Reprogrammation des neutrophiles en conditions inflammatoires
Étude du transcriptome des neutrophiles reprogrammés en présence de cytokinesMémoire
Flavia Ribeiro de Vargas
Maîtrise en Microbiologie-immunologie Maître ès Sciences (M.Sc.)
Québec, Canada
iii
Résumé
Le neutrophile est une cellule caractérisée par sa «plasticité» qui, dans des conditions pathologiques, lui permet d'acquérir des nouvelles fonctions qui sont impliquées dans la pathogenèse des maladies inflammatoires chroniques comme dans l’arthrite auto-immune (AAI). Les études suggèrent que le neutrophile peut être impliqué dans la résorption osseuse qui accompagne la maladie. Pour mieux analyser cette possibilité, nous avons mis des neutrophiles en présence d’un cocktail de cytokines présentes dans l’AAI afin d’analyser la modulation des gènes liés à la différenciation en cellule de type ostéoclaste. Les résultats ont démontré que le neutrophile est capable d’acquérir de nouvelles fonctions en présence de cytokines, notamment des fonctions importantes dans l’orchestration du système immunitaire et la réponse inflammatoire. Le neutrophile est également une source de molécules permettant la résolution de l’inflammation, notamment l’élafine, ce qui pourrait être un agent thérapeutique prometteur dans les maladies inflammatoires chroniques comme l’AAI.
v
Abstract
The plasticity of neutrophils is a very useful property that in pathological conditions allow these cells to acquire new functions involved in the pathogenesis of chronic inflammatory diseases such as autoimmune arthritis (AIA). Studies suggest that neutrophils may be involved in bone resorption accompanying chronic inflammatory diseases. To better analyze this possibility, we have exposed neutrophils to a cocktail of cytokines present in AIA and studied their transcriptome to decipher the modulation of genes related to the differentiation into an osteoclast cell type. The results demonstrated the ability of neutrophils to acquire new functions when exposed to cytokines, including important functions involved in the orchestration of the immune system and inflammatory response. We also demonstrate that the neutrophil is a source of molecules involved in the resolution of inflammation, especially elafin, which seems to be a promising therapeutic agent in chronic inflammatory diseases and perhaps in AIA.
vii
Table des matières
Résumé ... iii
Abstract ... v
Liste des tableaux ... ix
Liste des figures ... xi
Liste des abréviations ... xiii
Remerciements ... xv
Avant-propos... xvii
Chapitre I. Introduction... 1
I.1. Le neutrophile ... 1
I.1.1. Introduction ... 1
I.1.2. Production et régulation des neutrophiles dans la circulation ... 1
I.1.3. Les fonctions du neutrophile ... 3
I.1.4. Le neutrophile et l’inflammation chronique ... 13
I.2. Les Arthrites Auto-immunes (AAI) ... 15
I.2.1. Introduction ... 15
I.2.2. Épidémiologie de l’AAI ... 15
I.2.3. Étiopathogénèse ... 16
I.2.4. Présentation et évolution clinique... 19
I.2.5. La résorption osseuse ... 19
I.2.6. Traitement de l’AAI ... 22
I.2.7. Le rôle du neutrophile dans l’AAI ... 26
Chapitre II. Hypothèse et objectifs de l’étude ... 31
Chapitre III. Matériels et méthodes ... 33
III.1. Isolation de neutrophiles et incubation avec cytokines ... 33
III.2. Récupération de cellules viables après 48h d’incubation ... 34
III.3. Extraction d’ARN ... 35
III.4. Étude par Biopuce et analyse des résultats ... 35
Chapitre IV. Résultats ... 39
IV.1. Analyse génerale des résultats de l’étude par biopuce ... 39
IV.2. Analyse de la reprogrammation en ostéoclaste ... 41
IV.3. Analyse d’autres types de reprogrammation ... 46
IV.4. Analyse de gènes reliés aux fonctions d’inflammation et de réponse immune ... 47
Chapitre V. Human neutrophils reprogrammed into long-lived leukocytes express anti-inflammatory genes mainly against peptidases. Promotion of trappin-2/elafin. ... 49
V.1. Résumé ... 51
V.2. Abstract ... 53
V.3. Introduction ... 55
V.4. Materials and Methods ... 57
V.5. Results ... 63 V.6. Discussion ... 71 V.7. Abbreviations ... 75 V.8. Competing interests ... 77 V.9. Authorship ... 79 V.10. Acknowledgements ... 81 V.11. References ... 83
V.13. Tables ... 101 Chapitre VI. Discussion générale ... 105 Chapitre VII. Références ... 111
ix
Liste des tableaux
Table I.1. Les sérines-protéases du neutrophile et leurs cibles biologiques ………... 9
Table I.2. Cytokines pouvant être exprimées par les neutrophiles humains ………13
Table I.3. Incidence actuelle et future de la PAR par sexe au Canada de 2010 à 2040 selon le dernier rapport de l’Alliance Arthrite Canada ... 16
Table I.4. Principaux DMARDs utilisés dans les traitement de l'AAI ……… 24
Table I.5. Médiateurs inflammatoires présents dans le LS qui peuvent activer les neutrophiles ………. 27
Table IV.1. Table avec le résumé de l'analyse de l'étude ………...39
Table IV.2. Gènes reliés aux fonctions de résorption osseuse ……….. 43
Table IV.3. Gènes reliés à la différentiation et fusion en ostéoclastes ………. 44
Table IV.4. Gènes reliés à la formation de la bordure en brosse ……….. 45
Table IV.5. Gènes reliés à la reprogrammation en cellule dendritique ………. 46
Table V.1. List of the primers and conditions used for qRT-PCR ……….101
xi
Liste des figures
Figure I.1. Les granules du neutrophile et leur contenu. ... 2
Figure I.2. Voies de l'apoptose... 4
Figure I.3. Adhésion et migration du neutrophile ... 6
Figure I.4. Récepteurs impliqués dans le processus de phagocytose. ... 7
Figure I.5. Formation d’anions superoxydes par le neutrophile ... 10
Figure I.6. Voie canonique d’activation de NFκB. ... 12
Figure I.7. L’articulation normale versus l’articulation atteinte d’arthrite rhumatoïde ... 18
Figure I.8. La résorption osseuse physiologique... 20
Figure I.9. Mécanismes impliqués dans la destruction osseuse de l’AAI. ... 22
Figure I.10. Radiographies démontrant l’érosion osseuse avant et après le traitement ... 23
Figure I.11. Les actions biologiques du TNF-alpha dans l’AAI. ... 25
Figure III.1. Isolation de granulocytes par gradient de Ficoll.. ... 33
Figure III.2. Récuperation des cellules viables par gradients de PercollTM ... 35
Figure IV.1. Graphiques répresentant les résultats de la biopuce.. ... 40
Figure IV.2. « Heatmap » de gènes sur ou sous-exprimés plus de cinq fois de façon significative.. ... 41
Figure IV.3. Gènes signature de la reprogrammation en ostéoclaste. ... 42
Figure IV.4. Analyse fonctionnelle de gènes modulés dans la condition LL par DAVID 6.7. ... 47
Figure V.1. Gene expression signatures in human LL neutrophils evaluated by a two-way hierarchical clustering analysis... 91
Figure V.2. Genes clustered into known biologic functions expressed by human LL neutrophils. ... 92
Figure V.3. Human LL neutrophils expressed genes associated with inflammation.. ... 92
Figure V.4. Validation of microarray results by qRT-PCR. ... 94
Figure V.5. Measurement of selected chemokines produced by LL neutrophils.. ... 94
Figure V.6. Prodution of trappin-2/elafin by LL neutrophils. ... 95
Figure V.7. Regulation of trappin-2/elafin production by LL neutrophils... 96
Figure V.8. Measurement of trappin-2/elafin present in synovial fluids (SFs) and produced by SF neutrophils of auto-immune arthritis. ... 97
Figure V.9. Effects of elafin on neutrophil apoptosis and CXCL8/IL-8 production.. ... 98
xiii
Liste des abréviations
5-LO 5-lypoxigenas; 5 lipo-oxygénase AA Arachidonic acid; acide arachidonique
AAI Autoimmune arthritis; arthopathie auto-immune
AINS Nonsteroidal anti-inflammatory drug; anti-inflammatoires non stéroïdiens AP Psoriatic arthritis; arthrite psoriasique
AP Acid phosphatase; phosphatase acide
ARAL Slow-acting anti-rheumatic drugs; antirhumatismaux à action lente ARNm Messenger RNA; ARN messager
BLyS B-lymphocyte stimulator; molécule stimulatrice de lymphocyte B CG Cathesin G; cathepsine G
CR Complement receptor; récepteur du complément
CXCR2 C-X-C Chemokine receptor type 2; récepteur de chimiokine C-X-C 2 CXCR4 C-X-C Chemokine receptor type 4; récepteur de chimiokine C-X-C 4
DMARD Disease-modyfing antirheumatic drugs; médicaments anti-rhumatismaux modifciateurs de la maladie
EGF Epidermal growth factor; facteur de croissance épidermique
ERK Extracellular signal-regulated kinases; kinases régulées par des signaux extracellulaires FasL FasFas ligand; ligand du recépteur
FcR Fc receptor; récepteur Fc
fMLF Formyl-methionyl-leucyl phenylalanine
G-CSF Granulocyte colony-stimulating factor; facteur stimulateur des granulocytes
GM-CSF Granulocyte-macrophage colony-stimulating factor; facteur stimulateur des granulocytes et macrophages
GPCRs G protein-coupled receptors; récepteurs couplés aux protéines G HBSS Hank’s balance salt solution
IAP Inhibitor of apoptosis proteins; inhibiteurs des proteines de l’apoptose ICAM-1 Intercellular adhesion molecule 1; molécule d’adhésion cellulaire 1 Ig mmunoglobulin; immunoglobuline
IL Interleukin; interleukine
IκB-alpha Alpha IκB kinase; IκB kinase alpha
LFA-1 Lymphocyte function-associated antigen 1; antigène-1 associé à la fonction du lymphocyte LPS Lipopolysaccharide; lipopolysaccharide
LS Synovial lyquide; liquide synovial LTA4 Leukotriene A4; leucotriène A4 LTB4 Leukotriene B4; leucotriène B4;
MAP Mitogen-activated proteins; MAP kinases Mcl-1 Myeloid cell leukemia 1
MHCII Major hystocompatibility complex class II; complexe majeur d’histocompatibilité de classe II MMP Matrix metalloproteinase; métalloprotéinase
MRBs Biological response modifiers; médicaments modificateurs de la réponse biologique
NADPH Nicotinamide adenine dinucleotide phosphate; nicotinamide adénine dinucléotide phosphate hydrogène
NE Neutrophil’s elastase; elastase du neutrophile NLR Nod-like receptor; récepteur de type Nod OPG Osteoprotegerin; ostéoprotégérine
OSM Oncostatin M
PBS Phosphate buffered saline
PI Propidium iodide; Iodure de propidium PICD Phagocytosis-induced cell death PR3 Proteinase 3; protéinase 3
PS Phosphatydilserine; phosphatydilsérine
PTH Parathyroid hormone; hormone parathyroïdienne
qRT-PCR Real time polymerase chain reaction; réaction en chaine de polymérase en temps réel RANK Receptor activator of NF-κB; récepteur activateur de la voie NF-κB
RANKL RANK ligand; RANK ligand
ROS Reactive oxygen species; formes réactives d’oxygène SLPI Secretory leukocyte peptidase inhibitor
TGF-beta Transforming growth factor beta; facteur de croissance transformant beta TLR Toll-like receptor; récepteur Toll
TNF-alpha Tumor necrosis factor alpha; facteur de nécrose tumorale alpha TNFR TNF receptor; récepteur du TNF
TRAIL TNF-related apoptosis-inducing ligand
VCAM Vascular cell adhesion protein 1; protéine d’adhésion vasculaire 1 VEGF Vascular endothelial growth factor; facteur de croissance vasculaire
XIAP X-linked inhibitor of apoptosis protein; protéine inhibitrice de l’apoptose liée au chromosome X
xv
Remerciements
La science est passionnante. J’ai découvert ça pendant mes études universitaires quand j’ai pu avoir accès aux laboratoires et à la recherche. La passion avec laquelle les scientifiques développent leurs idées et leurs projets est contagieuse. Quand on est dans un domaine scientifique, c’est impossible de nier l’importance de la recherche fondamentale qui nous donne à chaque jour de nouvelles possibilités. Ainsi, à la première opportunité de réorienter mon cheminement professionnel, j’ai choisi la recherche fondamentale.
C’est difficile d’exprimer la chance que j’ai eue de pouvoir travailler dans le domaine qui m’intéressait avec des scientifiques du plus haut niveau. Le Dr. Patrice Poubelle est une référence dans l’étude des neutrophiles et de l'arthrite. Il est, en plus, un grand professeur, qui sait transmettre sa passion et ses connaissances à tous ceux qui s’y intéressent. La Dre. Isabelle Allaeys, dont les connaissances techniques solides lui permettent d’adapter n’importe quelle idée à un bon projet de recherche. On ajoute à cela la grande expérience en recherche de ces deux professionnels, leurs connaissances solides dans le domaine et sur le sujet et leur façon extrêmement éthique et logique de mener un projet de recherche. Tout cela m’a permis une expérience unique et la possibilité de vous présenter un projet de recherche qui comble bien plus que mes attentes.
C’est avec plaisir que je vous présente le projet de recherche sur lequel j’ai travaillé pendant les dernières années. Je suis fière du produit final et je suis certaine que cette étude ouvrira des portes à bien d’autres. À mon avis, cette étude est un bel exemple de la complexité de la démarche en recherche scientifique : c’est difficile de répondre à une question car les résultats nous en apportent toujours d’autres. On sort toujours transformé après un tel projet : on modifie ses idées, ses techniques et ses questions.
Je tiens encore à remercier mes plus grands exemples : le Dr. Patrice Poubelle et la Dre. Isabelle Allaeys. Je tiens à remercier aussi la Dre. Emanuelle Rollet Labelle pour tous les conseils (professionnels ou autres), Irina Gymninova pour le soutien et l'amitié. À tous les professeurs, qui m’ont inspirée et ouvert l'esprit à de nombreuses possibilités. À tous les collègues, toujours disponibles pour m’aider dans le travail (ou juste pour jaser…). À Lucas, mon conjoint, qui m’a comprise et soutenue pendant tout mon parcours. À Raphaël, mon fils, qui a enrichi ma vie et m’a donné une motivation extraordinaire d’être toujours meilleure. Un gros merci à tous!!
xvii
Avant-propos
Ce mémoire comporte un article inséré qui représente le résultat de tout le travail exécuté. Il a été soumis pour publication le 22 février 2016.
Concernant les coauteurs, Patrice Poubelle est médecin rhumatologue et professeur-chercheur au Centre de Recherche du CHUQ. Ses projets de recherches sont axés sur le rôle du neutrophile dans les arthropathies auto-immunes, notamment au cours de la résorption osseuse présente dans la maladie. Isabelle Allaeys est professionnelle de recherche dans le laboratoire. Elle participe à l’enseignement aux étudiants et les suit pendant leur projet de recherche.
Flavia Ribeiro de Vargas, l’étudiante qui vous présente ce mémoire, est l’auteure principale de l’article. Elle a été responsable de mener le projet de recherche ainsi que de rédiger l’ensemble du manuscrit avant sa correction par les coauteurs et sa soumission à des revues scientifiques.
1
Chapitre I. Introduction
I.1. Le neutrophile I.1.1. Introduction
Le neutrophile est le leucocyte le plus abondant dans la circulation sanguine, il représente environ 65% des leucocytes circulants [1]. Il est connu pour son rôle primordial dans la réponse immune innée et il est considéré comme la première ligne de défense de l’organisme contre les pathogènes [2]. Le neutrophile est une cellule clé du système immunitaire qui réagit aux signes de danger. Il s'agit de la cellule qui arrive le plus rapidement au site inflammatoire et qui a une fonction importante pour empêcher la propagation d’une infection [3]. Il est démontré que l’absence de neutrophiles dans un site enflammé nuit à la réponse à l’infection et que les individus atteints de neutropénie (une baisse de la quantité des neutrophiles dans le sang) sont vulnérables aux infections et considérés comme immunodéficients [4] [5]. Les traitements qui nuisent à la réponse des neutrophiles mènent également à un état d’immunodéficience. Tout cela démontre l’importance de cette cellule au maintien et au contrôle de la réponse immunitaire de l’organisme.
Une accumulation et une activation déréglées des neutrophiles dans les tissus enflammés peuvent être impliquées dans plusieurs maladies inflammatoires chroniques comme les maladies auto-immunes et les maladies pulmonaires chroniques [6] [7]. Dans ces maladies, la résolution de l’inflammation n’est pas accomplie et la présence continue des stimuli inflammatoires la perpétuent ainsi que le recrutement et l’activation des neutrophiles. L’homéostasie et le contrôle de la production et de l’activation des neutrophiles sont donc des mécanismes clés à une réponse inflammatoire efficace ainsi qu'à sa résolution.
I.1.2. Production et régulation des neutrophiles dans la circulation
Le neutrophile provient des cellules souches pluripotentes qui se différencient en cellules myéloïdes dans la moelle osseuse. Les stades de différenciation subséquents comprennent : le myéloblaste, le promyélocyte, le métamyélocyte, le neutrophile immature (« band cell ») et le neutrophile mature. La différenciation, jusqu’à la forme mature, prend environ trois jours et comprend l’acquisition des granules, caractéristique importante de cette cellule. Les granules peuvent être classifiés en quatre types, selon leur contenu. Les granules azurophiles ou primaires sont les plus précoces dans la différentiation de la cellule et riches en myeloperoxidase. Ils sont plutôt fusionnés aux phagosomes et sécrétés dans le milieu extra-cellulaire. Les granules formés ensuite sont appelés spécifiques. Ils ne contiennent pas de myeloperoxidase mais sont riches en substances antimicrobiennes et ils sont libérés plus facilement dans le milieu extracellulaire. Les granules acquis à la fin de la maturation sont les tertiaires, ou gélatinases. Ils sont riches en enzymes capables de dégrader la matrice extracellulaire. Les neutrophiles possèdent aussi les vésicules
sécrétoires : des granules contenant une variété de récepteurs membranaires. Ils sont les premiers à être libérés en réponse aux stimuli inflammatoires [8][9]. La figure I.1. représente les granules du neutrophile et leur contenu.
Figure I.1. Les granules du neutrophile et leur contenu. Figure représentant les différents types de
granules du neutrophile et leur contenu enzymatique. Ils se trouvent dans le cytoplasme et sont mobilisés en réponse à l’activation de la cellule. Les granules sécrétoires sont les premiers à le faire. Ensuite les granules contenant des gelatinases, les granules spécifiques et, en dernier, les granules azurophiles (Figure tirée de : Wright HL et al. Rheumatology 49:1618).
Dans des conditions normales, c’est le neutrophile mature qui est libéré dans la circulation sanguine où il a une survie de deux à trois jours, mais les neutrophiles immatures peuvent être libérés aussi, notamment en réponse à une infection. Après quelques jours dans la circulation, le neutrophile subit un processus d’apoptose spontanée qui est caractérisé par le changement de l’expression de molécules d’adhésion et de récepteurs : une augmentation de l’exposition de phosphatydilserine (PS), un lipide de la membrane cellulaire qui, lorsqu'exposé, permet la liaison des molécules d’annexine V, une augmentation de l’expression de « C-X-C chemokine receptor type 4 » (CXCR4) et une diminution de l’expression de « C-X-C receptor type 2 » (CXCR2) [10] [11]. Ces marqueurs sont ensuite reconnus par des macrophages qui phagocytent et dégradent les neutrophiles apoptotiques. La phagocytose des neutrophiles apoptotiques par des macrophages de la moelle osseuse stimule la production de « granulocyte colony-stimulating factor » (G-CSF) [12]. Le G-CSF joue un rôle primordial dans la production des neutrophiles : il induit la différenciation
3 myéloïde, la prolifération des précurseurs granulocytiques et mobilise les neutrophiles [13]. En plus de cet axe central de production des neutrophiles par stimulation du G-CSF, des études récentes ont démontré un possible axe périphérique de régulation de la production des neutrophiles par l’interleukine 17 et 23 (IL-17, IL-23) où la phagocytose des neutrophiles apoptotiques par des macrophages tissulaires pourrait supprimer la production d’IL-23 par ces cellules. La suppression d’IL-23 module négativement la production d’IL-17 par les cellules T et diminue la production de G-CSF dans la moelle osseuse [14] [15]. Cet axe de régulation négative paraît jouer un rôle central dans la granulopoïèse normale [16]. En réponse à une infection, il existe une granulopoïèse dite d’émergence où les produits bactériens stimulent la production de cytokines comme l’interleukine 1(IL-1), le « tumor necrosis factor alpha » (TNF-alpha) et des facteurs de croissance comme le « granulocyte-macrophage colony-stimulating factor » (GM-CSF) qui vont stimuler la production de neutrophiles par la moëlle osseuse et entraîner leur libération dans la circulation [17]. Dans ce cas, même des neutrophiles immatures ou des précurseurs peuvent être libérés dans la circulation et ensuite migrer au site inflammatoire où ils subiront les dernières étapes de différenciation [18].
I.1.3. Les fonctions du neutrophile I.1.3.1. Introduction
Les neutrophiles libérés dans la circulation ont une survie de un à trois jours. Ils sont maintenus en état de repos pour que leur contenu ne soit pas libéré dans la circulation. Ils constituent des cellules sentinelles du système immunitaire qui, en réponse aux médiateurs inflammatoires, seront mobilisées au tissu en danger. Le milieu inflammatoire du tissu est capable de moduler plusieurs fonctions de ces cellules et déclencher leurs fonctions effectrices contre les bactéries.
I.1.3.2. Modulation de l’apoptose
La régulation de l’apoptose du neutrophile est primordiale : l’augmentation de la survie de la cellule est importante pour une réponse immune efficace et, ensuite, son élimination par apoptose est nécessaire à la résolution de l’inflammation. Pour cela, le processus d’apoptose est un mécanisme étroitement contrôlé [19] . Des dérégulations dans ce processus peuvent conduire à une réponse inflammatoire accrue causant une résolution inefficace de l’inflammation accompagnée de dommages tissulaires importants [20].
Il existe deux voies intracellulaires classiques impliquées dans l’apoptose des neutrophiles. La voie extrinsèque est déclenchée par l’activation des récepteurs de mort cellulaire présents à la surface de la cellule, comme le récepteur de surface Fas. Ce récepteur est capable d’être activé par son ligand (FasL) et par d’autres molécules comme le TNF-alpha et le « TNF-related apoptosis-inducing ligand » (TRAIL). La voie intrinsèque est déclenchée par la rupture de la membrane mitochondriale et la libération intracellulaire de
cytochrome C et d’autres molécules apoptotiques [21]. Une troisième voie d’apoptose dans les neutrophiles est induite par la phagocytose (« phagocytosis-induced cell death » ou PICD)[22].
Peu importe la voie par laquelle le processus est déclenché, il est initié par l’activation des caspases (figure I.2.). Les caspase 8 et 9 sont des caspases qui débutent ce processus et la caspase 3 est la caspase exécutrice [23]. Les caspases sont des protéases présentes dans le cytosol sous forme inactive. Sa forme inactivée est maintenue par la liaison avec des molécules de la famille des inhibiteurs des protéines d’apoptose (les IAPs) [24][ 25].
Figure I.2. Voies de l'apoptose. La voie extrinsèque est déclenchée par l’activation des récepteurs de mort
cellulaire et l’activation ensuite des caspase 8 et caspase 3. La voie intrinsèque est déclenchée par une augmentation des protéines pro-apoptotiques de la famille Bcl-2 , Bax et Bak, et une diminution des molécules anti-apoptotiques comme le récepteur « myeloide cell leukemia 1 » (Mcl-1) et la molécule A1. Cela permet l’oligomérisation de Bax et Bak qui forment des pores sur la membrane mitochondriale entraînant la libération de cytochrome C dans le cytosol. Le cytochrome active les caspases 9 et 3. La PICD est activée par l’internalisation de particules bactériennes et la production des formes réactives d’oxygène (ROS), ce qui permet la libération des cathepsines des granules azurophiles dans le cytosol. Les cathepsines sont capables d’activer la caspase 8 et contribuent à l’activation de la voie intrinsèque en activant Bid, protéine responsable de l’activation de Bax et Bak, et en dégradant Mcl-1, favorisant l’apoptose. (Figure tirée de McCracken JM et al. J Cell Death. 2014 7:15).
Une fois recruté au tissu enflammé, le neutrophile est capable de survivre entre cinq à dix jours contrairement aux neutrophiles circulants qui ont une survie de un à trois jours. La présence de plusieurs
5 facteurs inflammatoires, comme le G-CSF et le GM-CSF, permet de moduler le mécanisme d’apoptose du neutrophile [26] . Ces molécules sont capables d’activer des kinases régulées par des signaux extracellulaires (« extracellular signal-regulated kinases » ou ERK) de la voie des protéines activées par la mitose (« mitogen acitvated proteins » ou MAP kinases) [27] [28]. Cette voie augmente la stabilité de Mcl-1 [29] et de « X-linked inhibitor of apoptosis » (XIAP) [30], des protéines anti-apoptotiques. Le « lypopolisaccharide » (LPS) est aussi capable d’augmenter la survie du neutrophile par le même mécanisme [28].
L’adhésion aux cellules épithéliales et la présence de CXCL8/IL-8, de TNF et d’IL-1 sont d’autres facteurs capables de moduler la survie des neutrophiles [31]. De plus, les tissus enflammés sont caractérisés par une baisse de la concentration d’oxygène. L’hypoxie est un facteur important qui contribue à la modulation de l’apoptose du neutrophile par la voie des MAP kinases [32]. La voie du « nuclear factor » NF-κB est une voie d’activation cellulaire classique dans l’inflammation responsable de la transcription de plusieurs gènes reliés aux fonctions d’inflammation et de réponse immune (cytokines, chimiokines et molécules d’adhésion) [33]. L’activation de la voie NF-κB par des médiateurs inflammatoires est aussi responsable de la transcription des gènes des IAP et d’autres protéines anti-apoptotiques [34].
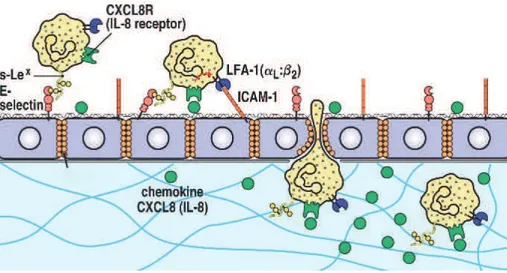
I.1.3.3. Adhésion et migration
En réponse à un gradient de chimiokine ou de cytokine, le neutrophile circulant est activé, ce qui le rend capable de se lier aux cellules endothéliales et de migrer vers le tissu enflammé. Dans un premier temps, le neutrophile se lie faiblement aux cellules endothéliales par les sélectines, ce qui permet le roulement de la cellule sur l’endothélium. La présence de facteurs inflammatoires active à la fois les cellules endothéliales et les neutrophiles et favorise l’expression des molécules d’adhésion de forte affinité, comme les intégrines, ce qui permet l’arrêt de la cellule [35] [36].
Une fois arrêté et en présence de facteurs inflammatoires, le neutrophile réorganise son cytosquelette et déclenche l’étape de diapédèse et de migration : il va s’insinuer entre les cellules endothéliales, perforer la membrane basale et pénétrer les espaces extravasculaires (Figure I.3). Les granules tertiaires jouent un rôle important dans cette étape en libérant des gélatinases qui dégradent la matrice extracellulaire et permettent la migration efficace du neutrophile vers le site inflammatoire [8] [37].
Figure I.3. Adhésion et migration du neutrophile. Schéma représentant les interactions cellulaires pendant
l’adhésion et la migration du neutrophile. En présence de médiateurs inflammatoires les cellules endothéliales expriment des sélectines qui peuvent se lier aux récepteurs des neutrophiles circulants et les faire rouler sur les cellules endothéliales. L’activation du neutrophile par les sélectines provoque l’activation des intégrines qui peuvent se lier à leur ligand. La « intercellular adhesion molecule 1 » (ICAM-1 ou CD54) des cellules endothéliales se lie au « lymphocyte function-associated antigen 1 » (LFA-1). Cette liaison LFA-1/ICAM-1 est une interaction de forte affinité qui permet l’arrêt de la cellule et qui déclenche des voies de signalisation permettant la réorganisation du cytosquelette et la migration du neutrophile à travers l’endothélium vers le tissu. (Figure tirée de Janeway CA et al. Garland Science, 2005).
Des études ayant analysé des neutrophiles activés par des facteurs inflammatoires ayant entamé le processus de migration démontrent que ces neutrophiles expriment plus d’intégrines, notamment l’ICAM-1[38], et que d’autres fonctions, comme la production de ROS [39] et la dégranulation [40] sont amplifiées en réponse aux médiateurs inflammatoires alors que la chimiotaxie est diminuée [20].
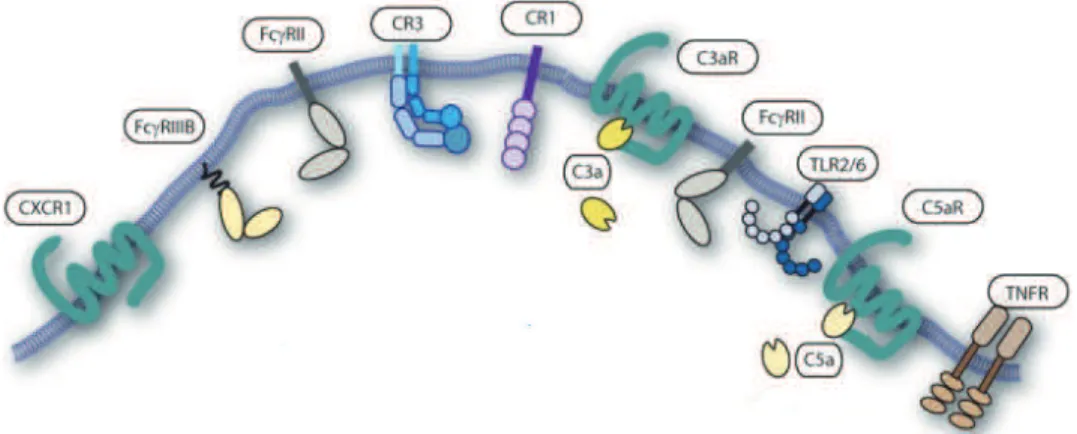
I.1.3.4. Phagocytose
La phagocytose est le processus permettant à une cellule d'englober et ensuite de digérer une substance étrangère. Ce phénomène joue un rôle de défense dans la fonction cellulaire. Rendu au site inflammatoire le neutrophile est une arme importante d’élimination des produits bactériens, viraux, des particules étrangères et mêmes des cellules nécrotiques, grâce à sa grande capacité de phagocytose. Il est capable de reconnaître ces molécules par des récepteurs membranaires (Figure I.4) et les enrober par l’activation de filaments d’actine et de myosine formant une vacuole, le lysosome. Ce lysosome est ensuite internalisé et fusionné à une vésicule intracellulaire avec des enzymes protéolytiques, formant le phagosome. Le contenu du phagosome est donc dégradé [41].
La présence d’immunoglobulines (Ig) et des molécules du complément dans le plasma sont les principaux facteurs qui déclenchent le processus de phagocytose. Ils entourent les bactéries (opsonisation)
7 et sont reconnus par les récepteurs Fc (FcR) et par les récepteurs du complément (CR) présents sur les neutrophiles, ce qui active le processus de phagocytose. Les immunoglobulines de type G (IgG) et les composants du complément sont les molécules responsables de l’opsonisation des bactéries. La phagocytose des bactéries opsonisées, reconnues par les FcR et les CR du neutrophile, est le mécanisme de phagocytose le plus classique et le plus efficace lors d’une infection bactérienne. Il dépend, en partie, de la réponse immune adaptative, qui implique la production d’anticorps de type IgG [42]. Les neutrophiles expriment aussi d’autres récepteurs qui peuvent reconnaitre les microbes ou les particules bactériennes de façon directe comme les « Toll-like receptors » (TLR) [43], les « Nod-like receptors » (NLR) [44] et Dectin-1 [45].
Les mêmes récepteurs qui sont impliqués dans le processus de reconnaissance des particules sont capables de moduler l’influx de calcium et l’activation de phospholipases et d’autres voies d’activation [46]. Ainsi, la phagocytose est capable de moduler l’apoptose, d’augmenter l’expression de certains marqueurs de surface et d’activer la dégranulation et la formation de ROS. À l’inverse, l’activation de la cellule par des médiateurs inflammatoires peut aussi moduler les processus de phagocytose et le rendre plus efficace [47] [48][49].
Figure I.4. Récepteurs impliqués dans le processus de phagocytose Plusieurs récepteurs du neutrophile
sont impliqués soit dans la reconnaissance de particules opsonisées, soit dans l’activation de la phagocytose. Les récepteurs impliqués dans la reconnaissance des particules opsonisées sont les FcRs, capables de reconnaître les IgG, et les récepteurs du complément CR1 et CR3, qui reconnaissent les molécules du complément 3a (C3a). Les récepteurs impliqués dans l’activation du processus de phagocytose sont les récepteurs du complément C3aR et C5aR, le récepteur de chimiokine CXCR1 qui se lie à CXCL8/l’IL-8 et le récepteur du TNF (TNFR) pour le TNF-alpha. Les récepteurs toll-like 2 et 6 (TLR2, TLR6) sont des récepteurs qui reconnaissent directement des lipoprotéines bactériennes. (Figure tirée de Kok P. M. van Kessel et al. Immunol. 2014. 5: 467).
I.1.3.5. Dégranulation
Tel que cité précédemment, la présence de granules intracellulaires est une caractéristique importante du neutrophile. Ces granules possèdent des enzymes importantes aux fonctions de migration et de phagocytose (table I.1). L’activité antibactérienne du neutrophile, comme cité dans la section précédente, est
très dépendante de cette fonction. Une fois libérés, ces granules sont des sources de molécules antibactériennes dans le milieu extracellulaire, ainsi que des sérines protéases comme la cathepsine G, l’élastase du neutrophile (NE) et la protéinase 3 (PR3), présentes dans les granules azurophiles [50] et l’azurocidine: une protéine de type sérine protéase inactive avec activité antibactérienne et de chimiokine [51]. Le neutrophile est reconnu comme une cellule capable de provoquer une lésion tissulaire par la libération d’enzymes protéolytiques qui se trouvent dans ses granules, tel que cité antérieurement. Étant donné que la dégranulation est une fonction modulée par l’activation du neutrophile, son activation exagérée ou déréglée est impliquée dans plusieurs maladies inflammatoires chroniques associés au remodelage et à la lésion tissulaire comme les maladies pulmonaires chroniques [52] [53], la polyarthrite rhumatoïde (PAR) [54] et la lésion tissulaire suite à l’ischémie-reperfusion [55]. En fait, la résolution de l’inflammation est caractérisée par une diminution de la libération de ces enzymes associée à une augmentation de la libération de molécules inhibitrices, les inhibiteurs de peptidases, comme l’élafine et le « secretory leukocyte peptidase inhibitor » (SLPI), ce qui favorise la réparation tissulaire. L’activation déréglée des neutrophiles associée aux maladies inflammatoires chroniques brise cet équilibre et favorise la lésion tissulaire [56].
Dans les dernières années plusieurs études ont démontré que les protéines présentes dans les granules du neutrophile n’ont pas juste des fonctions antimicrobiennes et de lésion mais sont aussi impliquées dans d’autres processus importants à la régulation de la réponse immune [57]. Padrines et al. ont démontré que les protéinases du neutrophile sont capables de cliver CXCL8/l’IL-8 pour la convertir en une forme plus puissante et résistante à la dégradation [58]. Richter et al. ont montré que la « cathepsin G » (CG) et la NE sont capables de cliver la chimiokine CCL15 et la rendre active, jouant un rôle dans le mécanisme d’activation de macrophages [59]. En plus de moduler la biodisponibilité de plusieurs cytokines et chimiokines, les sérines-protéases du neutrophile sont capables d’activer le récepteur du « epidermal growth factor » (EGFR) ce qui augmente la production de « transforming growth factor beta » (TGF-beta) [60]. Ces molécules jouent un rôle sur la croissance et le remodelage tissulaires. Les sérine protéases peuvent également activer d’autres récepteurs liés aux mécanismes pro-inflammatoires comme les intégrines [61] et les récepteurs activés par les protéases (PAR) [62] responsables de l’activation des voies de signalisation intracellulaires qui provoquent la libération des médiateurs inflammatoires. Preston et al. ont démontré que la PR3 et la NE peuvent induire la production de molécules anti-apoptotiques par la voie ERK, JNK et p38 MAPK et moduler la survie des cellules endothéliales [63]. Chez les souris, la CG peut moduler l’activation des lymphocytes et la réponse à l’antigène, comme démontré par Yamazaki et al. [64].
9
Table I.1. Les sérinesprotéases du neutrophile et leurs cibles biologiques. Le tableau résume les effets
de sérines-protéases du neutrophile sur leurs cibles biologiques. (Table tiré de Pham CT. Int J Biochem Cell Biol. 2008. 40:1317)
Toutes ces informations démontrent que les fonctions des sérineprotéases du neutrophile sont nombreuses et dépassent le rôle de molécules antimicrobiennes. Ce fait place le processus de
dégranulation du neutrophile dans le complexe ensemble de fonctions impliquées dans l’orchestration de la réponse immune.
I.1.3.6. Formation d’anions superoxydes
Les formes réactives d’oxygène, dont les anions superoxydes (O2-), sont des molécules
biologiquement toxiques et utilisées par les cellules immunitaires, et par le neutrophile, comme mécanisme de défense. Les anions superoxydes sont produits par l’enzyme NADPH oxydase [65]. Des mutations ou des dysfonctions de cette enzyme peuvent engendrer une forme d’immunodéficience, ce qui souligne l’importance de ce mécanisme contre les infections. La NADPH oxydase est située à la membrane cytoplasmique et également sur la membrane des vésicules. Elle permet la libération des anions à la surface externe de la membrane. Les anions peuvent ainsi agir soit à l’extérieur de la cellule, soit dans les phagosomes lors de son internalisation, comme montré dans la figure I.5.
Figure I.5. Formation d’anions superoxydes par le neutrophile. Schéma représentant la forme inactive et
active de la NADPH oxydase. La partie associée à la membrane comprend les sous-unités catalytiques gp91phox, p22phox. Les protéines phox p47, p67 et p40 forment la partie cytosolique. Pendant l’activation de l’enzyme, le complexe cytosolique se lie au complexe membranaire. RhoGDI stabilise la protéine G Rac qui est, alors, inactive. Lorsque la protéine G Rac est active, elle peut se transloquer à la membrane et se lier au complexe. L’assemblage de toutes ces sous-unités permet le transfert des électrons du cytoplasme à l’oxygène moléculaire par la NADPH oxydase et, par conséquent, la production de ROS. (Figure tirée de McCann SK et al. Brain Sci. 2013. 3:561).
Les anions superoxydes sont de précurseurs d’autres formes réactives d’oxygène plus toxiques comme le peroxyde d’hydrogène (H2O2), le radical hydroxyle (OH•) et l’oxygène singulet (1O2). La
phagocytose est le principal stimulus pour l’assemblage du complexe NADPH oxydase et la formation de ROS [66]. La libération de ROS dans le milieu extracellulaire est un des mécanismes responsables de la lésion
11 tissulaire. Elle provoque la peroxydation lipidique qui cause une désorganisation membranaire ou l’altération des protéines et d’acides nucléiques. Suite à l’activation de la cellule, la production de ROS peut aussi provoquer la carbonylation des protéines intracellulaires, notamment de la calprotectine, ce qui peut changer les fonctions de ces protéines et moduler des fonctions liées à l’activation du neutrophile et à l’inflammation [67].
La production augmentée de ROS est une des caractéristiques des neutrophiles activés. Plusieurs études ont déjà démontré la capacité du neutrophile prédisposé ou reprogrammé de produire une quantité de ROS plus élevée qu’un neutrophile circulant [68] [69] [70] [71]. La production de ROS, étant liée à la phagocytose et à l’inactivation des produits bactériens, il n’est pas étonnant qu’elle soit une des fonctions modulées par l’activation de la cellule.
I.1.3.7. Production des médiateurs inflammatoires
Le neutrophile est capable de produire plusieurs médiateurs inflammatoires : des médiateurs lipidiques comme les leucotriènes, des cytokines et des chimiokines.
Les leucotriènes sont des métabolites oxygénés de l’acide arachidonique (AA). La stimulation de la cellule provoque l’activation de la phospholipase A2 cytosolique (cPLA2) qui métabolise des lipides de la membrane cellulaire produisant l’AA. Par l’action de l’enzyme 5-lipoxygénase (5-LO), l’AA est tranformé en leucotriène A4 (LTA4) qui peut être metabolisé en leucotriène B4 (LTB4). La synthèse des leucotriènes est modulée par plusieurs stimuli comme le calcium, les cytokines, les peptides formylés comme le formyl-methionyl-leucyl phenylalanine (fMLF) et l’AA exogène. La production des leucotriènes implique un mécanisme de translocation de la 5-LO au noyau cellulaire [72] [73]. Les leucotriènes sont capables d’agir sur le neutrophile et d’autres types cellulaires de façon autocrine ou paracrine. Leurs récepteurs sont de type couplés aux protéines G (GPCRs) et sur le neutrophile ils sont capables de moduler sa chimiotaxie, son adhésion à l’endotélium, sa production de cytokines et d’anions superoxydes [74] [75].
Les neutrophiles sont capables de produire des cytokines et des chimiokines de façon constitutive ou induite par plusieurs stimuli comme le LPS, le TNF-alpha, le GM-CSF, etc. La voie de production des cytokines et chimiokines implique la modulation de la transcription de leurs gènes par l’activation du facteur de transcription NF-κB. Ce facteur de transcription est régulé dans le cytoplasme par un complexe protéique avec les IB-kinases : IKKa, IKKb et IKKg. IL est retenu dans le cytoplasme par la protéine inhibitrice IκB-alpha. La dégradation d’IκB-alpha par phosphorylation et ubiquitination permet la translocation de NF-κB jusqu'au noyau (figure I.6) et l'activation de la transcription des gènes cibles [76] [77] [78]. Son activation exagérée (lors de la présence de bactéries dans le sang, par exemple) peut provoquer un choc
septique. L'implication du facteur NF-κB dans les maladies inflammatoires chroniques, la cancérogénèse et les processus de tumorisation de certaines cellules a été démontrée dans des nombreuses publications. L’inhibition de la voie NF-κB peut diminuer ou même bloquer la production de cytokines ou de chimiokines par le neutrophile [79] [80].
Figure I.6. Voie canonique d’activation de κB. La voie canonique active le plus souvent les dimères
NF-κB comprenant les protéines Rel-A, c-Rel, Rel-B et p50. L’activation d’un des récepteurs capables d’activer la voie (les TLRs, par exemple) entraîne un recrutement et l’activation du complexe des IκB-kinases. Ce complexe phosphoryle IκB-alpha qui est ensuite envoyé vers le protéasome pour subir une dégradation. L’activation du neutrophile par des médiateurs inflammatoires provoque la translocation des protéines κB/Rel au noyau. Cet ensemble augmente l’affinité de fixation de NF-κB à l’ADN et permet la transcription des gènes des cytokines et chimiokines. (Figure tirée de Monaco C et al. Proc. Natl. Acad. Sci. U.S.A. 2004. 101: 5634).
Les cytokines et chimiokines sont des médiateurs inflammatoires qui agissent de manière paracrine ou autocrine sur les neutrophiles ou d’autres types cellulaires et sont capables de moduler plusieurs fonctions comme l’apoptose, la chimiotaxie et la production d’autres médiateurs inflammatoires. Le répertoire
13 des cytokines et de médiateurs inflammatoires exprimés par les neutrophiles est grand, comme montré dans la table I.2., et change en fonction des différents stimuli [81]. Les neutrophiles inflammatoires présentent une production augmentée de cytokines et de lipides bioactifs par rapport aux neutrophiles circulants [71], notamment de CXCL8/IL-8 et de LTB4 [70]
Table I.2. Cytokines pouvant être exprimées par les neutrophiles humains. Tableau résumant les
cytokines qui peuvent être exprimées par les neutrophiles humains. Les données ont été validées par techniques d’expression génique et par études du protéome. * validés seulement par étude d’expression d’ARNm. ? Données controversées. (Table tirée de Scapini P et al. Blood. 2014. 124:710)
I.1.4. Le neutrophile et l’inflammation chronique
Le neutrophile est souvent considéré comme une cellule complètement différenciée, incapable de changer de phénotype et d’acquérir de nouvelles fonctions. Dans les dernières années, plusieurs études ont démontré que le neutrophile circulant est différent du neutrophile présent dans les sites
inflammatoires [82]. Les différences comprennent des changements de phénotype et aussi de fonctions. En fait, toutes les fonctions décrites dans les sections antérieures sont modulées après l’activation du neutrophile. Une fois activé, le neutrophile inflammatoire passe par un changement de conformation et de polarisation de ses organelles, ce qui change sa forme et lui permet de migrer dans le tissu, de mobiliser ses granules et ses protéines de surface [36]. Il prend alors une forme plus allongée. Il est une cellule capable de survivre de cinq à dix jours dans le tissu enflammé, contrairement aux neutrophiles circulants qui ont une survie de un à trois jours [83]. Il possède des fonctions de dégranulation et de phagocytose augmentées. Il est aussi capable de produire beaucoup plus de formes réactives d’oxygène en réponse aux stimuli [68] [69] [70] [71]. Il exprime différents marqueurs de surface qui n’étaient pas présents sur le neutrophile circulant, notamment des marqueurs liés à la présentation d’antigène [84]. Il est capable aussi d’exprimer plusieurs cytokines que le neutrophile circulant n’exprime pas [81]. La capacité du neutrophile activé par des médiateurs inflammatoires d’exprimer des molécules responsables de la présentation d’antigène et de devenir une cellule présentatrice d’antigène professionnelle (APC) [85] [86] ou même de se différencier en macrophage en présence de combinaisons de cytokines [87] est déjà bien établie dans la littérature.
Étant donné les fonctions du neutrophile et ses capacités de lésion tissulaire, ce n’est pas étonnant qu’il soit une cellule jouant un rôle important dans la pathogenèse de plusieurs maladies inflammatoires, infectieuses ou pas. En fait, toutes ces modifications des fonctions décrites antérieurement permettent au neutrophile de s’adapter à un nouvel environnement (un tissu enflammé) et très importantes pour la réponse immune contre les pathogènes, sont les mêmes fonctions que, dans un contexte d’inflammation déréglée, peuvent être délétères pour les tissus.
Les maladies inflammatoires chroniques sont caractérisées par un dérèglement de la réponse immune combinée à une résolution inefficace de l’inflammation, ce qui produit sa perpétuation. Dans ce cas, l’activation déréglée et exagérée des neutrophiles est, en particulier, responsable de la lésion aux tissus et du remodelage tissulaire.
Le rôle du neutrophile dans les maladies inflammatoires chroniques est déjà bien connu : la présence de neutrophiles dans les tissus atteints d’une inflammation chronique due à une pathologie est un facteur déterminant de l’activité de la maladie. Par contre, la présence de peu de neutrophiles dans les tissus atteints est corrélée à une bonne réponse au traitement (rémission) ou à une maladie moins grave [88] [89] [90]. En plus, les neutrophiles sont une source de facteurs inflammatoires qui modulent la réponse immunitaire locale et provoquent l’activation d’autres cellules immunitaires et des cellules du tissu, provocant plus d’inflammation et un changement de la physiologie tissulaire, perpétuant l’état inflammatoire.
15 I.2. Les Arthrites Auto-immunes (AAI)
I.2.1. Introduction
Il existe plus de 100 types d’arthrite. Elle peut être causée par l’inflammation de l’articulation (arthropathie inflammatoire) ou par le dommage progressif du cartilage puis de l'os (arthropathie dégénérative ou arthrose). Peu importe sa cause, l’atteinte articulaire chronique est une des causes principales de handicap fonctionnel au Canada et atteint plus de quatre millions de personnes de différents âges, conditions physiques et contexte social.
L’Arthropathie Auto-Immune (AAI) est une maladie inflammatoire chronique des articulations couramment trouvée au cours des certaines maladies auto-immunes. Elle engendre une inflammation qui peut s’étendre à tout l’organisme et aboutir à la destruction des tissus atteints. C’est donc une maladie inflammatoire systémique et délétère dont une des principales caractéristiques est un tropisme pour le tissu synovial, tissu qui recouvre la partie interne de la capsule articulaire, causant ainsi une arthrite. La Polyarthrite Rhumatoïde (PAR) et l’Arthrite Psoriasique (AP) sont les principales maladies de ce genre. La PAR est la principale des AAI et peut atteindre plus d’une articulation, d’où son nom: polyarthrite. Elle est, en fait, la principale cause de polyarthrite chronique chez l’humain. L’AP apparait souvent au cours du psoriasis, une maladie auto-immune qui affect la régénération des cellules de la peau et provoque un épaississement cutanée localisé. Dans les deux maladies, l’arthrite est déclenchée par une inflammation dérégulée au niveau du tissu synovial et peut aboutir à des déformations articulaires importantes [91] [92].
I.2.2. Épidémiologie de l’AAI
La prévalence de l’AAI est variable selon les pays. Au Canada, la PAR touche un pour cent de la population, soit 300 000 personnes [91]. Elle peut se développer à n’importe quel âge mais son pic d’incidence est entre 25-50 ans. Comme d’autres maladies auto-immunes, la PAR est plus courante chez les femmes avec un ratio de 3:1. Selon le dernier rapport de l’Alliance de l’arthrite du Canada, les simulations qui concernent le taux de vieillissement de la population et la prévalence des facteurs de risque pour la PAR suggèrent que l’incidence de la maladie au Canada est en hausse, comme montre la table I.3.
Table I.3. Incidence actuelle et future de la PAR par sexe au Canada de 2010 à 2040 selon le dernier rapport de l’Alliance Arthrite Canada. L’incidence de la PAR devrait augmenter à cause du vieillissement de
la population et le nombre annuel de cas devrait être plus élevé chez les femmes que chez les hommes. D’ici 2040 le nombre de cas de PAR devrait atteindre 23732 contre 17916 en 2010. (Table tirée de www.arthritisalliance.ca. 2011)
Le psoriasis touche 2 à 3 pourcent de la population mondiale et l’AP entre 10 et 30% des personnes atteintes de psoriasis et autant d’hommes que de femmes, contrairement à la PAR. Elle apparaît entre 20 et 50 ans et, dans 85% des cas, après les symptômes cutanés. La gravité des symptômes cutanés n’est pas corrélée à la gravité de l’arthrite et inversement [93].
I.2.3. Étiopathogénèse
Comme toutes les maladies auto-immunes, les AAI sont caractérisées par un dérèglement du système immunitaire qui déclenche une réponse contre un tissu du soi. Dans le cas d’AAI cette réponse se dirige contre le tissu synovial présent dans les articulations, causant son inflammation. Ce tissu enflammé produit plusieurs facteurs inflammatoires et est capable de recruter continuellement des cellules immunitaires. Ces dernières vont aussi produire des facteurs inflammatoires et engendrer des réponses contre le tissu créant une inflammation chronique. Au niveau des articulations c’est le tissu synovial qui est touché.
La cause exacte de la PAR ou de l’AP, c’est-à-dire ce qui déclenche l’inflammation synoviale, est encore méconnue, mais la prédisposition génétique associée aux facteurs environnementaux jouent un rôle non négligeable. Dans le cas de la PAR, on observe une agrégation familiale de cas et la présence des allèles HLA-DR1 et -DR4 dans 60% des patients. La mutation de certains gènes est liée à un risque accru de développer la maladie (mutation du gène PTPN22) ou est corrélée avec des formes plus graves (mutation du gène TRAF1-C5) [91]. Les facteurs environnementaux et hormonaux semblent également jouer des rôles en interagissant avec les facteurs génétiques et favorisant le dérèglement de l’activité immunitaire. Les causes de l’AP sont aussi inconnues, mais environ un tiers des personnes atteintes de psoriasis signalent une histoire familiale de la maladie. Les études des jumeaux identiques suggèrent un
17 risque de développer la maladie de 70% si l'autre jumeau a la maladie, et un risque d'environ 20% pour les jumeaux non identiques, ce qui confirme une susceptibilité génétique et une réponse de l'environnement dans le développement de la maladie [94]. Les analyses du génome ont identifié neuf loci sur des chromosomes différents dont des variations sont associés au psoriasis. Ils sont appelés le « psoriasis susceptibility » 1 à 9 (PSORS1-PSORS9). Ces gènes sont liés aux voies d'inflammation et aux médiateurs inflammatoires qui affectent les cellules du système immunitaire, notamment les cellules T. Certains de ces gènes sont également impliqués dans d'autres maladies auto-immunes [95]. Récemment une mutation rare dans le gène codant pour la protéine CARD14 a été décrite comme suffisante pour causer le psoriasis en plaques (la forme la plus courante du psoriasis) [96]. Parmi les facteurs environnementaux, les infections chroniques, le stress et les changements de saison et du climat semblent jouer un rôle. L'eau chaude, le grattage des lésions cutanées du psoriasis, la sécheresse de la peau, la consommation excessive d'alcool, le tabagisme et l'obésité sont aussi des facteurs possiblement impliqués [97].
Pour comprendre la pathogénèse des AAI il faut d’abord connaitre l’articulation normale. Elle est composée de deux extrémités osseuses qui sont recouvertes par le cartilage et englobées par une membrane qui confère une individualité à l’articulation. Cette membrane est la capsule articulaire. Elle est tapissée par le tissu synovial qui est composé de synoviocytes, cellules responsables de la production du liquide synovial (LS). Ce liquide est formé principalement par l’acide hyaluronique et lubrifie l’articulation permettant un meilleur glissement entre les deux extrémités osseuses (figure I.7). Bien qu’il soit produit en très faible quantité, le LS fournit les molécules qui nourrissent et maintiennent le cartilage.
Dans l’AAI, une fois que la réponse immunitaire anormale est initiée contre le tissu synovial, les cellules B produisent des quantités anormales de facteurs et d’anticorps qui sont capables ensuite d’activer d’autres cellules immunitaires comme les macrophages et les neutrophiles par leur FcR. Les cellules immunitaires activées et recrutées dans le tissu synovial produisent plusieurs médiateurs inflammatoires comme les cytokines et les lipides bioactifs. Ces médiateurs entrainent la vasodilatation des vaisseaux provocant l’œdème et permettant le recrutement de cellules immunitaires. Les lymphocytes B, lymphocytes T, les cellules dendritiques, les macrophages et les neutrophiles jouent un rôle dans l’AAI: les lymphocytes B produisent des anticorps et activent d’autres cellules immunitaires, les lymphocytes T activés fonctionnent comme des cellules cytotoxiques, les macrophages et les neutrophiles comme des cellules effectrices et les cellules dendritiques comme des cellules présentatrices d’antigène. Ces cellules infiltrent toutes le tissu synovial [98].
L’inflammation du tissu synovial induit la production de facteurs de croissance comme le TGFbeta et le « vascular endothelial growth factor » (VEGF) par les cellules immunitaires et par le tissu même (les cellules synoviales, les chondrocytes, les ostéoblastes et les ostéoclastes). Ces facteurs induisent la
prolifération des cellules synoviales et endothéliales et entrainent l’épaississement et la néovascularisation du tissu synovial qui devient de plus en plus rigide et complètement infiltré par les cellules immunitaires. Ces cellules et les facteurs inflammatoires qu’elles produisent se retrouvent dans le LS qui, dans des conditions normales, est acellulaire. La production de LS est également accrue par l’activation continue des cellules synoviales. Dans la PAR, le remodelage du tissu synovial progresse et provoque la formation d’un tissu de granulation en bordure de la membrane synoviale, beaucoup plus rigide que le tissu synovial normal, appelé pannus, comme montré dans la figure I.7. La présence du pannus est une caractéristique de la pathologie. Ce pannus est comparé à un tissu pseudo-tumoral du fait de son caractère prolifératif, certes bénin, mais progressant inéluctablement dans toutes les structures adjacentes qu'il détruit. Dans l’AP, l’inflammation s’étend plus couramment aux tendons, causant l’enthésite, une inflammation des tendons, et aussi la dactylite, un gonflement important des doigts atteints par l’arthrite [99].
Figure I.7. L’articulation normale versus l’articulation atteinte d’arthrite rhumatoïde. L’articulation
normale est représentée à gauche. Le tissu synovial tapisse la partie interne de la membrane synoviale et le liquide synovial est acellulaire. L’articulation malade est représentée à droite où on peut remarquer la prolifération de synoviocytes, de nouveaux vaisseaux et une infiltration du tissu et du LS par plusieurs types de cellules immunitaires comme les macrophages, les lymphocytes, les neutrophiles, les cellules dendritiques et d’autres. Les résultats sont : l’inflammation, l’épaississement du tissu, la formation du pannus et aussi l’activation des cellules capables de dégrader la matrice osseuse causant des érosions dans l’os et la déformation de l’articulation. (Figure tirée de Strand V et al. Nature Reviews Drug Discovery 2007. 6:75)
19 I.2.4. Présentation et évolution clinique
Le patient atteint d’AAI présente des signes d’inflammation dans la majorité des cas dans plus d’une articulation. Dans la PAR, les petites articulations des doigts de la main et du poignet sont plus touchées et dans l’AP ce sont les articulations plus distales des mains et des pieds mais aussi les articulations du poignet, du dos et de la cheville. Dans l’AAI, les articulations atteintes sont gonflées, douloureuses, chaudes et rougeâtres. Il est caractéristique que les articulations aient moins de souplesse le matin, ce qui s’améliore au cours de la journée [99].
Les symptômes varient d’une personne à l’autre et d’un jour à l’autre. Dans certains cas, l’inflammation est mineure et ne touche qu’une articulation, tandis que dans d’autres, cas l’inflammation est aiguë et touche un grand nombre d’articulations. En général, l’AAI se présente de façon progressive: quelques articulations sont d’abord touchées et, au cours des semaines ou des mois suivants, d’autres sont atteintes. En général, les deux côtés du corps sont touchés selon un schéma symétrique caractéristique de la PAR, mais pas de l'AP [92].
L’AAI est caractérisée par des périodes de poussées inflammatoires et d’amélioration des symptômes. Les médiateurs inflammatoires libérés pendant la poussée atteignent la circulation sanguine et provoquent des symptômes systémiques comme la fièvre, la fatigue, des changements d’appétit, une perte de poids, etc. L’inflammation systémique augmente le risque des maladies cardiovasculaires chez les patients atteints de la maladie, ce qui entraîne un risque plus élevé de mortalité. L’espérance de vie d’une personne souffrant de PAR, par exemple, est en moyenne 10 ans inférieure à celle de la population générale [92] et chez les patients atteints d’AP les maladies cardiovasculaires sont la principale cause de décès [93]. D’autres maladies chroniques et/ou auto-immunes sont aussi plus courantes chez les patients d’AAI comme certains types de cancers, le diabète et le syndrome métabolique, la dépression et la maladie de Crohn [100]
I.2.5. La résorption osseuse
L’AAI est une maladie destructrice car l’inflammation chronique engendre des lésions tissulaires, notamment dans le tissu articulaire et provoquant sa déformation. La lésion tissulaire la plus grave de la maladie est la résorption osseuse excessive. Selon le dernier rapport de l’Alliance Arthrite Canada, au cours des dix années suivant le déclenchement de la maladie, jusqu’à 50% des patients atteints de PAR deviennent inaptes au travail s’ils ne sont pas traités [101].
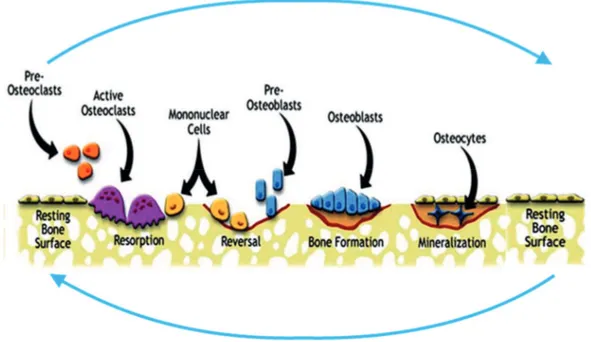
La résorption osseuse est un processus physiologique essentiel à l’homéostasie de l’os. Elle est caractérisée par un cycle équilibré entre résorption, par les ostéoclastes, et formation osseuse, par les ostéoblastes, cycles s’alternant en réponse aux facteurs de modulation, comme montré dans la figure I.8. La période de résorption osseuse dure environ 2 semaines et la phase de formation osseuse, beaucoup plus
longue, dure 4 mois [102]. Le système crucial de régulation de la résorption osseuse est constitué d’une triade de molécules: le récepteur activateur de la voie NF-κB (RANK), son ligand (RANKL) et l’ostéoprotégérine (OPG), un récepteur leurre de RANKL. RANKL est une cytokine qui appartient à la superfamille du TNF-alpha est présent surtout dans les tissus osseux et lymphoïdes. Elle est exprimée par les ostéoblastes, les cellules stromales, les cellules T activées et les neutrophiles [103] [104] [105]. RANK et OPG font partie de la superfamille du TNFR. RANK est un récepteur homotrimérique considéré comme une molécule “signature” des ostéoclastes et des précurseurs des ostéoclastes, cependant on le retrouve également sur les cellules dendritiques, les monocytes, les cellules T activées, les neutrophiles et dans certains organes [103] [106]. L’OPG est une protéine exprimée de façon ubiquitaire [105].
Figure I.8. La résorption osseuse physiologique. La résorption osseuse physiologique est un cycle
équilibré entre résorption et formation osseuse. La première phase est celle de résorption et le résultat de l’activation des ostéoclastes et leurs précurseurs. Elle est suivie par la phase de formation osseuse qui est déclenchée par l’activation des ostéoblastes et leurs précurseurs. La fin du processus est marquée par la différenciation des ostéocytes et minéralisation de la matrice, suivi par un état de quiescence. Cet état peut être brisé en réponse aux stimuli de cisaillement, ce qui provoque la libération des molécules capables d’activer des ostéoclastes et recommencer le cycle.
I.2.5.1. La résorption osseuse physiologique
Dans sa forme active, l’ostéoclaste est capable de former une zone d’étanchéité (« seeling zone ») où la dégradation de la matrice osseuse est facilitée grâce à l’attachement de la cellule à la matrice sous-jacente par des intégrines, comme l’intégrine alpha 5 beta 3 (αvβ3) [107]. La formation de podosomes caractérise la bordure en brosse des ostéoclastes, et l’anneau d’actine, l’endroit où la résorption osseuse a lieu. Dans cette ambiance fermée, l'ostéoclaste acidifie le milieu favorisant ainsi l’action des enzymes de
21 résorption, protéases et phosphatases présentes dans leurs lysosomes. Cette acidification est obtenue grâce à l’activité de l’anhydrase carbonique II, des pompes à protons et de l’ATPase vacuolaire [108]. Les enzymes les plus impliquées dans la résorption osseuse sont : la protéase acide (PA), la cathepsine K et les MMP, notamment la métalloprotéinase 9 (MMP9).
La liaison RANKL / RANK déclenche des voies de signalisation, dont la voie NF-κB et le facteur de transcription NFATC1 responsable de l’activation de la transcription des gènes liés à la différenciation en ostéoclaste et aux activités de résorption, comme les gènes du récepteur de calcitonine, de la MMP9, de la TRAP et d’autres. Après la phase de résorption, les cellules adjacentes produisent des facteurs qui vont permettre d’arrêter ce processus, notamment l’OPG, qui est capable de se lier à RANKL et empêcher son action sur le récepteur RANK à la surface des ostéoclastes, arrêtant ainsi leur différenciation et favorisant la formation osseuse par les ostéoblastes [106].
Le remodelage osseux est régulé au niveau systémique par des hormones dont les plus importantes sont l’hormone parathyroïde (PTH), les glucocorticoïdes, les hormones thyroïdiennes, les œstrogènes, le calcitriol (forme active de la vitamine D) et la calcitonine. La régulation locale est modulée par la présence de médiateurs inflammatoires comme le M-CSF, le TNF-alpha, l’IL-1beta, l’IL-6, l’IL-10, le TGF-beta et par divers lipides bioactifs (prostaglandines et leucotriènes), ainsi que par la triade RANK/ RANKL/ OPG [102].
I.2.5.2. La résorption osseuse pathologique
Comme décrite antérieurement, la différenciation et l’activation des ostéoclastes par RANKL est un facteur critique pour la résorption osseuse et sa modulation est contrôlée par un équilibre RANKL/ OPG. Dans l’AAI, la présence de plusieurs médiateurs inflammatoires provoquent une augmentation de la production de RANKL par les cellules tissulaires et par les cellules immunitaires présentes. La balance RANKL /OPG est alors déséquilibrée. Les cellules T activées CD4+ et CD8+, les fibroblastes synoviaux et les macrophages expriment RANKL [109] [110]. L’IL-1beta, le TNF-alpha et l’IL-17 secrétées par les macrophages activés stimulent l’expression de RANKL par les ostéoblastes pendant que l’IL-6, l’IL-17, l’IL-1beta et le TNF-alpha stimulent l’expression de RANKL par les synoviocytes. Le M-CSF augmente l’expression de RANK par les macrophages du pannus [111] [112]. La surexpression de RANKL dans les articulations enflammées aboutit à la formation excessive des ostéoclastes [113] [114]. Associé à ce fait, il y a un déficit de l’expression d’OPG par les cellules synoviales activées au cours de la PAR [115] [116].Toutes ces altérations favorisent l’activation et la différenciation des ostéoclastes et le recrutement de leurs précurseurs menant à une résorption osseuse anormalement accrue, comme montre la figure I.9.
Figure I.9. Mécanismes impliqués dans la destruction osseuse de l’AAI. La présence de plusieurs
médiateurs inflammatoires et leurs actions sur les cellules synoviales et immunitaires augmentent la production de RANKL et, sur les ostéoclastes, l’expression du récepteur RANK. La libération des cytokines pro-inflammatoires par les cellules immunitaires comme le TNF-alpha, l’IL-1beta, l’IL-6 et l’IL-17 sont capables d’induire l’expression de RANKL par les cellules synoviales. De plus, RANKL est exprimé par les fibroblastes synoviaux et par les cellules T activées. Les ostéoclastes formés en excès sont responsables d’une résorption accrue de l’os aboutissant à la destruction de l’articulation. (Figure tirée de Choi Y et al. Nat Rev Rheumatol 2009. 5: 543).
I.2.6. Traitement de l’AAI
L’AAI est une maladie qui ne peut pas être guérie mais que l’on peut contrôler. Dans les dernières années, le traitement de l’AAI s’est beaucoup amélioré et permet un très bon contrôle chez la majorité des patients. Cette amélioration est reliée au développement des nouvelles thérapies qui ciblent la caractéristique inflammatoire de la maladie : les « antirhumatismaux à action lente » (ARAL) et les « médicaments modificateurs de la réponse biologique » (MRB). Ces médicaments sont utilisés pour traiter l’AAI et d’autres affections rhumatismales par une réduction de l’inflammation et un ralentissement de l’évolution de la maladie avec un très bon contrôle de la résorption osseuse (Figure I.10). C’est pourquoi ils