (1/500; Millipore, MAB377), rabbit anti-STOML2 (SLP-2) (1/500; Abcam, ab191884). Appropriate secondary antibodies were used: Dylight 405 anti-rabbit (1/200; Jackson ImmunoResearch, 711-475-152), Dylight 405 anti-sheep (1/200; Jackson ImmunoResearch 713-475-147), Alexa Fluor 488 anti-mouse (1/400; Life Technologies, A21202), Alexa Fluor 488 anti-rabbit (1/400; Life Technologies, A21206), Alexa Fluor 488 sheep (1/400; Life Technologies, A11015), Alexa Fluor 488 anti-sheep (1/400; Jackson ImmunoResearch, 713-545-147), Alexa Fluor 647 anti-anti-sheep (1/400 Life Technologies, A-21448).
For human brains, the sections were provided and prepared by Martin Parent laboratory (CERVO, Quebec, Canada) for us as a collaboration. The slices were blocked for nonspecific antibody binding with PBS 1X-NDS2%-Triton 0.5% for 1 hour at room temperature, and subsequently incubated O/N at 4ºC with primary antibodies at optimized concentrations and agitation. The slices were then washed in PBS 1X and placed in a solution containing the secondary antibodies for 2 hours at room temperature. After immunofluorescence
23
histochemistry, place the sections in PBS 1X for 5 min and then immerse them in 70% ethanol for 5 min. After, immerse the sections in the Autofluorescence Eliminator Reagent (Millipore, #2160) for 5 min and when time passes, immerse them in three changes of 70% ethanol for 1 min. each time.
The following primary antibodies were used: sheep anti-TH (1/250; Pel-Freez Biologicals, P60101), rabbit anti-STOML2 (SLP-2) (1/500; Abcam, ab191884), mouse anti–phospho-Ser129 (1/2000; Wako Chemicals, 015-25191). Appropriate secondary antibodies were used: Alexa Fluor 647 anti-sheep (1/200 Life Technologies, A-21448), Alexa Fluor 488 rabbit (1/200; Life Technologies, A21206), Alexa Fluor 488 anti-mouse (1/200; Life Technologies, A21202).
All images were then acquired using a LSM710 PASCAL (Zeiss) confocal microscope, a TISSUEscope 4000 (Huron Technologies) and processed using Image J software. The same software was used to measure the corrected total cell fluorescence (CTCF) by using the following calculation:
CTCF = Integrated Density – (Area of selected cell * Mean fluorescence of background readings)
Stereotaxic injections
For stereotaxic injections mice were anesthetized with 4% isoflurane and they received presurgical care with subcutaneous injections of Carprofen (2 mg/ml) and Bupivacaine-Lidocaine (0,4 mg/ml – 0,8 mg/ml). Then, skulls were immobilized in a stereotaxic apparatus and prepared for virus injection with a capillary. The volume of injected virus was 1μL and they were injected unilaterally into the SNc (anteroposterior: − 3.5 mm from bregma; mediolateral: 1.1 mm; dorsoventral: 4.0 mm). The viruses were injected at a rate of 0.5-nL/sec using an automatic microinjector. The capillary containing the virus was left in place for 3 min before proceeding to virus delivery. Once virus was injected, capillary was also left in place for 5 min before proceeding to capillary slow removal in order to allow virus diffusion and prevent virus reflux. The mice were then sutured and provided with postsurgical care with subcutaneous injections of saline (0.9% NaCl) and Carprofen (2mg/ml) 48 hours post-surgery. Virus were custom ordered from Molecular Tools Platform (CERVO, Quebec, Canada) and used in the study. AAV expressing A53T- α-syn mutation is driven by a cytomegalovirus immediate-early (CMVie) and human synapsin 1 (hSyn) gene promoters and the viral titer used was 7.5E12 genome copy (GC)/mL (AAV2/9-CMVie-hSyn-synA53T). The AAV expressing m-kate fluorescent protein is driven by hSyn gene promoter and the viral titer used was 3E13 GC/mL (AAV2/9-hSyn-mKate). The AAV expressing SLP-2 is driven by hSyn gene promoter and it also express mCherry fluorescence protein depending on internal ribosome entry site (IRES) sequence; the viral titer used was 4.16E11 GC/mL