The role of mRNA metaboism in NPM-ALK-mediated oncogenesis
244
0
0
Texte intégral
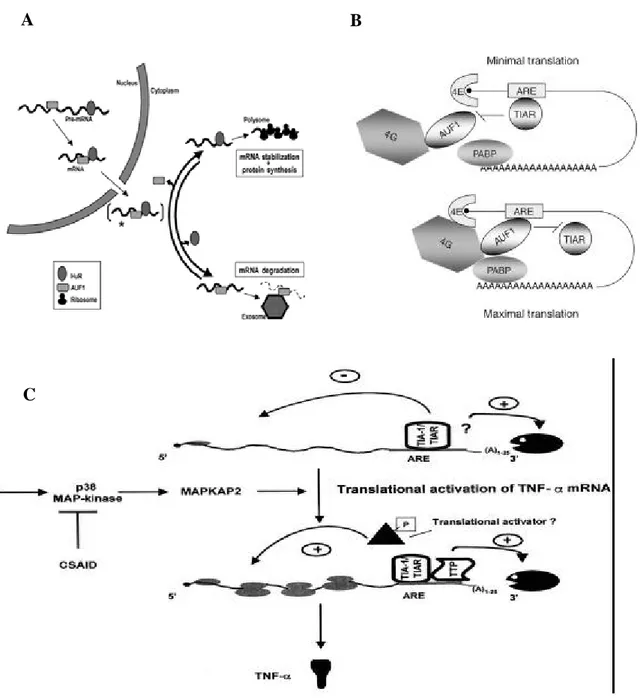
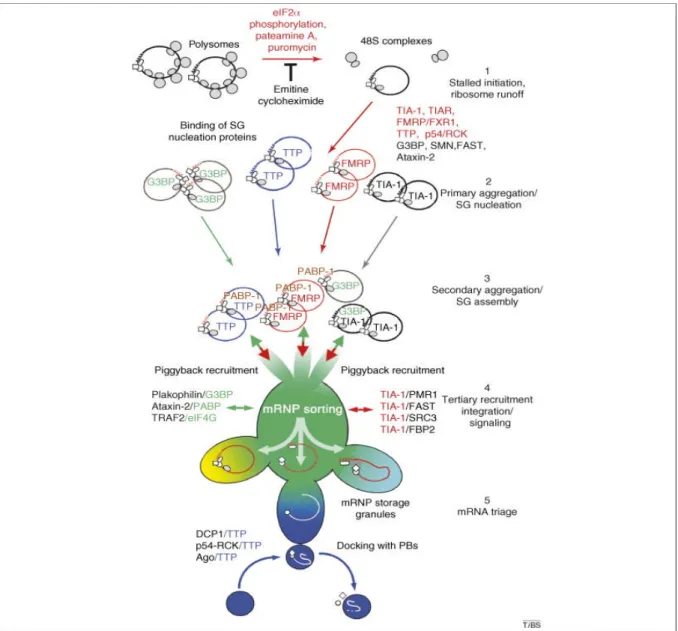
Figure

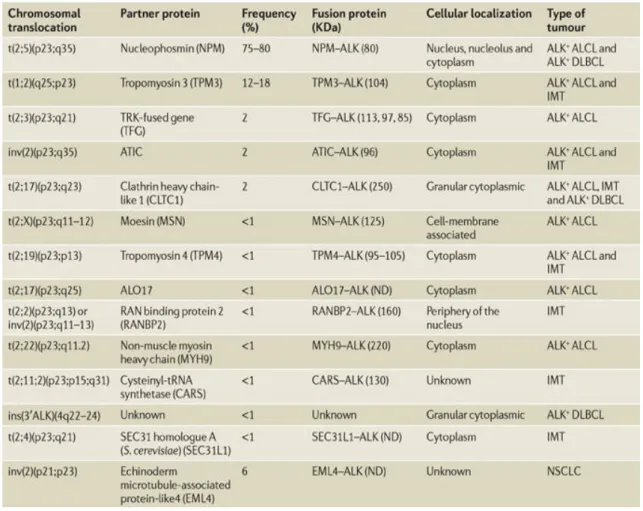

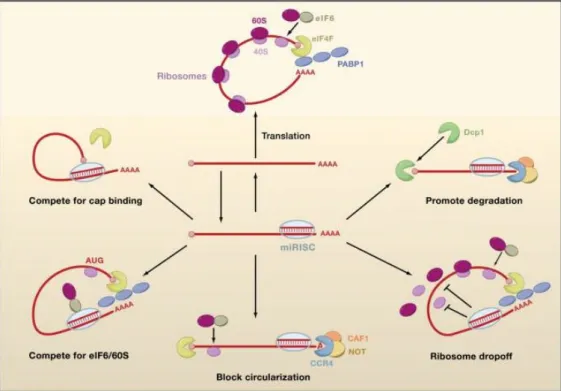
+7
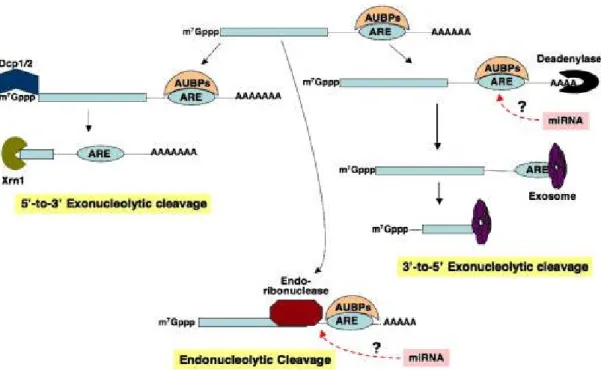


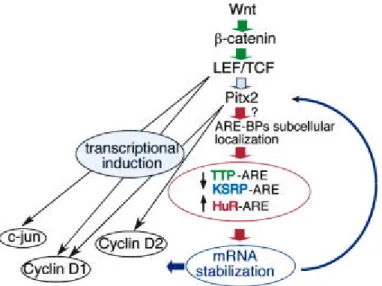
Documents relatifs