UNIVERSITE TOULOUSE III- PAUL SABATIER U.F.R PHYSIQUE CHIMIE AUTOMATIQUE
THESE
pour obtenir le grade de
DOCTEUR DE l'UNIVERSITÉ TOULOUSE III
Discipline :
CHIMIE-BIOLOGIE-SANTE
Présentée et soutenue
Par
Christelle DUPOUY
Le 21 septembre 2007
Directeur de thèse : Jean-Marc ESCUDIER
JURY :
M. Eric DEFRANCQ
Professeur, Grenoble Rapporteur
M. Jean-Christophe FRANCOIS Chargé de Recherche INSERM, Paris Rapporteur
M. Jean-Jacques VASSEUR Directeur de Recherche CNRS, Montpellier Examinateur
Mme Valérie SARTOR Chargée de Recherche CNRS, Toulouse Examinateur
M. Alain VIGROUX Professeur, Toulouse Président
M. Jean-Marc ESCUDIER
Chargé de Recherche CNRS, Toulouse
Directeur de thèse
Laboratoire de Synthèse et Physico-Chimie de Molécules d'Intérêt Biologique, UMR 5068 31062 TOULOUSE CEDEX 9(FRANCE)
! " # $ % & ' ( ) ' * + & , # ) ' - . ( & * %+ / * / $ + " * / ) 0 ' ' % 0 + / / % ) ' + / + $ + $ ) % + ) / " + % + % 1 2 ) + * % 333 + / ) ' * % + 33 4 + / / * % + $$ ) ' , 5 6 7, 8 + $ , 5 9:: ; <) ' $ 6 7, 8) ' = ; * 5 , % $ % ) ( + 4 + + + / $ % * ) ' 1 2 % $ > ( ? @+ A A ( A A # A ' B A A 5 A C ) + $ + 1 2 > + 0 + 5 + ! A 1 2 >
? @+ 4 ? $ % @+ - + 0 $ + & + / $ * ) ' $ > ) + , + + $ - + + $ 33 > - + - + 1 !( 2 1 !( 2 ? C @ % 333 ' 1 2 > A D A = < A A ' A $ A 5 4 A 1 $ $ 2 >4 + & + ( 4 ?1 4 < # E 33+ 33+ # 332@ A % / A < / $ + % % ** 33 ' + + * 333 + $ + $ % / ) 333333
Introduction
1
Chapitre I Les acides nucléiques contraints
A Généralités sur les acides nucléiques ... 3
I. Nucléosides et Nucléotides... 3
II. Conformation des nucléotides ... 4
II.1 « Plissement » du sucre ...4
II.1.1 Autour de la liaison β-N-glycosidique 5 II.2.1 Orientation de la liaison C-4’/C-5’ 5 III. Les acides nucléiques ... 6
III.1 Structure primaire des acides nucléiques ...6
III.2 Structures secondaires des acides nucléiques ...6
III.1.2 La structure hélicoïdale de l’ADN-B 7 III.2.2 Structure de type A 9 III.3.2 Structures secondaires non hélicoïdales 9 B Les acides nucléiques modifiés ... 12
C Concept de préorganisation : application à la synthèse de nucléotides contraints modifiés. ... 14
I. Concept de préorganisation... 14
II. Stabilisation des structures secondaires par des ponts disulfures interbrins... ... 15
III. Structuration du sucre ... 16
III.1 Les nucléosides dérivés des hexoses...17
III.1.1 Les pentopyranosyles 17 III.2.1 Les hexopyranosyles 18 III.3.1 Les hexitols 18 III.2 Les nucléosides avec un ribose contraint ...19
III.1.2 Les thréofuranosyles 19 III.2.2 Les nucléosides bicycliques de Leumann 19 III.3.2 Les nucléosides contraints en forme Sud 21 III.4.2 Nucléosides contraints en forme Nord : les LNAs 22 IV. Rigidité au niveau du lien internucléosidique... 23
IV.1 Les liaisons amides ...23
IV.2 Les PNA ...24
IV.3 Les modèles du U-Turn...25
D Notre approche ... 30
Bibliographie ... 33
Chapitre II Les
α,β
α,β
α,β
α,β
-D-CNAs
A Synthèse des α,β-D-CNAs XT (X = A,T,G,C) et des LNA/α,β-D-CNAs TT... ... 41II. Rappel sur la synthèse des α,β-D-CNA TT ... 41
III. Synthèse «one-pot» des dinucléotides XT de l’α,β-D-CNA ... 42
IV. Synthèse des LNA/α,β-D-CNAs ... 45
B Analyses conformationnelles des α,βα,βα,βα,β-D-CNAs XT et des LNA/α,βα,βα,βα,β-D-CNAs TT ... 49
I. Rappel sur les α,β-D-CNAs TT... 49
II. Les α,β-D-CNAs XT (X = G, C ou A) ... 51
III. Les LNA/α,β-D-CNAs TT... 52
C Conclusion ... 58
Bibliographie ... 59
Chapitre III Les
ε,ζ
ε,ζ
ε,ζ
ε,ζ
-D-CNAs
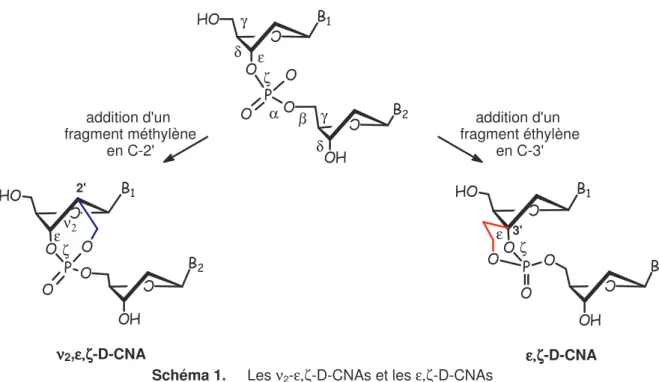
A Les νννν2-ε,ζε,ζε,ζε,ζ-D-CNAs et les (2’,5’)-νννν2-εεεε’,ζ,ζ,ζ,ζ’-D-CNAs... 62I. Rappel bibliographique ... 64
II. Synthèse des
ν
2-ε,ζ-D-CNAs et des (2’,5’)-ν
2-ε',ζ'-D-CNAs... 67II.1 Synthèse du 3-désoxy-3[(benzoyloxy)méthyl]-1,2-O-isopropylidène-α-D -allofuranose...67
II.2 Synthèse du 5-O-benzoyl-3-désoxy-3[(benzoyloxy)méthyl]-1,2-O-acétyl-D -allofuranose...67
II.3 Synthèse du 5-O-benzoyl-2-désoxy-2-[(benzoyloxy)méthyl]-3-O-formyl1-O-acétyl-D-allofuranose...68
II.4 Introduction de la base par N-glycosylation ...69
II.5 Synthèse des précurseurs acycliques (2’,5’)- ν2-ε',ζ'-D-CNAs et des ν2-ε,ζ -D-CNAs ...70
II.6 Synthèse des (2’,5’)- ν2-ε',ζ'-D-CNAs ...71
III. Etude conformationnelle des (2’,5’)-
ν
2-ε',ζ'-D-CNAs et desν
2-ε,ζ-D-CNAs 73III.1 Analyse structurale des (2’,5’)-ν2-ε'ζ'-D-CNAs ...73
III.2 Analyse structurale des ν2-ε,ζ-D-CNAs ...74
B Les ε,ζε,ζε,ζε,ζ-D-CNAs... 79
I. Rappels bibliographiques : choix des deux voies de synthèse ... 82
I.1 Addition nucléophile sur une cétone de nucléoside ...82
I.2 Addition nucléophile sur une cétone de sucre...83
I.3 Rétrosynthèse des xylo-ε,ζ-D-CNAs et des ε,ζ-D-CNAs...84
II. Synthèse des xylo-ε,ζ-D-CNAs... 85
II.1 Synthèse de la 5’,2’-O-tert-butyldiméthylsilyl-(3’S)-C-(acétate de méthyl)-uridine... ...85
II.2 Synthèse de la 5’,2’-O-tert-butyldiméthylsilyl-(3’S)-C-tosyloxyéthyl-uridine ...86
II.3 Synthèse des xylo-ε,ζ-D-CNAs ...86
III. Analyse conformationnelle des xylo-ε,ζ-D-CNAs... 88
IV. Approche de la synthèse des ε,ζ-D-CNAs (SC3’, RP) et (SC3’, SP) ... 93
IV.1 Synthèse du 5-O-(tert -butyldiphénylsilyl)-3-C-[(2-benzyloxy)éthyl]-3-O-benzyl-1,2-di-O-acétyl-D-ribofuranose...93
IV.2 Synthèse du nucléoside monomère substitué en 3’...94
IV.3 Approche de la réaction de couplage phosphoramidique ...96
C Conclusion ... 98
Bibliographie ... 99
Chapitre IV Propriétés des
α,β
α,β
α,β
α,β
-D-CNAs
A Synthèse des oligonucléotides ...102I. Synthèse des phosphoramidites des α,β-D-CNAs...102
II. Synthèse et purification des oligonucléotides...102
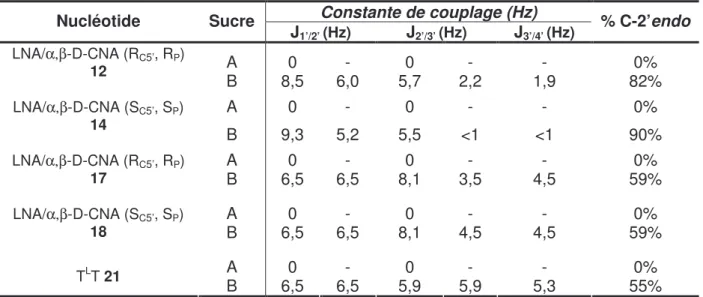
B Propriétés des α,βα,βα,βα,β-D-CNAs (g-, t) et (g+, t) ...107
I. Etude préliminaire de duplex ...107
II. Etude des effets de sels et de sélectivité...108
III. Etude structurale ...110
III.1 Dichroïsme circulaire...110
C Propriétés de l’α,βα,βα,βα,β-D-CNA (g-, t)...114
I. Etude de position ...114
II. Comparaison avec les propriétés des LNAs...116
D Propriétés de l’α,βα,βα,βα,β-D-CNA (g+,t) ...118
I. Mise en évidence de la stabilisation d’une tige-boucle ...119
II. Etudes de structures tige-boucles à 4 et 5 thymidines non appariées...121
II.1 Structures tige-boucles T4 ...121
II.1.1 Synthèse du tétramère TTTT 122 II.2.1 Structures tiges-boucles T4 123 II.2 Structures tige-boucles T5 ...126
II.3 Etude de l’effet de sel pour les structures tige-boucles T4 et T5 ...127
II.4 Dichroïsme circulaire...128
E Conclusion ...130
Bibliographie ...131
Conclusion
135
A Adénosine
Ac Acétate
AcOEt Acétate d’éthyle
ADN Acide désoxyribonucléique
ARN Acide ribonucléique
APTS Acide para-toluène sulfonique
Bz Benzoyle
Bn Benzyle
C Cytidine
CCM Chromatographie sur couche mince
D-CNA Dioxaphosphorinane-Constrained Nucleic Acid
COSY Correlation Spectroscopy
DCC Dicyclohexylcarbodiimide DMAP Diméthylaminopyridine DMF Diméthylformamide DMSO Diméthylsulfoxyde DMTr Diméthoxytrityle EP Ether de pétrole G Guanosine
HMBC Heteronuclear Multiple Bond Correlation
HMQC Heteronuclear Multiple Quantum Coherence
HPLC High Pressure Liquide Chromatography
LNA Locked Nucleic Acid
NOESY Nuclear Overhauser Enhancement Spectroscopy
Rf Rapport Frontal
Rdt Rendement
RMN Résonance Magnétique Nuclaire
SM Spectrométrie de masse
T Thymidine
TBDMS tert-butyldiméthylsilyle
TBDPS tert-butyldiméphénylsilyle
TFA Acide trifluoroacétique
THF Tétrahydrofurane
Ts Tosyle (para-toluène sulfonyl)
Introduction
-1-
Les nucléosides à conformation restreinte ont fait l’objet de nombreuses études ces dernières années en raison de l’importance des applications potentielles des oligonucléotides antisens. Cependant, la synthèse de ce type de nucléosides dans le but de mimer des structures secondaires hélicoïdales ou non d’ADN et d’ARN susceptibles de jouer un rôle biologique important n’a fait l’objet que de peu d’attention.
En effet, outre la conformation en double hélice, les acides nucléiques peuvent adopter de nombreuses structures secondaires telles que des boucles, des épingles à cheveux, des coudes ou des jonctions branchées. Ces structures secondaires contiennent des nucléotides non appariés ou non Watson-Crick et sont caractérisées par des conformations du squelette sucre-phosphate particulières qui diffèrent des états conformationnels réguliers établis pour les hélices double brins. Il est maintenant bien établi que ces structures jouent un rôle crucial dans les processus biologiques tels que les interactions ADN/protéines ou encore l’activité catalytique des ARNs. La détermination des rôles biologiques du squelette en fonction de la conformation est au centre de nombreuses études. Malheureusement les études structurales et fonctionnelles sont souvent compliquées en raison de la flexibilité et de la fragilité des fragments des simples brins. Avec des analogues stables de ces structures secondaires, nous pouvons espérer mieux comprendre leur rôle in vivo, l’information qu’ils portent et les éléments qui permettent leur reconnaissance par les autres macromolécules.
Notre approche générale consiste à la préorganisation d’un simple brin d’acide nucléique par l’introduction à différentes positions le long du squelette sucre-phosphate, de dinucléotides appelés D-CNA (dioxaphosphorinane Constrained Nucleic Acids) contraints par un cycle (1,3,2)-dioxaphosphorinane.
Le premier chapitre est une introduction générale aux acides nucléiques. Dans une première partie, nous rappellerons les notions de bases sur les acides nucléiques en vue d’une meilleure compréhension de la suite de ce travail. Puis nous nous intéresserons plus en détails aux acides nucléiques conformationnellement contraints permettant de stabiliser des structures secondaires par préorganisation du simple brin.
Les dinucléotides contraints par une structure dioxaphosphorinane : D-CNA feront l’objet des trois chapitres suivants. Nous expliciterons leurs synthèses, leurs structures conformationnelles et leurs applications.
Le deuxième chapitre est consacré aux dinucléotides α,β-D-CNAs dont les angles α et β sont contraints. Nous débuterons par l’optimisation de la voie de synthèse des α,β-D-CNAs
TT. Puis nous étendrons cette voie à la synthèse des α,β-D-CNAs XT (X = A, T, G ou C) en
faisant varier la base du nucléoside supérieur. Nous développerons également la synthèse et l’étude structurale des LNA/α,β-D-CNAs TT comportant un LNA sur le nucléoside supérieur.
Le troisième chapitre décrira la synthèse et la caractérisation de deux nouvelles familles de D-CNAs : les ν2-ε,ζ-D-CNAs et les ε,ζ-D-CNAs dans lesquels les angles de
torsion ε et ζ sont contraints. Nous commencerons par la synthèse des ν2-ε,ζ-D-CNAs et
d’un isomère de position, les (2’,5’)-ν2-ε’,ζ’-D-CNAs. Nous développerons ensuite la synthèse
et la caractérisation de dinucléotides dans lesquels le cycle dioxaphosphorinane sera en jonction spiro avec le sucre de configuration xylo (les xylo-ε,ζ-D-CNAs). Enfin, nous présenterons une approche de la synthèse de ces structures en configuration 2’-désoxyribose (les ε,ζ-D-CNAs).
Introduction
-2-
Le quatrième chapitre sera consacré aux propriétés induites par les α,β-D-CNAs pour
la stabilisation de structures secondaires d’acides nucléiques. Nous verrons d’abord que l’α,β-D-CNA (g-, t) présentant des valeurs d’angles α et β canoniques des formes A ou B de
l’ADN, favorise la formation de duplex de type-B. Nous développerons, ensuite les capacités de l’α,β-D-CNA (g+, t) dont l’angle α est contraint, dans une valeur atypique gauche (+), à
préorganiser une partie non appariée d’une structure secondaire épingle à cheveux (tige-boucle).
L’ensemble de ce travail sera résumé et des perspectives seront proposées. Enfin les protocoles expérimentaux seront décrits.
Chapitre I : Les acides nucléiques contraints
- 3 -
L'histoire de l'ADN débute en 1869 lorsqu'un médecin suisse, Miescher, découvre dans les noyaux extraits de cellules de pus une substance organique inconnue qu'il baptise "nucléine". Bientôt connues sous le nom d'acides nucléiques, ces molécules intriguent par leur richesse en phosphore, mais ne suscitent dans un premier temps qu'un intérêt passager. Il faudra attendre en effet 60 ans pour voir élucidées leurs structures primaires.
Les acides nucléiques ont depuis fait l'objet de très nombreuses études, et nous savons désormais qu'ils jouent un rôle central dans le stockage et l'expression de l'information génétique. Ceux-ci se distinguent en deux classes principales: les acides désoxyribonucléiques (ADN), dont le rôle est essentiellement la conservation de l'expression génétique, et les acides ribonucléiques (ARN), qui participent aux principales étapes de l'expression des gènes et à la biosynthèse des protéines.
A Généralités sur les acides nucléiques
[1-3]Les acides nucléiques sont des polymères de haut poids moléculaire obtenus à partir d'un enchaînement de motifs appelés nucléotides, eux-mêmes constitués par une base azotée, un sucre et un résidu phosphorique. Les acides nucléiques sont des édifices moléculaires complexes, il semble donc important dans un premier temps d'étudier les différents éléments qui les constituent.
I. Nucléosides et Nucléotides
Les nucléosides sont l’association par une liaison β-N-glycosidique d’une base purique ou pyrimidique et d’un sucre le ribose (pour l’ARN) ou le 2-désoxyribose (pour l’ADN). Cette liaison est formée entre l’azote 1 des pyrimidines ou l’azote 9 des purines et le carbone 1’ du sucre. La base est en position β c’est à dire qu’elle se situe au dessus du plan du cycle (en cis, du groupement 5’) (Figure 1).
Nucléosides à bases pyrimidines
Thymine Uracile Cytosine Guanine Adénine ADN : R = H ARN : R = OH
Nucléosides à bases purines
1' 8 4 3 2 5 1 7 6 9 2' 3' 5' 4' 1' 2' 4' 5' 3' 1 2 6 3 4 5 Nucléosides Nucléotides
Chapitre I : Les acides nucléiques contraints
- 4 -
Un nucléotide est un nucléoside portant un ou plusieurs groupements phosphates sur une des fonctions hydroxyles du sucre. Les ribonucléosides possèdent trois positions susceptibles d'être phosphorylées (les fonctions hydroxyles des carbones 2', 3' et 5') tandis que les désoxyribonucléosides ne peuvent être phosphorylés qu'en deux sites (3' et 5'). Pour la majorité des nucléotides, l’ester de phosphate se positionne sur l’hydroxyle porté par le carbone C-5’.
II. Conformation des nucléotides
Un nucléotide peut présenter diverses conformations liées à la géométrie du sucre, à la position de la base par rapport au sucre et aux valeurs des angles de rotation des groupements phosphates.
Les angles de rotation caractérisant un nucléotide sont appelés angles de torsion. Le squelette sucre - phosphate est défini par les angles de torsions α−ζ ; l'angle de rotation entre le sucre et la base est caractérisé par l'angle de torsion χ et enfin les angles de torsion du sucre sont notés de ν0 à ν4.
L’échelle utilisée en chimie organique pour définir ces angles a été proposée par Klyne et Prelog et fait appel aux termes suivants : gauche (+) (g+) = 60 ± 30°, anticlinal (+) (a+) =
120 ± 30°, trans (t) = 180 ± 30°, anticlinal (–) (a-) = 240 ± 30°, gauche (–) (g-) = 300 ± 30°
(Figure 2). χ ε δ γ β β α ζ α Squelette Sucre - Phosphate Sucre ADN ν ν ν ν ν 0° 180° 150° 90° 30° 330° 270° 210° Cis G auch e+ Gau che -A ntic lin al + A nt ic lin al -Trans g+ a+ a -g -cis trans
Figure 2. Nomenclature des angles de torsion du squelette sucre – phosphate et du sucre
II.1 « Plissement » du sucre
Le cycle furanose, contrairement aux bases, n’est pas plan, il adopte une structure plissée, susceptible de déformation. La forme privilégiée est celle dans laquelle trois atomes du cycle, dont l’oxygène sont coplanaires et les deux autres en dehors du plan.
Dans le cas des acides nucléiques, deux conformations principales sont observées : C-2’endo (forme Sud) et C-3’endo (forme Nord), le terme endo désignant le carbone du cycle qui est situé du même coté du plan que le carbone C-5’ et la base (Figure 3).
Chapitre I : Les acides nucléiques contraints
- 5 -
Dans les ADNs, les deux conformations sont observables mais c’est la forme C-2’endo
qui est la plus fréquente. En revanche, pour le cas des ARNs, la présence de l’hydroxyle en position C-2’, favorise la conformation C-3’endo.
3' 2'
C-3' endo
Nord C-2
' endo
Sud
Figure 3. Conformation du sucre ribose
II.1.1 Autour de la liaison β-N-glycosidique
La liaison entre la base et le sucre (β-N-glycosidique) définit un axe autour duquel la base est susceptible de tourner (angle de torsion χ). Les bases peuvent adopter deux positions : syn et anti (Figure 4). La conformation anti est la forme privilégiée pour l’ADN. Le conformère anti permet en effet aux plus petits atomes H6 (bases pyrimidiques) ou H8 (bases
puriques) de se situer au dessus du cycle du sucre minimisant les interactions répulsives entre les deux constituants. Dans le cas du conformère syn, ce sont les atomes O2 (bases
pyrimidiques) ou N3 (bases puriques) qui se placent dans cette position.
Position Syn de la guanine
Position Anti de la guanine
8
3 1' 1'
Figure 4. Conformation syn et anti de la liaison C-1’/N
II.2.1 Orientation de la liaison C-4’/C-5’
La liaison entre le carbone C-4' et le carbone C-5' définit un axe autour duquel le groupement phosphate peut tourner (angle de torsion γ) et ainsi se positionner différemment par rapport au sucre (Figure 5). Le phosphate en position 5' limitant le nombre de possibilités, les conformations les plus souvent observées dans les nucléosides sont les conformations synclinales (ou gauche) et antipériplanes (ou trans).
Chapitre I : Les acides nucléiques contraints - 6 - γ 5' 4' Synclinal (gauche (+)) Trans ’ ’ ’ ’ 4’ 5’ ’ ’ ’ ’ ’ 4’ ’ 5’
Figure 5. Conformations synclinales et antipériplanes autour de la liaison C-4’/C-5’
Pour les nucléosides pyrimidiques, la conformation synclinale est la plus souvent observée, dans ce cas le phosphate est au-dessus du sucre, tandis que pour les bases puriques les conformations synclinales et antipériplanes sont en proportions équivalentes. La conformation antipériplane est également favorisée lorsque la base porte des substituants encombrants.
III. Les acides nucléiques
III.1 Structure primaire des acides nucléiques
Les acides nucléiques sont des polymères de nucléotides ou de 2’-désoxyribonucléotides. Les liaisons phosphodiesters sont établies entre le groupement hydroxyle en position 3' d'un nucléotide et le groupement hydroxyle phosphorylé en position 5' du nucléotide suivant (Figure 6). Les chaînes sont donc lues dans le sens 5'-3'.
3’ 5’ Sen s 5’ 3’ -O -Adénine Guanine Cytosine Thymine Uracile 2’ 2’ 2’ 2’
Figure 6. Enchaînement des nucléotides dans l'ADN
Chapitre I : Les acides nucléiques contraints
- 7 -
La structure primaire des ARN diffère simplement par la présence d'un groupement 2'OH sur le sucre et le remplacement de la thymine par l'uracile (qui diffère de la thymine par l'absence de groupement méthyle en C5).
III.2 Structures secondaires des acides nucléiques
III.1.2 La structure hélicoïdale de l’ADN-B
L’ADN a été découvert en 1869 mais il ne suscita de l’intérêt que 60 ans plus tard avec l’élucidation de sa structure primaire. C’est grâce aux travaux d’Avery, MacLeod et McCarty, publiés pendant la Seconde guerre mondiale, en 1944, que l’on découvre le rôle des acides nucléiques dans le support de l’information génétique. Il restait alors à déterminer une structure susceptible de constituer un code et permettant d’envisager un mécanisme pour la réplication conforme du matériel génétique. La solution est apportée en 1953, dans la revue
Nature [4-6], dans laquelle on trouve dans le même volume trois publications consécutives
dont celle oubliée de Rosalind Franklin qui montre une magnifique diffraction des rayons X de l’ADN. Cette technique a largement contribué à l'étude de l'architecture moléculaire de l'ADN, dans un premier temps avec Astburry, puis Franklin et Gosling, et enfin Wilkins. C’est en utilisant ces informations que Watson et Crick établissent le modèle de la double hélice. Cette découverte, qui allait couronner Watson, Crick et Wilkins (auteur de la troisième publication) par un prix Nobel en 1962, ouvrait une nouvelle ère de l’étude du vivant : la biologie moléculaire. La conformation de l'ADN, structure en double brin antiparallèle, telle qu'elle est présente dans la cellule (encore appelée ADN B) est présentée de façon schématique dans la figure 7.
Grand sillon squelette sucre-phosphate Paire de bases Diamètre de l’hélice 20 Å Un tour d’hélice 10,5 pb/tour
Distance entre deux Distance entre deux nucléotides 3,4 Å nucléotides 3,4 Å
Petit sillon
Figure 7. Structure hélicoïdale de l’ADN de type B [7]
Dans ce schéma, le squelette sucre - phosphate est représenté par un ruban. Les bases sont situées à l'intérieur de la double hélice. Leurs cycles aromatiques sont empilés de façon perpendiculaire à l'axe de l'hélice. Chaque base subit une rotation de 35° par rapport à
Chapitre I : Les acides nucléiques contraints
- 8 -
celle qui la précède. Un tour complet de la double hélice (360°) contient donc environ 10 paires de bases.
La surface de la molécule est polaire et chargée négativement à cause des sucres et des résidus phosphates du squelette. Au contraire, l'intérieur de la double hélice est apolaire. Chaque base d'un brin est reliée par des liaisons hydrogène à une base complémentaire de l'autre brin. Une base purine est associée avec une base pyrimidique. Ainsi l'adénine est complémentaire de la thymine, et la guanine est complémentaire de la cytosine (Figure 8).
En plus, d’autres forces interviennent dans le maintien de la conformation bicaténaire hélicoïdale de l’ADN, telles que les interactions hydrophobes et électroniques entre les bases empilées les unes sur les autres
A
C T
G
Figure 8. Appariements pyrimidine – purines de type Watson –Crick
Les liaisons hydrogènes établies entre les bases de chacun des deux brins permettent de supporter une certaine flexibilité de la double hélice. Entre les deux brins se trouvent deux dépressions : une étroite appelée petit sillon et une large nommée grand sillon (Figure 9). Ce dernier est riche en atome d’azote et d’oxygène, il peut donc établir des liaisons hydrogènes (avec par exemple des chaines latérales d’acides aminés de protéines régulatrices de la transcription). En revanche, le petit sillon, tout aussi profond mais plus étroit est naturellement moins accessible.
grand sillon petit sillon Ω Sucre Pont phosphate Paire de bases Grand Sillon Petit Sillon
Figure 9. Schéma du petit et du grand sillon de l’ADN–B
En plus de l'appariement Watson-Crick que l'on trouve dans l'ADN, il existe d'autres types d'appariement entre les bases. Les plus fréquemment rencontrés sont les interactions Hoogsteen (Figure 10) observées entre un double brin d'ADN et un brin supplémentaire pour former des triples hélices.
C C+ G T A T Watson-Crick Hoogsteen Watson-Crick Hoogsteen
Chapitre I : Les acides nucléiques contraints
- 9 -
Les deux brins sont maintenus ensemble par des interactions non covalentes, on peut séparer la double hélice par chauffage en brins individuels (dénaturation). Par refroidissement lent des simples brins, l'hélice originale se reconstitue par appariement des bases (renaturation).
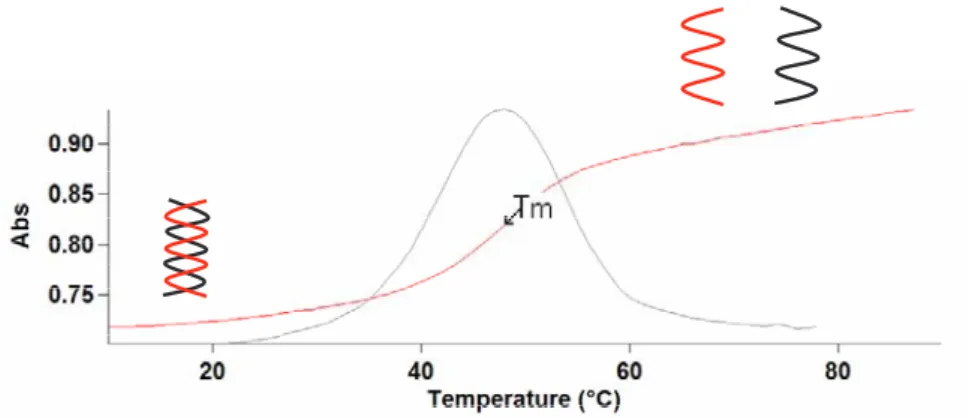
L'affinité d'hybridation de l'oligonucléotide pour la séquence complémentaire est caractérisée par la température de fusion Tm (melting temperature) du duplex (Figure 11).
Cette température est mesurée lors d'expériences d'hybridation: les brins complémentaires sont mis en présence et s'apparient pour former une double hélice. L'interaction - des bases empilées au niveau de l'hybride a pour effet de diminuer l'absorption UV à 260 nm (Figure 11). Lorsqu'on chauffe la cuve utilisée pour la mesure UV, les brins se désapparient, on observe une exaltation de l’absorbance à 260 nm. La température à laquelle 50% de l'acide nucléique est sous la forme de double hélice est appelée Tm.
Figure 11. Détermination du Tm : mesure de l’absorbance en fonction de la température
La structure secondaire de l'ADN représentée figure 7 (ADN-B) est la forme biologique la plus répandue. Cependant, l'ADN bicaténaire peut adopter au moins six formes (ADN-A à E et ADN-Z).
III.2.2 Structure de type A
La structure de type A est rencontrée souvent dans les ARNs. En effet, les doubles brins d'ARN ne peuvent pas avoir la structure de l'ADN-B car l'encombrement stérique provoqué par le groupement hydroxyle en position 2’ ne le permet pas. Il adopte une structure de forme A avec le sucre en conformation C-3’endo et le groupement hydroxyle est projeté à l’extérieur de l’hélice, loin des autres atomes. Sa structure est très similaire à celle de l'ADN de conformation A (Figure 12).
C’est une hélice droite avec un grand sillon très profond et un petit sillon quasi inexistant. Elle présente 11 paires de bases par tour d'hélice avec le sucre en conformation C-3’endo et la liaison glycosidique est en conformation anti.
Chapitre I : Les acides nucléiques contraints
- 10 -
ADN-A ARN-A
Figure 12. Comparaison des structures et des sillons de l’ADN-A et de l’ARN-A [7]
III.3.2 Structures secondaires non hélicoïdales
Outre la conformation en double hélice que nous avons présentée, les acides
nucléiques peuvent adopter de nombreuses autres structures secondaires [7] telles que des
boucles, des épingles à cheveux (ou tige-boucles), des coudes ou des jonctions branchées (Figure 13). Ces motifs sont une combinaison de paires Watson-Crick, non Watson-Crick et de paires non appariées et sont caractérisées par des conformations du squelette sucre-phosphate particulières qui diffèrent des états conformationnels réguliers établis pour les hélices doubles brins.
Les boucles et les épingles à cheveux se forment dans les doubles hélices d'ADN ou d'ARN en présence de nucléosides non appariés. Elles peuvent être constituées d'un ou de plusieurs nucléotides. Le classement en différentes catégories s'effectue selon leur localisation sur la séquence.
Epingle à Boucle cheveux Boucles Jonctions branchées Épingle à cheveux
Figure 13. Représentations schématiques des structures secondaires
Chapitre I : Les acides nucléiques contraints
- 11 -
Dans les ADNs, la formation de boucles peut provoquer des mutations du à des erreurs de réplication ou de transcription. Dans les ARNs, les boucles sont souvent présentes dans les régions importantes pour la reconnaissance protéine/ARN ou pour les activités catalytiques des ARNs tels que les ribozymes.
Les hélices branchées à trois ou quatre jonctions sont présentes dans l'ADN et l'ARN. Deux types d'hélices à quatre jonctions existent dans les ADNs: les cruciformes et les "Holliday". Dans les ADNs, les jonctions à quatre bras sont impliquées dans des processus biologiques importants tel que l'initiation de la réplication de l'ADN. Alors que dans les ARNs, ils constituent un élément important dans l'organisation des structures tertiaires responsables de l'activité catalytique et de la spécificité des ARNs.
Chapitre I : Les acides nucléiques contraints
- 12 -
B Les acides nucléiques modifiés
Le développement de la synthèse automatisée sur support solide depuis 30 ans a permis l’utilisation d’oligonucléotides dans la chimie, la biochimie et la médecine.
La production d'acides nucléiques modifiés a principalement été motivée par le développement de la stratégie antisens [8] et plus modestement par l'étude de l'étiologie de
l'ADN et de l'ARN [9-12] ainsi que par la recherche de modèle pour l'étude des structures non
hélicoïdale [13-15].
La stratégie anti-sens a été proposée il y a environ 30 ans par Zamecnick et Stephenson [16, 17]. L’approche est basée sur l’utilisation d’un oligonucléotide (antisens) qui
est complémentaire de l’ARN messager d’intérêt (sens) pour inhiber l’expression d’un gène au niveau de l’étape de traduction de la synthèse des protéines (Figure 14).
Noyau ADN Oligonucléotide antisens ARNm
X
Protéines CytoplasmeFigure 14. Schéma de la stratégie antisens
Cependant, ces oligonucléotides antisens doivent répondre à plusieurs critères : une bonne solubilité en phase aqueuse, une bonne biostabilité (stabilité vis-à-vis des exonucléases), biodisponibilité (bonne pénétration cellulaire) et une bonne affinité pour l’ARN cible. Ils doivent être également capables d’activer la ribonucléase RnaseH qui est une enzyme ubiquitaire qui coupe les duplex ADN/ARN. Pour obtenir de bonnes réponses, les oligonucléotides naturels sont loin d’apporter toutes les satisfactions aux critères. L’idée est d’utiliser des oligonucléotides présentant des modifications soit au niveau des bases, des sucres ou du lien phosphodiester pour améliorer les propriétés des acides nucléiques naturels et de développer des nouvelles applications (Figure 15).
Pour exemple, en 1997, Altmann et coll. ont étudié la stabilité de 200 acides nucléiques modifiés et ont montrés la faible proportion de modifications stabilisantes en particulier vis à vis des ARNs [18].
Chapitre I : Les acides nucléiques contraints - 13 - Modification au niveau du sucre Modification au niveau de la base Modification au niveau du lien phosphodiester
Figure 15. Différentes modifications possibles sur les acides nucléiques
Les phosphorothioates constituent la première génération d’analogue utilisé pour la stratégie anti-sens [19, 20]. Un atome d’oxygène du lien phosphodiester est remplacé par un
atome de soufre, créant ainsi un nouveau centre asymétrique. Ils présentent plusieurs avantages en particulier une meilleure résistance aux nucléases que l’ADN, une bonne solubilité dans l’eau et ils forment des duplex avec les ARNs reconnus par la RnaseH. Cependant, ils possèdent une moins bonne affinité d’hybridation avec l’ADN que l’ADN naturel.
La deuxième génération correspond aux nucléosides substitués au niveau de la position 2’ du sucre pour augmenter la résistance aux nucléases. Toutes les positions du sucre ont été ensuite modifiées. La synthèse de nucléosides modifiés au niveau du sucre a été également développée depuis une vingtaine d’année avec la découverte de l’AZT (3’-azido-3’deoxythymidine) et du ddC (2’,3’-dideoxycytidine) comme agents thérapeutiques pour le traitement du HIV [21, 22].
La découverte en 1990 de l’ARN interférent (ARNi) qui se lie spécifiquement avec l’ARN messager entrainant la dégradation de celui ci et donc l’inhibition de l’expression de la protéine correspondante a relancé l’intérêt pour la synthèse des acides nucléiques modifiés
[23-25].
Nous ne pouvons pas ici présenter l’ensemble des modifications apportées à la structure des acides nucléiques. Nous mettrons en avant les analogues d’acides nucléiques conformationnellement contraint au niveau du sucre ou du lien internucléosidique afin d’utiliser une préorganisation du simple brin pour atteindre une stabilité importante des structures de complexes.
Chapitre I : Les acides nucléiques contraints
- 14 -
C Concept de préorganisation : application à la
synthèse de nucléotides contraints modifiés.
I. Concept de préorganisation
Le concept de préorganisation fut introduit par Cram dans l’analyse et la recherche de petites molécules pour former l’association de complexe ligand-accepteur. La formation de ce dernier n’est pas favorisée par l’entropie puisque les trois degrés de rotation et de translation ainsi que la rotation interne des molécules séparées sont perdus. L’idée est de rigidifier la molécule dans la conformation qu’elle adoptera lors de la formation du complexe pour minimiser les pertes d’entropie.
L’ADN est un excellent candidat pour l’application de ce concept. En effet, la formation du duplex est entropiquement défavorable mais suffisamment enthalpiquement favorable pour permettre l’association [26] (Figure 16).
Facteurs favorisant
la formation des simples brins la formation des duplexFacteurs favorisant
• due aux liaisons hydrogènes • due à l’empilement des bases • de libre rotation des liaisons
• de libre translation et rotation
Entropie Enthalpie
Figure 16. Facteurs favorisant la formation des duplex et des simples brins
Ainsi une grande affinité et sélectivité peuvent être obtenues par une modification structurelle unique qui va rigidifier le simple brin d’oligonucléotide dans une configuration structurelle qui ressemble à celle qu’il adopte dans les duplex. L’analogue idéal de nucléotide doit avoir un simple brin d’oligonucléotide préorganisé c'est-à-dire qu’il doit posséder les valeurs des angles de torsion du squelette sucre – phosphate et le plissement du sucre similaire à ceux rencontrés dans les duplex de forme A ou B (Figure 17).
Simple brin
Preorganisation
Duplex Figure 17. Concept de préorganisation appliqué aux acides nucléiques
Chapitre I : Les acides nucléiques contraints
- 15 -
II. Stabilisation des structures secondaires par des ponts
disulfures interbrins
Pour stabiliser des structures secondaires non-hélicoïdales, de nombreuses équipes [14, 15, 27-29] ont eu l'idée de lier de manière covalente les brins des doubles hélices se trouvant à
proximité pour les stabiliser. Le choix de la liaison covalente s'est vite porté sur la fonction pont disulfure qui peut être formée à partir de fonctions thiols portées par chacun des brins.
Les groupements thiols ont été choisis en raison de la chimie redox douce qu'ils mettent en jeu pour la formation des liaisons disulfures. Celles-ci se forment avec un très bon rendement et sont stables dans de nombreux solvants.
Un nucléoside modifié portant un groupement thiol est introduit au cours de la synthèse automatisée sur support solide de l’oligonucléotide. Pour former le pont, il suffit d'amener sur le brin opposé de l’hélice un autre groupement thiol. Le duplex est ainsi lié de façon covalente par oxydation et devient stabilisé thermodynamiquement (Figure 18).
HS SH
Figure 18. Formation d'un pont disulfure
Les nucléosides les plus utilisés pour la formation de ponts disulfures sont représentés dans la figure 19.
n
n = 1-3
Chapitre I : Les acides nucléiques contraints
- 16 -
Le nombre de carbone de la chaîne alkyle peut être modulé pour faire varier la flexibilité du pont disulfure. C'est dans ce but, que des thymidines modifiées en position C-5 par des bras alkylthiols allant de 2 à 4 carbones ont été synthétisées [29].
Les applications des ponts disulfures sont nombreuses et variées. La première utilisation est la stabilisation de structures secondaires d'ADN non hélicoïdales (épingle à cheveux, boucle, ADN-Z...) [28] (Figure 20).
Site I
Site III
Site II
Figure 20. Stabilisation d'un ARNt par des ponts disulfures
Ces structures secondaires sont souvent difficiles à isoler et instables, ce qui rend les conditions de caractérisations physico-chimiques très délicates voire impossibles. L'insertion d'un pont disulfure permet de les stabiliser, sans pour autant perturber leurs géométries. Une telle approche a été utilisée pour stabiliser intramoléculairement une épingle à cheveux d'ADN. Cette approche pourrait se généraliser à l'étude de l'ensemble des structures des acides nucléiques.
Une autre application des ponts disulfures, est l'étude des changements de conformation de la structure de l'ADN lors de reconnaissance par une protéine [14]. La fixation
d'anticorps monoclonaux sur des analogues d'épingles à cheveux avec des ponts disulfures en est un bon exemple. L'utilisation d'épingles à cheveux modifiées par un pont disulfure va permettre de déterminer si la préorganisation ou la réorganisation est le facteur prédominant lors de la fixation des anticorps monoclonaux sur l'ADN. Si la réorganisation de la conformation des épingles à cheveux est requise pour la fixation de l'anticorps, les anticorps monoclonaux vont posséder une faible affinité pour la séquence modifiée par le pont disulfure qui est rigide et peu déformable. Par contre, si la préorganisation de la conformation est importante pour la complexation alors l'oligonucléotide le plus rigide aura une plus grande affinité que le ligand non modifié. Les ponts disulfures vont donc permettre une meilleure compréhension du processus de reconnaissance des anticorps monoclonaux sur l'ADN. Plus globalement, on peut espérer utiliser les ponts disulfures comme outil pour l'étude des changements de structures conformationelles observés sur l'ADN.
III. Structuration du sucre
Le cycle furanose des nucléosides ou des simples brins d’oligonucléotide est connu pour avoir une conformation assez flexible. Un moyen efficace pour préorganiser le simple brin et ainsi favoriser la formation du duplex est de réduire la flexibilité du sucre soit en
Chapitre I : Les acides nucléiques contraints
- 17 -
par la stratégie anti-sens, un grand nombre d’analogues de nucléotides conformationellement contraint au niveau du sucre ont été produits à partir de ce concept.
III.1 Les nucléosides dérivés des hexoses
Il est reconnu qu’un cycle furanose est plus flexible qu’un cycle pyranose. Les conformères d’un cycle à cinq chainons possèdent une barrière énergétique d’interconversion faible tandis que les cycles à six chainons sont beaucoup plus stables et rigides en forme chaise. L’extension du cycle furanose à six chainons devrait rigidifier et preorganiser le simple brin lors de la formation des duplex.
La stratégie d'Eschenmoser et coll.[10] a consisté à synthétiser des acides nucléiques
modifiés en remplaçant le cycle à 5 chainons par un cycle à 6 chainons. Ils s’en servent comme outils pour l'étude de l'étiologie de l'ADN et de l'ARN. En effet, compte tenu de la diversité biologique et chimique comment la nature est-elle arrivée au choix des acides nucléiques pentose et non hexose ? [30, 31]
III.1.1 Les pentopyranosyles
Le premier oligonucléotide synthétisé a été le pentopyranosyl-(2'-4') pRNA [9] (Figure
21). Cet analogue de l'ARN naturel contient le même nombre de liaisons covalentes internucléosidiques qu’un ARN, le cycle à cinq chaînons a simplement été remplacé par un cycle à six chaînons. Le cycle pyranose est en forme chaise avec la base en position équatoriale et le lien phosphodiester entre la position 2’ et 4’. Le p-ARN est attendu pour avoir une structure rigide puisque les angles de torsion δ et γ sont inclus dans le cycle pyranose. Il montre un appariement de type Watson-Crick plus fort et plus élevé avec les ARN que les acides nucléiques naturels (∆ Tm = +2 à 4 °C par paires de base). La structure du duplex s’apparente à une faible hélice twistée gauche.
δ γ
D-ββββ-ribo pRNA D-ββββ-xylo L-αααα-lyxo L-αααα-arabino
Figure 21. Famille des p-RNA
L’étude est étendue à des oligonucléotides contenant des isomères du pRNA : β-xylo, α-lyxo et α-arabino pentopyranosyles (2’-4’) avec la base toujours positionnée en équatoriale. Tous les membres de la famille montrent des résultats similaires au pRNA.
L'oligonucléotide construit avec le nucléotide α-arabino-pyranosyl est celui qui forme des duplex avec la plus grande stabilité en raison des contraintes stériques provoquées sur
le groupe phosphodiester par la base et le groupement hydroxyle en 3'. Les valeurs de Tm
obtenues varient cependant fortement en fonction de la séquence en raison de la forte inclinaison de l'axe des paires de bases [32]. En outre, pour tous les membres de la famille
des pentapyranosyles l’appariement des paires de bases de type Watson-Crick est orthogonal c'est-à-dire qu’ils sont capables de former des paires de bases entre eux mais pas avec des ARN ou des ADN naturels [33].
Chapitre I : Les acides nucléiques contraints
- 18 -
III.2.1 Les hexopyranosyles
En 1986, Eshenmoser introduit les Homo-ADN, composés hexopyranosyls 2'-3'-désoxygénés, premiers analogues de l’ADN avec comme sucre un cycle pyranose [10, 34, 35].
La base est positionnée au niveau du carbone anomérique et le lien phosphodiester est entre la position 4’ et 6’ (Figure 22).
"Homo-ADN" (R, R', R'', R'''=H) D-ββββ-Allo- (R, R''=OH et R',R'''=H) D-ββββ-Gluco- (R, R'''=OH et R',R''=H) D-ββββ-Altro- (R',R''=OH et R,R'''=H) '' ' ''' 4' 6' 2' 3' Figure 22. Homo-ADN
Ils ont été les premiers acides nucléiques modifiés montrant des paires de bases avec des interactions Watson-Crick supérieures à celles observées dans les ADN naturels. La grande stabilité thermodynamique des duplex Homo-ADN est attribuée à la rigidité du cycle pyranose comparée au cycle furanose, permettant ainsi un haut degré de préorganisation du simple brin lors de la formation des duplex. L’analyse par RMN montre que les duplex de
Homo–ADN adoptent une structure quasi-linéaire [36]. Comme pour les p-RNA l’appariement
des paires de bases est orthogonal aux bases naturelles. Les oligonucléotides correspondants sont incapables de former des duplex avec l’ADN ou l’ARN.
Les composés hydroxylés nommés β-allo-, β-gluco-, et β-altropyranosyl au contraire forment des paires de bases très faibles ou inexistantes [10]. Cette inaptitude s'explique par
l'encombrement stérique provoqué par le groupement hydroxyle en position 2'.
III.3.1 Les hexitols
Conduit par la stratégie anti-sens, Herdewijn a développé une série d’acides nucléiques héxitol [37, 38]. Le 1,5-anhydrohexitol-ADN (HNA) s’est avéré le plus prometteur.
Les HNA comportent une différence par rapport aux Homo-ADN, la base a bougé de la position 1’ à 2’ (Figure 23). Un groupement méthylène sépare le carbone portant la base et l’oxygène du cycle. Cette modification apparemment mineure provoque des changements importants sur les propriétés des paires de base dans les duplex avec les HNA, l'ADN et l'ARN.
Figure 23. HNA : 1,5-anhydrohexitol-ADN
Les oligonucléotides incorporant une ou plusieurs modifications HNA forment des duplex par appariement de type Watson-Crick plus stables avec les ARN qu’avec les ADN. HNA est sensible à la présence d’acides nucléiques non appariés (mismatch). L’analyse par dichroïsme circulaire des duplex HNA/ARN et HNA/ADN montre une structure similaire à l’ARN de type A. De plus, les angles de torsion du simple brin HNA dans un duplex
HNA/ARN adoptent une conformation de type A à l’exception de l’angle γ qui est contraint
Chapitre I : Les acides nucléiques contraints
- 19 -
III.2 Les nucléosides avec un ribose contraint
III.1.2 Les thréofuranosyles
Les oligonucléotides α-thréofuranosyl-(3'-2') appelés plus couramment TNA [40],
dérivent de sucres furanoses et constituent une des plus simples alternatives aux acides nucléiques (Figure 24). Les TNA s'hybrident efficacement avec les ARN et les ADN [11]. Les
duplex TNA/ARN possèdent une stabilité comparable à celle des duplex ARN/ARN. Par ailleurs, les oligonucléotides TNA/TNA forment des duplex avec des paires de bases de type Watson-Crick de stabilité semblable à celle des ARN.
3'
2' 1' TNA α(3'-NH)-TNA X=O,Y=NH-L-Tréofuranosyl X=O, Y=OH
2
(2'-NH)-TNA X=NH, Y=OH
Figure 24. Les TNA
Une étude aux rayons X et par dichroïsme circulaire de la structure cristalline d'un duplex d'ADN de type B possédant une modification TNA par brin montre que le nucléotide TNA s’accommode facilement dans une structure de type B sans modification de l’empilement des bases [41]. En revanche, un changement conformationnel important du
squelette sucre phosphate, au niveau de l’incorporation du TNA est observé. Ce dernier semble induit par la diminution du nombre de liaisons covalentes internucléosidiques et la conformation C-4’exo du sucre.
Les (3'-NH)-TNA et (2'-NH)-TNA [42], analogues des TNA dans lesquels le squelette
sucre-phosphate est substitué par un lien phosphoramidate ont ensuite été étudiés (Figure 24). Ils s'associent avec les ARN et les ADN par des paires de bases de type Watson-Crick comme les TNA.
III.2.2 Les nucléosides bicycliques de Leumann
En 1994, Leumann et coll. proposent les premiers nucléosides bicycliques : les bicyclo-ADNs qui possèdent un pont éthylène entre les carbones 3’ et 5’ [43-45]. Ce dernier permet de
figer la liaison entre les carbones 4’ et 3’, celle entre les carbones 4’ et 5’ et aussi il permet de contraindre les angles de torsion δ et γ (Figure 25).
3' 5' γ 2' C-2' endo 4' δ δ γ
Chapitre I : Les acides nucléiques contraints
- 20 -
L’analyse structurelle par RMN et diffraction des rayons X montre que le sucre est en conformation C-2’endo (Sud) et que l’angle de torsion γ adopte une valeur de +149° (a+) avec le groupement hydroxyle en 5’ en position équatoriale. Cette valeur d’angle diffère considérablement de celle pour les duplex de forme A (γ = + 66, g+) ou B (γ = + 54, g+). La valeur de l’angle de torsion δ (+ 118, a+) est en revanche similaire à celle rencontrée dans
les duplex d’ADN–B (+ 122°, a+).
Ces dérivés hybrident fortement avec l’ARN bien qu’ils adoptent une structure assez proche de celle observée dans les duplex de forme B. C’est le cas par exemple de l'homodécamère adénine bicyclo-ADN qui s'associe plus fortement avec un brin polyuridine (∆Tm = +13°C) que ne le fait un brin d'ADN polyadénine (∆Tm = -4°C). Lors de la formation du duplex, les bases s’associent par des interactions de type Hoogsteen plutôt que Watson-Crick. Ce changement peut être directement relié à la valeur atypique de l’angle γ.
La deuxième génération de ces composés sont les tricyclo-ADNs avec introduction d'un cyclopropane permettant de renforcer la stabilité de la conformation du nucléoside [46, 47].
Le cycle furanose est contraint en forme C-3’endo (nord) et l’angle de torsion γ est maintenu dans sa conformation anticlinale (a+) comme pour les bicyclo-ADNs (Figure 26).
4' 5' γ
β C-3' endo
δ β γ δ
Figure 26. Tricyclo - ADN
Les tricyclo-ADN offrent une bonne affinité d'hybridation avec l'ARN et l'ADN. Les
valeurs de ∆Tm obtenues pour des oligonucléotides décamères avec l'ADN et l'ARN sont de
respectivement +1,2 et +2,4°C par modification. Les duplex tricyclo-ADN/tricyclo-ADN présentent une plus grande stabilité avec des valeurs de ∆Tm de l'ordre de +3,1°C par modification par rapport aux ADN.
Les analyses spectroscopiques et la modélisation moléculaire montrent que les duplex tricyclo-ADN/ADN adoptent, contrairement aux bicyclo-ADN, une structure proche des ARN de type A avec des paires de bases de type Watson-Crick. Ce changement de conformation provient du cycle cyclopropane qui maintient le cycle furanose dans une conformation Nord (C3'-endo) et impose par contrainte stérique une valeur à l'angle β de 87° (g+) (ADN de type
A ou B β = t) et une valeur de + 92° (g+) pour l’angle δ (observée dans l’ADN de type A).. Ces
variations semblent être à l'origine de l'amélioration des interactions Watson-Crick. Les tricyclo-ADN sont également de bons candidats pour la formation de triplex avec l’association de paire de bases de type Hoogsteen [48].
Pour ajuster l’angle γ dans une conformation typique d’ADN-A ou B (g+), Imanishi et
coll. ont synthétisé le 5’-amino-3’5’-BNA (Bridged Nucleic Acid) [49] (Figure 27). Un pont
méthylène est ajouté entre les carbones 5’ et 3’ et l’hydroxyle en 5’ est remplacé par une amine. D’après une détermination de la structure par diffraction des rayons X, l’angle γ à une valeur + 28° (g+) suffisamment proche à la valeur rencontrée dans un duplex de type A ou B.
En revanche, le sucre adopte une conformation intermédiaire entre C-2’endo et C-3’endo
(O-4’endo). Ce dérivé, une fois incorporé dans un oligonucléotide, montre une bonne capacité
Chapitre I : Les acides nucléiques contraints - 21 - 3' 4' 5' γ Figure 27. 5’-amino-3’5’-BNA
III.3.2 Les nucléosides contraints en forme Sud
L’équipe de Poul Nielsen s’est intéressée aux composés bicycliques de Leuman en série ribose par réaction de métathèse des oléfines [50, 51] (Figure 28). Malgré la présence
d'une fonction hydroxyle en position 2', le sucre conserve une conformation de type ADN (C-2'endo). 3' 4' 5' δ γ C-2' endo, Sud γ δ 5' 4' 3'
Figure 28. Bicyclo – ADN en série ribose
A partir des composés bicycliques, l'équipe de Poul Nielsen a synthétisé des nucléosides tri-cycliques [52] (Figure 29) très contraints. Ils adoptent une conformation de type
Sud (C-2'endo). Mais la contrainte imposée à la liaison C-4'/C-5' (angle de torsion γ à +137°, a+) et l'impossibilité pour la base d'adopter une conformation anti à cause de l'encombrement
stérique provoquent une forte diminution de l'affinité des oligonucléotides modifiés pour des séquences d'ADN et d'ARN.
C-2' endo, Sud
γ γ
Figure 29. Tricyclo ADN de Nielsen
Pour pallier au problème de la contrainte imposée à la liaison C-4'/C-5', Imanishi et coll.
[53] ainsi que Nielsen et coll. [54, 55] développèrent en même temps le 3’,4’-trans nucléoside
bicyclique (trans-3’,4’-BNA) par introduction d’un cycle à six chainons entre le carbone 3’ et 4’ (Figure 30). La liaison C-4'/C-5' n’est plus contrainte et la conformation du sucre est
bloquée en sud. L’incorporation du nucléoside modifié trans-3’,4’-BNA avec le groupement
Chapitre I : Les acides nucléiques contraints
- 22 -
formation des duplex avec l’ADN et l’ARN [56] (∆Tm = -10°C en moyenne). Cette
déstabilisation peut être expliquée par l’encombrement stérique crée par la présence du groupement méthoxy en position C-2’ et de la valeur de l’angle δ (+ 175°, t) qui dévie de celle rencontrée dans les duplex de type B (δ = +123°, a+).
4' 5' 3' 2' γ δ 4' 5' 3' 2' γ δ
Figure 30. 3’ 4’-trans Bicyclo ADN (trans-3’,4’-BNA)
III.4.2 Nucléosides contraints en forme Nord : les LNAs
De nombreux nucléosides avec le sucre contraint en conformation C-3’endo ont montré
une augmentation de la stabilité des duplex.
Imanishi fut le premier à décrire la synthèse des LNAs en 1997 [57, 58]. Ses travaux
furent rapidement complétés par Wengel [59, 60]. Le LNA est un analogue de l’ARN dans
lequel le sucre est contraint par un pont méthylène entre l’oxygène en 2’ et le carbone en 4’ (Figure 31).
Les LNAs présentent une affinité d'hybridation avec les ADN (∆Tm +3 à +5°C) et les
ARN (∆Tm +4 à +8°C) sans précédent. Avec une température de fusion de 92°C, les duplex
LNA/LNA (5'-(GLTLGLALTLALTLGL MeCL)/3'-(MeCLAL MeCLTLALTLAL MeCLGL)) sont quant à eux les
duplex d'acides nucléiques les plus stables connus à ce jour [60].
La spectroscopie RMN et la diffraction par les rayons X du nucléotide LNA ont mis en
évidence une conformation Nord (C-3'endo) pour le sucre fortement bloquée. Sur
l’oligonucléotide seul ou hybridé avec de l’ADN, les analyses RMN confirment la conformation du sucre en Nord mais aussi mettent en évidence la capacité du LNA à faire basculer le sucre de ces proches voisins non modifiés de la conformation Sud en Nord [61-63].
L’insertion de LNA dans un simple brin préorganise le squelette sucre-phosphate de ce dernier dans une conformation favorisant la formation de duplex. Une plus grande stabilité est observée pour les duplex LNA/ARN que LNA/ADN car la conformation du sucre en Nord est caractéristique des duplex ARN/ARN et non des duplex ADN/ADN (le sucre préfère une conformation Sud). C-3' endo X = CH3 méthylphosphonate X = S- phosphorothioate 2'-thio-LNA X = S 2'-amino-LNA X = NH ENA X = CH2O PrNA X = CH2CH2O X = CH2CH2 4' 2' LNA
Chapitre I : Les acides nucléiques contraints
- 23 -
En raison de la très forte capacité d'hybridation des LNAs, des nombreux analogues avec le sucre contraint en forme Nord ont été préparés [64] (Figure 31). Les premières
modifications ont porté sur l'atome d'oxygène 2' qui a été remplacé successivement par un atome de soufre [65] puis d'azote [66] montrant des propriétés d’hybridation avec l’ADN et
l’ARN semblable au LNA original. Le 2’-amino-LNA est utilisé en fluorescence en fonctionnalisant la position N-2’ avec des groupements pyrènes [67].
Des analogues du LNA ont été synthétisés par modification du lien méthylène entre l’oxygène 2’ et le carbone 4’. Imanishi et coll. développa les ENA (ethylene-bridged nucleic acids) dans lequel le groupement méthylène est remplacé par un groupement éthylène [68, 69].
Les ENA possèdent une forte capacité d’hybridation avec les ARN (semblable au LNA) mais plus faible vis-à-vis de l’ADN. L’introduction d’un groupement propylène (PrNA) montre une déstabilisation des duplex avec l’ADN (∆Tm = -2,0°C) et l’ARN (∆Tm = -1,0°C).
Nielsen et coll. ont synthétisé des analogues du LNA avec l’insertion d’un groupement propyle entre l’oxygène en 2’ et le carbone 4’ [70]. Ils ont montré l’importance de l’oxygène
dans le bicycle lors de la formation et la stabilisation des duplex ADN/ADN de type A. Par contre dans la formation des hybrides ADN/ARN de type A, la présence de l’oxygène n’est pas nécessaire.
Des LNAs possédant des liens internucléotidiques phosphorothioate [65], amides [71] et
méthylphosphonates [72] sont ensuite apparus montrant pour tous une affinité d'hybridation
avec l'ADN et l'ARN inférieure à celle obtenue avec les LNA.
IV. Rigidité au niveau du lien internucléosidique
Une des premières approches est de limiter la rotation des liaisons autour du squelette sucre – phosphate de l’ADN ou de l’ARN en remplaçant le groupement phosphate par des liaisons possédant moins de degrés de liberté.
Ces modifications ont été réalisées au niveau des cinq liaisons internucléosidiques. Dans un grand nombre de cas, la charge négative a été supprimée permettant ainsi d’augmenter l’affinité d‘hybridation.
IV.1 Les liaisons amides
Les oligonucléotides dans lesquels le groupement phosphate a été substitué par une liaison amide [73-76] s'hybrident avec l'ARN complémentaire avec une bonne affinité (Figure
32).
ADN amide-3-ADN amide-4-ADN
Chapitre I : Les acides nucléiques contraints
- 24 -
Les duplex ARN/oligonucléotides modifiés par un lien amide-3-ADN présentent un ∆Tm
par rapport aux ARN naturels de +0,6°C par modification. Le ∆Tm correspond à la différence
entre le Tm du duplex formé par l'oligonucléotide modifié et le Tm du duplex formé à partir de la même séquence non modifiée. Un ∆Tm positif signifie donc que l'oligonucléotide modifié a
une plus grande affinité d'hybridation avec le brin complémentaire que l'oligonucléotide naturel.
IV.2 Les PNA
Les acides nucléiques peptidiques (PNA) sont une famille d’analogues non nucléosidiques (Figure 33). Ils ont été décrits pour la première fois en 1991 par l'équipe de Peter Nielsen [77].
Dans les PNA, le sucre est absent et c'est par un bras méthylène carbonyle que la base est rattachée au squelette. Le phosphore à quant à lui disparu au profit d'une chaîne polyamine composée d’une répétition de N-(2-aminoethyl)glycyl qui a pour avantage d'être neutre et achirale. Ces particularités permettent de contourner certains problèmes inhérents aux analogues des oligonucléotides tels que la répulsion électrostatique avec les acides nucléiques chargés négativement et la chiralité de l'atome de phosphore. Un avantage supplémentaire des PNA est l'extrême stabilité envers les nucléases, protéases et peptidases. De plus, les deux terminaisons des PNA sont faciles à conjuguer avec une grande variété de marqueurs: peptides, biotine, fluorescéine.
Les PNA s’hybrident aussi bien avec l’ADN qu’avec l’ARN et ce avec une meilleure affinité et spécificité que l’ADN naturel [78]. Le ∆T
m par rapport aux ADN naturels est de l'ordre
de +1°C par paires de bases. Un duplex ADN/PNA pentadécamère présente une valeur de Tm de 69,5°C alors que le duplex correspond ADN/ADN possède un Tm = 53,3°C.
ADN
Figure 33. Pseudo peptides non nucléosidique : PNA
Les PNA sont aussi susceptibles de former des triples hélices en s’hybridant avec l’ADN double brin si les séquences sont appropriées.
Chapitre I : Les acides nucléiques contraints
- 25 -
Les PNA présentent quelques inconvénients comme la faible solubilité à l’eau et le manque de sélectivité car ils s’hybrident aussi bien avec l’ADN que l’ARN de manière parallèle ou anti-parallèle. L’équipe de Ganesh synthétisa des analogues de PNA en appliquant le concept de préorganisation [79, 80]. En effet, le simple brin du PNA est acyclique
et flexible. Lors de la formation du duplex, l’enthalpie est augmentée par les interactions faibles (liaisons hydrogènes et empilement des bases) entre les simples brins et en même temps un changement conformationnel du simple brin du PNA est observé entrainant une diminution de l’entropie. L’idée est de synthétiser des PNA modifiés adoptant la conformation favorable lors de la formation du complexe.
Un des exemples consiste à synthétiser des analogues de PNA avec l’angle de torsion β contraint pour augmenter la sélectivité d’hybridation avec les ARN ou les ADN. En effet, l’analyse par RMN et rayon X comparée [81], montre que l’angle de torsion β adopte la valeur
60-70° dans les duplex PNA/ARN alors que dans les duplex PNA/ADN β a pour valeur 140°.
Pour contraindre l’angle β, un cyclohexane est introduit (Figure 34) : deux catégories
de composés de quatre diastéréoisomères chacun sont synthétisés : le trans
-cyclohexanyl-PNA (trans-cpPNA) et le cis-cyclohexanyl-PNA (cis-cpPNA). Pour le trans-cpPNA, les quatre diastéréoisomères déstabilisent les duplex avec l’ADN et l’ARN [82]. Cette déstabilisation peut
s’expliquer par la valeur incompatible de l’angle de torsion β (+180°) par rapport à celle
observée lors de la formation des duplex. En revanche, le cis-cpPNA forme des duplex plus
stables avec les ARNs qu’avec les ADNs [83]. Cette préférence pour les ARN est expliquée
par la valeur de β (+ 65°) qui correspond à celle observée dans les duplex PNA/ARN.
aeg-PNA α β γ δ ε β β β trans-cpPNA β cis-cpPNAβ cis-chPNAβ
Figure 34. Analogues des PNA
Le remplacement du cyclohexane par un cyclopentane modifie les propriétés d’hybridations. En effet, le cis-cyclopentylPNA (cis-chPNA), avec l’angle β contraint à une valeur + 25°, hybride aussi bien avec les ADNs qu’avec les ARNs [84]. Cela peut s’expliquer
par le fait que le cycle à cinq chainons étant plus flexible qu’un cycle à six, il peut donc plus facilement s’adapter conformationnellement lors de la formation des duplex.
IV.3 Les modèles du U-Turn
Le groupe de Sekine s'est intéressé à une structure particulière rencontrée dans les boucles des anticodons des ARNt [85] et plus récemment découverte dans le site actif des
ribozymes à tête de marteaux [86, 87] : le "U-Turn" qui est une structure coudée (Figure 35).
Les structures coudées naturelles sont généralement stabilisées par la fixation de protéines sur la séquence ou par la structure tridimensionnelle de la molécule. Dans ce cas,
Chapitre I : Les acides nucléiques contraints
- 26 -
la molécule comporte souvent un nucléoside rare permettant par exemple la stabilisation par liaison hydrogène de cette structure. Ce type de stabilisation a par exemple été observé dans l'ARNtarg de E. Coli. Une 5-méthylaminométhyl-2-thio-Uridine (mnm5s2U
34) est capable
de former une liaison hydrogène entre le groupement amino et l'hydroxyle en position 2' du nucléoside précédent (U33). Cette interaction stabilise d'une part la structure en coude de
l'acide nucléique mais aussi la conformation C-3'endo de l'uridine essentielle pour la reconnaissance spécifique de l'anticodon.
L'objectif des travaux de Sekine et coll. est de synthétiser des motifs coudés artificiels par modification chimique, qui pourront être stables indépendamment de ces interactions complexes. Ces composés pourront permettre d'étudier le rôle de ces conformations et d'obtenir des motifs uniques tels que des boucles très stables.
Sekine et coll. ont dans un premier temps choisi de mimer la structure U-Turn en introduisant un lien covalent entre les deux nucléosides sous la forme d'un macrocycle [13, 88]
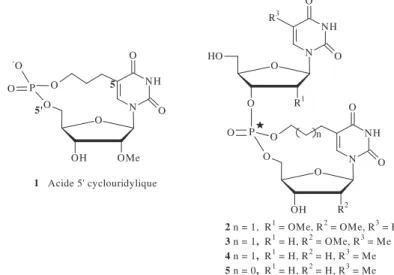
(Figure 35). Les dinucléosides diuridine monophosphate 1-3, sont cyclisés par un lien carbamate ou amide (n=0 ou 1). 1 n = 0 carbamate, R=OH 2 n = 1 amide, R=OH 3 n = 1 amide, R=H U-Turn mnm5s2U34 U33
Figure 35. U-Turn (coude naturel) et les mimes de Sekines
La spectroscopie RMN, le dichroïsme circulaire et la modélisation moléculaire ont permis d'obtenir de nombreuses informations sur ces structures. Il en ressort que le composé 2 contenant le lien amide présente la structure la plus proche des ARN coudés, la meilleure résistance aux nucléases et la plus grande rigidité. Par ailleurs, la conformation des sucres riboses dans cette structure est Sud (C-2'endo).
Le macrocycle 3 analogue en série 2'-désoxyribose du composé 2 a été synthétisé [89]
par la méthode phosphoramidite classique puis incorporé dans des oligonucléotides tétra- déca- et héxadéca-mères. Les études par dichroïsme circulaire ont permis de montrer là encore une augmentation de la rigidité conformationelle. Cependant celle-ci est inférieure à celle obtenue lors de l'incorporation du composé 2. La conformation majoritaire des sucres est toujours Sud (C-2'endo). La conservation de la structure coudée a été démontrée par la spectroscopie RMN et le dichroïsme circulaire. Elle a aussi été confirmée par la diminution de la stabilité thermique de ces duplex par rapport aux structures non modifiées ( Tm= -20°C pour le décamère).
Ces oligonucléotides coudés artificiellement pourraient servir de modèles pour l'étude des structures coudées naturelles présentes dans l'ADN lors de la fixation de protéines

![Figure 12. Comparaison des structures et des sillons de l’ADN-A et de l’ARN-A [7]](https://thumb-eu.123doks.com/thumbv2/123doknet/2177453.10359/24.892.250.650.108.387/figure-comparaison-structures-sillons-l-adn-l-arn.webp)