w
31156008425442
^5
3"^
ETUDE DE LA REACTION DE DIELS-ALDERTRANSANNULAIRE DE
CYCLOTETRADECATRIENONES DE GEOMETRIE TRANS'TRANS-CIS ET
77L4XSI-CZS-C/^COMPORTANT UN DIENOPHILE ACTIVE. APPROCHES EN ^^
VUE DE LA SYNTHESE DE DFTERPENES.
par
LOUIS BARRIAULT
these presentee au Departement dc chimie en vue de Pobtention du doctorat es sciences (Ph. D.)
FACULTE DES SCIENCES
UNIVERSITE DE SHERBROOKE
II manque les pages 21-281-283
Sherbrooke, Quebec, Canada, septembre 1997
395 Wellington Street Ottawa ON K1AON4 Canada 395, rue Wellington Ottawa ON K1AON4 Canada
Your file Volre reference
Our file Nolre reference
The author has granted a
non-exclusive licence allowing the National Library of Canada to reproduce, loan, distribute or sell copies of this thesis in microform,
paper or electronic formats.
The author retains ownership of the
copyright in this thesis. Neither the
thesis nor substantial extracts from it may be printed or otherwise
reproduced without the author's
permission.
L'auteur a accorde une licence non exclusive permettant a la
Bibliofheque nationale du Canada de
reproduire, preter, distiibuer ou vendre des copies de cette these sous
la forme de microfiche/fihn, de
reproduction sur papier ou sur format electronique.
L'auteur conserve la propriete du droit d'auteur qui protege cette these.
Ni la these ni des extraits substantiels de celle-ci ne doivent etre imprimes
ou autrement reproduits sans son
autonsation.
0-612-40508-7
President-rapporteur: Membre: Membre: Examinateur externe: M.Luc Ruest Departement de chimie M. Claude Spino Departement de chimie
M. Pierre Deslongchamps \^&^/
Departement de chimie Dr. Rene Roy
Universite d'Ottawa
Nous atteindrons notre but sansfaute.
(0 Sensei Chitose (1898-1984))
SOMMAIRE
La premiere et la troisieme parties de cette these decrivent la synthese
enantioselective de cyclotetradecatrienones de geometrie trans-trans-cis (TTC) et
trans-cis-cis (TCC), suivie de l^etude de la cycloaddition [4+2] transannulaire thermique et catalysee par les acides de Lewis. De plus, des resultats de calculs de modelisation moleculaire des etats de transition de la reaction y sont discutes.
La seconde et la quatrieme parties traitent des approches possibles en vue de la synthese enantioselective de l'(-)-acide fusidique et de derives non-naturels de la (+)-pyripyropene A et de la (-)-forskoline.
REMERCIEMENTS
Je desire remercier sincerement Ie professeur Pierre Deslongchamps pour m'avoir
accueilli dans son laboratoire et m*avoir foumi les outils n^cessaires au developpement de mon esprit scientifique.
Dans cette meme veine de pensee, je remercie Ie Dr. Pierre Soucy pour m'avoir
enseigner les rudiments de la chimie organique experimentale a mon premier stage d'ete ainsi que mes collegues de laboratoire particulierement mes amis Guillaume Belanger, Luc Ouellet, Rico Lavoie et Dr. Dennis Hall pour les nombreux echanges d*id6es et Ie bon temps passer dans Ie «trailer ». De plus, j'aimerais remercier Ie Pr Yves Dory pour les calculs de modelisation moleculaire ainsi que les etudiants d'ete qui m'ont aide dans cette aventure
scientifique: Samuel Fortin (ete 94), Alain Rouillard (et6 95) et Stephane G. Ouellet (etc 96).
Je tiens a remercier les employ6s de soutien tel que M. Real Dubuc au soufiflage de la verrerie, M. Gaston Boulay a la spectroscopie de masse, M. Marc Drouin a la diffiraction des rayons-X et Ie Dr. Nonnand Pothier a la spectroscopie RMN pour leur apport technique en
la matiere.
Je tiens tout specialement ^ remercier ma femme Julie pour son soutien moral tout au long de mes etudes et pour m'avoir aider a dactylographier ce travail. De plus, mes parents,
Evelyne et Bernard, reyoivent toute ma gratitude pour leurs encouragements incessants ainsi que mes beaux-parents, Roger et Jeanne.
Finalement, mes remerciements s*adressent aux Fonds pour la Formation de
Chercheurs et I* Aide a la Recherche (FCAR) et la compagnie Bio-Mega/Boehringer
REMERCffiMENTS...iv TABLE DESMATffiRES...^ LISTEDES TABLEAUX...^^ LISTEDES FIGURES...ix LISTEDESSCHEMAS...^ INTRODUCTION...! 1. GENERALITES...1
2. ACTIVATION DE LA REACTION DE DffiLS-ALDER...1
3. DffiLS-ALDERINTRAMOLECULAIRE...4
4. RESULTATS DE TRAVAUX DE XUETROUGHTON...8
5. DESCRIPTION DUPROJET ...12
RESULTATSET DISCUSSION...16
1. PREMIERE PARTffi: ETUDE DE LA REACTION DE DffiLS-ALDER
TRANSANNULAIRE D'UN MACROCYCLE DE GEOMETRffi
TRANS-TRANS-CIS(TTC) COMPORTANT UN DIENOPfflLE ACTIVE...161.1. Synthese enantioselective d'un cyclotetradecatrienone TTC... 16
1.1.1. Voie A...16
1.1.2. VoieB...25
1.1.3. VoieC...28
1.1.4. Voie D...36
1.1.5. VoieE...39
1.2. Reaction de Diels-Alder transannulaire du cyclotetradecatrienone TTC...43
1.2.2. Etude de la reaction par activation thermique et resultat
d'epimerisation..,...47
1.3. Resultatsdescalculs de modelisation...51
1.4. Conclusion...53
2. DEUXffiMEPARTffi: APPROCHES VERS LA SYNTHESE DE L'ACIDE
FUSIDIQUE...552.1. Introduction...55
2.2. Approche Uneaire en vue de la formation du bicyclo[12.3.0]heptadecatetraenone 142...55
2.2.1. Retrosynthese...55
2.2.2. Synthese 6nantioselective d'un cycloheptadecatrienone en utilisant la fonction chlorocetone comme agent de couplage...57
2.3. Approche convergente en vue de la formation du bicyclo[12.3.0]heptadecat6traenone 142...65
2.3.1. Retrosynth^se...65
2.3.2. Synthese enantios^Iective du fragment 150...65
2.3.3. Synthese enantioselective du fragment phosphonate 162...66
2.3.4. Essais de couplage des deux fragments 150 et 162.. ...70
2.4. Conclusion...72
3. TROISffiMEPARTffi: ETUDE DE LA REACTION DE DIELS-ALDER
TRANSANNULAIRE D'UN MACROCYCLE A 14 MEMBRES DE
GEOMETRffi TRANS'CIS-CIS (TCC) COMPORTANT UN DffiNOPISLE
ACTIVE...743.1. Synthese enantioselective d'un cyclotetradecatrienone TCC... ,.74
3.1.1. Voie A...74
3.1.2. Voie B...„...„...„..„...77
3.1.3. VoieC...79
3.2, Reaction de Diels-Alder transannulaire du cyclotetradecatrienone TCC ,...„,...„...,...,...„...,.,...,„.,,...,,,.,...„..,.,.,..,.„..,..,.83
3.2.1. Etude de la reaction par i'activation avec les acides de Lewis.. .83
3.2.2. Etude de la reaction par activation thermique...,,,..,.,,,....,,...86
3.3. Resultats des calculs de modelisation...89
3.4. Conclusion...91
4. QUATRffiME PARTffi: ETUDE AUTOUR DES SYNTHESES DE LA
14-DEOXAFORSKOLINE ET DE LA 14-DEOXAPYRIPYROPENE A...934.1. Approche vers la synthese de la 14-deoxapyripyropene A...93
4.1.1. Introduction...93
4.1.2. Retrosynthese...94
4.1.3. Approche pour la formation du cycle D...95
4.1.4. Etude autour de la fonctionnalisation du cycle B de la 14-deoxapyripyropene A... 106
4.2. Approche pour la synthese de la 14-deoxaforskoline...110
4.2.1. Introduction...110
4.2.2. Retrosynthese...Ill 4.2.3. Etude autour de la fonctionnalisation des cycles B et C de la 14-deoxaforskoline...ll3 4.3. Conclusion...117 CONCLUSION GENERALE...119 PARTffiEXPERIMENTALE...120 SPECTRES RMN...^ BBLIOGRAPFIffi...
LISTE DES TABLEAUX
Tableau 1. Resultats des variations de temperature de la reaction
de Wittig-Homer-Emmons pour Ie compose 49 ...22
Tableau 2. Resultats des differents essais de macrocyclisation du triene 55...25
Tableau 3. Resultats des essais de macrocyclisation de chlorocetone 60 et 66... .27
Tableau 4. Resultats de la reaction de Diels-Alder transannulaire de 56 avec differentsacides de Lewis...47
Tableau 5. Resultat de la reaction de Diels-ASder transannulaire de 56 a differentes temperatures...50
Tableau 6. Resultat de la macrocyclisation des trienes 133,134 et 135... .63
Tableau 7. Resultat de la reaction (Taldol transannulaire de 141...64
Tableau 8. Resultats des essais de couplage entre 162 et 150...72
Tableau 9. Resultats de la reaction de Diels-Alder transannulaire catalysee par des acides de Lewis sur Ie macrocycle 176...84
Tableau 10. R^sultats de la reaction de Diels-Alder transannulaire thermique sur 176...87
LISTE DES FIGURES
Figure 1. Disposition HOMO-LUMO catalys^e par les acides de
Lewis (ligne hachuree) et non-catalysee (ligne pleine)...3 Figure 2. Vue spectroscopique des etats de transitions A et B ...13 Figure 3. Etats de transition de la reaction de Wittig-Homer-Emmons
du compose 49...22
Figure 4. Mecanisme de deprotection et de lactonisation du macrocycle 87... ,35
Figure 5. La formation du tetraene 88 vs Ie macrocycle 87...36 Figure 6. Structure moleculaire etablie par 1'analyse de la diffraction des
rayons-X du tricycle 115...46
Figure 7. M^canisme d'isom^risation de 1'enone du macrocycle 56 ...53
Figures. La formation du cycle D de 142...66
Figure 9. Structure mol^culaire etablie par 1'analyse de la dif&action des
rayons-Xdumacrocycle 176...80
Figure 10. Etats de transition de la reaction de Diels-Alder transannulaire...85
Figure 11. Etat de transition d^conjugue de la reaction de Diels-Alder...86
Figure 12. Structure moleculaire etablie par 1'analyse de la diffraction des
rayons-Xdutricycle 190...88
Figure 13. Vue stereoscopiquedesetats de transition Cet D...90 Figure 14. Structures de la pyripyropene A 192 et de son analogue non-naturel 193 ..94 Figure 15. Structures en 3-D desisomeresZet£ 205 et 207...102 Figure 16. Intermediaire de la reaction de Pr^vost-Woodward ...106
Figure 17. Structures de la (-)-forskoline 227 et de la (-)-14-deoxaforskoline 228... 110
Figure 18. Derive non-naturel de la forskoline...Ill Figure 19. Vue en 3-D des cycles Get D ...116
Schema 2 ...2 Schema 3 ...2 Schema 4 ...4 Schema 5 ...5 Schema 6 ...6 Schema? ...7 Schema 8 ...8 Schema 9 ...9 Schema 10 ...10 Schema 11 ...11 Schema 12 ...12 Schema 13 ...15 Schema 14 ...17 Schema 15 ...18 Schema 16 ...20 Schema 17 ...24 Schema 18 ...26 Schema 19 ...29 Schema 20 ...30 Schema 21 ...31 Schema 22 ...32 Schema 23 ...33 Schema 24 ...34 Schema 25 ...37 Schema 26 ...38 Schema 27 ,.,...,...,.,,,...,.,...,.,...,,...,...,.,..,,....,...,....,,,...,.,.,...,,,,.,.,...,...,,,.40 Schema 28 ...,...,...,...,...,...,.,....,,..,,,...,,,...,,,...,,...,,..,...,..,....,...,.,41
Schema 29 ...42 Schema 30 ...44 Schema 31 ...45 Schema 32 ...48 Schema 33 ...49 Schema 34 ...50 Schema 35 ...52 Schema 36 ...56 Schema 37 ...58 Schema 38 ...59 Schema 39 ...60 Schema 40 ...62 Schema 41 ...67 Schema 42 ...68 Schema 43 ...69 Schema44 ...71 Schema 45 ...73 Schema 46 ...75 Schema 47 ...76 Schema 48 ...78 Schema 49 ...81 Schema 50 ...82 Schema 51 ...83 Schema 52 ...83 Schema 53 ...95 Schema 54 ...97 Schema 55 ...98 Schema 56 ..,...,...,...,,...,...,.,.,...,...,,.,.,....,,...,...,,,..,,...,...,...,,...99 Schema 57 ,...,..,,..,...,,.,,..,...,..,.,.,..,,,,,,.,..,,..,....,,...,,..,,,,...,,,..,...,,...,...,,,...,.101 Schema 58 ...,,,.,,,..,...,,.,..,,,..,.,.,..,.,...,.,.,...,.,,.,...,.,,..,..,.,,,.,..,,....,...,,..,.,103
Schema 59 ...,...,.,..,...,..,,..,,.,.,...,...,,...,,,...,..,,.,...,,,.,...,....,,...,....105 Schema 60 ...108 Schema 61 ...109 Schema 62 ...112 Schema 63 ...114 Schema 64 ...115 Schema 65 ...118
Depuis plusieurs decennies, la recherche de nouvelles methodes efficaces pour la
formation de liaison carbone-carbone a suscite beaucoup cTinteret aupres de nombreux chimistes organiciens. Des lors, la decouverte de nombreuses reactions tel que la simple
reaction d*aldol en passant par les alkylations 1,2 et 1,4, les cycloadditions. Ie couplage avec des metaux de transition et, jusqu'a recemment la metathese d'olefines ont permis de faire des pas considerables dans la science de la construction des composes organiques
relativement complexes. C'est Ie cas notamment de la reaction de Diels-Alder qui est une des reaction les plus etudiees et utilisees en synthese organique. Sa decouverte remonte en
1928 lorsque deux chimistes allemands, Kurt Alder et Otto Diels (1). Cette reaction necessite 1'interaction d*un diene conjugue 1 avec un dienophile 2, lequel peut ^tre sous forme d'une liaison double ou triple qui peut conduire, par un processus pericyclique a six electrons pi, a la construction du cycle a six membres 3 (schema 1).
Schema 1
De plus, la reaction peut etre hautement regio-, stereo- et/ou enantioselective et,
dependamment du degre de substitution du diene et du dienophile, la reaction de Diels-Alder
peut creer a la fois jusqu*a quatre nouveaux centres chiraux contigus (schema 2).
2. Activation de la reaction de DieIs-AIder
Dans plusieurs cas, la reaction de Diels-Alder a lieu soit a la temperature ambiante, soit a des temperatures plus elevees. Ce dernier cas aboutit quelques fois a la degradation
employees sont la catalyse soit, par la haute pression, les acides de Bronsted et de Lewis,
les radicaux cations, les anticorps catalytiques et meme, dans certains cas, par des enzymes
(2). D'autres methodes comme les ultrasons et la catalyse heterogene inorganique sont
moins utilisees mais n'en demeurent pas moins des methodes efficaces.
+
c
R? R4 Schema 2 R'6
r"
R2z'
4L
'R4Pour nos travaux, celle qui a retenu davantage notre attention a 6te la catalyse par les acides de Lewis. La premiere constatation d'une acceleration de la reaction de DieIs-Alder par les acides de Lewis a ete faite par Yates en 1960 (3). En effet, il a demontre que la vitesse de cycloaddition de Fanthracene et de Fanhyride maleique pour donner l*adduit 4 augmentait (Tun facteur 192 000 en presence de trichlorure d'aluminium dans Ie dichloromethane (schema 3).
0
.00
A1C1
CHiCb, 25°C
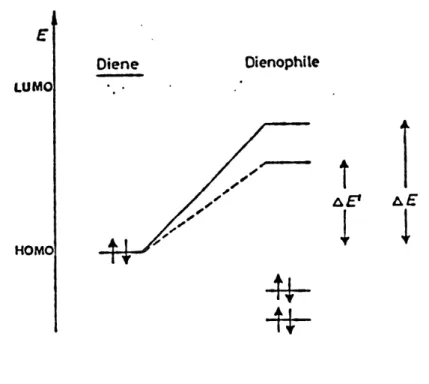
par Epiotis (4) et Branchadell (5) en appliquant la theorie des orbitales moleculaires frontieres (6). L'interaction donneur-accepteur entre Ie dienophile et Ie catalyseur diminue Penergie de la LUMO du dienophile. Pour Ie cas de la cycloaddition [4+2] avec une demande electronique normale, cela signifie que la difference (Tenergie entre les orbitales moleculaires du diene et du dienophile diminue, ce qui abaisse Ie niveau d'energie de Fetat
de transition (figure 1). Des calculs ont demontre, par ailleurs, que la grosseur de certains
coefficients orbitalaires et Ie renforcement des interactions orbitalaires secondaires augmentent (6) ce qui tend a expliquer Faugmentation de la regio- et de la stereoselectivite.
LUMO
HOMO
Diene Oienophde
Af
I
Figure 1. Disposition HOMO-LUMO catalysee par les acides de Lewis (ligne hachuree) et
dienophile sont attaches par une chatne de carbone qui peut contenir dans certains cas un ou plusieurs toeroatomes. La cyclisation donne lieu a des syst^mes bicycliques fusionnes ou pontes (schema 4). G6n6ralement, Ie processus de 5 vers 6 est largement favorise sur celui de 7 vers 8 lorque la chaTne est petite. Mais si la chalne comporte plus de dix atomes, alors il peut y avoir, dans certains cas, apparition de compose ponte (7) (schema 5).
8
Schema 4
Par centre, lorsque la chame est en position C2 tel que dans Ie diene 13, la cycloaddition [4+2] intramoleculaire foumit exclusivement un bicycle ponte 14 (8). La stereoselectivite de la reaction de Diels-Alder intramoleculaire depend des effets
conformationnels conditionnes par les effets steriques et electroniques a I'etat de transition. Contrairement a la reaction intermoleculaire, Ie processus intramoleculaire limite Ie nombre
10
11
(77%; 6.2:6.8:1)r^\
"^
13
395°C phase gazeuze Schema 5II a ete demontre par Roush (9) qu'avec un dienophile active de geometrie trans 15 (schema 6), la reaction de DieIs-Alder thermique conduit a la formation majoritalre du produit endo; Ie bicyclo[4.3.0] trans 16. Cependant, avec un dienophile cis 18, la reaction donne un melange de bicyclo[4.3.0] 19 et 20 dont Ie majoritaire est Ie produit exo 19, Dans Ie cas de la reaction de Diels-Alder catalysee par un acide de Lewis, Ie triene 15 a donne
exclusivement Ie produit efido 16. Par contre, en utilisant Ie triene 18, Ie ratio de la reaction n'a pratiquement pas change.
15
H
16
17
Toluenel50°C, 16/17=72:28 Acide de Lewis, 16/17 = 100 : 0 H3C02C. C02CH3COjCHa
18
H
1920
Toluene 180 °C, 19/20 = 67 : 33 Acide de Lewis, 19/20 = 68 : 32 Schema 619 (schema 7). ,.R3
R2-7^
/"^;
^"'\ ..••H Ri=CH(CH3)2 R2=C02CH3 R3=H endo16
Rl=CH(CH3)2Rz-CQzCHs R3S=H endo20
Rl=CH(CH3)2 R2=H R3:=C02CH3 exo19
Rl==CH(CH3)2R2==H R3=C02CH3 exo17
Schema 7La reaction de Diels-Alder intramoleculaire de niveau 2, c*est-a-dire transannulaire, est un autre volet tres interessant du processus intramoleculaire. Celui-ci comporte plus d'avantages que Ie niveau 1 du au fait que la reaction de Diels-Alder transannulaire, par sa
nature, rapproche Ie diene et Ie dienophile et, peut engendrer la reaction sans activation du dienophile. De plus, les degres de substitution du diene et du dienophile ne constituent pas
un obstacle a la reaction de Diels-Alder transannulaire. En effet, contrairement a la reaction intramoleculaire de niveau 1, ou un diene trisubstitue ou un dienophile tetrasubstitue rendent
OBZ X^OMOM E=C02CH3 OBz H + OBz 67:33
^T~
210°C,18 h Pas de reaction OBz Schema 84. Resultat de travaux de Xu et Roughton
Depuis 1984, Ie Pr Deslongchamps et ses collaborateurs ont developpe une approche convergente pour la synthese de molecules polycycliques via la reaction de Diels-Alder
transannulaire de trienes macrocycliques a treize, quatorze et quinze membres (11), Depuis 1990, se basant sur des resultats concluants provenant d'etudes modeles, des approches vers
1996 (16) (schema 9).
TBDPSO
TBDPSO'HO
(+)-Nargenicin A i Schema 9Beaucoup de produits naturels tels que dans la famille des diterpenes et quassinoides, component un methyle en position C8 (schema 10). II nous apparait alors possible de
transannulaire d'un cyclotetradecatriene comportant un methyle sur Ie diene. Xu et
Roughton (17) ont ete les premiers a explorer cette voie par 1'etude de la reaction de
Diels-Alder sur des modeles cyclotetradecatrienes de geometric cis-cis (TCC) 21,
trans-trans-trans (TTT) 22 et trans-trans-trans-cis (TTC) 23 (schema 11).
Schema 10
Les resultats de Roughton ont determine que la cycloaddition transannulaire [4+2] du macrocycle TCC 21 conduit uniquement au tricycle de stereochimie trans-syn-cis (TSC) 24. Par contre, les resultats de Xu ont ete mitiges, d'une part Ie macrocycle TTT 22 conduit exclusivement au tricycle 25 et, d'autre part. Ie macrocycle TTC 23 donne un melange de quatre tricycles 26, 27, 24 et 28 dans un rapport de 39:7:3:1 (schema 11). Ce melange de tricycles provient de Pinterconversion du macrocycle TTC 23 en d'autres macrocycles par
un rearrangement sigmatropique-1,5 du diene (schema 12). En efFet, il a etc demontre par
Xu (17) que Ie ^arrangement sigmatropique 1,5 du macrocycle TTC 23 se produit a 200°C
pour donner Ie macrocycle cis'trans-cis CTC 29. Ce demier s'interconvertit en d'autres macrocycles lorsque la temperature s'eleve et conduit eventuellement a un melange de
^\-^
21
\^ .^~VE
s^y^B
23
E = C02CH3 •iiiEE' VH
320°C toluene 200°C toluene 310°C toluene •E 2824 (6%)
Schema 11E. ..E
23
E'' >E E. .E r 'E En...29
Schema 12Les travaux de modelisation moleculaire fails par Dory (18) ont appuye fortement Phypothese de Pmterconversion de macrocycles et ont foumi une explication claire et concise du ratio obtenu et de la formation plut6t inhabituelle du tricyclo[5.3.2.0]decatetraene
28.
5. Description du projet
Les travaux de Xu et de Roughton ont d^montre que la presence d'un methyle sur un diene influence la temperature de reaction. Afin de realiser la reaction de Diels-Alder
transannulaire, la temperature doit atteindre 310°C et cela engendre dans certains cas, la
formation de plusieurs produits de reactions secondaires. De plus, la modelisation moleculaire tend a demontrer qu'une repulsion entre Ie methylene en alpha du dienophile et
Ie methyle sur Ie diene serait la cause du melange de tricycles trans-syn-trans TST 27 et cis-syn-cis CSC 26, Une superposition des etats de transition calcules de la reaction de
Diels-Alder avec et sans methyle sur Ie diene indique une deformation du cycle C dans Petat de
transition A (figure 2). Cette deformation causee, par la presence d'interactions non-liantes entre Ie methyle et methylene, augmenterait davantage Fenergie de 1'etat de transition A, favorisant Ie passage de la reaction par Petat de transition B,
B
Par consequent, notre projet consistait en 1'etude de la reaction de Diels-Alder
transannulaire de trienes macrocycliques chiraux TTC 30 et TCC 31 comportant tous deux un dienophile active (schema 13). Premierement, 1'activation du dienophile devrait diminuer la temperature de la reaction sous la barre des 200°C pour ainsi eviter Ie ^arrangement sigmatropique 1,5 dans Ie cas de la serie TTC. Deuxiemement, I'introduction d'une fonction cetone en alpha du dienophile devrait permettre une diminution des
interactions transannulaires a Petat de transition.
De plus, outre la possibilite d'une diminution de la temperature de reaction des macrocycles TTC 30 et TCC 31, la synthese enantioselective de ceux-ci fonctionnalises adequatement devrait pennettre des approches interessantes pour la synthese de produits naturels. D'une part, la reaction de Diels-Alder transannulaire du macrocycle TTC 30 conduirait au tricycle TST 32, ce qui nous ouvrirait une route pour une eventuelle synthese de Facide fusidique 35. D'autre pan. Ie macrocycle TCC 31 foumirait, par Ie m6me processus. Ie tricycle TSC 33 qui, par une epimerisation en C9, conduirait au tricycle de ster^ochimie trans-cmti-trans (TAT) 34. Ce demier serait un intermediaire snteressant en vue de la synthese de la quassine 36.
CH30
CH30
30
H
32
PCH-CHsO
COsH
OAc Schema 13CH30
H
34
QCHi
0
0
CH30 o" ^oRESULTATS ET DISCUSSION
1. PREMIERE PARTIE:
ETUDE DE LA REACTION DE DIELS-ALDER TRANSANNVLAIRE D'UN
MACROCYCLE DE GEOMETRIE TRANS'TRANS-CIS (TTC) COMPORTANT UN
DIENOPHILE ACTFVE
1.1 Synthese enantioselective d'un cyclotetradecatrienone TTC
1.1.1 Voie A
La synthese a debute par 1'epoxydation selective de Pacetate de neryle commercial en utilisant Pacide m-chloroperbenzoique dans Ie dichlorom6thane a OOC (schema 14). 1/epoxyde brut a etc traite en presence d'acide periodique dans Ie tetrahydrofurane a 0°C pour donner 1'aldehyde 37 avec 75% de rendement pour les deux etapes (19). La protection
de Faldehyde 37 a ete faite en utilisant la m6thode de Luche (20) pour fournir Ie
dimethylacetal 38 dans un excellent rendement de 94%. Par la suite, la methanolyse de Pacetate 38 suivie de Foxydation de 1'alcool 39 par la m6thode de Swern (21) ont foumi Paldehyde 40 de fayon quantitative. Une reaction d'alkylation 1,2 sur l*aldehyde 40 en utilisant Ie chloroiodomethane et Ie butyllithium dans Ie tetrahydrofurane a -78°C (22) nous a permis d'obtenir la chlorohydrine 41 (94%).
L'alcool secondaire 41 resultant a etc protege sous la forme de triisopropylsilylether en utilisant une methode developpee par Corey (23) pour nous fournir Ie silylether 42 (98%) (schema 15). L'hydrolyse en milieu acide de la fonction acetal de 42 a donne Pald^hyde 43 dans un rendement de 94%. I/introduction de deux carbones asymetriques controles et contigus a etc possible grace a la m^thode (Taldol asymetrique developpee par Evans (24).
En premier lieu, 1'enolate de geometric Z du (7?)-3-(l-oxopropyl)-4-benzyl-2-oxazolidinone
(25) a etc obtenu avec Paddition tres lente du triflate de di-w-butylborane suivie de la triethylamine a 0°C. Par la suite, 1'addition lente de 1'aldehyde 43 nous a foumi un seul
OAc l)m-ClC6H4C03H CH2C12, 0°C
2)H5l06
THF, 0°C OAc LaCl3 XHsO(CH30)3CH
CH30H
OAc CH30' ^OCHs38
K2C03CH30H
CHsO' ^OCH?39
1) (COC1)2, DMSO
CH2C12, -78°C 2)Et3N,0°C CH30' "OCHs40
ICHiCI, BuLi THF, -78°CHO^^,
CH3CT "OCH3 41a
Schema 1441 TIPSOTf, 2,6-lutidine CH2C12,0°C
APTS
acetone / eau 25°CTIPSO
43
9 oI) ,\\ , BuzBOTf
-t-(.-^—^
Et3N,CH2Cl2 0°Ca-78°COH
TIPSO.HN(CH3)OCH3HCI
A1(CH3)3, CH2C12 -l5°Ca25°C CH30. 45 Schema 15diastereoisomere d'adduit aldolique 44 avec un rendement 59% ou les deux nouveaux centres chiraux sont de configurations absolues IR^S. L'adduit d'aldol 44 a subit une reaction de transamidation dans les conditions de Weinreb (26) avec Ie trimethylaluminium et Fhydrochlomre de N,0-dimethylhydroxylamine dans Ie dichloromethane a ~25°C pour nous founir I'amide 45 (87%).
L'alcool secondaire 45 a ete prot^ge sous forme de methylether 46 (schema 16). Ainsi, Falcoolate forme a 1'aide de 1'hydrure de sodium a ete piege par 1'iodomethane dans un melange de tetrahydrofurane et de dimethylformamide (4:1)^ 0°C dans un rendement de 98% (27). La reduction de Famide 46 avec Fhydmre de diisobutylaluminium dans Ie tetrahydrofurane a ~78°C a fourni 1'aldehyde 47 (100%). Une homologation de quatre carbones par une olefination de Wittig-Homer-Emmons a ete possible grace au phosphonate 48 (27-28). Dans ce cas-ci, Finfluence de la temperature de reaction joue un r61e crucial au niveau du ratio des isomeres geometriques et du rendement de la reaction. Nous avons
remarque, lorsque la temperature de la reaction est augmentee, que Ie pourcentage
trans-trans (TT) par rapport au cis-trans-trans (CT) augmente regulierement jusqu*a -10°C (tableau 1). Par centre, au-deU de cette temperature, des produits de degradation commencent a apparaitre. Etant donne que nous utilisons un anion de phosphonate stabilise, la reaction favorise un etat de transition tardif, c*est-a-dire un etat de transition qui ressemble au produit
final. Done, en augmentant la temperature, nous favorisons Ie passage par Fetat de
transition A plut6t que B (figure 3).
La fonction ester du melange inseparable des isomeres TT et CT 49 a ete reduite avec Phydrure de diisobutylaluminium dans Ie dichloromethane a -78°C. L'alcool 50 pur obtenu avec un rendement 83% a ete protege a 1'aide du 2-methoxypropene et de Pacide pyridinium /?-toluenesulfonique dans Ie dichloromethane. L'acetal 51 obtenu (85%) a ete traite avec Ie fluorure de tetrabutylammonium dans Ie tetrahydrofurane a -20°C afin de generer la chlorohydrine correspondante 52 dans un rendement moyen de 69%. II est a
noter que la basse temperature permet d'eviter la formation cTun epoxyde a partir de la
45
CH3I, NaH CH30. THF/DMF,0°C TEPSO. 0 OCH3 DIBAH, THF -I -78°CTIPSO
CHsO
47
(CH30)2-48
BuLi, Et20 -20°C 0 OCH3CHsO
TIPSO
Cl
OCH3
DfflAH, CHzCli -78°CTIPSO
CH30
2-Methoxypropene PPTS, CH2C12 -^ o°c CH30 TIPSO,^
51 TBAF, THF -^ -20°C OMOP CH30 HO,x^l
52OMOP
Schema 16Tableau 1. Resultats des variations de temperature de la reaction de Wittig-Horner-Emmons pour Ie compose 49.
Essai
1
2
3
4
5
Temperature °C -78 -40 -25 -20 -10 Ratio* TT/TC 3: 1 8: 1 11:1 16: 1 16: 1 Rendement %41
76
71
80
801) Ratio determine par RMN 1H. 2) Rendement isole. 47+48
/
\
C02CH3
CH302C0
°^Ert(9CH3)2
RfT-H
'HB
» R^^^^OCH3
TT 0
. r^Y°"
R CT 6
Figure 3. Etats de transition de la reaction de Wittig-Horner-Emmons du compose 49.
L'oxydation de Palcool secondaire 52 avec Ie perruthenate de tetrapropylammonium (29) en quantite catalytique et Ie N-oxyde de N-methylmorpholine dans 1'acetonitrile a
fourni la chlorocetone 53 (83%) (schema 17). L/insertion du connecteur malonate a etc effectuee avec Panion du malonate de m6thyle et Fiodure de sodium dans Ie dim6thylformamide a 25°C suivi d'une hydrolyse en milieu acide de 1'acetal MOP pour nous conduire a Palcool 54 (62%). Lors de cette reaction d'alkylation, nous avons observe que la polarite du solvantjoue un role important au niveau de 1'isom^risation de l*enone 54. Lorsque nous avons utilise Ie tetrahydrofurane comme solvant, il y a eu isomerisation partielle de Fenone en faveur de 1'olefine E dans un rapport de 2:1.
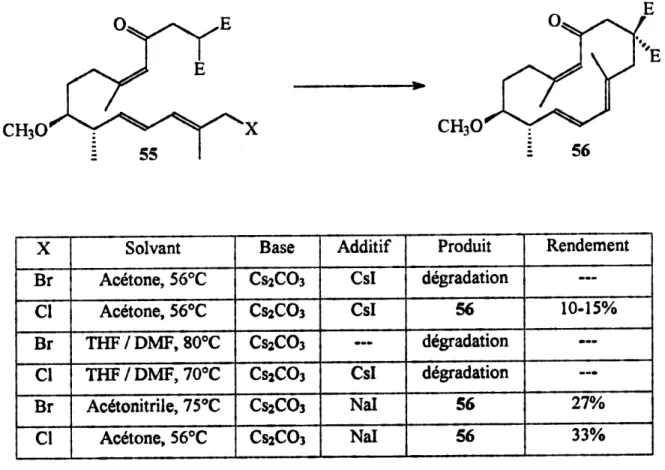
Par centre, en utilisant un solvant beaucoup plus polaire, soit Ie dimethylformamide, la reaction secondaire d'isomerisation a ete diminuee de fa?on substantielle. La geometrie Z de Fenone 54 a pu etre conserv^e dans un ratio de 23:1 et la vitesse de reaction a ete augment6e d'un facteur 4. La transformation quantitative de Palcool allylique 54 en chlorure 55 a ete possible en utilisant Phexachloroacetone, la 2,6-Iutidine et la triph6nylphosphine dans Ie t^trahydrofurane a -40°C (30). La macrocyclisation du triene 55 a ete effective en presence de carbonate de cesium et d'iodure de sodium dans 1'acetone a une concentration de 1.5 x 10'3 M pour nous conduire au macrocycle TTC 56 avec de tres faibles rendements variant de 10% a 33%. L'isolation de ce demier a et6 difficile ^ cause de la quantite importante de produits de degradation observes. Le produit isol6 est un melange inseparable du macrocycle TTC 56 et de sous-produits de la reaction. D'autres essais de macrocyclisation ont etc tentes mais sans amelioration du rendement (tableau 2).
Les faibles rendements de la macrocyclisation peuvent etre expliques par la rigidite de la molecule. Le triene 55 compone sept des quatorze carbones du futur cycle sous forme d'hybridation sp2. Ainsi, en ayant un nombre important de carbones spl conjugues, on augmente la rigidite de la molecule; d*autant plus que Ie diene de geometric trans'trans n'aide guere a faciliter I'approche du connecteur malonate vers Ie chlorure allylique. La
fonction enone de la molecule pourrait favoriser aussi, par son caractere electrophile, la
formation de produits d*addition de Michael. Toutefois, les produits de ces reactions
52
TPAP, NMO
tamis mol. 4A°
-»
CH3CN
CH30
Cl
1) NaCH(C02CH3)2, Nal, DMF, 25°C -»OMOP 2)HC1(0.1N)
acetone / eauCH30
Hexachloroacetone PPhs. 2,6-lutidine -^ THF, -40°COH CHsO
E=C02CH3 CS2C03, Csl acetone, 56°C -^ addition lente []=1.5xlO-3M CHsO56
Schema 17Tableau 2. Resultats des differents essais de macrocyclisation du triene 55
CH30
CH30
56
x
Bra
BrCl
BrCl
Solvant Acetone, 56°C Acetone, 56°C THF/DMF,80°CTHF/DMF770°C
Ac^tonitrile, 75°C Acetone, 56°C Base CS2C03 CsaCOs CS2C03 CsiC03 CssCOs Cs2COsAdditif
Csl Csl Csl Nal Nal Produit degradation56
degradation degradation56
56
Rendement 10-15%27%
33%
En vue d'ameliorer la synthese du macrocycle 56, d'autres approches ont ete
effectuees en modifiant Ie precurseur macrocyclique. Ces approches derivent toutes de celle
decrite ci-dessus.
1.1.2 Voie B
Cette approche implique Ie remplacement du chlorure allylique par un autre type
(Telectrophile, soit la chlorocetone. La sequence a debute avec l*alcool 50 qui a subi une
reaction de bromination en utilisant Ie tetrabromure de carbone et la triphenylphosphine dans Ie dichloromethane avec 98% de rendement (schema 18). Par la suite. Ie bromure 57 a ete traite avec 1'anion du malonate de dimethyle dans un melange de dimethylformamide et
50
CBr4, PPh3 CH2C12, 25°C CH30NaCH(C02CH3)2
-• DMF / THF, 25°C TIPSO.^.
CH30^S;^^VV1
58
TBAF, THF -20°CCH30
TPAP, NMOtamis mol. 4A°
CH3CN
CH30
^
''x
60
de tetrahydrofurane. Le silylether 58 obtenu (78%) a ete mis en presence d'ions Huomres pour nous fournir la chlorohydrine 59 (82%). L'oxydation de cette derniere en chlorocetone en utilisant Ie perruthenate de t6trapropylammonium a donne Ie produit 60 avec 50% de
rendement. Maintenant que la molecule est adequatement fonctionalisee, de nombreuses tentatives de macrocyclisation ont etc essayees mais sans succes. Par contre, d'autres
produits de reactions secondaires out ete observes (tableau 3).
Tableau 3. Resultats des essais de macrocyclisation des chlorocetones 60 et 66
a
CH30^S><^V^C02CH3
x
CHsO
X=C02CH3 etY=Cl 60 X=S02Ph etY=Cl 66 X=SC02CH3 56 X=S02Ph 67 Chlorocetone 6060
60
60
60 60 60 60 66 Solvant THF/DMF,77°C THF/DMF, 25°C Acetonitrile, 60°C Et20/MeOH,25°C MeOH, 25°C Acetone, 56°C Acetone, 56°C Acetone / eau, 56°C Acetonitrile, 45°C Base CsiCOs NaH CS2C03 NaH MeONa CS2C03 CssCOj CSlCOl CszCOiAdditif
Csl Csl Produit 60 etdeg. Degradation Degradation Degradation Degradation 60,Y=H Degradation 60, Y= OH Degradation Rendement75%
N.D.La synthese d'un autre type de chlorocetone ayant comme conneeteur Ie
phenylsulfonacetate de methyle est decrite aux schemas 19 et 20. Encore une fois, aucun produit de macrocyclisation n'a pu etre observe, Le probleme serait du a la rigidite de la molecule qui confererait un acces plus facile vers des etats de transition de reactions
secondaires comme dans ce cas-ci ; i.e. la reduction de la chlorocetone plutot que ta macrocyclisation.
1.1.3 VoieC
Les prochaines sequences decrites dans les schemas 21 et 22 utilisent 1'intermediaire 39 comme reactifde depart et suivent la strategic de synthese asymetrique deja employee a la section 1.1.1. Une serie de protections, reductions et deprotections telle que decrite aux schemas 21 et 22 nous a foumi Falcool 79 dans un excellent rendement. L'oxydation de 79 suivie (Tune alkylation-1,2 avec Ie CIMgCH2(CH3)2SiOCH(CH3)2 a conduit vers un intermediaire silylalkyle tres instable (schema 23). Ce demier a ete immediatement traite en presence de peroxyde (Thydrogene 35% et de bicarbonate de sodium dans 1c methanol. Cette condition oxydative a permis Ie bris de la liaison carbone-silicium afin d'obtenir Ie dial 81 avec 72% de rendement pour les deux etapes (31).
La tosylation de 81 en utilisant Ie chlorure de p-toluenesulfonyle fi-aTchement recristallis6, la dimethylaminopyridine et la triethylamine dans Ie dichloromethane a 0°C (32) a donne Ie tosylate 82 tres instable (59%). La protection de Palcool 82 sous forme de MOPO 83 suivie (Tune reaction de type SN2 avec Penolate de potassium du malonate de dimethyle en presence cTiodure de potassium et (Tether lS-couronne-6 dans Ie toluene a reflux ont foumi Ie triene 84 (72%). Une deprotection avec les ions fluorures du silylether 84 a donne Falcool 85 avec 71% de rendement. La conversion de l*alcool 85 en chlorure 86, effectuee avec la methode de Schreiber (30) (schema 24), suivie de la macrocyclisation
du triene 86, dans I'acetonitrile a 7S°C avec Ie carbonate de cesium et I'iodure de cesium, nous ont conduit vers deux produits qui ont ete isoles. Une analyse approfondie des spectres
Hexachloroacetone PPh3, 2,6-Iutidine
94
(voir section 1.1.4) THF, -40°CCHsO
KH, CH2(C02CH3)2 KI,18-C-6 -^ toluene, 60°CCHsO
62
AcOH, H20 25°C CH30SOiPh
TsCl, DMAP Et3N, CHzCli o°c CHaO SOzPh Schema 1964
Dess-Martin CH2C12 CH30 OTsLiCI
DMF, 25°CS02Ph
CH30
66
CS2C03-^
CHsCN, 45°CCH30
0
67
Schema 2039
TBDPSC1, imidazole THF, 0°C•^
CH3CT "OCH3 68OTBDPS
APTS
acetone / eau (4 : 1)OTBDPS
0 QQ 0
l)oJL?'S,Bu2BOTf
^,.-_^ ^^
Q''- \-/
EtaN, CH2C12 0°Ca-78°C 'BnOH
70
OTBDPS
A1(CH3)3 Q
HN(CH3)OCH3HC1 CH30^
CH2Cl2,-150C&250COH
OTBDPS
CHsI, NaH THF/DMF,0°CQ QCH3
CHsO,OTBDPS
DffiAH
THF, -90°C CH30OTBDPS
Schema 2173
(H3CO)2-BuLi, 11 /p.48
THF, -10°c
*OCH30
CHsOTBAF
THF
OCH3
TESC1, imidazole CH30 ^OHQ
THF, 0°C "OCHa CH3075
OTES
0
OCHs DffiAH, CH2C12 -78°CCH30
OTES
TBDPSC1, imidazole -i THF,0°COTES
CH30PPTS
CH30H, 0°COTBDPS CH30
OTBDPS
Schema 22TPAP, NMO
tamis mol. 4A°
79 CH2CI2
CH30
1) ClCH2Si(CH3)20CH(CH3)2
Mg(0),THF,0°COTBDPS
2)H20235%,NaHC03 CHsOH, reflux CHsOOH
TsCl, DMAP Et3N, CHzCl:, 0°C HO, SOTs-f
OTBDPS CH30
82
OTBDPS
MOPO
2-Methoxypropene PPTS, CH2C12 o°c CH30 OTsOTBDPS
KCH(C02CH3)2
KI, 18-C-6 toluene, UO°CMOPO
CHaOMOPO
TBAF, THF -^ 25°COTBDPS CH30
OH
Schema 23MOPO
85
Hexachloroacetone PPh3, 2,6-tutidine -i THF, -40°C CH30 C&2C03, Csl -^ CHsCN, 75°C []=1.5xlO-3M CH30MOPO
CHsO
87
Schema 24RMN 'H demontre clairement que les deux composes obtenus sent la macrolactone 87
(13%) et Ie tetraene 88 (15%).
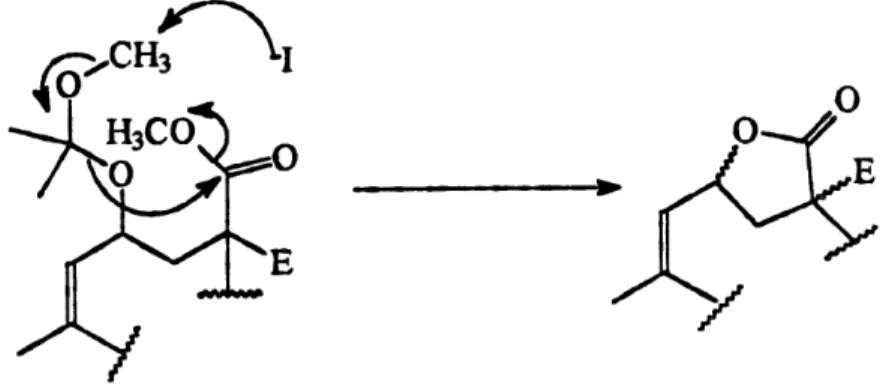
L'obtention de la macrolactone 87 pourrait provenir de la perte du groupe protecteur MOP avant la macrocyclisation favorisee par une decompression sterique, generant un
alcoolate qui, ^ son tour, a lactonise avec un des deux esters du connecteur malonate (figure
4) pour donner 87. Le tetraene 88, quant a lui, provient d'une reaction d'elimination du chlorure ou de Piodure allylique. Cette reaction secondaire pourrait etre la consequence
cfune decompression sterique du lien C8-C9.
CH3 YI
^
HsCO:
^>r°
:E
<»W»NN»
Figure 4. Mecanisme de deprotection et de lactonisation du macrocycle 87.
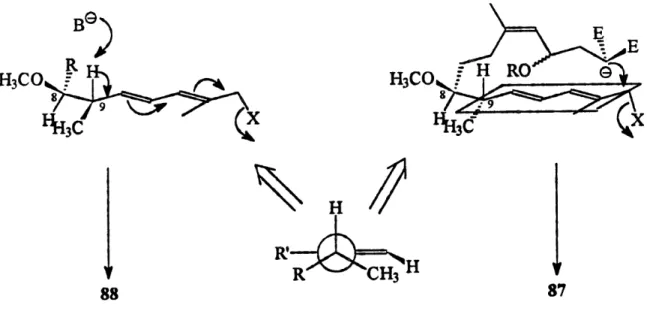
En regardant la projection de Newman du lien C8-C9 lors de 1'approche du nucleophile vers Ie chlorure allylique (figure 5), nous notons qu'il y a une interaction allylique 1,3 entre Ie methyle et 1'hydrogene de 1'olefme. Cette interaction est due au fait que, lors du repliement de !a chame, les deux substituants doivent etre en position pseudo equatoriale pour minimiser les effets steriques. Ceci pourrait avoir comme consequence
d'augmenter un peu plus Penergie de 1'etat de transition de la macrocyclisation.
l/elimination peut se produire seulement lorsque 1'hydrogene en C9 et Ie groupe partant sont paralleles aux orbitales p du diene. Cela impliquerait alors la formation du
interaction, Ie systeme peut passer a un autre rotamere ou bien conduire au produit (TeHmination 88. En tenant compte de ce resultat, un changement de connecteur pourrait
eventuellement ameliorer la synthese de 87. Le phenylsulfonylacetate de methyle a ete
utilise comme connecteur. En repetant la sequence de la voie C (schema 25), cela nous a permis d'obtenir Ie chlorure 91 avec de bans rendements. Par centre, la reaction de
macrocyclisation utilisant Ie chlorure 91 n* a pas donne Ie macrocycle 92 mais seulement de la degradation.
HsCO
88
Figure 5. La formation du tetraene 88 vs Ie macrocycle 87.
1.1.4 Voie D
Une autre methode de macrocyclisation a ete etudiee: la macrocyclisation en presence de paIladium(O). Cette methode necessite un connecteur ph6nylsulfonylacetate de methyle et un acetate allylique, lequel est deplace par Ie palladium qui s'insere ensuite sur Ie diene (33). La sequence a debute par la protection quantitative du dial 81 sous forme
d'acetonide 93 avec Ie 2,2-dimethoxypropane en presence d'acide ^-toluenesulfonique dans Pacetone a 0°C (schema 26). L'alcool 94 a ete regenere gr^ce au traitement avec Ie fluorure
83
MOPO
KCH(C02CH3)S02Ph
KI, 18-C-6 -i toluene, 110°CCH30
TBAF
OTBDPS
THF
MOPO
CHsO
Hexachloroacetone PPh3,2,6-lutidine THF, -40°COH CHsO
MOPO
CS2C03, Csl -^-CHsCN, 75°CMOPO
CHsO
S02Ph
92
Schema 2581 2,2-dimethoxypropane APTS, acetone o°c CH30
TBAF
THF
OTBDPS
CHsO
PvCl, pyridine -^-CH2a2.40°COH CHsO
OPv HCl(l.ON) THF, 25°C HO, "OHCH30^T^^Y^OPv
96
Schema 26resultant en pivaloate 95 en utilisant Ie chiorure de pivaloyle et la pyridine dans Ie dichloromethane a 40°C. L*acetonide 95 obtenu a 99% a etc traite en milieu acide aqueux pour nous donner Ie diol 96 avec 80% de rendement. Par la suite. Ie diol 96 a subi une monotosylation en utilisant les conditions usuelles pour nous conduire au tosylate 97, un intermediaire tres instable (schema 27). L*alcool 97 a ete aussitot protege sous forme de silylether 98 avec Ie triHate de triethylsilane en presence de 2,6-lutidine dans Ie dichloromethane a 25°C. Le produit brut a ete mis dans les conditions d'alkylation usuelles, soit avec Ie sel de potassium du phenylsulfonylacetate de methyle, Fiodure de potassium et
P ether lS-couronne-6 dans Ie toluene a reflux. Cette reaction d'alkylation nous foumit
deux produits qui ont ete separes: I'ester de methyle 99 (29%) et la sulfone 100 (31%). La sulfone 100 provient de la d^carboxylation de 60. N^anmoins, la quantite obtenue de 99 a
ete suflRsante pour poursuivre la sequence.
En utilisant les conditions d6velopp6es par Marshall (34), Ie silyl6ther d'^nol fomie a
partir de Fester 99 et du N,0-bis(trimethyl)ac6tamide dans Ie tetrahydrofiirane, a et6 ajoute sur une p6riode de 10 h dans une solution de palladium(O) dans Ie tetrahydrofurane. Malheureusement, aucun produit de macrocyclisation 101 n'a ete observe; seul Ie t6tra6ne 102 a ete isole avec 86% de rendement. Ceci demontre encore une fois que lorsque nous sommes en presence d'un tres bon groupe partant, tel qu'un allyle palladium(II), la reaction d*elimination est grandement favorisee par rapport a la macrocyclisation.
1.1.5 Voic E
Un changement du groupement protecteur MOP du synthon 86 par un groupement plus solide pourrait empecher la formation de la lactone a cinq membres 87. Le choix du
groupe protecteur s'est arrete sur Ie triethylsilane TES. La synthese a commence avec
Palcool 94 qui a subi une etherification avec Ie chlorure de /?-methoxybenzyle, I'iodure de sodium et 1'hydrure de sodium dans Ie tetrahydrofurane a reflux (schema 28) avec 99% de rendement. Par la suite, suivent des reactions d'hydrolyse (83%), de tosylation (49%), de protection (91%) et (Talkylation (73%) qui ont etc effectuees dans les conditions deja
TsCl, DMAP Et3N
96 —^
CH2CI2, 0°C OTs CHaO OPv TESOTf, 2,6-lutidine CH2C12, 25°CCH30
TESO
OTsKCH(C02CH3)S02Ph
KI.18-c-6,toluene, 110°C OPvCHsO
TESO
S02Ph
OPv CHsOTESO
S02Ph
Pd(PPh3)4
N,0-BTSAPh2P(CH2>3PPh2
THF, reflux CHsO101
TESO
SOiPh
OPv + CH30TESO
SOiPh(100%)
Schema 2794 PMBC1, Nal THF, 65°C CH30
OPMB
AcOH, HsO 25°COH
CH30
TsCl, DMAP Et3N, CHiCli, 0°COPMB
CH30
HO, "OTsOPMB
105
TESOTf, 2,6-lutidine -9 CH2C12, 0°CTESO
CH30^s/^^^-^
KHCH(C02CH3)2
KI, 18-C-6 -» toluene, UO°C 106OPMB
CH30OPMB
Schema 28107
DDQ
CH2C12, H20(18 : 1) CH3°
.E.
8'<108
0
LiAl(0/Bu)3H
THF, 0°CCH30
TESO^, ^^ ^E •>( Hexachloroacetone PPh3, 2,6-lutidine -»• THF, -40°COH CHaO
TESO
109
TESO
CS2C03, Csl CHaCN, 75°C CH30Ill (0%)
CH^O112 (100%)
Schema 29mentionnees auparavant pour nous fournir Ie ^-methoxybenzyl ether 107. Une reaction de deprotection en utilisant Ie 2,6-dichloro-3,5-dicyano-l,4-benzoquinone (DDQ) a conduit au
produit de suroxydation 108 avec un Sres faible rendement de 32% (schema 29), La reduction selective de Paldehyde 108 avec Fhydmre de tri-/-butoxyalumimum de lithium dans Ie tetrahydrofurane a 0°C a fourni Falcool 109 (100%). La chloration dans les conditions usuelles, suivie de la macrocyclisation dans Pacetomtrile a 75 °C en presence de
carbonate de cesium et d'iodure de cesium, a conduit seulement au tetraene 112 mais avec un rendement bas de 23%. II est interessant de constater que, lorsque nous employons Ie
groupe protecteur MOP, Ie produit de macrocyclisation apparait. Cela nous laisse croire que la lactonisation s'effectue avant la macrocyclisation, ce qui faciliterait peut-etre la reaction
attendue.
Malgre les insucces rencontres au cours de la synthese, il a ete neanmoins possible de synthetiser une quantite suffisante de macrocycle 56 en vue de 1'etude de la reaction de
DieIs-Alder transannulaire.
1.2 Reaction de Diels-Alder transannulaire du cydotetradecatrienone TTC
S .2*1 Etude de la reaction par activation avec les acides de Lewis
II est connu dans la litterature que les acides de Lewis diminuent la barriere energetique de la reaction de Diels-Alder tout en favorisant 1'etat de transition endo par Faugmentation des efFets orbitalaires secondaires (6). En regardant ie macrocycle TTC 56,
nous pouvons remarquer qu'il y a deux etats de transition possibles: endo et exo (schema
30). L'etat de transition exo 56 A nous conduit vers Ie tricycle 113 de stereochimie en
jonction des cycles A.B.C. trans'syn-trans (TST). Par contre, 1'etat de transition endo 56 B
nous dirige vers Ie tricycle 114 de stereochimie cis-syn-cis (CSC). A prime abord. Ie produit attendu par 1'activation des acides de Lewis serait Ie tricycle 114 via I'etat de transition endo
CH30
56
DATA
CH30
CHsO56 A
56 B
exo ^ CHsO AE* s= 0 kcal/mol endo ^ CH30 AE*==2.8lkcal/mol 0. 0.114
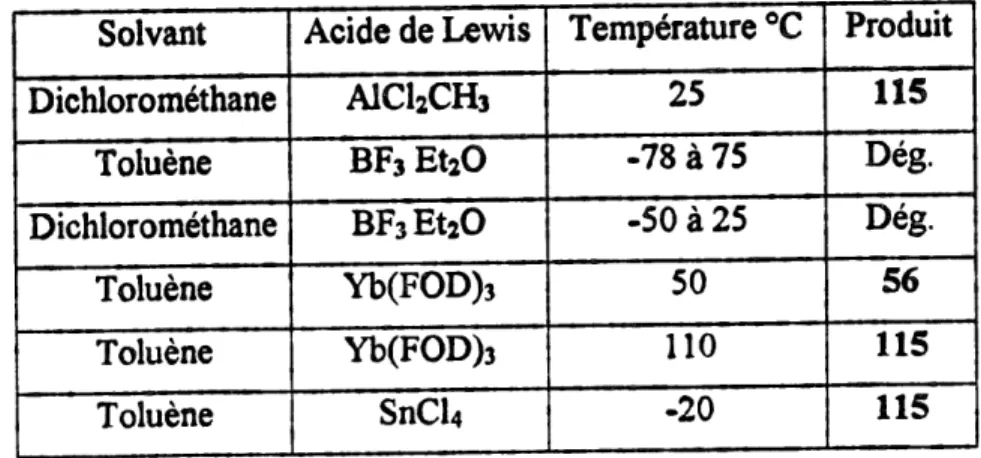
Schema 30Le macrocyle TTC 56 a etc mis en presence du tetrachlorure d'etain (IV) dans Ie toluene a -20°C pour nous fournir un seul tricycle (schema 31). Apres une analyse exhaustive des spectres RMN 'H, COSY, NOESY et J-Resolved de la molecule, nous sommes arrives a la conclusion que la stereochimie relative en jonction des cycles ABC est trans-anti-cis (TAC) 115. La dif&action des rayons-X confirme de fa?on absolue la stereochimie des centres chiraux du tricycle (figure 6). D'autres acides de Lewis ont ete essayes et conduisent tous au meme resultat (tableau 4). Le tricycle TAC 115 est 1'epimere
en position C9 du tricycle TST 113 (schema 32). Des calculs de modelisation (35) ont
demontre que les tricycles TAG 115 et TST 113 ont des AH de formation de 13.20 kcaVmol et 24.60 kcal/mol respectivement. Alors, Ie tricycle TAC 115 est d'environ 11 kcal/mol plus stable que son epimere TST 113, ce qus nous laisse croire a une possible epimerisation du trieycle TST 113 en TAG 115 apres que la reaction de Diels-Alder transannulaire ait eu lieu.
E
CHsO
SnCU, tobene-20°C
CH30
56
CHsO
Les resultats experimentaux obtenus nous suggerent deux routes possibles. La
premiere est Ie passage par 1'etat de transition exo 56 A pour obtenir Ie tricycle TST 113 qui lui s'epimerise pour aboutir au tricycle TAC 115 (schema 32). Si nous considerons ce chemin reactionnel, nous tenons compte de l*importance des effets steriques iors de 1'etat de
transition qui domineraient sur les interactions orbitalaires secondau-es. Par contre, si nous
considerons Pimportance de l^effet endo^ il y a une deuxieme possibilite; celle ou il y aurait (Tabord une isomerisation du macrocycle TTC 56 en macrocycle TTT 116. Ce demier opterait, par la suite pour Ie passage par Fetat de transition endo, ce qui nous fournirait Ie tricycle TAG 115. Pour approfondir la question mecanistique, des essais sans acide de Lewis ont ete faits de m^me que des caSculs de modelisation des etats de transition impliqu^s.
Tableau 4. ResuStats de la reaction de Diels-Alder transannulaire de 56 avec differents acides de Lewis. Solvant Dichloromethane Toluene Dichloromethane Toluene Toluene Toluene Acide de Lewis AlClzCHs BF? EtiO BFsEtiO
Yb(FOD)3
Yb(FOD)3
SnCU
Temperature °C25
-78 a 75 -50 a 2550
110 -20 Produit115
Deg. Deg.56
115
115
1.2.2 Etude de la reaction par activation thermique et resultat d'epimerisation
La reaction de Diels-Alder transannulaire thermique a ete faite dans Ie toluene degaze a des temperatures variant de 125°C a 160°C (tableau 5). Peu importe 1'ajout
CH30
114
AH=13.94kcaVmolDATA
(endo) CH30 AH=9.54kcal/moIDATA
(exo)CHsO
CH3056
DATA
(exo)i H U3
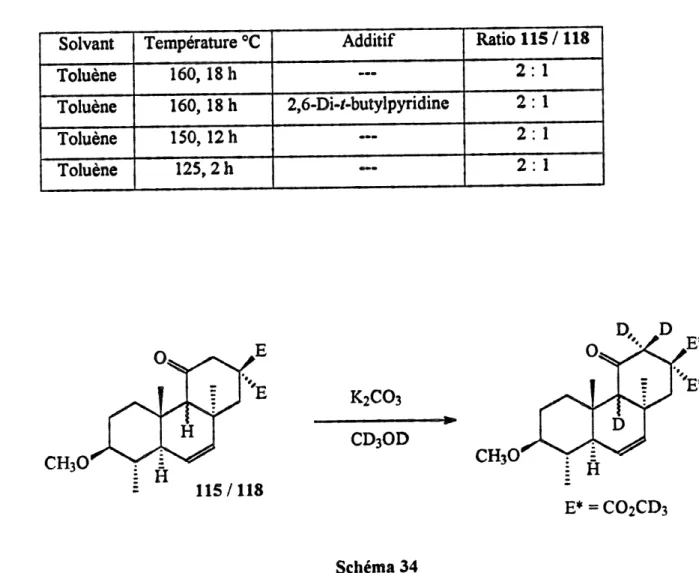
AH = 24.60 kcal/mol CH30 DATA (endo) AH = 13,20 kcal/mol Schema 32reaction, nous avons toujours obtenu un melange inseparable (2:1 ) de deux tricycles (schema 33). Le tricycle majoritaire a ete caracterise par la spectroscopie RMN H et correspond au tricyle TAG 115. Par centre, la determination de la stereochimie en jonction des cycles A, B et C du tricycle minoritaire 118 n'a pu etre possible par l*analyse du spectre RMN 1H. Toutefois, des preuves chimiques et des calculs de modelisation vont nous aider a
determiner Ie chemin parcouru par la reaction.
CHsO toluene
CH30
56
CH30 Schema 33 118A premiere vue. Ie melange pourrait etre constitue des tricycles TAC 115 et TST 113
resultat d'un equilibre thermodynamique apres la reaction de Diels-Alder. Atm de prouver s'il s'agissait bien d'un equilibre thermodynamique, nous avons place Ie melange en presence de carbonate de potassium dans Ie methanol deutere a reflux pendant quelques
heures (schema 34). Selon Ie spectre RMN H, les positions C9 et C 12 ont ete deuterees
ainsi que les esters en C5 mais il n'y a eu aucun changement du ratio des tricycles. Nous
avons poursuivi les eludes d'epimerisation en playant Ie tricycle TAC 115 pur dans les
memes conditions que precddemment. Apres quelques heures a reflux, il n'y a aucune trace
d'epimerisation selon Ie spectre RMN !H. Ces resultats d*epimerisation nous indiquent tres clairement et sans ambiguite que Ie tricycle minoritaire n'a pas la st6reochimie TST 113 et ne provient pas d*un equilibre thermodynamique des tricycles TAC 115 et TST 113.
Tableau 5. Resultat de la reaction de Diels-Alder transannulaire de 56 a differentes
temperatures. Solvant Toluene Toluene Toluene Toluene Temperature °C 160,18h 160,18 h 150,12h 125,2h
Additif
2,6-Di-^-butylpyridine «•• Ratio 115 ,118 2: 1 2:1 2: 1 2:1CHsO
115/118
K2C03CDsOD
CH30
Schema 341.3 Resultats des calculs de modelisation
Atm de determiner Ie chemin reactionnel de la reaction de Diels-Alder transannulaire de la s^rie TTC et de proposer une stereochimie en jonction des cycles du tricycle minoritaire 118, des calculs de modelisation des etats de transition ont ete realises en collaboration avec Ie professeur Dory (36). Les calculs de 1'energie a 1'etat de transition de la reaction de Diels-Alder des macrocycles TTC 56 et TTT 116 ont demontre que les etats de transition exo 56 Aet 116 B sont plus bas en energie que les etats de transition endo correspondant de 2.81 kcal/mol et de 3.20 kcal/mol respectivement (schemas 30 et 35). Ce resultat nous indique que les etats de transition endo 56 B et 116 A conduisant respectivement aux tricycles CSC 114 et TAC 115 sont defavorises a cause probablement des contraintes steriques transannulaires. Par consequent, la domination des effets steriques sur les interactions orbitalaires secondaires favorise la formation exclusive des tricycles TST
113 et CAT 117.
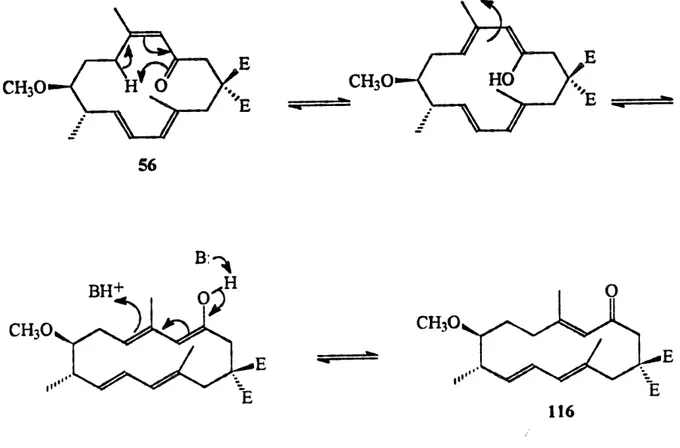
D'apr^s Fensemble des resultats experimentaux et theoriques obtenus, il nous apparait evident que la reaction de Diels-Alder transannulaire de la cyclot6tradecatri6none TTC 56 activee par les acides de Lewis, pass® par un processus de cyclisation exo 56A pour foumir Ie tricycle TST 113 (schema 30). Par la suite, celui-ci stepim6rise facilement en position C9 pour conduire au tricycle TAC 115 qui est beaucoup plus stable. Par centre, Ie melange des tricycles TAG 115 et 118 (2:1) obtenu par Ie processus thermique indique indeniablement qu'une isomerisation de l*enone a lieu en premier pour obtenir un melange thermodynamique des macrocyles TTC 56 et TTT 116 dans un rapport de 2 :1. D'autant plus que Penergie fondamentale calculee des macrocycles TTC 56 et TTT 116 indique que Ie macrocycle TTC 56 est plus stable de 1.51 kcal/mol.
Cette isomerisation pourrait s'expliquer par 1'arrachement d'un hydrogene
intramoleculaire suivi d'une rotation du lien carbone-carbone et (Tune reprotonation
intermoleculaire (figure 7). Par la suite, la cycloaddition [4+2] transannulaire exo des macrocycles TTC 56 et TTT 116 conduit vers un melange des tricyctes TAG 115 et CAT
CHsO
CHsO
CH30DATA
116 A
116 B
endoCHaO
AE*=3.20kcal/mol exo CH30 AE* == 0 kcaVmolH
115
117 Schema 35117 (schema 32). Done, nous pouvons suggerer sur cette base que la stereochimie en
jonction des cycles A, B et C du compose minoritaire 118 est cis-anti-trafis (CAT) soit Ie tricycle 117.
1.4 Conclusion
Bref, toutes les voies examinees jusqu'a present ont demontre clairement la difficulte de la macrocyclisation d'un triene TTC fonctionnalise en position C3, C8 et C9. A notre avis, la principale cause des echecs de la macrocyclisation sont la tension allylique-1,3 du diene due au repliement de la chalne au moment de la cyclisation et Ie nombre de carbones d'hybridation sp qui favoriseraient davantage les reactions secondaires. Toutefois, malgre la faible quantite de macrocycle 56 obtenu, il nous a ete possible de realiser une etude assez d^taill^e de la reaction de Diels-Alder transannulaire activee.
CHsO
CHsO
56
CH30. •••• BHf.^
B:-\ <H°p"
CH30L*etude a demontre, entre autres, que 1'insertion de la fonction cetone en alpha du
dienophile a permis une baisse considerable de la temperature de reaction de 340°C. Par contre, malgre I'utilisation des acides de Lewis comme catalyseur, les contraintes steriques a Petat de transition favorisent largement te passage de la reaction par Petal de transition exo. Cette hypothese est appuyee par les calculs de modelisation moleculaire des etats de transition de la reaction. Cela nous a permis de connaTtre Ie chemin reactionnel thermique et catalytique et fmalemant de proposer la stereochimie du tricycle minoritaire.
2. DEUXIEME PARTDE:
APPROCHES VERS LA SYNTHESE DE L'ACIDE FUSIDIQUE
2.1 Introduction
L'acide fusidique 35 a ete isole de la fermentation d'un champignon Ie Fusidium coccineum et caracterise en 1962 par Ie groupe de Godtfredsen (37) (schema 36). Parmi les triterpenes tetracycliques de la s6rie dammarane, 1'acide fusidique 35 comporte 1'activite biologique anti-bacterienne la plus puissante centre les infections causees par la bacterie Staphylococci (38). Son mecanisme (Faction consiste en Finhibition de la synthese de proteines bacteriennes par 1'interference du transfert des acides amines a partir de I'aminoacyi-sARN vers la proteine sur les ribosomes. Depuis sa decouverte en 1962, son importance clinique a et6 dau-ement etablie (39) et une foule de connaissances ont etc acquises en ce qui conceme ta modification et la degradation de sa structure (38). AujourcThui Pacide fasidique 35 est utilise comme antlbiotique en m^decine et il est vendu par la compagnie pharmaceutique Leo sous Ie nom de Fucidin.
2,2 Approche Sineaire en vue de la formation du bicyclo[12.3«0]heptadecatetraenone
142
2.2.1 Retrosynthese
I/analyse de la structure de Pacide fusidique 35 nous revile une st^reochimie
trans-syn-trans pour la jonction des cyles A, B et C, ce qui confere au cycle B une conformation bateau. Chez les diteq)enes, cette conformation est tres rare et ne se retrouve que dans la
famille des fusidanes tel que la cephalosporine Pi 119 (40) et 1'acide helvolique 120 (41) (schema 36), De plus, il est difficile de synthetiser ce genre de tetracycles par les votes classiques (42). L/excellente diastereoselectivite obtenue lors de la reaction de Diels-Alder
transannulaire du cyclotetratrienone 56 catalysee par un acide de Lewis, nous a permis d'elaborer une strategic de synthese totale de 1'acide fusidique 35, II est a noter, meme s'il
n'est pas possible presentement d'empecher I'isomerisation du tricycle TST 113 en tricycle TAC 115, nous pensons que ce!a ne sera pas un probleme lors de la synthese de Facide fusidique 35. En effet, il a etc demontre lors de la degradation de I*acide fusidique 35 (43) et de sa synthese en 1982 par Dauben (44) qu'il est possible de changer la stereochimie en jonction des cycles A, B etC de TAC a TST par seulement quelques transformations. La
seule difficulte majeure est Ie bas rendement de la macrocyclisation qui nous empeche d'obtenir une bonne quantite du macrocycle 56. Toutefois, ce probleme pourrait etre surmonte en minimisant les interactions defavorables par la formation d'un plus grand
macrocycle.
C02H OAc
COzH
OAc HO'' V1'Y' ^OH
H |
OAc n9 COsH
OAc
Pour ce faire, 1'acide fusidsque 35 proviendrait du tetracycle 145 par I'addition de la chame olefmique en C 17 (schema 37). Le tetracycle 145, quant a lui, proviendrait d'une addition conjuguee 1,4 d*un methyle en C 14 sur 144. Ce dernier pourrait etre obtenu par la reaction de Diels-Alder transannulaire catalysee du macrocycle 142. Le cyclotetradecatriene 142 serait Ie fi-uit d'un aldol transannulaire d'un derive du macrocycle TTC 139 a 17 membres. L'ouverture du cycle conduirait au triene 127 et ce demier proviendrait de 1'ester
connu 74.
2.2.2 Synthese enantioselectlve d9 un cycloheptadecatrienedione en utilisant la
fonction chlorocetone comme agent de couplage
La synthese a demarre par la reduction de Fester 74 avec I'hydrure de diisobutylaluminium dans Ie dichloromethane a -78°C (schema 38). L'alcool 121, obtenu dans un rendement de 80%, a ete oxyde en presence du periodinane de Dess-Martin (45).
Ainsi, Faldehyde 122 (86%) a subi une alkylation 1,2 avec Ie chlorom^thyllithium forme m
sstti par la metallation du chloroiodom6thane en utUisant Ie butyllithium dans Ie tetrahydrofurane a -78°C. La chlorohydrine 123 (86%) a ete protegee sous forme d'ac^tal MOP dans les conditions usuelles pour nous donner Ie compose 124 avec 83% de rendement. Un traitement aux ions fluorures dans les conditions habituelles a regener^ Falcool 125 a 89%. Une reaction d'oxydation avec Ie perruth^nate de tetrapropylammonium (94%) suivie d'une alkylation de Grignard avec Ie magnesien
ClMg(CH2)30MgCl (46) dans Ie tetrahydrofurane a -78°C a conduit au diol 127 (84%)
(schema 39). Une monoprotection du diol 127 avec Ie ^-butylchlorodimethylsilane en presence d*imidazole dans Ie tetrahydrofurane a fourni Palcool 128 avec 95% de rendement.
Une deuxieme protection, cette fois-ci, sur l*alcool secondaire 128 en utilisant Ie
chlorure de p-(trimethytsilyl)ethoxymethylether (47), la diisopropylethylamine et Piodure de
tetrabutylammonium comme catalyseur dans Ie dichloromethane a 40°C, a donne Ie
H
CH30
35
RO
('.. OAc •I..HO
r*'SEMO
CHaO
142
OAcCH30
CH30SO^Ph
0
OTBDPS
0
OCH,CHjO
Schema 3774
DIBAH
CH2C12, -78°C CH30OTBDPS
Dess-Martin CH2CI2, 25°COTBDPS
CH30
ICHzCl, BuLi -^-THF, -78°C0 CHsO
2-Methoxypropene, PPTS -i CH2C12, 0°C .OTBDPSOMOP
/C1 124TBAF
THF
.OH TPAP, NMOtamis mol 4 A°
QMOP
CHsO ,C1
125
ClMg(CH2)30MgCl
126 —^
THF. -78°C CH30OH
TBSC1, imidazole -»• THF, 0°CCHaO
OTBS
SEMC1, DIPEA TBAI, CH2C12 a 400C CH,0SEMO
128
a
TBAF
THF
SEMO
CHsO
130
OH
TPAP.NMOtamis mol. 4A°
-»• CH2C12
SEMO
CH^O ^0 l)CH3S02Ph,BuLi THF, -78°CQMOP 2)H30+
SEMO
a
CH30S02Ph
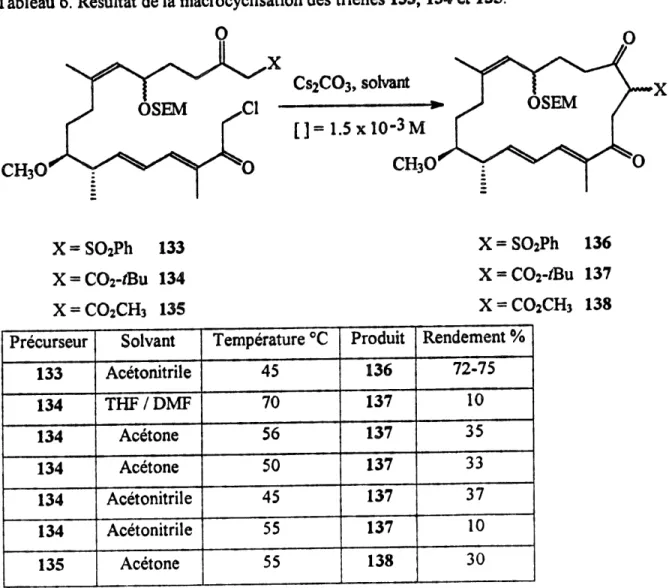
Schema 39tetrabutylammonium dans les conditions deja employees a permis d'obtenir 1'alcool primaire 130 (86%). La formation du connecteur P-cetosulfone a etc possible grace a Foxydation de 130 avec Ie perruthenate de tetrapropylammonium (88%), suivie de Falkylation avec 1'anion du benzylmethylsulphone dans Ie toluene a -78°C, et finalement cTune hydrolyse en milieu acide qui a genere Ie dial 132 avec 87% de rendement pour les deux etapes. L'oxydation des deux alcools secondaires de 132 avec Ie periodinane de Dess-Martin est venue completer la synthese du precurseur macrocyclique 133 (85%) (schema 40).
Nous nous sommes rapidement aper^us que la temperature joue un role extremement
important lors de la macrocyclisation (tableau 6). En effet, !a fonction chloroc^tone se degrade tres rapidement lorsque la temperature du milieu reactionnel depasse 50°C; au-dela de cette temperature, les rendements chutent de fayon dramatique. Afin d'etudier I importance de la nature du connecteur, deux autres precurseurs macrocycliques 134 et 135 out ete synth6tis6s par la m^thode employee pour obtenir 133 (schema 39 et 40) et ee, avec des rendements semblables. Les reactions de macrocyclisation ont ete faites a une concentration de 1.5 x 10M en presence de carbonate de c^sium avec un temps d'addition
de 2 heures.
Les resultats obtenus sont eloquents: 1'utilisation de la P-cetosulfone comme
connecteur surpasse de fa?on considerable 1'emploi des P-cetoesters. 11 se peut que des
produits de 0-alkylation aient ete formes lors de la macrocyclisation; probablement que ceux-ci se sent degrades lors du traitement de la reaction. Ceci poun-ait en panic expliquer
les faibles rendements de la reaction avec les P-cetoesters comme connecteur. Nous avons aussi observe que Ie solvant a tr6s peu d'mfluence sur Ie componement de la reaction.
Done, nous avons reussi a ameliorer grandement la macrocyclisation de tri^ne TTC par
Paugmentation du nombre de carbones de la chaTne et I'emploi d'un connecteur efficace. Revenons a la synthese, normalement une simple desulfonylation, en utilisant des conditions standard, aurait du nous conduire directement au macrocycle 141.
SEMO
Dess-Martin 132 CH2C12CHsO
S02Ph CS2C03 CH3CN, 45°CCHsO
SOsPh DBU
0
139
Pd(PPh3)4. (Bu)3SnH
C^, CH3C02H CHsO You" tableau 7SEMO
<r0 CH30'143 I S 142
Schema 40Malheureusement, toutes les tentatives ont echoue. Alors, nous avons envisage un processus en deux etapes: I'elimination de la sulfone et la reduction de Penedione formee.
D'abord I'elimination de la sulfone 139 a ete effectuee en presence du 1,7-diazabicyclo[4.5.0]undecene (DBU) dans Ie tetrahydrofurane pour obtenir Penedione 140 avec 90% de rendement. D'enormes difficultes au niveau de la reduction de 140 sont
apparues; plusieurs agents reducteurs ont ete utilises, mais sans succes. Apres un effort
considerable, nous avons trouve que Ie tetrakis(triphenylphosphine)palladium(0) avec Phydmre de tributyletain en presence decide acetique dans Ie benzene degaze (48) conduit au produit desire 141 avec 91% de rendement.
Tableau 6. R^sultat de la macrocyclisation des trienes 133,134 et 135.
0
.X CS2C03, sok^ant.a —»•
CH30
[]=1.5xlO-3M0 CHaO
0
X=S02Ph 133 X=C02-fBu 134 X=C02CH3 135 X=S02?h 136 X=C02-/Bu 137 X-COzCHs 138 Precurseur 133 134 134 134134
134 135 Solvant AcetonitrileTHF/DMF
Acetone Acetone Acetonitrile Acetonitrile Acetone Temperature °C45
70
56
50
45
55
55 Produit136
137
137137
137
137
138 Rendement % 72.7510
35
3337
10 30Ayant en mains une bonne quantite du macrocycle 141, nous avons essaye une des etapes clefde la synthese, soit la reaction cTaldoI transannulaire. De nombreuses conditions ont ete essayees et, a notre grand dam, aucune n'a fourni Ie produit escompte 142 (tableau 7). Dans certains cas, nous avons retrouve Ie produit depart, dans d*autres, de la degradation
et aussi Ie produit elimine 143. II semble que 1'acidite plus elevee des protons en alpha de la dienone, favorisant ainsi un enol conjugue avec Ie diene et un mauvais alignement lors de Pattaque transannulaire, expliquerait notre perte. Cette voie Kit mise de cote pour un certain
temps.
Tableau 7. R^sultat de la reaction d'aldol transannulaire de 141.
R6actif
NaOH(6N)
MeONa MeONa f-BuOK KzCOj BF30Et2/(AcO)20 K2C03PPTS
...SnCU
SolvantTHF
MeOH
MeOHTHF
MeOH AcOHEtOH
Benzene Benzene Benzene Toluene Temperature °C80
25
50
25
50
0
72
80
270
200
0
Produit141
141
Deg. Deg. 141 Deg. 143 Deg. Deg.141
Deg.2.3 Approche convergente en vuc de la formation du
bicyclo[12.3.0j-heptadecatetraenone 142
2.3.1 Retrosynthese
Suite aux difficultes rencontrees pour fonner Ie cycle D par aldol transannulaire afin d'obtenir 142, nous avons envisage la formation du cycle a cinq membres par une reaction de Wittig transannulaire. Ainsi, Ie bicyclo[12.3.0]heptadecatriene 142 pourrait provenir du macrocycle 164 (schema 41). Ce dernier serait obtenu par la macrocyclisation au palladium(O) du triene 163. Puis, en utlisant une avenue convergente, nous poumons obtenir Ie triene 163 grace aux retrons 148 et 156. l/iodure vinylique 148 proviendrait en quelques etapes de 1'aldehyde connu 73 et Fetain vinylique 156, quant a lui, serait obtenu a partir de Falcool propargytique commereial. Cette approche tire avantage de I'irr^versibilite de la reaction de Wittig. Contrairement a la reaction d'aldol ou 1'adduit fonne peut revenir en arrive, la formation de 1'oxaphosphetane ne peut que fi-agmenter vers Ie produit d*elimination, soil la formation du cycle D (figure 8).
2.3.2 Synthese ^nantioselective du fragment ISO
La sequence a debut6 par une olefination par la methode de Takai (49) sur Paldehyde 73 avec Ie chlorure de chrome (II) et 1'iodoforme dans un melange de 1,4-dioxane et de tetrahydrofurane (10:1) suivi du traitement aux ions fluorures. Cette reaction nous a permis d'obtenir Piodure vinylique trcms 146 avec un rendement de 66% pour ces deux etapes (schema 42). II est interessant de noter que la procedure originate n'a pas fonctionne; une modification du nombre d^quivalents des reactifs et du ratio du melange de solvants a ete necessaire atm (TameHorer 1'homologation. L'oxydation de I'alcool 146 en aldehyde 147 avec Ie periodinane de Dess-Martin (97%) et, par la suite, une hydroxymethylation avec des conditions dej^ employees precedemment ont donne Ie diol 148 avec 83% de rendement. La transformation de 1'alcoot primaire en groupe partant tosylate 149 (71%) et la protection de
Palcool residuel, sous forme de triisopropylsilylether 150 (99%), ont ete effectues dans les conditions deja utilisees anterieurement.
Y
0
o ,ro(^
PO(OCH3)
Figure 8. La formation du cycle D de 142.
2.3.3 Synthese enantioselective du fragment phosphonate 162
Premierement, une reaction d'addition d8un methyle sur la triple liaison de Falcool propargylique a Paide du trimethylalumium et du dichlorure de bis(cyclopentadienyl)-zirconium dans Ie dichloromethane a 25°C a conduit a 1'intermediaire vinylzirconocene qui a ete trappe avec de l*iode moleculaire (schema 43) (50). L'alcool libre de iodure vinylique
obtenu, etant tres instable, a ete immediatement protege sous forme de