N-HALOGÉNOVINYLATION INTRAMOLÉCULAIRE D'AMIDES ET DE CARBAMATES CATALYSÉE PAR LE CUIVRE : SYNTHÈSE DE
P-HALOGÉNOÉNAMIDES HÉTÉROCYCLIQUES
MÉMOIRE PRÉSENTÉ
COMME EXIGENCE PARTIELLE
DE LA MAÎTRISE EN CHIMIE EXTENSIONNÉE DE L'UNIVERSITÉ DU QUÉBEC À MONTRÉAL
PAR
NICOLAS GILBERT
UNIVERSITÉ DU QUÉBEC À MONTRÉAL Service des bibliothèques
Avertissement
La diffusion de ce mémoire se fait dans le respect des droits de son auteur, qui a signé le formulaire Autorisation de reproduire et de diffuser un travail de recherche de cycles supérieurs (SDU-522 - Rév.1 0-2015). Cette autorisation stipule que «conformément
à
l'article 11 du Règlement no 8 des études de cycles supérieurs, [l'auteur] concèdeà
l'Université du Québecà
Montréal une licence non exclusive d'utilisation et de publication de la totalité ou d'une partie importante de [son] travail de recherche pour des fins pédagogiques et non commerciales. Plus précisément, [l'auteur] autorise l'Université du Québec à Montréalà
reproduire, diffuser, prêter, distribuer ou vendre des copies de [son] travail de rechercheà
des fins non commerciales sur quelque support que ce soit, y compris l'Internet. Cette licence et cette autorisation n'entraînent pas une renonciation de [la] part [de l'auteur]à
[ses] droits moraux nià
[ses] droits de propriété intellectuelle. Sauf entente contraire, [l'auteur] conserve la liberté de diffuser et de commercialiser ou non ce travail dont [il] possède un exemplaire.»F
Je tiens, en premier lieu, à remercier mon directeur de recherche le professeur Benoit Daoust. Au-delà d'un simple mentor, je le considère comme un exemple du niveau de connaissances et de tigueur que j'espère atteindre un jour.
Bien évidemment, ces deux années m'auraient semblé nettement plus longues sans personne avec qui partager le laboratoire. Je me dois donc d'offrir mes remerciements à Pam, Frank et Caro qui furent, pour moi, bien plus que des collègues de travail. À Pierre, qui a su porter avec moi une partie du fardeau que représentait un tel projet et sans qui le chapitre VI de ce travail n'existerait pas. Enfin, à Simon: nos délires au labo ont certainement évité à ma santé d'esprit d'y passer complètement. Merci aussi à Sabrina, et encore plus à Vincent, qui ont dû cohabiter avec 1 'humeur difficile de quelqu'un qui passe beaucoup trop d'heures dans un laboratoire.
Je dois aussi remercier ma famille, particulièrement mes parents, ma sœur, Marraine : vous rn' encouragez, sans nécessairement le savoir, à poursuivre des ambitions qui semblent a priori hors d'atteinte. Sachez qu'à travers cette nouvelle étape qui commence, même à 1 'autre bout de la terre, je continue mon histoire avec vous.
Je remercte aussi le Conseil de recherches en sciences naturelles et en génie du Canada (CRSNG) et le Fonds de recherche du Québec- Nature et technologie (FRQNT) qui ont rendu tout ça possible.
Enfin, à tous les autres qui ont marqué cette étape de ma vie, et qu'il me serait impossible de faire tenir sur 1 'unique page de remerciements que je rn 'étais fixée : merci.
TABLE DES MATIÈRES
INTRODUCTION ... 1
CHAPITRE I LA FONCTION B-HALOGÉNOÉNAMIDE ... 3
1.1 Les éna mi des ... 4
1.1.1 Intérêt et réactivité ... 4
1.1.2 Méthodes de synthèse ... 14 · 1.2 Les P-halogénoénamides ... 20
1.2.1 Intérêt et réactivité ... 20
1.2.2 Méthodes de synthèse ... 26
1.3 Objectif du projet ... 31
CHAPITRE II LA RÉACTION DE COUPLAGE CATALYSÉE PAR LE CUIVRE ... 35
2.1 Généralités et historique des couplages croisés ... 35
2.2 La fmmation de liens C- ... 37
2.3 Le palladium versus le cuivre ... 39
2.4 Le mécanisme du couplage au cuivre entre un amide et un halogénure insaturé ... 43
2.5 Le rôle du Iigand ... 45
CHAPITRE III LE COUPLAGE INTRAMOLÉCULAIRE D'HALOGÉNURES VINYLIQUES CATALYSÉ PAR LE CU1VRE ... 49
3.1 Généralités des réactions de cyclisation ... .49
3.2 Le couplage de mono halogénures vinyliques ... 52
3.3 Le couplage de dihalogénures vinyliques ... 58
3.3.1 Cyclisations 4-e.xo versus 5-endo ...... 58
3.3.2 Cyclisations 5-e.xo versus 6-endo ···---····---··--···--·--61
3.3.3 Cyclisations 6-exo versus 7-endo ... 62
CHAPITRE IV
PRÉPARATION DES DIHALOGÉNURES VINYLIQUES MODÈLES ... 65
4.1 Synthèse des modèles d'amides ... 65
4.1. J Synthèse de pent-4-ynamides et hex-5-ynamides ... 66
4.1.2 Synthèse des amides di iodés ... 68
4.1.3 Synthèse des amides dibromés ... 78
4.1.4 Synthèse et diiodation de but-3-ynamides ... 87
4.2 Synthèse des modèles de carbamates ... 88
4.2.1 Synthèse des 0-prop-2-ynyl et 0-but-3-ynylcarbamates ... 89
4.2.2 Synthèse des carbamates di iodés ... 90
4.2.3 Synthèse des carbamates et des alcools dibromés ... 92
4.2.4 Synthèse du N-phényl-0-éthynylcarbamate ... 93
4.3 Synthèse des modèles d'acétamides et de benzoylamides ... 97
CHAPITRE V COUPLAGE C-N INTRAMOLÉCULAIRE DES DIHALOGÉNURES VINYLIQUES MODÈLES ... 104
5.1 Cyclisatioos 5-exo vs 6-endo ....... 105
5 .1.1 Couplage des amides di iodés ... 105
5.1 .2 Couplage des amides dibromés ... l10 5.1.3 Couplage des carbamates diiodés ... 117
5 .1.4 Couplage des carbamates dibromés ... 121
5.2 Cyclisations 6-exo vs 7-endo ...... 125
5.2. J Couplage des amides di.iodés ... 125
5.2.2 Couplage des amides dibromés ... 127
5.2.3 Couplage des carbamates di iodés ... 131
5.2.4 Couplage des carbamates dibromés ... 132
5.3 Cyclisations 4-exo vs 5-endo ...... 133
5.4 Tests de bullage à l'azote ... l34 CHAPITRE VI OPTIMISATION DU COUPLAGE C-N INTRAMOLÉCULAIRE ... 137
6.1 Optimisation du ligand ... 138
6.3 Optimisation de la source de cuivre ... 142
6.4 Optimisation du solvant ... 144
6.5 Optinrisation de la concentration ... 145 6.6 Optimisation du ratio cuivre/substrat.. ... l46 6. 7 Optimisation du ratio base/substrat ... 14 7 6.8 Optimisation de la température ... l48 6.9 Étendue de la méthode ... 150
CHAPITRE VII FONCTIONNALISATION DE LA (E)-4 -(IODOMÉTHYLÈNE)-3-PHÉNYLOXAZOLIDIN-2-0NE ... 153
7.1 Couplages catalysés par le palladium ... 154
7.2 Couplages catalysés par le cuivre ... 157
CONCLUSION ... 160
ANNEXE A PARTIE EXPÉRIMENTALE ... 162
A.J Remarques générales ... 162
A.2 Modes opératoires ... 163
ANNEXEE SPECTRES RMN 1H ET 13C ........................................................................ 210
ANNEXEC MESURE DES RENDEMENTS PARRMN QUANTITATIVE ... 319
Figure Page
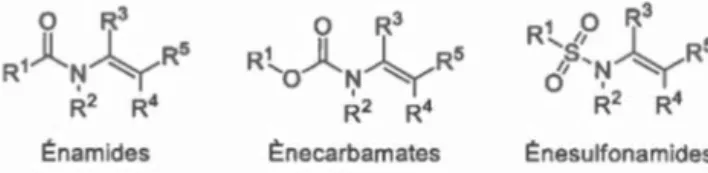
1.1 Structure générale des énamides, ènecarbamates et ènesulfonamides ... 3
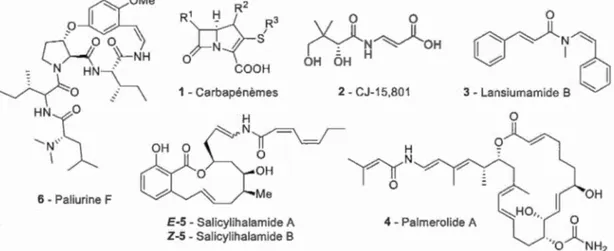
1.2 Structure chimique de produits naturels contenant des énamides ... 5
1.3 Structure générale et sites réactifs des énamides ... 6
1.4 Structure générale d'hétérocycles synthétisés à partir d' énamides ... 8
1.5 Intermédiaire cyclique à 6 membres impliqué dans l'activation C-H d'énamides ... 13
1.6 Structure générale d'un ~-halogénoénamide ... 20
3.1 Angle de Bürgi-Dunitz ... 49
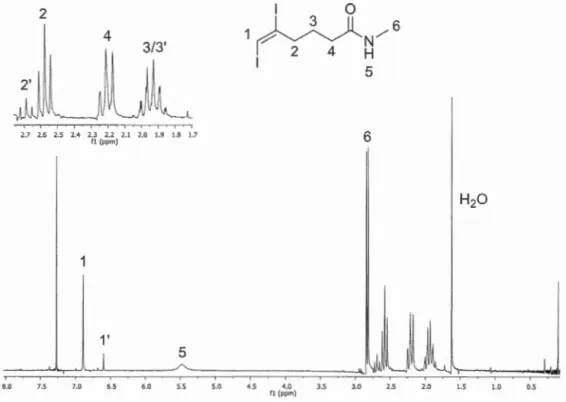
4.1 Analyse du spectre RMN 1H de 1 'amide 266b, contaminé par un bromoiodure ... 73
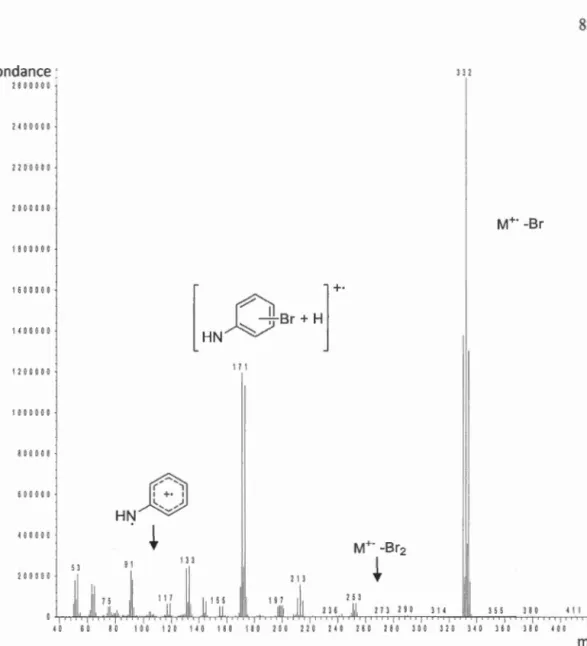
4.2 Analyse du spectre de masse du sous-produit tribromé 275 ...... 82
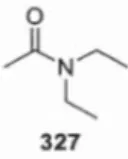
4.3 Structure chimique du sous-produit d'acétylation 327 . ...... 100
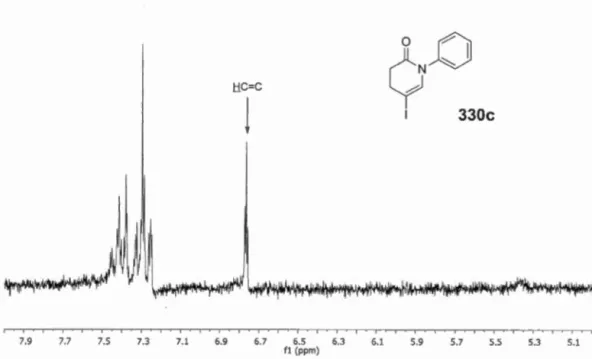
5.1 Spectre RMN 1H du produit cyclisé 6-endo 330c ... 109
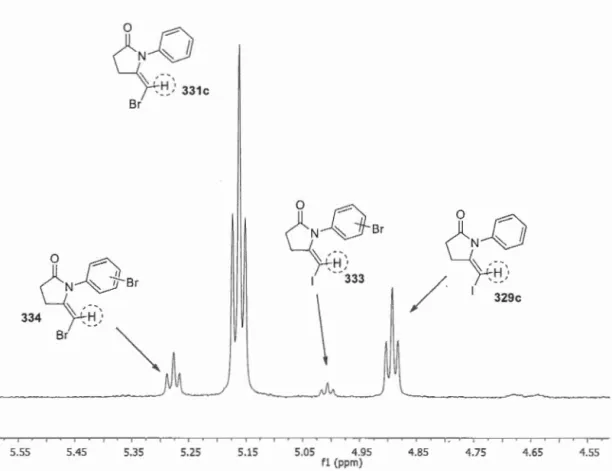
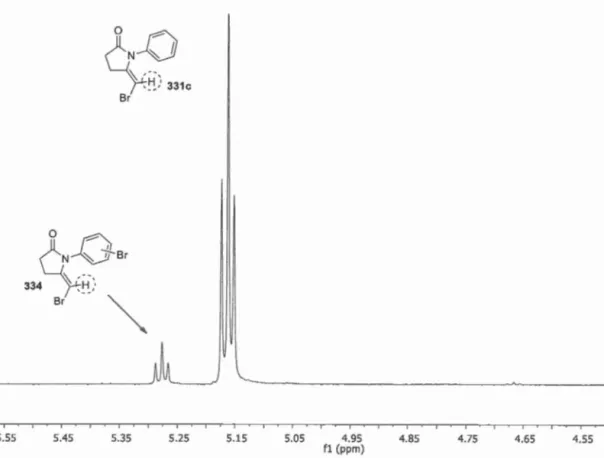
5.2 Analyse du spectre RMN 1H du produit brut obtenu lors de la synthèse de 331c en présence de Cul. ... 113
5.3 Analyse du spectre RMN 1H du produit brut obtenu lors de la synthèse de 331c en présence de CuBr ... 115
5.4 Structure chimique du sous-produit d'hydrogénolyse 337 ... 0000119
5.5 Chromatogramme par CPG-SM de la réaction menant à E-338. ···o···o····•ooooo 123 5.6 Chromatogramme par CPG-SM de la réaction menant à Z-338. ooooooooooooooooooooooooooo. 124 601 Résultats de l'optimisation du ligand dans le couplage de 274c. OOOOOOOOoooooooooooooooooooo 139
6.2 Résultats de l'optimisation de la base dans le couplage de 274c .. ooooooooooooooo ... oo.oooooo 141
6.3 Résultats de l'optimisation de la source de cuivre dans le couplage de 274c .. oooooooo 142
6.4 Résultats de l'optimisation du solvant dans le couplage de 274c. 000000000000000000000000000 144
605 Résultats de l'optimisation de la concentration dans le couplage de 274co oo.oo ... oooo. 145
606 Résultats de l'optimisation du ratio cuivre/substrat dans le couplage de 274coooooooo 146
6.7 Résultats de l'optimisation du ratio base/substrat dans le couplage de 274c à 50% de cuivre .. 0 0 0. 0 .... 0 0 0 0 00.0. 0 0 0 0. 0 00 0. 0 0 0 0 0 0 00 0 0 00 ... 00 .... 00 ... 00.00 0. 0. 0 0 0 0 00 ... 0 0 0. 0 0 0 0 14 7
6.8 Résultats de l'optimisation du ratio base/substrat dans le couplage de 274c à 30% de cuivre ... 0 .... 0. 0 ... 0 ... 0 ... 148
LISTE DES TABLEAUX
Tableau Page
3.1 Résumé des règles de Baldwin pour les cyclisations trigonales ... 50
4.1 Rendement pour la synthèse des amides 261a-b et 262a-b ... 66
4.2 Rendement pour la synthèse des amides 261c et 262c ... 67 4.3 Rendement pour la synthèse des diiodures 265a-c en présence de bromure de
magnésium éthérate ... 69
4.4 Rendement pour la synthèse des diiodures 266a-c et 265b sans acide de Lewis ... 70
4.5 Rendement pour la synthèse des diiodures 266b-c en présence de bromure de magnésium éthérate ... 71 4.6 Rendement pour la synthèse des di iodures 265b-c en présence de divers acides de
Lewis ... 74
4.7 Résultat de la synthèse du diiodure 265c avec divers protocoles de génération in situ d'iodure de magnésium éthérate ... 75
4.8 Rendement pour la synthèse des diiodures 265c et 266c en présence d'iodure de magnésium éthérate généré in situ ......................................................... 77
4.9 Rendement pour la synthèse des diiodures 265b-c et 266b en présence d'iodure de magnésium éthérate isolé ... 78
4.10 Rendement pour les premiers essais de synthèse des di bromures 273a, 273c et 274a ... 79
4.12 Résultat de la synthèse du dibromure 273c en variant les conditions de bromation
··· 83
4.13 Ratio des produits 273c, 275 et 276 avec divers acides de Lewis ... 84
4.14 Résultat de la synthèse du dibromure 273c avec diverses conditions réactionnelles ... 85
4.15 Rendement pour la synthèse des di bromures 277 et 278 ... 86
4.16 Rendement pour la synthèse des amides 273c et 274c ... 86
4.17 Rendement pour la synthèse des di iodures 296 et 297 ... 90
4.18 Rendement pour la synthèse des alcools dibromés 301 et 302.. ... 92
4.19 Rendement pour la synthèse des di bromures E-303, Z-303 et 304 ... 93
4.20 Résultat de la synthèse de l'acétylamide 322a en passant par l'azoture 325 ... 99
4.21 Rendement pour la synthèse des di iodures 323a-b ... 103
5.1 Résultat du couplage C-N intramoléculaire des amides diiodés 265a-c ... 106
5.2 Résultat du couplage C-N intramoléculaire des amides diiodés 265b-c ... 108
5.3 Résultat du couplage C-N intramoléculaire des amides dibromés 273a-c en présence de Cul ... 111
5.4 Résultat du couplage C-N intramoléculaire des amides dibromés 273b-c ... 114
5.5 Résultat du couplage C-N intramoléculaire de l'amide dibromé 273b dans différentes conditions réacti01melles ... 116 5.6 Résultat du couplage C-N intramoléculaire du carbamate diiodé 296 ... 117
5.8 Résultat du couplage C-N intramoléculaire des carbamates dibromés E-303 et
Z-303 ··· 121
5.9 Résultat du couplage C-N intramoléculaire des amides diiodés 266b-c ... 126
5.10 Résultat du couplage C-N intramoléculaire des amides dibromés 274b-c en présence d'iodme de cuivre ... 127
5.11 Résultat du couplage C-N intramoléculaire des amides dibromés 274b-c en présence de bromure de cuivre ... 128
5.12 Résultat du couplage C-N intramoléculaire du carbamate diiodé 297 ... 131
5.13 Résultat du couplage C-N intramoléculaire du carbamate dibromé 304 ... 132
5.14 Résultat du couplage C-N intramoléculaire des amides diiodés 323a-b ... 133
5.15 Résultat des tests de bullage à l'azote ... 135
6.1 Conditions optimales pour le couplage C-N intramoléculaire de 274c ... 150
6.2 Résultat du couplage sur les modèles 274b, 274c, 304, 273c, 303 et 296 dans les conditions optimisées ... 151
7.1 Rendement du couplage de Stille sur le P-iodoènecarbamate 335 ... 154
7.2 Rendement du couplage de Heck sur le P-iodoènecarbamate 335 ... 155
7.3 Rendement du couplage de Sonogashira sur le P-iodoènecarbamate 335 ... 156
7.4 Rendement du couplage de Suzuki sur le P-iodoènecarbamate 335 ... 157
Schéma Page
1.1 Hydrogénation catalytique énantiosélective d'un acide déhydroaminé ... 7
1.2 Substitution élech·ophile d'un énamide sur un indole ... 7
1.3 Substitution nucléophile d'un énamide sur un N-acyliminoester et réactions subséquentes ... 8
1.4 Couplage de Heck intramoléculaire de l'énamide 27 ... 9
1.5 Couplage de Heck intramoléculaire énantiosélectif et migration ... 10
1.6 Cyclisation d'un alcyne sur un énamide, catalysée par l'argent. ... 10
1. 7 Méta thèse intramoléculaire d'un énamide, catalysée par Je ruthénium ... 10
1.8 Couplages de Heck intermoléculaires sur des énamides ... 11
1.9 Fonctionnalisations C-H d'énamides ... 12
1.10 Vinylation de l'ènesulfonamide 51 par fonctionnalisation C-H ... 13
1.11 Condensation entre un amide et un aldéhyde ... 14
1.12 Condensation enh·e un carbamate primaire et un aldéhyde en présence d'un sulfinate ... 15
1.13 Attaque d'un réactif de Grignard sur un nitrile et acylation de 1 'iminomagnésien résultant. ... 15
1.15 Hydroamidation E-et Z-sélective d'alcynes ... 16
1.16 Fmmation d'un azoture d'acyle a,p-insaturé et réarrangement de Curtius ... 17
1.17 Synthèse d'un acide déhydroaminé par élimination ... 17
1.18 Synthèse d'un énamide par oléfination de Wittig ... 17
1.19 Synthèse d'un énamide par oléfination de Horner-Wadsworth-Emmons ... 18
1.20 Synthèse d'un éna mi de par oléfination de Peterson ... 18
1.21 Analyse rétrosynthétique de la préparation d'énamides par couplage C-N ... 19
1.22 Synthèse d'énamides par couplages C-N catalysés par le palladium et le cuivre ... 19
1.23 Synthèse d'un pyrrole à partir d'un P-halogénoénamide ... 21
1.24 Synthèse d'un imidazole à partir d'un P-halogénoénamide ... 21
1.25 Synthèse d'un oxazole à pmtir d'un P-ha1ogénoénamide ... 21
1.26 Fonctionnalisation d'un P-halogénoénamide par couplage de Heck, Sonogashira et Suzuki ... 22
1.27 Fonctionnalisation d'un P-halogénoénamide par couplages de Stillet et Nozaki -Hiyama-Kishi ... 22
1.28 Fonctionnalisation d'un P-halogénoénamide par couplage avec des composés hétéroatomiques ... 23
1.29 Séquence couplage C-0 1 réarrangement de Claisen pour la synthèse d'aldéhydes a-azotés ... 24
1.30 Fonctionnalisation d'un P-iodoènecarbamate acyclique ... 25
1.31 Bromation stéréosélective d'un acide déhydroaminé ... 26
1.33 Iodation d'un ènecarbamate ... 27
1.34 Iodation et acétylation d'oximes ... 27
1.35 Iodocyclisation entre un carbamate et un alcyne ... 28
1.36 ~,~-Dibromation et réduction énantiosélective de formamides ... 28
1.3 7 Réduction énantiosélective de ~,~-dihalogénoénamides ... 29
1.38 Schéma général du couplage au cuivre entre un amide et un dihalogénure vinylique ... 29
1.39 Couplage au cuivre entre un amide et un dibromme vinylique, et formation d'un oxazole in situ ... 30
1.40 Couplage au cuivre entre des amides et le diiodoéthène ... 30
1.41 Couplage au cuivre entre des carbamates et des diiodures vinyliques ... 31
1.42 Schéma général de la N-halogénovinylation intramoléculaire d'amides et de carbamates catalysée par le cuivre ... 32
1.43 Synthèse d'énamides insatmés portant w1 carbonyle en position exocyclique ... 33
2.1 Équation générale d'une réaction de couplage croisé ... 35
2.2 Ce1tains couplages au cuivre historiques ... 36
2.3 La réaction de Buchwald-Hartwig ... 37
2.4 La réaction de Chan-Lam ... 37
2.5 Première vinylation d'amides, catalysée par le CuTC ... 38
2.6 L'arylation et la vinylation d'amines et d'amides, catalysées par un système cuivre/ligand N,N ... 39
2.8 La sélectivité du palladiwn et du cuivre dans la vinylation intramoléculaire d'alcools ... 41
2.9 Une modulation de la chimiosélectivité d 'w1e arylation catalysée par le cuivre ... 41
2.10 L'arylation sélective du trans-diaminocyclohexane catalysée par le cuivre ... .42
2.11 Une application du cuivre dans la synthèse de duocarmycines ... 43
2.12 Mécanisme proposé de la réaction entre un amide et un bromure aromatique, catalysée par une source de cuivre I. ... 44
2.13 Équilibres impliqués dans la complexation du cuivre par le ligand dans la réaction d 'Ullmann ... 46
2.14 Un exemple de 1 'influence du ligand sur la régiosélectivité du couplage C-N ... 4 7
2.15 Une modulation de la chimiosélectivité, induite par le ligand, de 1 'arylation catalysée par le cuivre ... 47
3.1 Étapes de la vinylation intramoléculaire d'amides menant au produit 5-endo ..... 51
3.2 Étapes de la ~-halogénovinylation intramoléculaire d'amides menant aux produits
5-endo et 4-exo . ...... 52
3.3 Vinylation intramoléculaire d'amides catalysée par le cuivre ... 53
3.4 Vinylation intramoléculaire d'un amide primaire et migration ... 54
3.5 Vinylation inh·amoléculaire d'amides menant au produit 5-endo .... 54
3.6 Vinylation intramoléculaire de sulfonamides menant au produit 4-exo et
ozonolyse de l'oléfine exocyclique ... 55
3.7 Vinylation intramoléculaire d'un amide menant au produit 4-exo . ... 55
3.8 Expériences de compétition entre la cyclisation 4-exo et divers autres modes de cyclisation ... 56
3.9 Système catalytique Cul/N,N-diméthylglycine ne permettant pas la cyclisation
5-exo ... 57
3.10 Vinylation intramoléculaire 4-exo d'w1 dihydrazide ... 57
3.11 Fonnation d'un cycle à 13 membres dans la synthèse de la paliurine F ... 58
3.12 ~-lodovinylation intramoléculaire 4-exo d'un dicarbazate ... 59
3.13 ~-Halogénovinylation intramoléculaire 4-exo vs 5-endo d'un dicarbazate ... 59
3.14 Diiodation et ~-iodovinylation intramoléculaire 5-endo sur un carbamate ... 59
3.15 ~-Halogénovinylation intramoléculaire 5-endo d'un lactame ... 60
3.16 ~-Iodovinylation intramoléculaire de lactames ... 60
3.17 ~-lodovinylation intramoléculaire 6-endo d'w1 sulfonamide ... 61
3.18 Comparaison de la ~-iodovinylation intramoléculaire 6-endo et de 1 'iodocyclisation sur un triazole ... 62
3.19 ~-Chlorovinylation intramoléculaire 7-endo d'un lactame ... 62
3.20 Schéma général de la bromoiodation d'alcynes ... 63
4.1 Schéma général de la synthèse des modèles d'amides ... 65
4.2 ·Mécanisme menant aux sous-produits 263 et 264 ... 68
4.3 Synthèse des di iodures 265a-c et 266a-c ... 69
4.4 Couplage du produit 266c, menant aux produits 267c et 268c, et aux sous-produits bromés 269c et 270c ... 71
4.5 Diiodation et hétérohalogénation de l'amide 262 ... 72
4.7 Dibromation de l'amide 261c, menant au dibromure 273c et au sous-produit tribromé 275 .... 81
4.8 La réaction entre 1 'acide but-3-ynoïque et une source d'imidazole ... 87
4.9 Synthèse de 1' amide 286 . ..... 88
4.10 Synthèse du di iodure 288 ............. 88
4.11 Schéma général de la synthèse des modèles de carbamates ... 89
4.12 Synthèse du carbamate 294 par la méthode de Oh ... 89
4.13 Synthèse du carbamate 295 . ..... 90
4.14 Synthèse de l'alcool diiodé 300 . .... 91 4.15 Synthèse du carbamate di iodé 296 . .... 91 4.16 Mécanisme supposé de formation de 1 'alcool dibromé Z-301.. .... 93
4.17 Synthèse de 0-éthynylcarbamates par la méthode de Gralla ... 94
4.18 Synthèse du carbamate 309 .... 94
4.19 Mécanisme supposé pour la formation du produit 310 ........................................ 95
4.20 Mécanisme de la déprotection d'un N-méthylamide par la méthode de Beyersbergen ... 96
4.21 Acétylatlon de l'amine 315 et tentative de déprotection de l'amide 316 ............ 96
4.22 Trifluoroacétylation du carbamate 309 ..... 97 4.23 Trifluoroacétylation du carbamate 309 et tentative d'élimination ... 97
4.25 Schéma général de la synthèse de l'acétylamide 322a en passant par l'azoture 325 .
... 98
4.26 Schéma général de la synthèse de l'acétylamide 322a en passant par le phtalimide 328 ... lOO 4.27 Synthèse du phtalimide 328 ... 101
4.28 Déprotection du phtalimide 328 ... 101
4.29 Acétylation de l'amine 326 ... 101
4.30 Déprotection et acétylation du phtalimide 328 ... 102
4.31 Déprotection et benzoylation du phtalimide 328 ... 102
5.1 Couplage C-N intramoléculaire de Z-303, menant au produit Z-338 et au sous-produit 294 ... 122
5.2 Couplage C-N intramoléculaire des amides 266a-c ... 125
Spectre Page
B.1 N-Méthyl-N-phénylacétamide (316) ... 211
B.2 Pent-4-ynamide (261a) ... 212
B.3 N-Méthylpent-4-ynamide (26lb) ... 213
B.4 N-Phénylpent-4-ynamide (261c) ... 214
B.5 Hex-5-ynamide (262a) ... 215
B.6 N-Méthylhex-5-ynamide (262b) ... 216
B.7 N-Phénylhex-5-ynamide (262c) ... 217
B.8 (E)-4,5-Diiodo-pent-4-énamide (265a) ... 218
B.9 (E)-4,5-Diiodo-N-méthylpent-4-énamide (265b) ... 219 B.1 0 (E)-4,5-Diiodo-N-phénylpent-4-énamide (265c) ... 220
B.11 (E)-5,6-Diiodo-hex-5-énamide (266a) ... 221
B.12 (E)-5,6-Diiodo-N-méthylhex-5-énamide (266b) ... 222
B.13 (E)-5,6-Diiodo-N-phénylhex-5-énamide (266c) ... 223
B.14 (E)-6-(Iodométhylène )-1-phénylpipéridin-2-one (267c) ... 224
B.16 6-Bromo-1-phényl-4,5-dihydro-1H-azépin-2(3H)-one (270c) .... 226 B.17 (E)-4,5-Dibromo-pent-4-énamide (273a) ... 227 B.18 (E)-4,5-Dibromo-N-méthylpent-4-énamide (273b) ... 228 B.l9 (E)-4,5-Dibromo-N-phénylpent-4-énamide (273c) ...... 229 B.20 (E)-5,6-Dibromo-N-méthylhex-5-énamide (274b) ..................................... 230 B.21 (E)-5,6-Dibromo-N-phénylhex-5-énamide (274c) .... 231 B.22 Acide (E)-4,5-dibromopent-4-énoïque (277) ... 232
B.23 Acide (E)-5,6-dibromopent-5-énoïque (278) .... 233
B.24 (E)-3,4-Diiodo-N-phénylbut-3-énamide (288) ... 234
B.25 0-Prop-2-ynyl-N-phénylcarbamate (294) ... 235
B.26 0-But-3-ynyl-N-phénylcarbamate (295) ... 236
B.27 (E)-0-2,3-Diiodoprop-2-ényl-N-phénylcarbamate (296) ...... 237
B.28 (E)-0-3,4-Diiodobut-3-ényl-N-phénylcarbamate (297) ... 238
B.29 2,3-Diiodo-prop-2-én-l-ol (300) ....... 239 B.30 (E)-2,3-Dibromo-prop-2-én-l-ol (E-301) ....... 240 B.31 (Z)-2,3-Dibromo-prop-2-én-l-ol (Z-301) ...... 241 B.32 3,4-Dibromobut-3-én-l-ol (302) ....... 242 B.33 (E)-0-2,3-Dibromoprop-2-ényl-N-phénylcarbamate (E-303) ................. 243 B.34 (Z)-0-2,3-Dibromoprop-2-ényl-N-phénylcarbamate (Z-303) ..... 244
Bo35 (E)-0-3,4-Diiodobut-3-ényl-N-phénylcarbamate (304) ooooooooooooooooooooooooooooooooooooo 245
Bo36 0-2,2,2-Tribromoéthyl-N-phénylcarbamate (309) ooooooooooooooooooooooooooooooooooooooooooooo 246
Bo37 N-But-3-ynylacétamide (322a)ooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooOOooooooooooo 247
Bo38 N-But-3-ynylbenzamide (322b) OOooooOOOooOOooOOOOooOOoooooooooooooooooooooooooooooooOOooooooooooooooooo 248
Bo39 (E)-N-3,4-Diiodobut-3-énylacétamide (323a) oooooooooooooooooooooooooooooooooooooooooooooooooooo 249
B.40 (E)-N-3,4-Diiodobut-3-énylbenzamide (323b) OOoooooooooooooooooooooooooooooooOOOOoooooooOOoooo250
B.41 2-(But-3-yn-1-yl)isoindoline-1 ,3-dione (328) 000000 .... 00 .. oo ... oooooooooooooooooooooooooooooo 251
B.42 (E)-5-(Iodométhylène )-1-méthylpyiTolidin-2-one (329b) 0000 00000000 ... 00 0 ooooooo 00 252
B.43 (E)-5-(Iodométhylène )-1-phénylpyiTolidin-2-one (329c) 0000000000000 0000 000 00000 0000000 00 253
B.44 (E)-5-(Bromométhylène )-1-méthylpyiTolidin-2-one (331 b) oooooooooooooooooooooooooo 0000 254
B.45 (E)-5-(Bromométhylène )-1-phénylpyrrolidin-2-one (331c) 0000000000000 00000 000 oooooo 0000 255
B.46 (E)-4-(Iodométhylène )-3-phényloxazolidin-2-one (335) 0000000000000000 0000000000 000000000 256
B.4 7 (E)-4-(Bromométhylène)-3-phényloxazolidin-2-one (E-338)0ooooooooooo0oooooooooooo 000 257
B.48 (Z)-4-(Bromométhylène)-3-phényloxazolidin-2-one (Z-338) OOOOOOooOOoooooooooooooooooo 258
B.49 (E)-1-(2-(Iodométhylène )azétidin-1-yl)éthanone (344a) .. 00 oooooooooooooooooooooo 000000000 259
Bo50 1-( 4-Iodo-2,3-dihydro-1H-pyiTol-1-yl)éthanone (345a) 000000 .. 00000000 0000000000000000 00 00 260
Bo51 (E)-4-Allylidène-3-phényloxazolidin-2-one (346) OOoooooooooooooooooooooooooooooooooOOoooooooo 261
Bo52 (2E,4E)-tert-Butyl-4-(2-oxo-3-phényloxazolidino-4-ylidène)but-2-énoate
(347) ooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooooo262
B.54 (E)-4-Benzylidène-3-phényloxazolidin-2-one (349) ... 264 B.55 N-Méthyi-N-phénylacétamide (316) .................. 265 B.56 Pent-4-ynamide (261a) ... 266 B.57 N-Méthylpent-4-ynamide (261 b) ...... 267 B.58 N-Phénylpent-4-ynamide (261c) ... 268 B.59 Hex-5-ynamide (262a) ... 269 B.60 N-Méthylhex-5-ynamide (262b ) ... 270 B.61 N-Phénylhex-5-ynamide (262c) ... 271 B.62 (E)-4,5-Diiodo-pent-4-énamide (265a) ... 272 B.63 (E)-4,5-Diiodo-N-méthylpent-4-énamide (265b) ... 273 B.64 (E)-4,5-Diiodo-N-phénylpent-4-énamide (265c) ....................... 274 B.65 (E)-5,6-Diiodo-hex-5-énamide (266a) ... 275 B.66 (E)-5,6-Diiodo-N-méthylhex-5-énamide (266b) ... 276 B.67 (E)-5,6-Diiodo-N-phénylhex-5-énamide (266c) ...................... 277 B.68 (E)-6-(Iodométhylène )-1-phénylpipéridin-2-one (267c) ... 278 B.69 (E)-6-(Bromométhylène)-1-phénylpipéridin-2-one (269c) ... 279 B. 70 6-Bromo-1-phényl-4,5-dihydro-IH-azépin-2(311)-one (270c) ...... 280
B.71 (E)-4,5-Dibromo-pent-4-énamide (273a) ...... 281
B. 73 (E)-4,5-Dibromo-N-phénylpent-4-énamide (273c) ....... 283
B.74 (E)-5,6-Dibromo-N-méthylhex-5-énamide (274b) ........................................ 284
B.75 (E)-5,6-dibromo-N-phénylhex-5-énamide (274c) ..... 285
B.76 Acide (E)-4,5-dibromopent-4-énoïque (277) ... 286
B.77 Acide (E)-5,6-dibromopent-5-énoïque (278) ...... 287
B.78 (E)-3,4-diiodo-N-phénylbut-3-énamide (288) ... 288 B.79 0-Prop-2-ynyl-N-phénylcarbamate (294) ... 289 B.80 0-But-3-ynyl-N-phénylcarbamate (295) ... 290 B.81 (E)-0-2,3-Diiodoprop-2-ényl-N-phénylcarbamate (296) ..... 291 B.82 (E)-0-3,4-Diiodobut-3-ény1-N-phénylcarbamate (297) ... 292 B.83 2,3-Diiodo-prop-2-én-1-ol (300) ...... 293
B.84 (E)-2,3-Dibromo-prop-2-én-1-ol (E-301) ...... 294
B.85 (Z)-2,3-Dibromo-prop-2-én-1-ol (Z-301) ..... 295
B.86 3,4-Dibromobut-3-én-1-ol (302) ..... 296
B.87 (E)-0-2,3-Dibromoprop-2-ényl-N-phénylcarbamate (E-303) ............ 297
B.88 (Z)-0-2,3-Dibromoprop-2-ény1-N-phénylcarbamate (Z-303) ....... 298
B.89 (E)-0-3,4-Diiodobut-3-ényl-N-phénylcarbamate (304) ...... 299
B.90 0-2,2,2-Tribromoéthyl-N-phénylcarbamate (309) ... 300
B.92 N-But-3-ynylbenzamide (322b) ... 302 B.93 (E)-N-3,4-Diiodobut-3-énylacétamide (323a) ... 303 B.94 (E)-N-3,4-Diiodobut-3-énylbenzamide (323b) ... 304 B.95 2-(But-3-yn-1-yl)isoindoline-1 ,3-dione (328) ... 305 B.96 (E)-5-(Iodométhylène )-1-méthylpyrrolidin-2-one (329b) ... 306 B.97 (E)-5-(Iodométhylène)-1-phénylpyrrolidin-2-one (329c) ... 307 B.98 (E)-5-(Bromométhylène )-1-méthylpyrrolidin-2-one (331 b) ... 308 B.99 (E)-5-(Bromométhylène)-1-phénylpyrrolidin-2-one (331c) ... 309 B.1 00 (E)-4-(Iodométhylène )-3-phényloxazolidin-2-one (335) ... 310 B.1 01 (E)-4-(Bromométhylène )-3-phényloxazolidin-2-one (E-338) ... 311 B.1 02 (Z)-4-(Bromométhylène )-3-phényloxazolidin-2-one (Z-338) ... 312 B.1 03 (E)-1-(2-(Iodométhylène )azétidin-1-yl)éthanone (344a) ... 313 B.104 1-(4-Iodo-2,3-dihydro-1H-pyrrol-1-yl)éthanone (345a) ... 314 B.1 05 (E)-4-AIIylidène-3-phényloxazolidin-2-one (346) ... 315 B.1 06 (2E,4E)-tert-Butyl-4-(2-oxo-3-phényloxazolidino-4-yl idène )but-2-énoate
(347) ··· 316 B.l 07 (E)-3-Phényl-4-(3-phénylprop-2-yn-1-ylidène)oxazolidin-2-one (348) ... 317 B.l 08 (E)-4-Benzylidène-3-phényloxazolidin-2-one (349) ... 318
Ac aq. Ar BINAP Bn Boe Bu Bz CCM CLHP cod CPG-SM Cy
d
dba DBU DCC DCM dcypbdd
Acétyle Solution aqueuse Aromatique2,2'-Bis( diphénylphosphino )-1, 1 '-binaphtyle Benzyle
tert-Butoxycarbonyle
Butyle Benzoyle
Chromatographie sm couche mince
Chromatographie liquide à haute pression
Cycloocta-1 ,5-diène
Chromatographie en phase gazeuse couplée à un spectromètre de masse
Cyclohexyle Doublet (RMN) Dibenzylidèneacétone 1 ,8-Diazabicyclo[5.4.0]undéc-7-ène N,N' -dicyclohexylcarbodiimide Dichlorométhane
1 ,4-Bis( dicyclohexylphosphino )butane
DMA DMAP DME DMEDA DMF DMSO e.e. Et éq. f GEA Hex HOMO HTIB i-IR L LDA LUMO m m Diméthylacétamide 4-Diméthylaminopyridine Diméthoxyéthane N,N' -Diméthyléthylènediamine N,N-Diméthylformamide Diméthylsulfoxyde Excès énantiomérique Éthyle Équivalent Faible (IR) Groupement électroattracteur Hexyle
Orbitale moléculaire occupée de plus haute énergie (Highest occupied molecular orbital) Hydroxy(tosyloxy)iodobenzène Intense (IR) Iso Spectroscopie infrarouge Ligand
Diisopropylamidure de lithium (Lithium diisopropylamide)
Orbitale moléculaire inoccupée de plus basse énergie (Lowest unoccupied molecular orbital)
Moyen (IR)
Multiplet (RMN)
Me met Ms n-NBS NIS NMP Nu Pent Ph ph en Ph th PMA Pr q quint R RMN s s(l) SMHR tt Méthyle Méthylallyle Mésyle ou méthanesulfonyle Normale N-Bromosuccinimide N-Iodosuccinimide N-Méthyl-2-pyrrolidone Nucléophile Pentyle Phényle 1,1 0-Phénanthroline Phtalimide (Phthalimide)
Acide phosphomolybdique (Phosphomolybdic acid)
Propyle
Quadruplet (RMN)
Quintuplet (RMN)
Groupement carboné
Résonance magnétique nucléaire
Singulet (RMN) Singulet large (RMN)
Spectrométrie de masse à haute résolution
Triplet (RMN)
Triplet de triplets (RMN)
t
-TC
Tf TFA THF TMG TMS t.p.uv
tert-Thiophènecarboxylate Triflate ou trifluorométhylsulfonateAcide trifluoroacétique (Trifluoroacetic acid)
Tétrahydrofuranne
1,1 ,3,3-Tétraméthylguanidine
Triméthylsilyle Température pièce
A
8 g h Hz J L M m/z mg mm mL mmol mol ~LL %ml v %mol Angstrom Degré Celsius Déplacement chimique Gramme Heure Hertz Constante de couplage (RMN) Litre Molaire ou mol/litre Masse sur charge Milligramme Minute Millilitre Millimole Mole MicrolitrePourcentage masse sur volume (ou g/lOOmL) Pourcentage molaire
RÉSUMÉ
Les énamides présentent un intérêt tout particulier en synthèse, autant par leur présence dans de nombreux produits naturels, que par leur réactivité remarquablement polyvalente. Toutefois, la formation stéréosélective d'énamides complexes dememe un défi. L'utilisation de ~ halogénoénamides constitue une solution prometteuse à ce problème. Nous nous sommes donc intéressés à la synthèse de ces derniers par le biais d'un couplage croisé catalysé par le cuivre entre un amide et un dihalogénure vinylique.
Ce projet se concentre plus particulièrement sur 1 'étude de la version intramoléculaire de cette réaction. Une variété de molécules modèles, portant à la fois un groupement amide ou carbamate et une insaturation dihalogénée ont d'abord été synthétisées.
Les énamides présentent un intérêt tout particulier en synthèse, autant par leur présence dans de nombreux produits naturels, que par leur réactivité remarquablement polyvalente. Toutefois, la formation stéréosélective d'énamides complexes dememe tm défi. L'utilisation de ~
halogénoénamides constitue une solution prometteuse à ce problème. Ce type de composé peut facilement être formé par couplage croisé catalysé par le cuivre entre un amide et un dihalogénure vinylique. Ce projet se veut une étude systématique de la version intramoléculaire de cette transformation, peu étudiée jusqu'à maintenant.
Pour arnver à cette fin, une variété de molécules modèles ont été préparées. Celles-ci comportent w1e fonction amide ou carbamate portant différents substituant sur 1 'azote, ainsi qu'une insaturation diiodée ou dibromée, séparées par une chaîne carbonée de longueur variable. La cyclisation de ces produits, catalysée par le cuivTe, permet d'accéder à une variété de ~-halogénoénamides cycliques. Étant donné l'utilisation d'un dihalogénure comme substrat de départ, possédant donc deux sites électrophiles non-équivalents, le couplage C-N fournit un mélange de deux produits, exo et endo, avec une sélectivité plus ou moins élevée. Les substrats étudiés ont permis d'évaluer l'influence de diverses variables sur la sélectivité de la réaction.
Nous avons ensuite procédé à une optimisation de la réaction, afin d'évaluer l'impact de
chacune des composantes réactionnelles sur cette sélectivité. Le paramètre ayant le plus grand impact s'est révélé être, sans surprise, le choix du ligand. Enfin, la fonctionnalisation de 1 'un des ~-iodoènecarbamates synthétisés a pu être explorée de façon préliminaire. Nous avons pu montrer que l'iodw-e vinylique restant sur ce substrat fait preuve d'une réactivité élevée vis-à-vis des réactions de couplage catalysés par les métaux de transition.
Mots-clés : Couplage croisé intramoléculaire, Catalyse au cuivre, Énamides, ~ halogénoénamides
Les énamides présentent un intérêt tout particulier en synthèse, autant par leur présence dans de nombreux produits naturels, que par leur réactivité remarquablement polyvalente. Ils peuvent participer à une foule de réactions chimiques, ouvrant ainsi la voie à une variété de
fonctionnalités azotées intéressantes et de motifs hétérocycliques. Toutefois, la
fonctionnalisation d'énamides dans 1 'optique d'en former des dérivés complexes et énantiopurs demeure un défi. Notre groupe s'est intéressé à l'utilisation de composés ~-halogénoénamides
pour remédier à ce problème, puisque ceux-ci peuvent facilement être transformés en énamides hautement fonctionnalisés. L'intérêt des énamides et P-halogénoénamides, ainsi que leurs méthodes actuelles de synthèse seront donc présentés au chapitre 1.
Dans le but de former ces P-halogénoénamides, nous nous sommes tournés vers le couplage croisé, catalysé par le cuivre, entre des amides et des dihalogénures vinyliques. li est connu que les réactions de couplage catalysées par les métaux de transition figurent parmi les méthodes de synthèse d'énamides les plus efficaces. Le cuivre constitue un catalyseur de choix pour ce type de réaction en raison de sa tolérance élevée envers différents groupements fonctionnels et de son faible coût par rapport aux autres métaux usuels (chapitre II).
La plupart des exemples de couplages entre des amides et des dihalogénures vinyliques retrouvés dans la littérature ont été réalisés de façon intramoléculaire. Ces réactions mettent en compétition deux modes de cyclisation, menant aux produits exo et endo, tout dépendant du site d'insertion du cuivre sur le dihalogénure. Toutefois, comme il sera présenté au chapitre III,
il est actuellement difficile de mettre en évidence des tendances quant à la sélectivité de la réaction entre ces deux modes. Les études trouvées dans la littérature sont rarement réalisées avec les mêmes conditions réactionnelles, et aucune étude systématique de cette réaction n'avait été entreprise jusqu'à maintenant.
Le but du présent projet est donc, d'abord, de préparer une variété de modèles pottant à la fois une fonction de type amide et une insaturation dihalogénée (chapitre IV). En soumettant ces molécules à des conditions de couplage constantes, nous avons pu obtenir une meilleure connaissance des facteurs influençant la préférence de la réaction pour la cyclisation exo ou endo, et ce pour la formation de cycles allant de 4 à 7 membres (chapitre V).
Pour compléter cette étude méthodologique, nous avons ensuite mené une optimisation des conditions réactionnelles (chapitre VI). Cette étape a pennis de confinner qu'il était possible, en variant les réactifs présents dans le milieu, d'influencer la sélectivité de la réaction. Enfin, nous avons exploré la possibilité de fonctionnaliser l'un des ~-halogénoénamides obtenus par le biais de diverses réactions de couplage (chapitre VII). Cela avait pour but de montrer l'intérêt synthétique et la polyvalence de ce type de substrats.
CHAPITRE 1
LA FONCTION ~-HALOGÉNOÉNAMIDE
Les énamides constituent une famille de composés chimiques présentant un intérêt tout particulier en synthèse, qui découle non seulement de leur présence dans plusieurs produits naturels, mais aussi de leur réactivité remarquablement polyvalente. En plus de participer aux réactions typiques des amides et des alcènes, ils possèdent un caractère ambivalent, agissant parfois comme électrophile, parfois comme nucléophile, leur permettant de former plusieurs fonctionnalités azotées intéressantes. Les énamides ouvrent aussi la voie à une variété de motifs hétérocycliques. II n'est donc pas surprenant de constater qu'ils aient été utilisés comme intermédiaires dans w1e foule de synthèses et que plusieurs méthodes aient été mises au point pour les préparer. Les premières sections de ce mémoire (1.1.1 et 1.1.2) serviront donc d'abord à exposer plus en détail l'intérêt des dérivés d'énamides puis, brièvement, les réactions permettant leur formation. II est important de noter que, parmi les composés de type énamide,
on retrouve aussi les ènecarbamates et les ènesulfonamides (voir figure 1.1) ; pour plus de simplicité, et à moins de mention contraire, l'appellation « énamide » désignera ces trois familles similaires de composés.
0 R3
R1Jl~
~
R
s
R2 R4 0 R3R
1___0)lN~Rs
R2
R4Énamides Énecarbamates Ènesulfonamides
Figure 1.1 Structure générale des énamides, ènecarbamates et ènesulfonamides.
Cette première discussion laissera transparaître certaines faiblesses inhérentes aux méthodes de synthèse et de fonctionnalisation d' énamides dans 1' optique d'en former des dérivés complexes et énantiopurs. Il ressortira d'abord que la plupart des synthèses d'énamides souffre d'inconvénients majeurs à cet effet, mais que la vinylation d'amides catalysée par des métaux
de transition se démarque clairement du lot par les nombreux avantages qu'elle présente. Nous mettrons aussi en évidence que, bien qu'ils participent à tout un éventail de transformations,
peu de réactions permettent la ~-fonctionnalisation directe d'énamides tout en conservant
l'intégrité de la liaison double carbone-carbone. L'utilisation d'une classe spécifique
d'énamides, les ~-halogénoénamides, se veut une solution à ce dernier problème, puisqu'ils permettent une fonctionnalisation simple et stéréosélective pow- la formation d'énamides hautement fonctimmalisés. La section 1.2.1 servira à établir plus en détail 1 'intérêt de ce type de composés. Les ~-halogénoénamides peuvent évidemment être préparés de manière plus «traditionnelle», c'est-à-dire par des réactions d'halogénation d'énamides ou de leurs
précw-sew-s. Toutefois, notre groupe a développé une méthode nettement plus intéressante d'iodovinylation catalysée par le cuivre pern1ettant leur synthèse directe à partir d'amides. La section 1.2.2 présentera donc ces deux avenues.
À ce point dans le texte, nous aurons mis en évidence que ces réactions ont surtout été explorées
de façon intermoléculaire, alors que les exemples intramoléculaires sont quasi-inexistants. La section 1.3 servira donc à établir clairement 1 'objectif du présent projet : 1 'étude de la réaction de N-halogénovinylation intramoléculaire d'amides et de carbamates catalysée par le cuivre. Nous exposerons, d'un point de vue méthodologique, notre intention de démontrer la viabilité
de cette réaction pow- la synthèse de ~-halogénoénamides cycliques, autrement difficiles à
préparer. L'atteinte de cet objectif passera inévitablement par une étude systématique de la réaction, dont la présentation servira à clore ce premier chapitre.
1.1 Les énamides
1.1.1 Intérêt et réactivité
L'intérêt du motif énamide passe d'abord par le fait qu'il constitue une cible synthétique, en ce sens qu'il apparaît dans une foule de produits natw-els et pharmaceutiques. La figw-e 1.2 rassemble certains exemples d'énamides présentant une importance biologique. Les carbapénèmes sont une famille d'antibiotiques à large spectre (Bimbaum et al., 1985), tandis
der Westhuyzen et al., 2012). Ces antibiotiques ont été découverts respectivement dans des
cultures de la bactérie Streptomyces cattleya et du fongus Seimatosporium sp. Les
lansiumamides sont une classe d'antihistaminiques extraits de l'arbre à agrumes Clausenia lansium (Matsui et al., 2013). Le palmerolide A, isolé de l'invertébré antarctique de la famille des tunicates Synoicum adareanum, et les salicylihalamides, provenant de 1 'éponge de mer Haliclona sp. (Snider et al., 2001), ont des propriétés cytotoxiques ciblant particulièrement les mélanomes. Enfin, la paliurine Fest un alcaloïde sédatif (Toumi et al., 2007) isolé de l'arbre Paliurus ramosissimus (Cordell, 2009). On remarque ainsi que les énamides sont produits par tme variété d'organismes vivants et qu'ils présentent des effets biologiques divers. Il est aussi important de noter la diversité et la complexité des patrons de substitution de 1' oléfine de ces énamides. De di- à tétrasubstitués, ils sont cycliques ou linéaires, E ou Z, et substitués par des
groupements alkyles, aryles, vinyles, carboxyles ou même hétéroatomiques.
6 - Paliurine F
1 - Carbapénèmes
E-5- Salicylihalamide A
Z-5 - Salicylihalamide B
0 0
P
N
~
OH
OH OH H 2- CJ-15,801 0~
N
è
u
1 ~1 3 - Lansiumamide B =::::--.. 0 0 OH 4 - Palmerolide AFigure 1.2 Structure chimique de produits naturels contenant des énamides.
D'un point de vue méthodologique maintenant, l'attrait principal des énamides provient de leur capacité à participer à une variété de réactions. Les énamides comportent principalement trois sites réactifs : le carbonyle de la fonction amide, qui possède un caractère électrophile, ainsi que chacun des carbones alcéniques. La délocalisation des électrons de 1' azote dans la liaison double fait que la position la plus nucléophile de l'énamide est le carbone~ (voir figure 1.3). Le carbone a, quant à lui, est plutôt électrophile en raison de l'effet inductif électroattracteur de l'hétéroatome adjacent (Matsubara et al., 2008). Une variété de réactions permet la
fonctionnalisation sur chacun de ces sites. Sans en faire une revue exhaustive, nous parlerons d'abord de l'hydrolyse et de l'hydrogénation catalytique des énamides, pour ensuite aborder
les réactions de substitution auxquelles ils participent, puis leur utilité dans la synthèse
d'hétérocycles azotés. Nous terminerons avec des exemples de réactions catalysées par les métaux de transition.
Figure 1.3 Structure générale et sites réactifs des énamides.
Hydrolyse
L'une des réactions les plus simples des énamides consiste en l'hydrolyse du lien amide pour
former une amine. En effet, les énamides sont, en quelque sorte, des énamines protégées et ces
deux classes de composés participent généralement aux mêmes types de réactions. Cependant,
les énamines ont l'inconvénient d'être instables: les énamides, moins réactifs et donc plus stables, peuvent au contraire facilement être conservés sur la paillasse et purifiés par chromatographie sur silice. Il peut donc être avantageux d'utiliser des énamides, sur lesquels
on procède à différentes transformations chimiques, pour ensuite générer un groupement amine
par hydrolyse.
Hydrogénation catalytique
L'hydrogénation catalytique est l'une des réactions des énamides les plus largement étudiées.
On en retrouve énormément d'exemples, autant sur des énamides «traditionnels» que sur des
acides déhydroaminés (Najera et al., 2007; Zhou et al., 2014). Il s'agit en effet de l'w1e des façons les plus fréquemment employées pour la synthèse d'acides aminés énantioenrichis. Une grande variété de systèmes ont été utilisés, ceux-ci impliquant généralement le rhodium comme
catalyseur ainsi que des ligands phosphines chiraux. Notons un exemple simple par Junge, représenté au schéma 1.1 (Junge et al., 2004). La réduction de substrats 7 de type
plus de 99% de conversion et des excès énantiomériques en faveur de l'isomère R de 94% et 95%, respectivement. 9 (2 %mol) [Rh(cod}2)BF4 (1 %mol) toluène, t.p. H2 (1 atm.) NHZ R~ C02Me 8 >99% conv. (R1=H, 94% e.e.) (R1=Ph, 95% e.e.) P-R 9, R=3,5-(t-Bu)2-phényl
Schéma 1.1 Hydrogénation catalytique énantiosélective d'un acide déhydroaminé.
Substitutions
On retrouve quelques exemples de réactions dans lesquelles la position a. agit comme électrophile, notamment dans l'acylation de Friedei-Crafts d'indoles, dont une verswn asymétrique est rapportée au schéma 1.2 (Terada et al., 2007). Pom ce qui est de la position ~'
les réactions les plus communes sont, au contraire, principalement des substitutions
nucléophiles. Celles-ci sont cependant limitées aux énamides secondaires ; les énamides dont l'azote ne porte pas d'atome d'hydrogène ne réagissent pas sur des électrophiles (Matsubara et
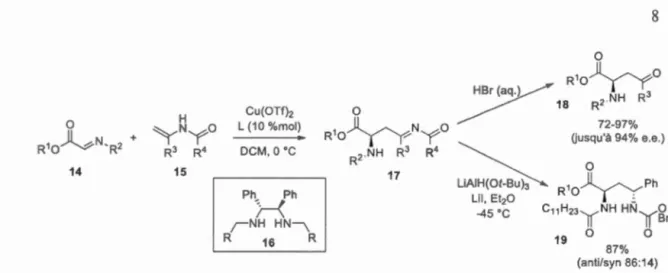
al., 2008). Kobayashi rapporte plusieurs exemples de synthèses énantiosélectives catalysées par des acides de Lewis coordinés par des ligands chiraux (schéma 1.3). L'attaque d'énamides
15 sur des N-acylimino esters 14 produit des acylimines chirales 17, avec des excès énantiomériques allant jusqu'à 94%. Il est intéressant de ·noter que les énamines correspondantes fournissent plutôt un mélange racémique du produit final. L'acylimine 17 peut être directement hydrolysée en cétone 18 ou réduite de façon asymétrique en amide 19. Les
énamides peuvent donc être utilisés autant pour introduire facilement une unité de deux carbones qu'w1 groupement azoté protégé.
00
+~
NHBoc
H 10 11 13 (2 %mol) MeCN -20 oc, 12 h:
HJ-lJ_J
H 12 R R 13, R=2,4,6-(i-Pr)J-phényl14 15 Cu(OTf)2 L (10 %mol) DCM, 0 •c Ph Ph
>---<
;NH HN\ R 16 R 17 0R,O~O
HBr (a~ 18 Rz.NH R3~
72-97% Uusqu'à 94% e.e.)~R'J
L1l, Et20 -A
-
"'(
..
Ph
_45 •c C,,H23f(NH HNf(~n 19 0 0 87% (anti/syn 86:14)Schéma 1.3 Substitution nucléophile d'un énamide sur un N-acyliminoester et réactions
subséquentes.
Formation d'hétérocycles
Les énamides brillent par leur aptitude à former des composés cycliques, dont une multitude d 'hétérocycles azotés. Cette capacité est d'autant plus intéressante que les hétérocycles sont omniprésents dans les produits naturels et présentent généralement une activité biologique
élevée (Evano et al., 2014). Certains hétérocycles accessibles à partir d'énamides sont
représentés à la figure 1.4. Panni ceux-ci, on compte des pipéridines 20 (B1izgys et al., 2012),
des pyrroles 21 (llivero et al., 2007), des 4-hydroxypyridines protégées 22 (Leche! et al., 201 0), des pyrimidines 23 (Es trad a et al., 2011 ), des pyrimidin-4-ones 24 (Ramanjulu et al., 201 0), des pyrid-2-ones 25 (Imase et al., 2008) et des isoquinolin-1 (2H)-ones 26 (Chen et al., 2009).
0 )lNH
l.J
N24
21 22 23
Réactions catalysées par les métaux de transition
Les énamides participent aussi à des réactions catalysées par les métaux de transition, formant principalement des liens C-C en a ou en~· L'avantage principal de ces réactions est qu'elles préservent, pour la plupart, l'intégrité de l'oléfine du substrat de départ, et servent essentiellement à obtenir des énamides substitués à partir d'énamides plus simples. On retrouve d'abord plusieurs exemples de réactions intramoléculaires catalysées par le palladium, en grande majorité des couplages de Heck. Un exemple particulièrement intéressant, rapporté par Rigby, est présenté au schéma 1.4 (Rigby et al., 1995). La régiosélectivité du couplage entre l'énamide et l'iodure aromatique peut être contrôlée en fonction des conditions utilisées.
,CsH11 ,CsH11 ,CsH11
HN HN HN
0
TBSO
~
Pd(OAch (10 %mol) Pd(OAch (1 0 %mol)
TBSO (o-toi)JP (20 %mol) KOAc (5,5 éq.) TBSO
Et3N (2,0 éq.) nBu4CI (2,0 éq.)
CH3CN/H20, 80 'C TBSO 1 DMF, 100 'C
~
27 ~
h
28MeO
MeO OMe OMe
32%
58%
Schéma 1.4 Couplage de Heck intramoléculaire de 1 'éna mi de 27.
La fonctionnalisation en a mène évidemment à la migration de la liaison double dans ce cas ; ce phénomène est cependant récurrent dans le cas des couplages en a, même avec des énamides disubstitués. Dès que la migration est possible, le couplage en a a donc l'inconvénient de ne pas mener à un énamide comme produit, et peut même produire un mélange d'isomères (voir schéma 1.5)(Sato et al., 1994).
30 [Pd2(dbalJ]CHCI3 (4 %mol) 33 (9,6 %mol) Zéolite (6,0 éq.) CaC03 (2,2 éq.) DMSO-DMF, 0 oc, 5 j. 0
ln
.
(o
31 H 1.4 H 32 94% (86% e.e.) th>-Ph-P~ OH 'Fe
' 33~
P-Ph pl{Schéma 1.5 Couplage de Heck intramoléculaire énantiosélectif et migration.
Mises à part les réactions au palladium, Dake rapporte aussi plusieurs exemples de cyclisations sur des alcynes catalysées par des sels d'or ou d'argent, menant à une variété de composés
bicycliques. Un exemple de cette réaction est représenté au schéma 1 .6; on y voit bien, encore
une fois, que les cyclisations en~ n'ont pas tendance à causer la migration de l'énamide (T. J.
HaiTison et al., 2004).
~
C02Me
AgOTf (2 %mol)c
J
DCM-THF, 60 oc~ 34
~co,Mo
~ 35
Ts Ts 99%
Schéma 1.6 Cyclisation d'un alcyne sur un énamide, catalysée par l'argent.
Enfin, les énamides participent aussi à la métathèse intramoléculaire d'oléfines catalysée par le ruthénium, qui constitue une forme de fonctionnalisation en ~(schéma 1.7) (Kindennan et al., 2001). R = H, Me GÉA
=
Ts, Bz, C02Et n=
1, 2 PCy3 Cl,. 1 CI,..Ru~ (1-5 %mol)~Cy
3
Ph
37 DCE, 40-80 oc, 2-16 h R GEA,N--\ 38~
n 25-93%Au niveau intermoléculaire, les réactions de couplage semblent nettement moins flexibles, et
limitées à un ensemble plus restreint de réactions. La plupart des exemples rencontrés utilisent,
encore une fois, principalement la réaction de Heck, qui cible spécifiquement le site a.. Pour ce
qui est de la position ~' le moyen quasi-exclusif de la fonctionnaliser est de passer par des méthodes d'activation C-H.
La majorité des substrats sur lesquels la réaction de Heck a été menée de façon intennoléculaire
sont des énamides dont 1 'alcène est terminal : dans le cas contraire, le couplage en a. mène, encore une fois, à la migration de la liaison double. Le schéma 1.8 illustre ces deux cas de
figure, par le couplage entre un énamide 39 et un triflate aromatique 40 (P. Harrison et al., 2004; Tu et al., 2003). La synthèse du composé 44 passe par la fom1ation d'un énamide et la migration de sa liaison double. Cela a l'avantage de former un centre asymétrique dont la
configuration est déterminée par le ligand ferrocène chiral 43 utilisé.
0
t:r
Meo) - . Û 44 39-76% (e.e. jusqu'à 97%) Pd(OAch (5 %mol) 43 (10 %mol) iPr2NEI (1 ,2 éq.) benzène, 80 oc GÉA-~~ + ArOTf '---' 39 40 Pd(OAch (1 %mol) 41 (1, 1 %mol) Et3N (1 ,2 éq.) DMF, 100 oc, 1.5 h Ph,p~p,Ph 1 1 Ph Ph 41Schéma 1.8 Couplages de Heck intermoléculaires sur des énamides.
0 Ar
)lN
~
H 42 24-69%
On retrouve quelques exemples de fonctionnalisation intermoléculaire en ~ d'énamides, mais
cette voie de synthèse dememe limitée. D'ailleurs, selon Gigant, ce type de réaction n'avait
pratiquement pas été investigué jusqu'en 2009 et constitue un important défi qui recèle un
grand potentiel synthétique (Gigant et al., 2014). En effet, la majorité des produits naturels
présentés en début de section sont substitués uniquement en~-D'un point de vue pragmatique, la plus grande préoccupation de la fonctionnalisation d'énamides devrait donc logiquement
être l'introduction régio- et stéréosélective de divers groupements en ~- Quelques réactions catalysées par des métaux de transition ont été employées à cet effet. Le schéma 1.9 regroupe
certains exemples d'activation C-H sur des énamides cycliques 45. Loh a rapporté l'arylation directe d'énamides à l'aide d'acides boriques aromatiques, permettant de fonner les composés 46 avec des rendements modérés à bons (Zhou, Chung, et al., 2009). La réaction conduit à la formation minoritaire d'un sous-produit aromatisé 47, en proportions plus ou moins élevées selon les substrats. Une autre méthode d'arylation a été développée par le même groupe, et consiste en w1 couplage de type Hiyama entre l'énamide et un aryltriméthoxysilane menant au produit 48 (Zhou, Xu, et al., 2009). Loh a aussi mis au point une méthode de sulfonylation en
~ d'énamides catalysée par le palladium afin d'obtenir des sulfones aromatiques 49 avec des rendements modérés (Xu et al., 2013). Enfin, Wang a réalisé l'acylation d'énamides à l'aide d'acides arylglyoxyliques, fonnant ainsi divers ~-acylénamides 50 (Wang et al., 2012). À travers ces exemples, on remarque cependant des lacunes flagrantes : ils se limitent à des substrats aromatiques, dont la portion énamide est toujours cyclique.
c6
AcHN Ar R1-46 +cO
AcHN Ar R1 -ArB(OHh (2,0 éq.) K2C03 (2,0 éq.)Pd(OAc)2 (10 %mol) dioxane, 80 •c, 16 h
Cu(OTfh (2,0 éq.) 47 57-82% (71 :29 à 98:2) ArSi(OMe)J (3,0 éq.) Pd(OAch (1 0 %mol) AgF (3,0 éq.) dioxane, 80 ·c, 16 h ArS02CI (1,5 éq.) K2C03 (50 %mol)
Pd(OAch (1 0 %mol) 02, dioxane, 120 ·c, 24 h
33-68%
Schéma 1.9 Fonctiormalisations C-H d'énamides.
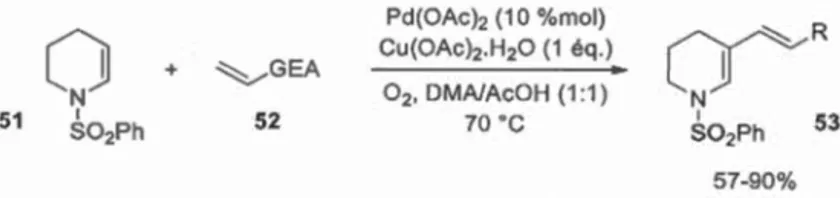
La méthode a, à tout le moins, été étendue à la vinylation d'énamides, mats limitée à
1 'utilisation d'alcènes pauvres en électrons (voir schéma 1.1 0). On remarque aussi que, malgré la présence d'un hydrogène en a de l'azote cette fois, la réaction présente une régiosélectivité complète en favem de la ~-fonctionnalisation (Gigant et al., 2012).
0
N + 1 S02Ph 51 ~GEA 52 Pd(OAch (1 0 %mol) Cu(0Ach.H20 (1 éq.) 02 , DMA!AcOH (1 :1) 70 ·c 57-90%Schéma 1.10 Vinylation de l'ènesulfonamide 51 par fonctionnalisation C-H.
Le mécanisme proposé par Zhou pour 1 'activation C-H d' énamides semble apporter un élément
de réponse justifiant cette régiosélectivité (Zhou, Chung, et al., 2009). Celle-ci est vraisemblablement due à la participation de l'intermédiaire cyclique à 6 membres 54, qui ne peut se former avec la position a. Cet intermédiaire semble expliquer pourquoi les réactions intermoléculaires d'activation C-H développées pom les énamides se cantonnent à leur
fonctiotmalisation en
p.
R1 oANHAc0-id~R2
L: R3 54Figure 1.5 Intermédiaire cyclique à 6 membres impliqué dans l'activation C-H d'énamides.
La fonctionnalisation en
p d'énamid
es reste une avenue limitée, en ce sens qu'elle est circonscrite à un ensemble de réactions d'activation C-H très spécifiques. Si l'on pense auxréactions de couplage catalysées par les métaux de transition dans leur ensemble, on peut donc
considérer que les énamides ne sont pas des partenaires très généraux dans les réactions de ce type. L'on pourrait toutefois envisager d'introduire un groupement activant en position
oléfinique, comme un halogène, ce qui transformerait essentiellement 1 'énamide en halogénure
qui ouvrirait la voie à tme multitude de réactions de fonctiormalisation d'énamides. Encore selon Gigant, toutefois, la préfonctionnalisation en ~est complexe et aucw1e méthode générale n'avait été rapportée pomy procéder (Gigant et al., 2014). À la section 1.2, nous présenterons une solution, récerrm1ent mise au point par noh·e groupe, permettant la synthèse d'énamides préfonctionnalisés. Nous nous devons donc d'aborder, d'abord, les diverses méthodes actuellement disponibles pour la préparation d'énamides.
1.1.2 Méthodes de synthèse
Les méthodes h·aditiormelles de préparation d'énamides incluent, de manière non exhaustive, la condensation d'amides avec des aldéhydes, l'hydroamidation d'alcynes, l'acylation d'imines et d'oximes, le réarrangement de Cwtius d'azotures a,~-insaturés, ainsi que les oléfinations de Peterson et de Wittig.
Condensation
La synthèse directe d'énamides par condensation entre un amide 55 et un composé carbonylé 56 se limite à l'utilisation d'amides secondaires (schéma 1.11) (Genovino et al., 2012). Puisqu'il s'agit d'un processus foncièrement thermodynamique, la formation de l'énamide E
(57), plus stable, est généralement favorisée. La méthode a été indirectement étendue aux carbamates primaires 58 par le groupe de Petri ni (schéma 1.12) (Mecozzi et al., 2000). Dans leur synthèse, 1 'intem1édiaire de type imine formé après la condensation est piégé sous forme d'acétal N,S 60. Contrairement à la réaction de condensation conventionnelle présentée au schéma 1.11, 1 'élimination du groupement sulfonyle de 60 donne accès plutôt à 1 'ènecarbamate
Z (61), avec une sélectivité E/Z allant jusqu'à 1 : 9. 0
H
~
R4
56 R3 1.1 éq. TiCI4, Et3N o ·cà t.p., 2-16 h Jusqu'à 75% (si R3=H, E seulement)DBU
THF, t.p.
61
60 64-90%
(E/Z de 1:2.3 à 1 :9)
Schéma 1.12 Condensation entre un carbamate primaire et un aldéhyde en présence d'un
sulfinate.
N-Acylation d'imines et d'oximes
Une autre avenue logique consiste à faire la N-acylation de composés dérivés d'imines. C'est
1 'une des approches employées par Kobayashi pour synthétiser les molécules modèles de son
étude de substitution nucléophile par les énamides (schéma 1.13) (Matsubara et al., 2008). La
réaction entre un nitrile 62 et un réactif de Grignard benzylique 63 (R2=Ph) forme un intermédiaire iminomagnésien 64 qui, lors de l'addition d'un chlorure d'acyle (65, R3=alkyle) ou d'un chloroformiate (65, R3=0-alkyle), fournit un énamide ou ènecarbamate 66. La réaction
mène à un mélange d'isomères E et Z, en un ratio non mentionné dans l'article. De manière
similaire, l'acylation réductrice d'oximes 68 en présence de fer métallique a été appliquée à la
synthèse de quelques énamides 69 (schéma 1.14). Le seul exemple ~-substitué 69a montrait
une faible stéréosélectivité EIZ de 3 : 2 (Burk et al., 1998).
Schéma 1.13 Attaque d'un réactif de Grignard sur un nitrile et acylation de 1 'iminomagnésien
résultant. 0 Ex: 0 Fe (poudre) _)lNH _ ) lNH 0 NH20H NOH Ac20 J YR3 J Y R3 ~R3
~
R1 R1 AcOH (3 éq.) R1 R2 R2 toluène, 75 ·c R2 69 69a 67 68 40-85% EIZ = 3:2Hydroamidation d'alcynes
L'hydroamidation d'alcynes a pom prem1er inconvénient de nécessiter l'utilisation de catalyseurs dispendieux de ruthénium (schéma 1.15) (Huang et al., 2015). Elle mène à la
fom1ation de l'énamide E 71 avec des sélectivités généralement assez peu élevées. L'isomère
Z 72 est accessible en ajoutant un co-catalyseur d'ytterbium, avec une séléctivité EIZ plus
élevée que 1 : 20 pour la majorité des substrats testés. Un autre inconvénient de cette méthode
est qu'elle se limite à l'utilisation d'alcynes terminaux.
(cod)Ru(meth (2 %mol) 0 n-Bu3P (6 %mol) 0 R)lt:JH ~ DMAP (4 %mol)
R1JlN
~
R3
Jusqu'à 99% + R3toluène, 1 00 oc, 15 h R2 (EIZ 2:1 à 30:1)
R2 55 70 71 (cod)Ru(meth (2 %mol) 0 dcypb (2,25 %mol) 0 R1JlNH ~ Yb(OTfb (4 %mol)
R1
Jl
t:J
~
Jusqu'à 99% + R3 chlorobenzène, 60 oc, 15 h (EIZ 1:1 à 1:>20) R2 R2 R3 55 70 72Schéma 1.15 Hydroamidation E-et Z-sélective d'alcynes.
Réarrangement de Curtius
Les azotures d'acyle a,~-insatmés 74, lorsque chauffés, procèdent à un réarrangement de
Curtius pour fonner des isocyanates d'alcényle 75 avec des rendements allant jusqu'à 92% (schéma 1.16) (Sato, 1961). L'attaque nucléophile d'un alcool sur l'isocyanate formé peut mener à une variété d'ènecarbamates 76. Sato ne rapporte pas de rendements pour la formation des carbamates, mais l'attaque d'un nucléophile sm un isocyanate se fait généralement avec
des rendements assez élevés (Oh et al., 2004). La configuration stéréochimique de l'oléfine
dépend de celle de l'acide carboxylique a,~-insaturé 73 utilisé au départ. Certains acides
insaturés posent toutefois 1 'inconvénient d'être difficiles à synthétiser (Matsubara et al., 2008), du moins plus que les réactifs très simples impliqués dans les autres méthodes présentées.
0 HOY R1 R2 73 1) Chloration 2) NaN3 O~C- R30H 'Ny R1 R2 75 17-92%
Schéma 1.16 Fmmation d'un azoture d'acyle a,~-insaturé et réarrangement de Curtius. Élimination
76
Une méthode largement employée dans la synthèse d'énamides dérivés d'acides aminés s'effectue à partir d'acides ~-hydroxyaminés 77 (schéma 1.17). Le groupement hydroxyle est
d'abord acylé, puis éliminé à l'aide de la 1,1,3,3-tétraméthylguanidine (TMG). Ferreira a employé cette séquence pour préparer divers dérivés déhydroalanines, déhydrophénylalanines
et d'acides déhydroaminobutyriques (Ferreira et al., 2008). Les composés portant une chaîne carbonée en~ de l'amide mènent uniquement à l'énamide Z.
H GEA,...NX C02Me R OH 77 1. Boc20/DMAP 2. TMG H ,...NJ(C02Me GEA
l
R 78 R=H, 50-72%R=Me, 71-88% (Z seulement) R=Ph, 87-91% (Z seulement)
Schéma 1.17 Synthèse d'un acide déhydroaminé par élimination.
Une approche différente consiste plutôt à former le double lien C-C de l'énamide par une réaction d'oléfination. La réaction de Wittig (schéma 1.18) entre 1 'ion phosphonium 79, dérivé de la glycine, et le trifluoropyruvate de méthyle 80 fournit l'énamide 81 en proportions égales
des isomères E et Z (Mazurkiewicz et al., 2002). La synthèse d'énamides par réaction de Wittig
sur des formamides a aussi été rapportée (Villa et al., 2007). L'oléfination de
Horner-Wadsworth-Emmons d'un phosphonate 82, dérivé de la glycine, a quant à elle 1 'avantage de favoriser la formation de l'ènecarbamate Z (84), avec des sélectivités EIZ allant jusqu'à 5: 95 (schéma 1.19) (Wang et al., 2002).
0 C02Me
) l ),._
+ 1-Bu N PPh3 H 79 + t.p., 6 hSchéma 1.18 Synthèse d'tm énamide par oléfination de Wittig.
95% (E/Z=1:1)