Faculté des Sciences, 4 Avenue Ibn Battouta B.P. 1014 RP, Rabat.
Tel : + 212 (0) 37 77 18 34/35/38, Fax : + 212 (0) 37 77 42 61, http://www.fsr.ac.ma
N° d’ordre : 2636 THESE DE DOCTORAT Présentée par JEDDANE Leïla Discipline : Biologie Spécialité : Immuno-génétique
Déficits Immunitaires Primitifs au Maroc :
Epidémiologie et Exemple d’étude sur le syndrome
d’Ataxie Télangiectasie
Soutenue le 4 mai 2013
Devant le Jury Président :
Pr Saaïd AMZAZI : PES, Faculté des Sciences, Rabat Examinateurs :
Pr Hicham BELLAOUI : PES, Faculté des Sciences, Rabat
Pr Ahmed Aziz BOUSFIHA: PES, Faculté de Médecine et Pharmacie, Casablanca Dr Abdelhamid BARAKAT : Docteur en Génétique, Institut Pasteur, Casablanca Pr Youssef BAKRI: PH, Faculté des Sciences, Rabat
Avant Propos
Les travaux présentés dans cette thèse ont été effectués au Laboratoire de Biochimie et Immunologie, dans l’unité de formation et recherche Biochimie-Immunologie de la Faculté des Sciences de Rabat, sous la direction du Professeur Hicham BELLAOUI.
Les travaux ont été codirigés par le Professeur Ahmed Aziz BOUSFIHA, au niveau de l’Unité d’Immunologie Clinique de la Faculté de Médecine et Pharmacie de Casablanca. De plus, le Professeur Abdelhamid BARAKAT, du Laboratoire de Génétique Humaine Moléculaire à l’Institut Pasteur de Casablanca, ainsi que le Professeur Dominique STOPPA-LYONNET, du Service de
Génétique de l’Institut Curie, m’ont fait l’honneur de m’accueillir au sein de leur laboratoire au cours de ma thèse.
Je tiens à remercier chaleureusement les professeurs qui m’ont encadré au cours de cette
thèse et les membres du Jury :
Monsieur le Professeur Saaïd AMZAZI, Doyen de la Faculté des Sciences de Rabat, qui a accepté de présider le jury.
Monsieur le Professeur Hicham BELLAOUI, qui a dirigé cette thèse au sein de la
Faculté des Sciences.
Monsieur le Professeur Ahmed Aziz BOUSFIHA, qui m’a proposé ce sujet.
Monsieur le Professeur Abdelhamid BARAKAT, qui m’a accueilli dans son laboratoire.
Monsieur le Professeur Youssef BAKRI,
qui m’a fait l’honneur d’être le rapporteur de ma thèse.Madame le Professeur Hanane SALIH ALJ, coordinatrice de la recherche au sein de la
MSPID, qui a suivi l’ensemble de mes travaux, et a accepté d’être mon rapporteur externe.
Madame le Professeur Dominique Stoppa-Lyonnet, qui m’a gentillement accueillie
lors de mon stage.
Madame le Professeur Fatima Ailal, qui a révisé les dossiers des patients avec moi, et
m’a grandement aidée sur la partie clinique.
Enfin, l’Association HAJAR d’aide aux enfants atteints de Déficits Immunitaires Primitifs a soutenu financièrement ce projet de thèse. La Moroccan Society for Primary Immunodeficiencies, quant à elle, a participé activement dans la construction scientifique de ce projet.
Je tiens également à remercier Monsieur le Professeur Abdelaziz BENJOUAD, qui m’a
introduit auprès de mes directeurs de thèse.
Ce travail n’aurait jamais pu être terminé sans l’aide technique et intellectuelle du Dr
Omar Abidi (Institut Pasteur), Madame Catherine Dubois-d’Enghien (Institut Curie) et du Dr
Ibtihal Benhsaien (UIC). Sans oublier l’aide précieuse de mes collègues doctorants ou autre
au sein de la MSPID (Zahra Aadam, Laila Aït Baba, Ayoub Aglaguel et Hamsa Aït Mimi), du
laboratoire de Génétique Moléculaire Humaine à l’Institut Pasteur (Majida Charifa, Houda
Benrahma, Safaa Bounaceur, Abdelmajid El Oualid, …), l’Association Hajar (Ibtissame Naïme,
Khadija Aadam, Abdellah Moudden…) et de l’Unité d’Immunologie Clinique (tous les
médecins se reconnaitront).
Enfin, lorsque je regarde en arrière, la liste des personnes à remercier est longue, de
la maternelle à la faculté… à commencer par mes parents et ma famille. En vrac et sans
hiérarchie, je remercie : Mme Jacqueline, Mme C. Liétart, Mme Ouassat, Mme Edith Sow,
Romina, Paola et leurs parents, Marie, Inès, Rossy, Youness, Mehdi, Maki, Maïmouna, Rabi,
Arthémond, Yasser, Fadwa, Houda E., Aouatif, Nisrine, Tarek, M. Vilain, M. Lahmidi, Julie et
sa famille, Khadija, … Je m’excuse auprès de ceux que j’ai pu oublier.
Dédicace
Je dédie ce travail à mes parents, Myriam et Ahmed, qui m’ont
soutenu tout au long de mes études, dans mes choix et mes erreurs, et
m’ont donné la force de persévérer. Grâce à eux, j’ai pu profiter du
meilleur des deux cultures et je leur en serai toujours reconnaissante.
A mon petit frère, Naïm, qui répond toujours présent.
Au reste de ma famille, grand-parents, tantes, oncles et
cousins, que ce soit au Maroc ou en Belgique. Je vous aime.
A mes amis, d’ici et de là-bas, qui m’ont aidé tout au long de
ce parcours. Je ne vous oublie pas.
A mes professeurs, qui nous ont toujours donné le meilleur
d’eux-mêmes pour nous apprendre au-delà de ce qui est écrit dans les
livres, l’école de la vie.
i
Table des matières
Table des matières ...i
Résumé ... vi
Abstract... vi
Liste des Abréviations ... vii
Liste des Illustrations ... x
Liste des tableaux ... xi
Glossaire ... xii
Etude Bibliographique ...1
I. Le système immunitaire ...2
I.1. Généralités ...2
I.2. Réponses immune innée et adaptative ...2
I.2.1. Réponse innée ...3
I.2.2. Réponse adaptative ...8
I.3. Régulation de la réponse immune ... 12
II. Les Déficits Immunitaires Primitifs ... 15
II.1. Définition... 15
II.2. Epidémiologie ... 16
II.2.1. En occident ... 16
II.2.2. Au Moyen-Orient ... 17
II.2.1. Au Maghreb ... 17
II.3. Classification de l’IUIS ... 20
II.4. Diagnostic ... 22
II.4.1. Evoquer un DIP... 22
II.4.2. Explorer un DIP ... 26
II.4.3. Diagnostiquer un DIP ... 29
II.5. Traitement... 34
II.5.1. Antibioprophylaxie ... 36
II.5.2. Traitement de substitution des Immunoglobulines ... 36
II.5.3. Greffe de cellules souches hématopoïétiques ... 37
i. Greffe de moelle osseuse ... 38
ii. Greffe de cellules issues du sang de cordon ... 39
ii
II.5.4. Autres traitements ... 40
i. Traitement des tumeurs malignes ... 40
ii. Traitement de l’autoinflammation ... 42
iii. Traitement de l’autoimmunité ... 43
iv. Thérapie génique ... 45
II.6. Diagnostic moléculaire ... 47
II.6.1. Diagnostic anténatal ... 47
i. Techniques de prélèvement ... 47
a. Amniocentèse ... 48
b. Choriocentèse ... 48
ii. Analyse de l’ADN fœtal ... 49
a. Caryotype fœtal ... 49
b. FISH ... 49
c. Analyse des marqueurs génétiques ... 50
d. Analyse directe de la mutation ... 51
iii. Considérations éthiques ... 51
II.6.2. Diagnostic pré-implantatoire ... 53
III. L’Ataxie Télangiectasie ... 54
III.1. Généralités ... 54
III.2. Epidémiologie ... 54
III.3. Diagnostic ... 57
III.4. Diagnostic différentiel ... 60
III.4.1. Défauts de réparation de l’ADN ... 60
III.4.2. Ataxies cérébelleuses autosomiques récessives ... 67
III.5. Physiopathologie ... 68
III.5.1. Gène de l’Ataxie Télangiectasie ... 68
III.5.2. Protéine ATM ... 73
III.5.3. Physiopathologie ... 76
i. Système nerveux ... 76
ii. Système immunitaire ... 77
iii. Néoplasies ... 77
iv. Atteinte cutanée ... 79
III.6. Traitement... 79
iii
MATERIELS ET METHODES ... 83
I. Etude épidémiologique ... 84
II. Registre ... 87
II.1. Réseau MSPID... 87
II.2. Collection des données ... 87
II.3. Critères de diagnostic et classification... 88
II.4. Base de données ... 88
II.5. Circuit d’enregistrement ... 88
II.6. Analyse des données et statistiques ... 88
III. Etude de l’Ataxie Télangiectasie ... 90
III.1. Patients ... 90
III.1.1. Recrutement ... 90
III.1.2. Critères d’inclusion ... 90
III.1.3. Examen clinique ... 90
III.2. Extraction de l’ADN ... 91
III.2.1. Méthode d’extraction au phénol-chloroforme ... 91
III.2.1. Méthode rapide d’extraction par les sels ... 92
III.2.2. Méthode d’extraction automatisée ... 92
III.3. Amplification de l’ADN par PCR ... 96
III.3.1. Amorces du gène ATM ... 96
III.3.2. Approche amplicon par amplicon ... 96
III.3.3. Approche en 2 phases ... 97
i. Mise en plaque des amorces ... 97
ii. PCR ... 98
III.4. Séquençage ... 101
III.4.1. Purification du produit PCR ... 101
i. Purification à l’Exo-SAP ... 101
ii. Purification sur membrane... 101
III.4.2. Quantification des matrices ... 102
III.4.3. Réaction de séquençage ... 102
III.4.4. Précipitation de l’ADN ... 103
III.5. Analyse des résultats ... 104
RESULTATS ET DISCUSSION ... 105
iv
I. Résultats ... 107
I.1. Estimations de l’épidémiologie à travers le monde ... 107
I.2. Couverture des registres ... 107
I.3. Estimations de l’incidence par groupe d’âges ... 111
I.4. Couverture des registres par groupe d’âge... 114
II. Discussion ... 116
II.1. Estimations de l’épidémiologie à travers le monde ... 116
II.2. Facteurs de sous-estimation ... 117
II.2.1. Sous-diagnostic des DIP... 117
II.2.2. Effet de la consanguinité ... 118
II.2.3. Révision de la définition des DIP ... 119
II.3. Couverture des registres ... 121
II.4. Effets de l’âge ... 122
II.5. Comparaison avec d’autres maladies ... 123
2ème Partie : Profil étiologique des Déficits Immunitaires Primitifs au Maroc ... 124
I. Résultats ... 125
I.1. Fréquence et Distribution des patients ... 125
I.2. Caractéristiques des patients ... 128
I.2.1. Age et sexe des patients ... 128
I.2.2. Consanguinité et antécédents familiaux ... 130
I.2.3. Date de diagnostic ... 130
I.2.4. Origine des patients ... 130
I.3. Manifestations cliniques ... 132
I.4. Tests génétiques ... 132
I.5. Prise en charge ... 134
I.6. Mortalité ... 134
II. Discussion ... 136
II.1. Résultats biaisés ... 136
II.2. Effet de la consanguinité ... 136
II.3. Retard de diagnostic ... 137
II.4. DIP chez l’Adulte ... 139
II.5. Profil étiologique ... 139
II.6. Manifestations cliniques ... 141
v
II.8. Evolution ... 144
3ème Partie : Etude clinique et moléculaire de l’Ataxie Télangiectasie ... 146
I. Résultats ... 147
I.1. Données épidémiologiques ... 147
I.2. Clinique ... 148
I.3. Analyse moléculaire ... 152
II. Discussion ... 158
II.1. Données épidémiologiques ... 158
II.2. Analyse moléculaire ... 162
II.2.1. Spectre de mutation au Maroc ... 162
II.2.2. Origine des mutations ... 162
II.3. Analyse des données cliniques ... 163
II.3.1. Corrélation génotype-phénotype ... 163
II.3.2. Familles multiplexes ... 168
II.3.3. Délai diagnostic ... 170
II.4. Polymorphisme et cancer ... 170
Conclusion et perspectives ... 173
I. Etude épidémiologique ... 174
II. Registre marocain ... 175
III. Ataxie Télangiectasie ... 176
IV. Conclusion générale ... 177
Médiagraphie ... 179
Bibliograhie ... 180
Liste des adresses URL ... 193
Annexes ... 194
Annexe 1 : Fiche de recrutement des malades atteints d’Ataxie Télangiectasie ... 195
Annexe 2: Préparation des solutions pour l’extraction de l’ADN ... 201
Annexe 3 : Séquence des amorces sens et antisens pour le gène ATM ... 203
Annexe 4 : Pedigree des patients atteints d’Ataxie Télangiectasie ... 205
vi
Résumé
Les Déficits Immunitaires Primitifs (DIP) représentent un groupe de maladies prédisposant à des infections récurrentes ou spécifiques. Au Maroc, peu de données sont disponibles dans la littérature concernant les DIP, que ce soit sur le plan épidémiologique, clinique ou génétique.
Nos principaux objectifs étaient d’estimer l’impact des DIP à travers le monde, en nous basant sur des études épidémiologiques récentes; de déterminer le profil étiologique des DIP au Maroc et de réaliser une étude clinique et génétique en prenant l’exemple du syndrome d’Ataxie Télangiectasie.
Ainsi, nous avons estimé que plus de 6 millions de personnes vivraient avec un DIP dans le monde, dont plus de 27 000 au Maroc. Toutefois, notre registre n’a comptabilisé que 351 patients en 2011, avec une surreprésentation des phénotypes sévères.
D’autre part, l’analyse de 19 cas index a révélé 14 mutations différentes sur le gène responsable ATM (Ataxia-Telangiectasia Mutated), dont la plus fréquente était portée par 26,32% des allèles.
Les Déficits Immunitaires Primitifs ne sont donc pas aussi rares qu’il n’est généralement admis. Ces pathologies sont largement sous-diagnostiquées au Maroc. Enfin, le diagnostic moléculaire est réalisable au Maroc et permettrait la mise en place d’un conseil génétique.
Mots-clés : Déficits Immunitaires Primitifs ; Ataxie Télangiectasie ; Epidémiologie ; Diagnostic moléculaire ; Registre marocain
Abstract
Primary immunodeficiencies (PID) are a group of disorders characterized by susceptibility to recurrent or specific infections. In Morocco, few data are available in the littérature concerning PID, being epidemiological, clinical or genetic profile.
Our main objectives were to estimate PID impact in the world, based on recent epidemiologic studies; to determine PID etiologic profile in Morocco and perform a clinical and molecular study on the example of ataxia telangiectasia.
So, we estimated than ore than 6 millions people would live with a PID in the world, 27,000 of them in Morocco. However, our registry accounted for only 351 patients in 2011, with surrepresentation of severe phenotypes.
On the other hand, analysis of 19 AT index cases detected 14 unique mutations différentes on ATM (Ataxia-Telangiectasia Mutated) gene, whose most common was carried out by 26,32% of alleles.
Thus, PID are not as rare as generally thought. These disorders are largely underdiagnosed in Morocco. At last, molecular diagnosis is possible in Morocco and would allow establishment of a genetic counseling.
Keywords : Primary immunodeficiencies; Ataxia Telangiectasia ; Epidemiology ; Molecular diagnosis ; Moroccan registry
vii
Liste des Abréviations
ADN AFP AGAR ALPS AP50 ARCA ARN ASCIA ASID AT ATLD ATM CD CEREDIH CH50 CHU CINCA CSH DIC DICV DICS DIP DHR EDA EDTA ESID FCAS Acide DésoxyriboNucléique Alpha-foetoprotéine
Agammaglobulinémie Autosomique Récessive Syndrome Autoimmun Lymphoprolifératif Alternative Pathway activity 50%
Autosomal Recessive Cerebellar Ataxia Acide RiboNucléique
Australasian Society of Clinical Immunology and Allergy African Society for ImmunoDeficiencies
Ataxie Télangiectasie
Ataxia-Telangiectasia Like Disorder Ataxia-Telangiectasia Mutated Cluster de Différentiation
Centre de Recherche sur les Déficits Immunitaires Héréditaires Classical pathway Haemolytic complement activity 50% Centre Hospitalier Universitaire
Chronic Infantile Neurologic Cutaneous and Articular syndrome Cellules Souches Hématopoïétiques
Déficit Immunitaire Combiné
Déficit Immunitaire Commun Variable Déficit Immunitaire Combiné Sévère Déficit Immunitaire Primitif
DiHydroRhodamine-1,2,3
Dysplasie Ectodermique Anhidrotique Ethylène diamine tétraacétique
European Society for ImmunoDeficiency Familial Cold Auto-inflammatory Syndrome
viii FISH FMF GMO GSC HCC HGMD HGT HIDS HLA II ICF IFNγ Ig IPIDR ITG IV JMCN JMF LAD I LAGID MSMD MSPID MW NBS NBT NCS NFS NK
Fluorescence In Situ Hybridization Fièvre Méditerranéenne Familiale Greffe de Moelle Osseuse
Granulomatose Septique Chronique Hypoplasie Cartilage-Cheveux Human Gene Mutation Database HypoGammaglobulinémie Transitoire Hyper-IgD Syndrome
Human Leukocyte Antigen type II (Complexe majeur d’histocompatibilité) Immunodeficiency with Centromeric instability and Facial anomalies Interféron-gamma
Immunoglobuline
Iranian Primary Immunodeficiency Registry Interruption Thérapeutique de grossesse Intraveineuse
Jeffrey Modell Center Network Jeffrey Modell Foundation
Leukocyte Adhesion Deficiency type I
Latin American Group for ImmunoDeficiencies Mendelian Susceptibility to Mycobacteria Disease Moroccan Society for Primary Immunodeficiencies Muckle-Wells syndrome
Nijmegen Breakage Syndrome NitroBleu de Tétrazolium Neutropénie Congénitale Sévère Numération Formule Sanguine Natural Killer
ix PBMC PIDJ PMS2 RAPID RIDDLE SC SCID SDS SPL TCR TE TSU TRAPS TREC UIC USIDnet WAS XLA XL-DKC
Peripheral Blood Mononuclear Cells Primary Immunodeficiency in Japan Postmeiotic segregation increased 2
Resource of Asian Primary ImmunoDeficiency
Radiosensitivity, ImmunoDeficiency, Dysmorphic features and Learning difficulties
Sous-cutané
Severe Combined ImmunoDeficiency Sodium Dodecyl Sulfate
Sous-Populations Lymphocytaires T-Cell Receptor
Tris- EDTA
Triméthoprime-Sulfaméthoxazole
TNFα Receptor-Associated Periodic Syndrome TCR Receptor Excision Circle
Unité d’Immunologie Clinique
United States Immunodeficiency Network Syndrome de Wiskott-Aldrich
Agammaglobulinémie liée à X Dyskératose congénitale liée à X
x
Liste des Illustrations
Figure 1. Schéma d'activation du complément ...5
Figure 2.Carte représentant les taux de consanguinité dans le monde ... 19
Figure 3. Schéma simple permettant l’identification d’un DIP chez un enfant présentant des infections sévères, inhabituelles ou récurrentes... 25
Figure 4. Examens de première intention en cas de suspicion d’un DIP ... 30
Figure 5. Explorations en cas de normalité des examens de première intention ... 31
Figure 6. Démarche diagnostique en 4 étapes en cas de suspicion d’un déficit immunitaire ... 33
Figure 7. Correction d’un gène par une nucléase (A) ou par l’utilisation d’un transponson (B) ... 46
Figure 8. Exemple d'analyse de marqueurs génétiques chez une famille porteuse de l’Ataxie Télangiectasie ... 50
Figure 9. Répartition des déficits immunitaires primitifs en 2009. ... 56
Figure 10. Photographie de télangiectasies bulbaires chez un patient AT ... 58
Figure 11. Proportion de l’incidence des néoplasies en fonction de l’âge chez les patients AT . ... 61
Figure 12. Ligne de temps montrant l’identification du gène ATM et de sa fonction ... 69
Figure 13. Représentation schématique du locus du gène ATM ... 71
Figure 14. Répartition des mutations et des polymorphismes sur le gène ATM ... 71
Figure 15. Schéma d’activation de la protéine ATM suite à une cassure double-brin et cascade de signalisation ... 74
Figure 16. Interactions de la kinase ATM avec ses différents substrats après activation ... 75
Figure 17. Schéma de la recombinaison isotypique des Immunoglobulines, où intervient la protéine ATM lors de la réparation de l’ADN ... 78
Figure 18. Schéma montrant le défaut du Réseau de Surveillance des dégâts à l’ADN chez les patients AT ... 78
Figure 19. Circuit d’enregistrement d’un patient DIP ... 89
Figure 20. Schéma du processus d'extraction de l'ADN au phénol chloroforme ... 93
Figure 21. Schéma du processus d'extraction de l'ADN par la méthode rapide par les sels ... 94
Figure 22. Préparation des échantillons pour l'extraction automatisée... 95
Figure 23. Schéma des plaques d’amorces ATM pour la PCR et le séquençage ... 98
Figure 24. Exemple d'une photo de gel d'électrophorèse après une PCR de Phase 1 ... 100
Figure 25. Taux de couverture (%) des registres sélectionnés ... 109
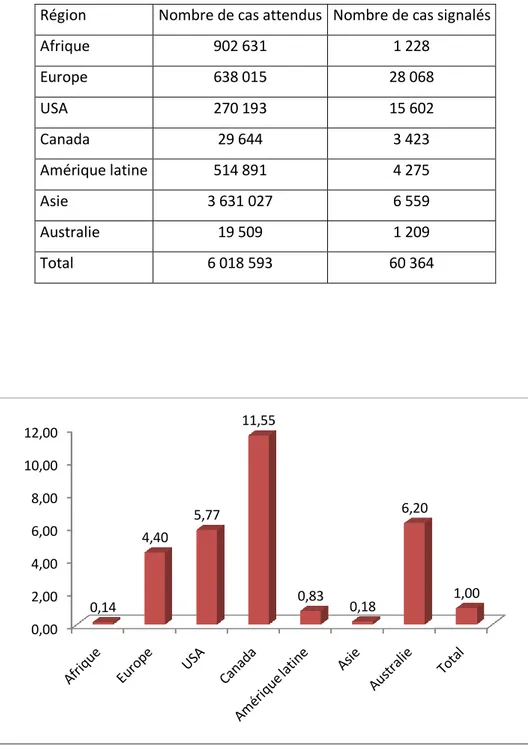
Figure 26. Taux de couverture par région dans le sondage JMF ... 110
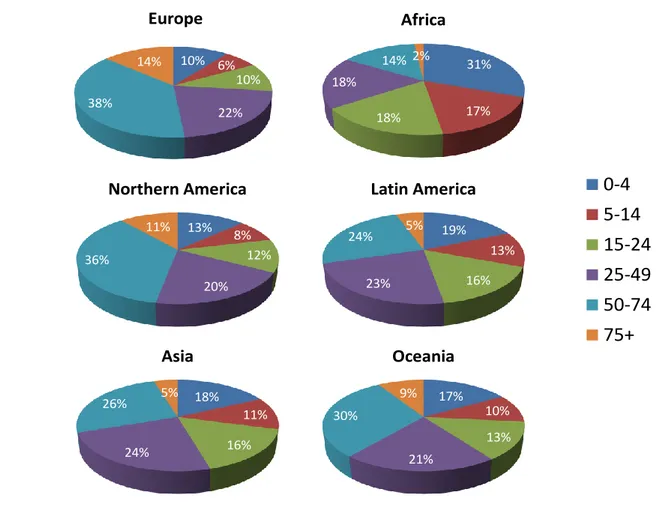
Figure 27. Distribution mondiale des nouveaux cas de DIP en 2012 par groupes d’âge ... 112
Figure 28. Distribution continentale par groupe d’âge des nouveaux cas de DIP en 2012 ... 113
Figure 29. Estimation des taux de couverture des registres ESID, PIDJ et marocain par groupe d’âge ... 115
Figure 30. Distribution des patients selon les principaux groupes de la classification IUIS ... 126
Figure 31. Distribution des patients marocains par maladie ... 127
Figure 32. Croissance du registre de 1998 à 2011 ... 131
Figure 33. Origine géographique des patients ... 131
Figure 34. Comparaison des âges moyen/médian au diagnostic entre les différentes séries ... 138
Figure 35. Comparaison des proportions (%) de patients adultes entre les différentes séries ... 140
Figure 36. Comparaison de la distribution des patients DIP dans les groupes majeurs de la classification IUIS entre le Maroc et d’autres séries ... 140
xi
Figure 38. Comparaison du taux de mortalité entre les différentes séries ... 145
Figure 39. Electrophorégramme montrant la mutation 5644C>T au niveau de l’exon 39. ... 154
Figure 40. Origine géographique des mutations retrouvées dans notre cohorte marocaine ... 156
Figure 41. Comparaison des âges médians au diagnostic avec les séries publiées ... 159
Figure 42. Comparaison des délais médians de diagnostic pour les différentes séries ... 159
Figure 43. Comparaison des taux de consanguinité dans la population et dans les séries ... 161
Liste des tableaux
Tableau I. Prévalence des DIP dans le monde ... 18Tableau II. Catégories de la classification IUIS des DIP ... 21
Tableau III. Valeurs normales des Immunoglobulines A, G, M en fonction de l’âge selon l’IFCC ... 27
Tableau IV. Valeurs normales des sous-populations lymphocytaires (103/µL) en fonction de l’âge... 27
Tableau V. Stratégies pour le traitement et la prise en charge des patients atteints de DIP ... 35
Tableau VI. Types et fréquences des tumeurs malignes rapportées chez les patients DIP ... 41
Tableau VII.Manifestations autoimmunes courantes dans les DIP ... 44
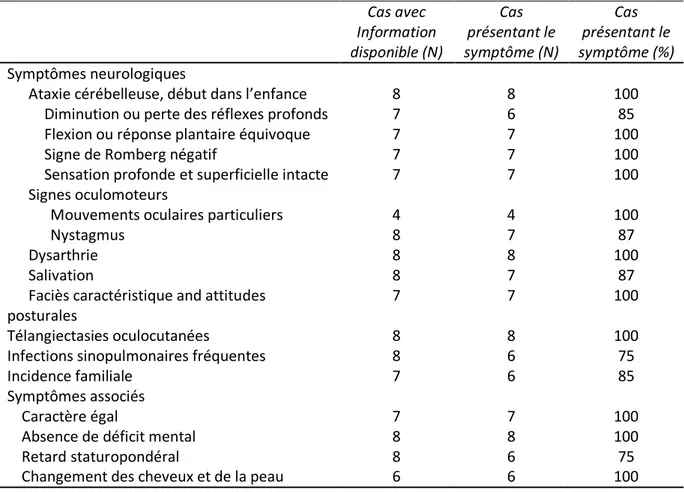
Tableau VIII. Manifestations cliniques chez 8 patients présentant une ataxie télangiectasie ... 55
Tableau IX. Epidémiologie de l’Ataxie-Télangiectasie à travers le monde ... 55
Tableau X. Extrait des Syndromes bien définis avec déficit immunitaire dans la classification IUIS ... 62
Tableau XI. Comparaison Ataxie Télangiectasie, Ataxia Telangiectasia-Like Disorder et Nijmegen Breakage Syndrome ... 64
Tableau XII. Comparaison des phénotypes cliniques des 2 patients RNF168-/-... 66
Tableau XIII. Exemples de mutations à effet fondateur du gène ATM dans différentes ethnies... 72
Tableau XIV. Méthodologie utilisée dans les publications sélectionnées ... 85
Tableau XV. Estimation de la fréquence des DIP dans le monde ... 108
Tableau XVI. Comparaison entre le nombre de cas attendus et le nombre de cas enregistrés dans les registres sélectionnés ... 109
Tableau XVII. Comparaison entre le nombre de cas attendus et le nombre de cas signalés dans le sondage de la JMF ... 110
Tableau XVIII. Estimation du nombre de nouveaux cas par groupe d’âge en 2012 ... 112
Tableau XIX. Estimation et distribution du nombre de cas en 2012 par groupe d’âge pour les registres d’ESID, de la PIDJ et du Maroc... 115
Tableau XX. Comparaison des fréquences aux Etats-Unis entre les DIP et d’autres maladies ... 123
Tableau XXI. Caractéristiques des patients DIP dans notre série ... 129
Tableau XXII. Manifestations cliniques des patients enregistrés ... 133
Tableau XXIII. Conditions des greffes de moelle osseuse des patients marocains... 135
Tableau XXIV.Signes cliniques observés chez nos patients au moment du recrutement ... 150
Tableau XXV. Résultats des analyses de laboratoire pour les patients marocains ... 151
Tableau XXVI. Spectre des mutations du gène ATM dans notre série ... 153
Tableau XXVII. Polymorphismes du gène ATM retrouvés chez nos patients AT. ... 157
Tableau XXVIII. Corrélation génotype-phénotype pour notre série et la série française ... 165
Tableau XXIX. Comparaison entre les premiers cas index et les cas diagnostiqués ... 169
Tableau XXX. Comparaison entre les patients présentant un délai diagnostic inférieur à 3 ans et ceux présentant un délai diagnostic supérieur à 3 ans ... 171
xii
Glossaire
Ataxie : - Troubles de la coordination des mouvements volontaires avec conservation de la force musculaire
-
Trouble moteur non paralytique caractérisé par une mauvaise coordination par des
mouvements, il se manifeste soit dans la station debout ou pendant la marche, soit lors de
l'exécution d'un segmentaire ou au maintien d'une attitude.
Télangiectasie : Dilatation pathologique et permanente de certains petits vaisseaux de la peau et des muqueuses, dont le trajet devient visible à l'oeil nu, sous forme de traînées linéaires (chevelu capillaire), de fins réseaux, de plaques circonscrites, ou d'étoiles vasculaires et qui disparaît à la vitropression.
Prévalence : Nombre de cas nouveaux et/ou anciens d'une maladie, au sein d'une population donnée, à un moment donné, soit un instant, soit un intervalle de temps. Ce n'est pas un taux mais une proportion.
Incidence : Nombre de nouveaux malades ou de nouvelles maladies dans une population au cours d’une période déterminée (la plupart du temps un an). Elle peut être exprimée sous forme d’incidence cumulée ou d’incidence instantanée.
Incidence cumulée : Rapport entre le nombre de nouveaux cas survenus pendant la période d'observation et le nombre de personnes en observation et susceptibles de devenir des cas au début de l'étude. Il s'agit d'une proportion et d'une mesure du risque qui doit toujours être accompagnée de la mention de la durée d'observation.
2
I.
Le système immunitaire
Notre travail de thèse repose sur l’étude des déficits immunitaires primitifs. Pour mieux appréhender cette notion, il convient de faire un petit rappel sur le système immunitaire.
I.1. Généralités
Dans l’environnement, l’organisme va être confronté à de nombreux agents extérieurs, potentiellement pathogènes, telles que des bactéries, virus, parasites, allergènes ou toxines. La fonction du système immunitaire est de défendre l’organisme contre ces agents pathogènes.
Le principe du système immunitaire repose sur le concept du Soi et du non-Soi. Le système immunitaire doit être capable de reconnaitre les antigènes du non-Soi, grâce à des récepteurs à l’antigène, afin de déclencher une réponse aboutissant à la neutralisation de l’agent pathogène et à l’intégrité du Soi.
L’immunité est donc définie comme l’ensemble des mécanismes qui permettent à un organisme de reconnaitre et de lutter contre les agents potentiellement pathogènes de l’environnement.
Le dysfonctionnement du système immunitaire se traduit par un déficit immunitaire quand il concerne un défaut de distinction du non-Soi, et par une auto-immunité quand il concerne un défaut de distinction du Soi.
I.2. Réponses immune innée et adaptative
La réponse immunitaire est l’ensemble de mécanismes qui mènent à la neutralisation de l’antigène. On distingue deux types de réponse immunitaire :
3
-
La réponse innée : immédiate, sans mémoire et non spécifique à l’antigène ;
-
La réponse adaptative : non immédiate, douée de mémoire et spécifique à
l’antigène.
Ces réponses mettent en jeu différents types de cellules et de protéines particulières.
I.2.1.
Réponse innée
L’immunité innée est la première ligne de défense de l’organisme contre les agents externes. Elle met en jeu des processus physico-chimiques et des cellules immunitaires non spécifiques. Elle est active immédiatement et est fonctionnelle 4 jours (96 heures).
La première défense de l’organisme est la barrière physico-chimique que constituent la peau et les muqueuses. Toutefois, de nombreux antigènes échappent à cette barrière, notamment au niveau de plaies, d’où la nécessité de déclencher une réponse inflammatoire, grâce aux cellules de l’immunité innée entre autres.
Ainsi, la réponse immunitaire innée est induite par un signal de danger, suite à l’interaction spécifique entre des récepteurs du Soi, appelés PRR (Pattern Recognition Receptors), et des molécules du Non-Soi présentes au niveau des micro-organismes, qu’ils soient pathogène ou non, appelés PAMP (Pathogen Associated Molecular Patterns). On distingue 3 types de PRR, selon leur localisation : les PRR solubles, les PRR membranaires et les PRR cytoplasmiques.
Parmi les PRR solubles, on retrouve notamment les composants du complément. Le complément est un système d’une vingtaine de protéines qui vont réagir en cascade les unes avec les autres. Le but du complément est d’activer la réponse inflammatoire, de faciliter la phagocytose des bactéries virulente non phagocytable directement et plus particulièrement de détruire la cellule cible.
4 On distingue 3 voies d’activation du complément (Figure 1) :
La voie classique, activée par la fixation de la protéine C1q soit directement
sur l’agent infectieux, soit sur la protéine CRP (pour « C-Reactive Protein »),
soit sur une paire d’anticorps déjà fixé à la surface de l’antigène.
La voie MBP (ou voie MBL, ou encore voie du mannose), activée par la fixation
de la protéine MBP au niveau de résidus mannose présent à la surface de
l’agent infectieux.
La voie alterne, activée par la fixation de la protéine C3b à la surface de
l’agent pathogène.
Le but de ces trois voies est l’activation d’une protéine zymogène, la C3 convertase, et la libération de différents composants du complément :
Les molécules C3a, C4a, et C5a sont des anaphylatoxines, molécules
responsables de l’activation de l’inflammation.
Les molécules C3b ont une triple action en permettent tout d’abord
l’opsonisation de l’agent pathogène en se fixant directement à sa surface, puis
en activant la suite de la cascade d’activation entrainant la formation du
complexe d’attaque membranaire, et finalement en amplifiant l’activation du
complément par la voie alterne.
Les molécules C5b, C6, C7, C8 et C9 permettent la destruction des agents
pathogènes par la formation du complexe d’attaque membranaire.
Parmi les PRR membranaires, on retrouve notamment les récepteurs TLR (Toll-like receptor) à la surface de quasiment toutes les cellules, mais plus particulèrement au niveau des macrophages et cellules dendritiques. Ces récepteurs jouent un rôle important dans la réponse immunitaire innée : phagocytose, réponse inflammatoire et reconnaissance des composantes parasitaires, bactériennes et virales.
5 Figure 1. Schéma d'activation du complément [cours-pharmacie, voir URL]
6 On a identifié une dizaine de TLR qui ont été séparés en deux groupes principaux :
Les TLR 1, 2, 4, 5, 6 situés au niveau de la membranaire plasmique et impliqués dans
la reconnaissance des composants de la paroi des agents infectieux.
Les TLR-3, 7, 8, 9 situés au niveau des endosomes et reconnaissant les composants
viraux et bactériens, surtout les acides nucléiques.
Les PRR cytoplasmiques correspondent aux récepteurs NLR et RLR.
Les récepteurs NLR sont une famille d’une vingtaine de protéines situées dans le cytoplasme et reconnaissant presque exclusivement des composants bactériens. Ils se subdivisent en 3 sous-familles : NOD, NALP et NAID. Les trois sous-sous-familles recrutent des caspases qui clivent un certain nombre de cytokines, en particulier les cytokines inflammatoires comme l’interleukine 1 se trouvant sous la forme inactive dans le cytoplasme et étant ainsi activé. Les caspases font partie de l’inflammasone (activation de cytokines).
Les récepteurs RLR reconnaissent essentiellement des composant viraux, principalement des acides nucléiques viraux, et vont activer toutes les voies de signalisation : NF-κB, MAP-kinases et interféron.
Enfin, une fois reconnu, l’agent infectieux sera phagocyté. La phagocytose est un phénomène induit qui permet d’endocyter des bactéries ou cellules mortes. Parmi les cellules phagocytaires, on retrouve les polynucléaires, les macrophages et les cellules dendritiques. La phagocytose se réalise en différentes étapes :
L’opsonisation (non obligatoire) correspond à l’attache des opsonines tout autour de
la bactérie.
Le chimiotactisme permet d’attirer les macrophages vers la bactérie opsonisée, et
ceci grâce aux chimiokines.
7
La phase d’adhérence correspond à la reconnaissance spécifique des opsonines
présentes à la surface de la bactérie par des récepteurs de la membrane plasmique
des macrophages. Cette phase déclenche la phagocytose proprement dite.
La phase rhéologique correspond à la formation de prolongements cytoplasmiques
que l’on appelle des pseudopodes enveloppant entièrement la bactérie. Il y a ainsi
formation d’une vacuole dans laquelle se trouve la bactérie ; on appelle cette vacuole
le phagosome.
La phase de destruction correspond à la digestion de la bactérie par fusion du
phagosome avec des lysosomes, formant ainsi le phago-lysosome. La digestion sera
réalisée par différents mécanismes : acidification, hydrolysation par des enzymes
hydrolytiques (lysozyme, protéase), production de dérivés toxique de l’oxygène (ions
superoxydes), production de dérivés nitrés.
Enfin, la réponse inflammatoire correspond à la sécrétion de facteurs solubles qui permettent le recrutement de cellules au site de l’inflammation :
Les cytokines pro-inflammatoires : le TNF-α, les chimiokines et les interleukines IL-1,
IL-6, IL-12 et IL18.
Les substances vasodilatatrices : le monoxyde d’azote (NO) et les prostanoïdes.
Les cytokines anti-inflammatoires : l’interleukine-10 et le TNF-β, jouant un rôle de
régulation de la réaction inflammatoire, permettant ainsi qu’elle ne devienne pas
exagérée et donc pathologique.
8
I.2.2.
Réponse adaptative
La réponse immunitaire adaptative est la seconde ligne de défense contre les agents infectieux et existe uniquement chez les vertébrés. Elle se met en place au bout de 4 jours environ et est caractérisé par la participation des lymphocytes qui ont un rôle majeur. Les lymphocytes sont de deux types, les lymphocytes B (LB) et les lymphocytes T (LT).
L’immunité adaptative fait intervenir les récepteurs BCR présents sur les LB, et les récepteurs TCR présent sur les LT ; ces récepteurs vont reconnaître un seul ligand uniquement. En effet, un lymphocyte est programmé pour répondre à un antigène, il présente donc un seul type de récepteur. Cette forte affinité pour un seul antigène est rendue possible grâce à la structure des récepteurs lymphocytaires, composés principalement par un dimère Igα-Igβcomposé de deux chaines lourdes et deux chaines légères, formant une fraction constante permettant la fixation à la membrane plasmique et une partie variable permettant la reconnaissance de l’antigène. La variabilité de ces chaines légères est permise par des réarrangements géniques lors de la recombinaison V(D)J.
Les lymphocytes T seront responsables de la réponse cellulaire et les lymphocytes B de la réponse humorale.
Toutefois, les lymphocytes doivent être préalablement activés grâce à une interaction avec les cellules de l’immunité innée, notamment les « cellules présentatrices de l’antigène » (CPA). En effet, les macrophages et cellules dendritiques peuvent, après phagocytose d’un pathogène, exprimer à leur surface des PAMP associés au complexe majeur d’histocompatibilité de classe II (CMH-II). Les CPA vont ensuite passer dans les organes lymphoïdes cible où ils pourront activer les lymphocytes T.
Une fois présentes au niveau des organes lymphoïdes secondaires, les CPA seront véritablement scannées par les lymphocytes T-CD4 naïf qui chercheront à reconnaître de manière spécifique le fragment antigénique dont ils sont spécifiques. Si le TCR reconnaît un antigène, le lymphocyte T s’arrêtera, permettant la formation d’une zone de contact particulière que l’on appelle une « synapse » et ceci par des réarrangements protéiques au niveau de celle-ci.
9 Suite à la formation de la synapse s’effectuera l’activation des lymphocytes T-CD4 et ceci par deux types de signaux :
Des signaux de stimulation permis par des kinases qui phosphoryleront les
motifs ITAM des régions intra-cytoplasmique des chaînes du CD3 associées au
TCR.
Des signaux de costimulation, indispensable à une activation totale du
lymphocyte, qui sont induit par l’interaction entre le cluster de différenciation
CD28 présent à la surface du lymphocyte T-CD4 et le récepteur B7 présent à la
surface de la cellule présentatrice d’antigène, ainsi que l’interaction entre le
ligand du récepteur CD40 (CD40-ligand) présent à la surface du lymphocyte et
le cluster de différenciation CD40 présent à la surface de la cellule
présentatrice d’antigène.
Une fois ces cellules activées, on observera une phase de prolifération et de différenciation.
Les lymphocytes T-CD8 sont activés par des fragments antigéniques présentés par des molécules du CMH-I, eux-mêmes exprimées par les cellules nucléées de l’organisme. En effet, les lymphocytes T-CD8 circulent à l’état pré-cytotoxique et reçoivent des signaux d’activation pour devenir cytotoxique. Ces signaux leurs sont donnés suite à leur interaction, également sous forme de « synapse », avec la cellule présentant le fragment antigénique associé au CMH-I.
Les lymphocytes T cytotoxiques sont responsables de l’immunité cellulaire aboutissant à la mort de la cellule cible. On observe une libération des granules cytotoxiques (lysosomes particuliers) qui contiennent deux catégories de molécules que l’on appelle des cytotoxines :
La perforine est une protéine qui en se polymérisant forme des pores dans la
membrane de la cellule cible.
Les sérine-estérases ont pour but de détruire l’ADN en activant des caspases
qui iront fragmenter l’ADN afin d’induire l’apoptose.
10 Pour la réponse humorale, l’activation des lymphocytes B peut se faire de différentes manières suivant l’implication des lymphocytes T : thymo-dépendante ou thymo-indépendante.
L’activation thymo-dépendante est la plus couramment utilisée et tout comme pour l’activation des lymphocytes T on distingue deux types de signaux qui sont induit par l’interaction antigène-BCR :
Les signaux de stimulation sont responsables d’une part de l’internalisation du
complexe antigène-BCR, permettant ainsi la dégradation de l’antigène dans le
système endosomale. Les fragments peptidiques obtenus seront associés à
des molécules du CMH-II, procurant au lymphocyte B le statut de cellule
présentatrice d’antigène. D’autre part, ces signaux sont responsables de
l’activation des tyrosines kinases qui phosphoryleront les motifs ITAM des
régions intra-cytoplasmiques du dimère Igα-Igβ associé au BCR, entraînant
ainsi l’activation de facteur de transcription qui permettront l’expression de
nombreuses molécules.
Les signaux de costimulation sont indispensables à une activation totale du
lymphocyte et sont permis par un certain nombre de corécepteurs (CD19,
CD21 et CD81) qui vont amplifier le signal.
D’autre part, les LB activés reçoivent encore des signaux de prolifération, qui ne sont cette fois-ci pas induit par l’interaction antigène-BCR mais par les Th2.
Suite à cette activation, les lymphocytes obtenus se multiplieront intensément et certains d’entre eux donneront des plasmocytes qui produiront alors des IgM de basse affinité pour l’antigène ; ces plasmocytes ne quitteront pas les organes lymphoïdes secondaires.
11 Les autres cellules continueront de se multiplier dans les follicules primaires afin de former des centres germinatifs, ces cellules sont alors appelées des centroblastes. Ces derniers n’expriment plus de BCR car des mutations s’effectuent au niveau des gènes codant pour les parties variables des chaines lourdes et des chaines légères, au fur et à mesure des divisions ; on parle d’hypermutation somatique.
Les centroblastes vont ainsi devenir des centrocytes qui ne se divisent plus et qui ré-expriment à leurs surface un BCR qui reconnaît toujours le même antigène de départ mais avec une affinité modifiée. Ces centrocytes vont être sélectionnés par des complexes antigène-BCR présent au niveau de cellules dendritiques folliculaires, et de cette manière seul ceux exprimant des BCR ayant une forte affinité pour l’antigène recevront le signal de survie.
Les centrocytes sélectionnés vont à ce stade de nouveau interagir avec les TH2 permettant ainsi la formation de deux types de cellules :
Des plasmocytes qui vont produire des anticorps (IgM) de haute affinité pour
l’antigène. La sécrétion d’interleukines va permettre la commutation de
classe, et de cette manière il n’y aura plus de sécrétion d’IgM mais d’IgA, d’IgE
ou d’IgG. On observera une latence de 4 à 8 jours entre la production
d’immunoglobulines de faible affinité et celles de haute affinité.
Des lymphocytes B mémoires qui vont quitter les follicules secondaires pour
aller dans la circulation et ceci afin de faciliter la rencontre avec l’antigène.
Ces cellules ont la caractéristique de pouvoir sécréter directement, sans
temps de latence, des anticorps de haute affinité lors d’une deuxième
infection par le même antigène. La réponse obtenue se produit pour des taux
beaucoup plus faible d’antigène et est considérablement plus importante en
intensité.
12 Contrairement à l’activation thymo-dépendante, les activations thymo-indépendantes ne nécessitent pas l’aide des TH2 pour produire les anticorps. On les classe en 2 catégories :
L’activation thymo-indépendante de type 1 entraîne une stimulation
polyclonale des lymphocytes B. Cette activation ne passe pas par le BCR mais
par des récepteurs communs à tous les LB qui reconnaissent les pathogènes
que l’on appelle des mitogènes.
L’activation thymo-indépendante de type 2 entraîne une stimulation
monoclonale des lymphocytes B. Cette activation passe cette fois-ci par le BCR
qui reconnaît des déterminants sucrés répétitifs. On observera cependant
essentiellement une production d’IgM.
Enfin, la cellule NK (cellule « Natural Killer ») fait partie des lymphocytes car elle découle du progéniteur lymphoïde au niveau de la moelle osseuse ; elle fait partie des grands lymphocytes granuleux (GLG). Elle ne correspond cependant ni à un lymphocyte B ni à un lymphocyte T, ne présentant respectivement ni le dimère Igα-Igβ ni le cluster de différentiation CD3, mais le cluster de différentiation CD56. La spécificité de ces cellules est d’être capable de lyser des cellules malades sans nécessiter d’activation préalable et sans rentrer en contact avec l’agent pathogène, grâce à une balance de signaux entre ses récepteurs activateurs et inhibiteurs.
I.3. Régulation de la réponse immune
Une régulation de la réponse immune est nécessaire pour éviter une réponse exacerbée et maintenir l’intégrité du Soi. Nous avons cité auparavant que la réponse innée, notamment la réponse inflammatoire, est régulée par des cytokines anti-inflammatoires inhibant cette réponse. L’activation ou l’inhibition de la réponse inflammatoire est donc la résultante d’une balance entre les cytokines pro-inflammatoires et les cytokines anti-inflammatoires.
13 La réponse adaptative est également régulée à différents niveaux.
En premier lieu, une sélection des lymphocytes T a lieu au niveau du thymus, au cours de leur maturation afin de sélectionner uniquement les lymphocytes T présentant une faible affinité pour les molécules de CMH (sélection positive), mais également éliminer les lymphocytes T présentant une forte affinité pour les antigènes du Soi (sélection négative) grâce à l’expression du facteur de transcription AIRE. Toutefois, certains LT auto-réactifs échapperont à cette double sélection, mais seront silencieux grâce à un phénomène de tolérance périphérique.
Enfin, les lymphocytes T régulateurs (Treg, ou lymphocytes T suppresseurs) sont une sous population de lymphocytes T CD4+ ayant la propriété d’inhiber la prolifération d’autres lymphocytes T effecteurs. Ils sont nécessaires au maintien de la tolérance immunitaire, ils participent donc au maintien de l'homéostasie. Ils sont essentiels pour la tolérance aux antigènes du soi, et aux antigènes non dangereux.
Les lymphocytes T régulateurs ne sécrètent pas d’IL-2 et prolifèrent peu lorsqu’ils sont activés par leur récepteur des cellules T suite à leur rencontre avec leur antigène, mais ils inhibent les réponses des autres lymphocytes T CD4+ et T CD8+. Ils inhibent les réponses des lymphocytes T effecteurs ou les font rentrer en apoptose, par différents mécanismes encore mal connus :
En sécrétant des cytokines suppressives (IL-10, TGF-β ou IL-35)
En consommant l’IL-2, ce qui limite la prolifération des autres lymphocytes
par un effet de compétition
Par cytolyse directe (destruction des lymphocytes cibles)
Via l’expression à leur surface de molécules inhibitrices (Galectin-1)
Les T régulateurs ont aussi un rôle de suppresseur vis-à-vis des cellules présentatrices d’antigènes, par exemple en envoyant un signal inhibiteur via la molécule de surface CTLA-4 reconnue sur la cellule présentatrice d’antigène par CD80 ou CD86.
14 On peut distinguer 3 types de lymphocytes T régulateurs :
Les Treg naturels (nTreg) sont naturellement généré dans le thymus. Ces
lymphocytes T présentent une forte avidité pour le complexe CMH-peptide du soi
présenté par les cellules dendritiques ou les cellules épithéliales thymiques : ils se
différencient en nTreg et après leur sortie dans le thymus, ils bloquent en périphérie les
réactions auto-immunes, potentiellement dangereuses.
Les Treg inductibles (iTreg) sont issus de la différenciation de lymphocytes T
CD4 naïfs en périphérie, par exemple au niveau des plaques de Peyer : leur rôle est
particulièrement important où le système immunitaire est stimulé en permanence par la
flore commensale. Ils sécrètent des cytokines telles que le TGF-β (transforming growth
factor-β) et de l’IL-35 qui inhibent la réponse des lymphocytes effecteurs.
Les Treg producteurs d’IL-10 (Tr-1) sont également issus de la différenciation
des lymphocytes T CD4 naïfs. Ils sécrètent principalement de l’IL-10, cytokine
immunorégulatrice majeure.
15
II.
Les Déficits Immunitaires Primitifs
II.1. Définition
Dans la dernière classification par le comité d’expert de l’International Union of Immunological Societies (IUIS) [Al-Herz et al., 2011], plus de 170 maladies ou syndromes ont été reconnus comme déficit immunitaire primitif (DIP). Toutefois, une définition précise et unilatéralement acceptée d’un DIP n’existe pas encore et reste un sujet de controverse [Notarangelo & Casanova, 2009].
En effet, au cours des dernières années, la définition des DIP a été considérablement révisée avec la découverte de nouveaux phénotypes dérivant d’un défaut congénital de l’immunité [Casanova & Abel, 2007]. Ainsi, les DIP furent longtemps considérés comme des maladies rares, familiales, monogéniques et de transmission récessive, résultant en un défaut dans le développement ou la fonction d’une ou plusieurs lignées leucocytaires, d’où le développement d’infection multiples, récurrentes, opportunistes et souvent fatales durant l’enfance. Toutefois, de nombreux syndromes n’entrant pas dans cette définition se sont révélés être dus à un défaut du système immunitaire. A commencer par l’épidermodysplasie verruciforme, une prédisposition aux verrues pouvant se révéler fatale, qui n’a été considérée comme un DIP qu’en 2002, lors de la découverte de deux gènes causals [Orth, 2006].
De plus, de nouveaux DIP ont été décrits qui confèrent une prédisposition spécifique à un pathogène [Bousfiha et al., 2010+, notamment une prédisposition génétique à l’Epstein-Bar Virus [Bassiri et al., 2008], à Neisseria [Mathew et Overturf, 2006], aux papillomavirus [Orth, 2006], à
Streptococcus pneumoniae [Picard et al., 2003], aux mycobactéries faiblement virulentes [Altare et al., 1998 ; de Beaucoudrey et al., 2010], au virus de l’Herpes simplex [Casrouge et al., 2006] et au Candida albicans [Puel et al., 2011]. Une prédisposition mendélienne à la tuberculose a également
été décrite [Boisson-Dupuis et al., 2011 ; Tabarsi et al., 2011+.D’autres phénotypes non-infectieux ont
été associés à un DIP, comme une autoimmunité, un angioedème, un granulome, une autoinflammation, un syndrome d’activation macrophagique ou des microangiopathies
16 thrombotiques [Casanova & Abel 2007]. De même, quelques DIP présentent une prédisposition aux allergies ou aux tumeurs [Casanova & Abel 2007].
En définitive, les DIP ne peuvent être clairement définis. Les experts émettent désormais le concept que toute maladie ou presque résulte d’un défaut spécifique et plus ou moins complexe du système immunitaire. Dans ce travail, nous utiliserons la définition générale de Notarangelo précisant qu’un DIP est un désordre qui affecte le développement et/ou le fonctionnement du système immunitaire [Notarangelo, 2010].
II.2. Epidémiologie
Etant donné ce manque de définition des DIP, déterminer leur fréquence se révèle difficile. Si, généralement, les DIP sont considérés comme des maladies rares (<1/2 000), que ce soit individuellement ou collectivement, avec une incidence de 1 sur 5 000 naissances vivantes [IDF, 1995 (voir URL) ; Chan & Lau, 1995; Ten RM, 1998], la découverte de ces nouveaux DIP, présentant des phénotypes plus courants, pourrait changer la donne. Dans ce travail, nous ne considérerons que les maladies incluses dans la classification de l’IUIS [Al-Herz et al., 2011].
II.2.1. En occident
Toutefois, concernant la prévalence des DIP, les chiffres varient beaucoup d’une étude à l’autre et d’un registre à l’autre. Ne serait-ce que dans la base de données de l’European Society of ImmunoDeficiencies (ESID, voir URL), la prévalence varie de 0.06/100 000 en Roumanie à 5.63/100 000 en France (voir URL). En Australie, la prévalence a été estimée à 5.6/100 000 en 2007 [Kirkpatrick & Rimington, 2007]. Le Tableau I reprend les prévalences rapportées dans différentes publications.
D’autre part, des études épidémiologiques sur les DIP ont été conduites récemment aux Etats-Unis. En effet, en 2007, Boyle & Buckley ont conduit un sondage téléphonique pour estimer la véritable prévalence des DIP. Cette étude, commanditée par l’Immune Deficiency Foundation (IDF), a révélé que 86.3/100 000 personnes [0.051-0.1215%] présenteraient un DIP aux Etats-Unis, ce qui correspondrait à 1 personne sur 1200 diagnostiquée avec un DIP au cours de sa vie [Boyle & Buckley, 2007].
17 Une autre étude épidémiologique, conduite par Joshi et al. à la Clinique Mayo, a déterminé l’incidence des DIP dans le comté d’Olmsted, Minnesota, USA, sur une période de 30 ans. Ainsi, Joshi
et al. ont observé une croissance de l’incidence au cours des 30 dernières années avec une incidence
globale de 4.6/100 000 personnes-années, et une incidence maximale de 10.3/100 000 personne-années pour la période 2001-2006 [Joshi et al., 2009].
II.2.2. Au Moyen-Orient
Etant donné que de nombreux DIP sont de transmission autosomique récessive, il est probable que la fréquence de ces affections soit plus importante dans des pays à fort taux de consanguinité, comme c’est le cas au Moyen-Orient (voir Figure 2).
Ainsi, en Iran, où le taux de consanguinité est estimé à 38.6% [Saadat et al., 2004+, l’incidence des DIP a été estimée à 6/100 000 naissances vivantes. L’incidence cumulée sur 10 ans a été estimée à 11.9/1 000 000 [Rezaei et al., 2006].
De même au Koweït, où le taux de consanguinité est estimé à 54% [Al Awadi et al., 1985], la prévalence a été estimée à 11.98/100 000 enfants, avec une incidence de 10.6/100 000 enfants par an *Al Herz, 2007+. Al Herz a donc estimé l’occurrence des DIP au Koweït à 1/1 000 naissances vivantes.
II.2.1. Au Maghreb
Au Maghreb, la véritable prévalence des DIP est inconnue et n’a pu être estimée en absence de registre national. Toutefois, on pourrait s’attendre à des prévalences proches de celles du Moyen-Orient, étant donné le taux de consanguinité élevé dans cette région : 15.25% au Maroc [Cherkaoui, 2009+, 34% en Algérie *Benallegue & Kedji, 1984+, 19.24% en Tunisie *Ben M’Rad & Chalbi, 2006], 32.8% en Egypte [Mohamed, 1995], 47.2% en Mauritanie [Hammami et al., 2005] et 48.4% en Libye [Broadhead & Sehgal, 1981].
18 Tableau I. Prévalence des DIP dans le monde
Pays Prévalence (/100 000 habitants)
Référence
Roumanie 0.06 ESID
Lituanie 0.09 ESID
Taiwan 0.78 Lee et al., 2011
Allemagne 1.83 ESID
Royaume-Uni 1.94 ESID
Italie 1.95 ESID
Irlande 1.99 ESID
Japon 2.3 Ishimura et al., 2011
Singapour 2.65 Lim et al., 2003
Espagne 4.3 ESID
France 4.4 CEREDIH, 2010
Israël 4.9 Golan et al., 2002
France 5.63 ESID
Australie 5.6 Kirkpatrick & Rimington, 2007
Iran 6 Rezaei et al., 2006
Koweït 11.98* Al Herz, 2007
Chine (Shanghai) 35.08** Wang et al., 2011 Etats-Unis 86.3 Boyle & Buckley, 2007
*Koweit : prévalence estimée chez les enfants **Chine : un seul centre
19 Figure 2.Carte représentant les taux de consanguinité dans le monde [Bittles, 2009]
20 En 2010, nous avons reporté un total de 1016 patients répartis sur 3 pays du Maghreb : 206 en Egypte, 290 au Maroc et 520 en Tunisie [Barbouche et al., 2011], ce qui correspondrait à des prévalences de 0.25, 0.91 et 4.96/100 000 habitants, respectivement. Toutefois, cette estimation est largement sous-estimée, puisqu’elle ne comptabilise pas tous les cas diagnostiqués dans ces pays, mais seulement les cas enregistrés dans quelques centres.
II.3. Classification de l’IUIS
En 1973, l’Organisation Mondiale de la Santé (OMS) a fondé un comité d’expert sur les DIP, repris dans les années 90’ par l’Union Internationale des Sociétés d’Immunologie (IUIS), dont le principal objectif est de décrire et d’établir une classification des DIP connus. Ainsi, lors de réunions biennales, ce comité d’expert se réunit pour la mise à jour de la classification. La dernière réunion a eu lieu à New-York, en mai - juin 2011.
Dans cette dernière classification [Al-Herz et al., 2011], 176 maladies ont été reconnues comme étant des DIP. Ces DIP sont classés dans 8 catégories, en fonction du type de déficit immunitaire (voir Tableau II).
Parmi les 176 DIP, 58 n’ont pas été rapportés chez plus de 10 patients, soit parce que la découverte du DIP est encore récente, soit parce qu’ils sont, de fait, extrêmement rares. La classification de l’IUIS fournit également des données concernant la biologie, les symptômes, la génétique et la pathogénèse de ces maladies.
21 Tableau II. Catégories de la classification IUIS des DIP
Catégorie
Fonction affectée
Exemples
I. Déficits immunitaires combinés
Défauts de développement ou de fonction des lymphocytes T
RAG 1/2 ; DOCK8 ; HLA-II ; ADA; syndrome d’Omenn II.Syndromes bien définis avec
déficit immunitaire
Déficits de l’immunité cellulaire dans le cadre d’un syndrome extra-immunitaire
Ataxie-télangiectasie ; syndrome Hyper-IgE ; syndrome de Wiskott-Aldrich ; syndrome de DiGeorge
III. Déficits prédominants en anticorps
Défauts de l’immunité humorale Maladie de Bruton ; AID ; CD40 ; déficit immunitaire commun variable
IV.Maladies de dérégulation immunitaire
Défauts de régulation de la réponse immune : autoimmunité ou lymphoprolifération Syndrome de Chediak-Higashi type 2 ; syndrome lymphoprolifératif auto-immun V.Défauts congénitaux
quantitatifs et/ou qualitatifs des phagocytes
Défauts de développement ou de fonction des neutrophiles ou monocytes/macrophages Granulomatose septique chronique ; neutropénie congénitale sévère ; déficit d’adhésion leucocytaire VI.Défauts de l’immunité innée Prédisposition à des pathogènes
spécifiques NEMO ; IRAK-4 ; epidermodysplasie verruciforme VII. Maladies autoinflammatoires
Syndromes de fièvre périodique Fièvre
méditerranéenne familiale ; syndrome de Muckle-Wells VIII.Déficits en complément Déficits en une fraction du complément Angiooedème
héréditaire ; C5 ; hémoglobinurie paroxystique nocturne
22
II.4. Diagnostic
Etant donné la méconnaissance des médecins concernant ces pathogénies, suspecter et diagnostiquer un DIP peut se révéler difficile. Ainsi, le délai entre les premiers symptômes et le diagnostic varie entre 73 jours et 6 ans en France, suivant la maladie [CEREDIH 2010]. La sensibilisation des médecins et l’amélioration des techniques a permis de réduire ce délai de diagnostic qui avoisinait 3.7 ans avant 1980 et est passé à 0.4 an après 2000 [CEREDIH 2010].
Au cours des années, des outils et des démarches diagnostiques ont été proposés afin d’évoquer, explorer et diagnostiquer un déficit immunitaire [Bousfiha et al., 2005].
II.4.1. Evoquer un DIP
Certains signes d’alerte peuvent nous amener à suspecter un DIP chez un enfant ou un adulte. En effet, la Jeffrey Modell Foundation (JMF, voir URL) a proposé 10 signes d’alerte permettant l’évocation d’un DIP. Ainsi, un DIP doit être suspecté chez un enfant présentant au moins deux des signes suivants :
1. Au moins 4 otites par an
2. Au moins 2 sinusites par an.
3. Des traitements par antibiotique d’au moins 2 mois, avec peu d’effet.
4. Deux pneumonies ou plus par an.
5. Ralentissement de la croissance.
6. Des abcès de la peau ou des organes récurrents.
7. Infection par champignon persistante de la bouche ou de la peau.
8. La nécessité d’un traitement antibiotique par voie intraveineuse.
9. Au moins deux infections sévères, y compris septicémie.
23 Certains DIP n’apparaissent qu’à l’âge adulte. Ainsi, un DIP doit être suspecté chez l’adulte, si ce dernier présente au moins deux des signes suivants :
1. Au moins 2 otites en un an
2. Au moins 2 sinusites en un an, en l’absence d’allergie
3. Une pneumonie par an depuis plus d’un an
4. Des diarrhées chroniques avec perte de poids
5. Des infections virales récurrentes (rhumes, herpès, verrues, condylome)
6. La nécessité récurrente d’un traitement antibiotique par voie intraveineuse
7. Des abcès profonds et récurrents de la peau ou des organes internes
8. Infection persistante par champignon de la peau ou autre
9. Infection par des bactéries similaires à la tuberculose, normalement bénignes
10. Cas connus de DIP dans la famille
Ces 10 signes d’alerte ont été promus par de nombreuses organisations. Toutefois, jusqu’à 2011, leur efficacité n’avait pas été rigoureusement testée. En 2011, une équipe britannique a évalué l’efficacité de ces 10 signes à prédire un DIP [Subbarayan et al., 2011], sur une cohorte de 563 enfants. Cette étude a montré que le signe d’alerte le plus prédictif était une histoire familiale de DIP, 18 fois plus fréquent chez les enfants qui ont présenté un DIP que ceux où on n’a pas pu définir un DIP. Les 2 autres signes les plus utiles étaient la nécessité d’un traitement antibiotique intraveineux pour soigner les infections bactériennes chez les enfants présentant un déficit des phagocytes, et le retard de croissance chez les enfants présentant un déficit immunitaire combiné. L’utilisation de ces 3 signes avec la recherche d’anomalies congénitales du syndrome de Di George aurait permis l’identification de 93% des enfants avec DIP. A partir de ces résultats, les auteurs ont proposé un diagramme d’identification d’un DIP chez un enfant présentant des infections sévères, récurrentes ou inhabituelles (Figure 3).
24 Les résultats de cette étude, combinés avec la découverte de nouveaux DIP présentant un phénotype non infectieux (autoimmunité, autoinflammatoire, tumeur), nous conduit à une redéfinition des signes d’alerte au 21e siècle [Arkwright & Gennery, 2011]. Etant donné que l’histoire familiale est le signe-clé pour identifier un DIP, Arkwright & Gennery préconisent de se focaliser sur l’éducation, la formation et le conseil des médecins du milieu hospitalier et des familles de patients.
Etant donné que de nombreux DIP se manifestent dans la première année de vie et sont généralement des urgences pédiatriques, Carneiro-Sampaio et al. ont proposé 12 signes d’alerte pour les nourrissons dans leur première année de vie [Carneiro-Sampaio et al., 2010] :
1. Infections fongiques, virales ou bactériennes sévères et/ou persistantes
2. Réaction défavorable aux vaccins vivants, particulièrement au BCG
3. Diabète sucré persistant ou autre manifestation autoimmune et/ou inflammatoire
4. Symptômes évoquant un sepsis sans isolement de germe
5. Lésions cutanées étendues
6. Diarrhée persistante
7. Malformations
cardiaques
congénitales
(principalement
des
anomalies
conotroncales)
8. Retard de chute du cordon ombilical (>30 jours)
9. Cas connus de DIP dans la famille ou de décès précoces suite à une infection
10. Lymphopénie persistante (2500 cellules /mm
3) ou autre cytopénie, ou leucocytose
en absence d’infection
11. Hypocalcémie avec ou sans convulsions
25 Figure 3. Schéma simple permettant l’identification d’un DIP chez un enfant présentant des
infections sévères, inhabituelles ou récurrentes [d’après Sabbarayan et al., 2011]. Notez que ce diagramme n’est pas exhaustif. Igs : Immunoglobulines ; UTI : Infection urinaire.
![Tableau IV. Valeurs normales des sous-populations lymphocytaires (10 3 /µL) en fonction de l’âge [Shearer et al., 2003]](https://thumb-eu.123doks.com/thumbv2/123doknet/2190860.11447/43.892.131.762.286.532/tableau-valeurs-normales-populations-lymphocytaires-fonction-age-shearer.webp)
![Figure 8. Exemple d'analyse de marqueurs génétiques chez une famille porteuse de l’Ataxie Télangiectasie [Bayat et al., 2007] Suite à la PCR du marqueur D11S1343, un gel d’électrophorèse met en évidence l’haplotype de ce dernier pour chaque membre de la](https://thumb-eu.123doks.com/thumbv2/123doknet/2190860.11447/66.892.103.796.653.914/marqueurs-genetiques-porteuse-telangiectasie-marqueur-electrophorese-evidence-haplotype.webp)

![Figure 18. Schéma montrant le défaut du Réseau de Surveillance des dégâts à l’ADN chez les patients AT [D’après Meyn, 1999]](https://thumb-eu.123doks.com/thumbv2/123doknet/2190860.11447/94.892.282.667.112.477/figure-schema-montrant-defaut-reseau-surveillance-degats-patients.webp)